

Abstract
Background
Recent reports demonstrate that multiple forms of cardiovascular stress, including pressure overload, chronic ischemia, and infarction-reperfusion injury, provoke an increase in autophagic activity in cardiomyocytes. However, nothing is known regarding molecular events that stimulate autophagic activity in stressed myocardium. Because autophagy is a highly conserved process through which damaged proteins and organelles can be degraded, we hypothesized that stress-induced protein aggregation is a proximal trigger of cardiomyocyte autophagy.
Methods and Results
Here, we report that pressure overload promotes accumulation of ubiquitinated protein aggregates in the left ventricle, development of aggresome-like structures, and a corresponding induction of autophagy. To test for causal links, we induced protein accumulation in cultured cardiomyocytes by inhibiting proteasome activity, finding that aggregation of polyubiquitinated proteins was sufficient to induce cardiomyocyte autophagy. Furthermore, attenuation of autophagic activity dramatically enhanced both aggresome size and abundance, consistent with a role for autophagic activity in protein aggregate clearance.
Conclusions
We conclude that protein aggregation is a proximal trigger of cardiomyocyte autophagy and that autophagic activity functions to attenuate aggregate/aggresome formation in heart. Findings reported here are the first to demonstrate that protein aggregation occurs in response to hemodynamic stress, situating pressure-overload heart disease in the category of proteinopathies.
Keywords: autophagy, heart failure, hypertrophy, protein aggregation, remodeling
Under conditions of stress, the heart undergoes a compensatory hypertrophic growth response that serves to normalize wall stress and to diminish myocardial oxygen demand.1 In the chronic state, cardiac hypertrophy is an independent risk factor for heart failure and lethal arrhythmia, leading causes of morbidity and mortality in Western society. Numerous pathways have been causally implicated in the transition from myocyte hypertrophy to failure,2 including programmed cell death, cellular atrophy, and very recently, autophagy.3,4 However, mechanisms governing the transition from compensated hypertrophy to heart failure are incompletely characterized.
Autophagy is a highly conserved process of protein degradation involved in turnover of mitochondria and peroxisomes and nonselective degradation of cytoplasmic components during periods of starvation or stress.5 The autophagic reaction begins with formation of the autophagosome, a double-membrane structure of unknown origin that engulfs cytoplasmic contents without clearly defined substrate specificity.6 As the autophagosome matures, it fuses with a lysosome to form an autolysosome, leading to proteolysis of engulfed materials.
Autophagic activity is associated with the pathogenesis of various diseases, including neurodegenerative disorders, skeletal myopathy, cancer, and microbial infection.7 Recent reports demonstrate that multiple forms of cardiovascular stress, including pressure overload, chronic ischemia, ischemia-reperfusion, and diphtheria toxin–induced injury, provoke an increase in autophagic activity in cardiomyocytes.4 Our group demonstrated recently that in pressure-overload heart failure, a common form of clinical heart failure, induction of autophagic activity is maladaptive.3
A prominent feature among neurodegenerative diseases associated with autophagy, including Huntington's disease, amyotrophic lateral sclerosis, parkinsonism, and Alzheimer disease, is deposition of proteins within intracellular aggregates.8 The prevailing notion is that autophagic pathways serve a salutary function by facilitating removal of aggregates too large for efficient proteasome-mediated clearance.9 In fact, in several neurodegenerative diseases, including Huntington's disease10 and amyotrophic lateral sclerosis,11 a strong association exists between induction of autophagy and the presence of protein aggregates. In the absence of basal levels of autophagic activity in brain, abnormal aggregates of intracellular proteins develop,12,13 and pharmacological or genetic induction of autophagy is sufficient to reduce polyglutamine-induced cytotoxicity in animal models of Huntington's disease.14 Taken together, these data suggest that intracellular protein aggregates are capable of stimulating autophagic activity, which serves, in turn, to facilitate clearance of the aggregates.
Methods
Pressure-Overload Hypertrophy
Male C57BL6 mice (6 to 8 weeks old) were subjected to pressure overload by severe thoracic aortic banding (sTAB), a model of pressure-overload heart failure.15
Primary Culture of Neonatal Rat Ventricular Myocytes
Cardiomyocytes were isolated from the ventricles of 1- to 2-day-old Sprague-Dawley rat pups and plated as described16 at a density of 1250 cells per 1 mm in medium containing 10% FCS with 100 μmol/L bromodeoxyuridine. Forty-eight hours after plating, cells were transferred to medium supplemented with 1% FBS and 1 μmol/L bromodeoxyuridine, at which point treatment began.
Immunohistochemistry
We performed antigen retrieval by microwave heat-induced epitope retrieval using 1× Biogenex Citra (Biogenex, San Ramon, Calif; 10 minutes at 95°C). Primary antibody dilutions were as follows: 1:30 α-B-crystallin (CryAB; Vector Laboratories, Burlingame, Calif; VP-A103), 1:50 anti–MAP-LC3 (Santa Cruz Biotechnology, Inc, Santa Cruz, Calif; sc-16756), 1:50 anti-vimentin (Santa Cruz; sc-5565), 1:50 anti-ubiquitin (Santa Cruz; sc-9133), 1:1000 anti-ubiquitin (Abcam; ab7254), or 1:50 γ-tubulin (Santa Cruz; sc-10732).
Immunocytochemistry
Cultured cardiomyocytes were washed 3 times in PBS supplemented with calcium (CaCl2 0.1g/L) and magnesium (MgCl2-6H2O 0.1g/L). Cells were then fixed in 4% paraformaldehyde, permeabilized for 2 minutes in 0.1% Triton-X 100, and then blocked for 15 minutes in PBS with 3% normal goat serum and 1% BSA.
Transient Transfection of Cultured Cardiomyocytes
Twenty-four hours after plating, neonatal rat ventricular myocytes (NRVMs) were transfected with a green fluorescent protein (GFP)–tagged LC3 construct as previously described.3
Chymotrypsin-Like Activity
Cells were harvested in buffer H (20 mmol/L Tris-HCl, 20 mmol/L NaCl, 1 mmol/L EDTA, 5 mmol/L bromomercaptoethanol, pH 7.6 at 4°C) and sheared by repeated passage through a 27-gauge needle, and the supernatant was collected after sedimentation of insoluble material (centrifugation at 16 000g for 10 minutes). Then, 10 μg supernatant protein was added to 300 μL activity assay buffer (50 mmol/L Tris-HCl, 5 mmol/L bromomercaptoethanol, 50 μmol/L Suc-LLVY-AMC, pH 8.0 at 37°C).
Statistical Methods
Averaged data are reported as mean±SEM. Statistical significance was analyzed (StatView) with Student's unpaired, 2-tailed t test (comparison of 2 groups) or 1-way ANOVA (comparison of ≥2 independent groups), followed by Bonferroni's method for post hoc pairwise multiple comparisons.
The authors had full access to and take full responsibility for the integrity of the data. All authors have read and agree to the manuscript as written.
Results
Pressure Overload Induces Proteotoxic Stress in Ventricular Myocytes
We have shown previously that cardiomyocyte autophagy contributes to pathological remodeling of the left ventricle (LV) induced by severe afterload stress.3 To define proximal events that trigger autophagy, we subjected mice to sTAB, a procedure that induces pressure-overload heart failure.15 As reported previously,15 7 days of sTAB was sufficient to induce clinical heart failure as demonstrated by development of robust cardiac hypertrophy, pulmonary edema, and diminished systolic function (Figure 1).
Figure 1.

sTAB induces clinical heart failure. A, Representative 4-chamber sections of mouse hearts 7 days after sham or sTAB surgery. Scale bar=1 mm. B, Seven days after sTAB, animals demonstrated signs of heart failure with significant increases in heart mass, lung mass (indicative of pulmonary edema), and impairment in systolic performance. Mean data from sham (n=5) and sTAB (n=5) mice subjected to surgery as listed. HW/BW indicates heart weight/body weight; LWeight, lung weight. *P<0.05.
Elevated afterload imposes oxidative, biomechanical, and neurohumoral stress on the heart, each of which is capable of eliciting proteotoxic injury. Given this, we examined pressure-overload–stressed LV for accumulation of damaged, misfolded proteins. Damaged proteins are covalently coupled to ubiquitin, which targets the tagged substrate for proteolysis by the proteasome. If accumulation of ubiquitinated proteins outpaces proteasomal degradation, buildup of intracellular ubiquitinated protein occurs, resulting in formation of protein aggregates, large heterogeneous complexes that are poor substrates for proteasome-mediated proteolysis.17
To test for increases in ubiquitinated proteins and protein aggregates, we immunostained sections of pressure-stressed heart for ubiquitin. Here, we detected dramatic accumulation of ubiquitin-positive inclusion bodies distributed throughout the LV 72 hours after sTAB (Figure 2A). No significant ubiquitin-like immunoreactivity was detected in regions not subjected to biomechanical stress such as right ventricle or either atria. Of interest, we detected a preponderance of ubiquitin staining in the basal septum, a region previously reported to be a “hot spot” for load-induced autophagic activity.3 Western blot analysis confirmed the robust induction of ubiquitinated protein, detected as diffuse, high-molecular-weight bands on immunoblot, in pressure-stressed LV (Figure 2B).
Figure 2.
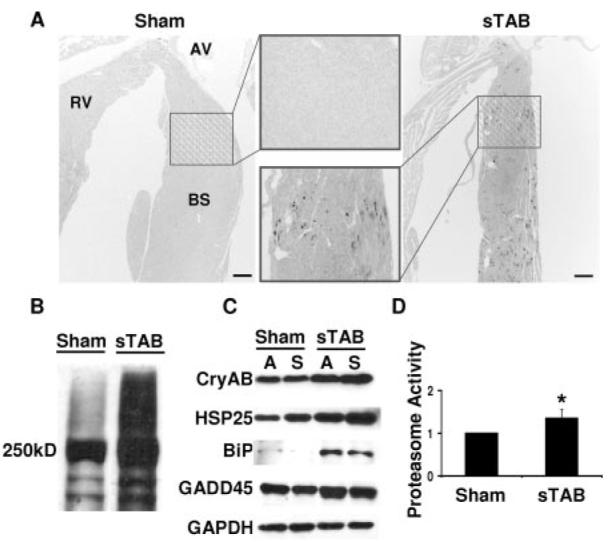
Proteotoxic stress in load-induced heart failure. A, Representative immunohistochemical staining revealing accumulation of ubiquitin-conjugated proteins in basal interventricular septum 72 hours after sTAB (scale bar=200 μm). B, Representative immunoblot probed for ubiquitin demonstrating induction of high-molecular-weight, ubiquitinated protein in pressure-stressed LV. C, Representative immunoblot of LV lysates 72 hours after sTAB or sham operation and probed for multiple protein chaperones, including CryAB, HSP25, BiP, and GADD45. D, Proteasome activity assayed 1 week after sTAB is increased (1.35±0.2; n=4) relative to sham-operated controls (1.0±0.3; n=4). RV indicates right ventricle; AV, aortic valve; and BS, basal septum. *P<0.05.
In the context of cellular stress, the abundance of chaperone proteins, molecules that play a critical role in protein targeting and stability, increases as an adaptive response. Consistent with this notion, we detected significant increases in the abundance of multiple heat shock proteins (HSPs) in pressure-stressed myocardium 7 days after surgery (Figure 2C). Steady-state abundances of CryAB, HSP25, and GADD45 were modestly increased and that of BiP was dramatically increased. Together, these findings are consistent with a model in which the intracellular milieu within load-stressed cardiomyocytes is conducive to protein damage and misfolding.
Accumulation of ubiquitinated proteins is generally believed to occur as a consequence of 1 of 3 mechanisms: an increase in protein ubiquitination, a decrease in deubiquitination, or a decrease in proteasome-mediated clearance. To determine whether the accumulation of ubiquitinated proteins in pressure-stressed LV was due to a decrease in proteasome activity, we assayed ventricular lysates for chymotrypsin-like activity (CTL-A). One week after sTAB, basal septum CTL-A was increased 35±2% compared with sham-operated controls (P<0.05; n=4; each lysate run in triplicate; Figure 2D). To test for a component of ATP-dependent proteasome activity, CTL-A was measured in lysates (n=3) supplemented with ATP. In these experiments, CTL-A was not significantly different from that measured without the addition of exogenous ATP (data not shown). Together, these data suggest that accumulation of ubiquitinated protein is not a consequence of diminished proteasome activity but rather is due to increased flux that exceeds proteasome capacity.
Autophagic Activity Increases in Pressure-Stressed Ventricle
Autophagic activity was monitored by morphological and biochemical means. In sTAB hearts, we detected an increased abundance of multilamellar autophagosomes by 72 hours after surgery (Figure 3A), findings indicative of autophagic activity. Additionally, during the autophagic response, LC3 (microtubule-associated protein 1 light chain 3), an 18-kDa homologue of Atg8 in yeast, is processed and lipid conjugated.18 The resulting 16-kDa active isoform migrates from the cytoplasm to isolation membranes and autophagosomes. Recently, intracellular migration of LC3 to vesicular membranes has emerged as a reliable marker of autophagic activity.3,19 Consistent with increased autophagic activity, we detected robust increases in autophagosome-localized LC3 in pressure-stressed cardiomyocytes (Figure 3B). These findings were corroborated by immunoblot analysis that demonstrated increases in LC3 processing and elevated levels of Beclin 1 protein, 2 markers of autophagic activity (Figure 3C, data not shown).
Figure 3.

Severe pressure stress induces cardiomyocyte autophagy. A, Representative electron micrograph revealing a multilamellar autophagosome in basal septum of LV 48 hours after sTAB (scale bar=500 nm). B, Anti-LC3 immunofluorescence reveals redistribution of LC3 into punctate dots, indicative of autophagosome accumulation (scale bar=10 μm). C, Representative immunoblot demonstrating that severe pressure stress triggers significant LC3 processing.
Intracellular Aggresome Formation in Pressure-Stressed Ventricle
In certain contexts, protein aggregates coalesce as perinuclear structures called aggresomes.8,20 Aggresomes are cytoplasmic inclusion bodies located in the perinuclear region near the microtubule-organizing center that result from active, dynein-dependent transport of small aggregates from other parts of the cell. Given our observations of robust increases in chaperone protein levels in the setting of accumulation of ubiquitin-positive high-molecular-weight proteins, we hypothesized that aggresome formation was occurring. To test for their presence, we costained pressure-stressed LV tissues for CryAB and vimentin, a structural component of the aggresome.9 After 7 days of severe pressure-overload stress, we detected juxtanuclear organization of CryAB-associated proteins with an adjacent vimentin-containing shell (Figure 4A, arrow), a finding consistent with aggresome formation. Further evidence of aggresome formation was found when we coimmunostained for CryAB and ubiquitin. Again, after 7 days of pressure stress, we observed an increase in both ubiquitin and CryAB staining, with colocalization of signal in the perinuclear region (Figure 4B, arrow). Thus, ubiquitin-like immunoreactivity in pressure-stressed cardiac myocytes accumulated as large perinuclear aggregates that colocalize with CryAB and vimentin (additional images in Figure I of the online Data Supplement). From these findings, we conclude that severe pressure overload triggers protein damage, protein ubiquitination, and formation of perinuclear aggresomes.
Figure 4.
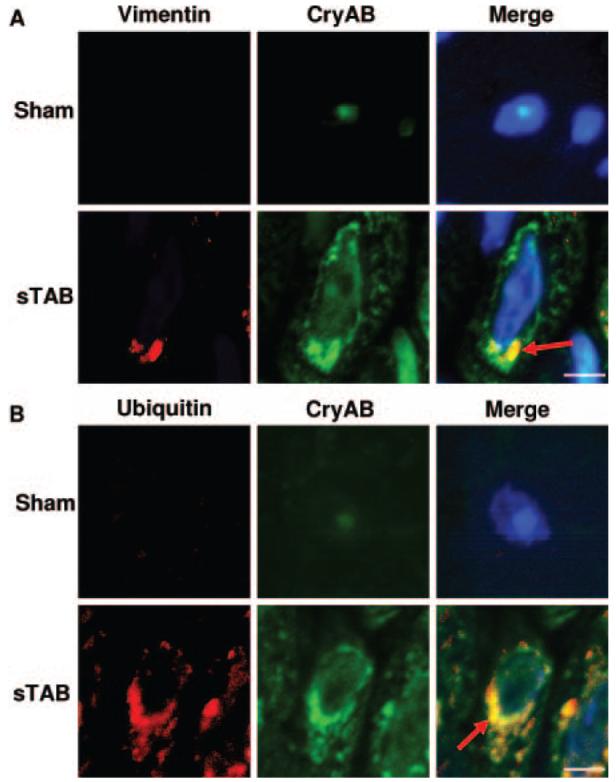
Aggresome formation in pressure-stressed LV. A, Representative immunohistochemical sections 7 days after sTAB immunostained for vimentin and CryAB (A) or ubiquitin and CryAB (B) (scale bar=25 μm; n=4 each group). Confocal imaging revealed perinuclear colocalization consistent with aggresome formation (arrow).
Protein Aggregation Triggers Autophagy
These findings led us to hypothesize that accumulation of ubiquitinated proteins is a proximal step leading to protein aggregation and subsequent activation of autophagic clearance mechanisms. To test this, we treated NRVMs with a proteasome inhibitor (MG-132, 5 μmol/L), isolated protein from the insoluble fraction, and probed for ubiquitin. In these studies, we found that insoluble, ubiquitin-tagged protein accumulation was detectable as early as 4 hours after proteasome inhibition (Figure 5A). At subsequent time points, accumulation of ubiquitin-tagged protein within the insoluble fraction was progressive and robust. No evidence of cytotoxicity was detected out to 16 hours (Figure 5B), although a modest degree of cell death was detected on exposure to MG-132 for ≥24 hours. These findings suggest that the cardiomyocyte cytosol has a limited capacity to retain ubiquitinated protein in the soluble fraction, with excessive accumulation resulting in protein precipitation as an insoluble matrix.
Figure 5.

In vitro aggresome formation is secondary to accumulation of insoluble ubiquitinated protein. A, NRVMs were loaded with polyubiquitinated protein by selective proteasome inhibition (MG-132, 5 μmol/L). Representative immunoblot demonstrating that ubiquitinated protein accumulates in the insoluble fraction beginning at 4 hours. B, Exposure to rapamycin, 3-MA, MG-132, or epoxomycin did not induce significant cell death (MTS assay). C, Representative immunoblot demonstrating that LC3-II levels, indicative of autophagic activation, are increased by rapamycin and diminished by 3-MA. Proteasome inhibition by MG-132 or epoxomycin elicited modest increases in LC3 processing. D, Mean data from 3 independent experiments. E, Representative immunocytochemical staining after 16 hours of proteasome inhibition revealing perinuclear coalescence of ubiquitin-tagged protein (E2). Inhibition of autophagy by 3-MA (5 mmol/L) amplified aggresome formation (E4) (scale bar=10 μm). Ctrl indicates control.
Next, we set out to confirm that autophagy is activated in cardiac myocytes under conditions of protein aggregation. To do this, we induced protein aggregation in NRVMs by proteasome inhibition (MG-132 or epoxomicin); autophagic capacity was reduced pharmacologically (3-methyladenine [3-MA]). 3-MA inhibits autophagy at the stage of sequestration in which a double-membrane structure forms around a portion of the cytosol and sequesters it from the rest of the cytoplasm to form an autophagosome.21 In these experiments, we observed increased LC3 processing in response to proteasome inhibition, consistent with an increase in autophagic activity (Figure 5C and 5D). Suppression of autophagy (3-MA) elicited decreases in LC3 processing, as expected, and a robust accumulation of ubiquitinated substrates. Rapamycin, an activator of autophagy, triggered modest increases in LC3 processing above that elicited by proteasome inhibition (Figure 5C and 5D). Together, these data are consistent with a model in which protein aggregation is sufficient to induce cardiomyocyte autophagy, which then functions as an aggregate clearance mechanism.
To determine the subcellular localization of ubiquitinated proteins that accumulate in response to proteasome inhibition—and to test further for aggresome formation—we studied NRVMs exposed to prolonged inhibition of the proteasome (16 hours). Here, we detected perinuclear coalescence of ubiquitin-conjugated proteins (Figure 5E) similar to that seen in pressure-stressed LV in vivo. To test whether autophagic activity can reduce the abundance of aggregated proteins, we cultured cells in medium supplemented with 3-MA (5 mmol/L). As expected, we detected no change in ubiquitin levels or subcellular organization of aggregates in control cells treated with a short course of 3-MA alone (Figure 5E, image 3). However, in cells in which proteasome activity was inhibited (MG-132) and autophagy was blocked (3-MA), we observed a dramatic increase in the size of perinuclear aggresomes (Figure 5E, image 4). These data suggest that autophagy antagonizes aggresome formation by providing an alternative clearance pathway.
This model was further tested by suppressing autophagy by siRNA-mediated knockdown of Beclin 1. First, robust knockdown of Beclin 1, a protein required for recruitment of Atg12-Atg5 conjugates to preautophagosomal membranes,22 was confirmed in control experiments (supplemental Figure II). Knockdown of Beclin 1 led to significant decreases in constitutive autophagic activation in NRVMs, and it was not associated with cytotoxicity (supplemental Figure II). Next, we tested the effects of Beclin 1 knockdown on autophagic activity. First, it is important to note that Beclin 1 is not required for LC3-I to LC3-II conversion (ie, lipidation) but is required for LC3 association with membranes.23 Given this, tracking LC3 processing is not a reliable means of quantifying autophagy in the setting of Beclin 1 knockdown. Thus, we tracked p62 abundance because this protein is degraded in autophagosomes.24 As expected, Beclin 1 knockdown at both 24 hours (Figure 6) and 48 hours (not shown) elicited increases in the abundance of p62. Proteasome inhibition with MG-132 decreased p62 levels, consistent with degradation of p62 by activated autophagy (Figure 6). Thus, these data lend further credence to the notion that protein aggregation is sufficient to induce cardiomyocyte autophagy.
Figure 6.

Levels of p62, an autophagy substrate, decline with proteasome inhibition. A, Representative immunoblot demonstrating that p62, a protein component of the autophagosome, increases with Beclin 1 knockdown–induced suppression of autophagy. Proteasome inhibition (MG-132, 5 μmol/L for 16 hours) antagonizes these increases in p62, consistent with activation of autophagy by protein aggregate accumulation. B, Mean data from 2 independent experiments. *P<0.05 vs control.
Parallel Activation of Proteasomal and Autophagic Clearance Pathways
In neurons, protein aggregates, similar to those reported here, are capable of inducing autophagy.25 Thus, we tested whether the presence of aggregates in cardiomyocytes is sufficient to trigger autophagy. To do this, we monitored autophagic activity in vitro by tracking the localization of GFP-tagged LC3 in transiently transfected cardiac myocytes. With this approach, autophagic activity can be detected as punctate, autophagosome-localized GFP signal, as opposed to the diffuse, cytosolic distribution seen under resting conditions.3,19 In cells treated with a proteasome inhibitor (MG-132, 5 μmol/L for 16 hours), we detected an abundant increase in autophagosome-localized LC3 (Figure 7A). In these experiments, autophagic activity, quantified as the number of cells with autophagic vacuoles divided by the total number of transfected cells, was increased 1.8-fold (P<0.05; Figure 7B). Of note, induction of cardiomyocyte autophagy in response to proteasome inhibition was similar in magnitude to that triggered by rapamycin (10 nmol/L), a powerful activator of autophagy (Figure 7B). Together, these data are consistent with the induction of autophagy in response to accumulation of protein aggregates and argue strongly against nonspecific effects resulting from MG-132 toxicity.
Figure 7.

Aggresomes are a proximal trigger of cardiomyocyte autophagy. A, NRVMs were transfected with GFP-tagged LC3 and subjected to proteasome inhibition (16 hours), resulting in redistribution of LC3 in an autophagosome-specific punctate pattern. Images of representative cells are depicted (scale bar=10 μm). B, Quantification of the numbers of cells manifesting autophagic activity (*P<0.05 relative to control). More than 300 cells were evaluated in each treatment condition in 2 independent experiments. C, Cells treated with 3-MA for 7 days were assayed for proteasome activity, revealing a robust increase in activity. *P<0.05.
Given our in vivo findings demonstrating that both proteasomal and autophagic clearance pathways are activated in response to pressure-overload stress, combined with our in vitro data demonstrating that inhibition of the proteasome leads to an increase in autophagic activity, we set out to determine whether the converse pertains, ie, whether inhibition of autophagy results in an increase in proteasome activity. NRVMs were cultured for 7 days in the presence of 3-MA or vehicle, with proteasome activity determined by measuring CTL-A. In these experiments, we observed a 73±17% increase (n=3; each lysate run in triplicate; P<0.05) in CTL-A after 7 days of 3-MA treatment relative to control (Figure 7C). These results are consistent with our finding of coordinated regulation of proteasomal and autophagic mechanisms in pressure-stressed heart. Furthermore, they lend support to a model in which autophagic and proteasomal clearance mechanisms function in parallel in cardiac myocytes.
Discussion
Recent reports reveal that cardiomyocyte autophagy is activated in response to multiple forms of cardiac injury.4 However, proximal triggers of autophagic activity in the heart are unknown. The major findings of this study are that (1) pressure-overload hemodynamic stress elicits changes in the cardiomyocyte milieu conducive to protein damage and misfolding with consequent accumulation of polyubiquitinated proteins and increases in chaperone protein levels; (2) protein aggregation takes place, leading to formation of perinuclear aggresomes; (3) clearance is activated via both autophagic and proteasomal protein quality control pathways; (4) protein aggregate and aggre-some formation are sufficient to trigger cardiomyocyte autophagy, and (5) autophagic activity in the cardiomyocyte participates in clearance of aggregated proteins. Finally, the findings reported here are the first to demonstrate that protein aggregation occurs in response to hemodynamic stress, situating pressure-overload heart disease in the category of proteinopathies.
Stress-Induced Cardiomyocyte Proteinopathy
Proteinopathy, toxic aggregations of misfolded proteins, is a growing family of human disorders that includes Alzheimer disease, parkinsonism, amyotrophic lateral sclerosis, and both polyglutamine and polyalanine expansion disorders.26,27 In heart disease, abnormal protein aggregation and accumulation of ubiquitinated proteins have been detected in human hearts with idiopathic or ischemic cardiomyopathies.28,29 In both brain and heart, however, relatively little is known regarding whether these intracellular inclusions are toxic themselves or whether they represent a compensatory mechanism that sequesters harmful, soluble proteins within the cytoplasm.
Insoluble protein aggregates are processed by pathways that are just now being deciphered. First, misfolded proteins are delivered to the microtubule-organizing center by dynein-dependent retrograde transport along microtubules. When the degradative capacity of the proteasome is exceeded, protein aggregates accumulate in perinuclear inclusions called aggresomes,20 organized structures surrounded by vimentin filaments that recruit chaperones, ubiquitin, proteasomes, and mitochondria. In pressure-stressed myocardium, we have detected an abundance of large perinuclear inclusions marked by ubiquitin that colocalize with chaperone proteins and vimentin, fulfilling the definition of aggresomes. In liver, aggresomes (Mallory bodies) form in response to a variety of stresses, including proteasome inhibition.30 Aggresomes also have been detected in a model of desmin-related cardiomyopathy, a disorder characterized by abnormal amyloid deposition owing to dysfunctional chaperone protein function.31 The findings reported here, however, are the first to describe aggresome formation in a common cardiovascular disorder, ie, load-induced heart disease.
Cardiomyocyte Autophagy Contributes to Protein Quality Control
Autophagy occurs in a wide range of eukaryotic cells and is the sole pathway for organelle turnover. Autophagy, the basic mechanisms of which are largely conserved from yeast to mammals,5 is a vital pathway for degrading normal and aggregated proteins, particularly under stress or injury conditions. Autophagy can be induced by diverse stimuli, including starvation, hypoxia, intracellular stress, and developmental signals. It accomplishes a host of functions critical to normal homeostasis and development. In the context of nutrient deprivation, autophagic activity is adaptive in that degradation of cytosolic components releases substrates for intermediary metabolism. Autophagy is also a mechanism for eliminating damaged proteins and organelles that might otherwise be toxic or trigger apoptotic death. For example, targeted inhibition of Atg7, a gene required for autophagic activity, leads to accumulation of ubiquitin-positive aggregates.32 During development, autophagy is a means of remodeling tissues or removing unneeded cells.33 In other settings, however, autophagic activity is associated with the pathogenesis of disease, and unrestrained autophagic activity can cause cell death.7
In the present study, we establish a link between protein aggregation and induction of cardiomyocyte autophagy. In vivo, we describe the accumulation of ubiquitinated proteins in a spatial and temporal pattern consistent with our recent report of pressure overload–induced autophagy.3 We further characterize the organization of damaged proteins into vimentin-associated, perinuclear, aggresome-like structures. Because aggresome formation is a general response of cells that occurs when proteasome capacity is exceeded by the production of aggregation-prone misfolded proteins,20 we inhibited the proteasome in cultured cardiomyocytes to test directly for a causal link between aggregate formation and cardiomyocyte autophagy. In these experiments, we found that accumulation and aggregation of ubiquitinated protein is a powerful inducer of autophagy, capable of robust activation of autophagy to levels comparable to pharmacological induction. Together, these findings are consistent with a model in which autophagic activity is a general response to protein aggregation in the heart and point to a potential role for autophagy in cardiomyopathies of diverse origin.
Interestingly, we and others do not detect inclusions that are membrane bound. The inclusions we detect are much larger than mammalian autophagosomes. Together, these findings suggest that autophagy serves to clear monomeric and oligomeric precursors of aggregates rather than the large inclusions themselves.
To test the involvement of proteasomal activity, we measured chymotryptic activity acting on a small fluorogenic peptide substrate Suc-LLVY-AMC. Owing to its small size, this peptide diffuses easily into the proteolytic chamber of the 20S proteasome34; hence, it provides a readout of 20S proteasome activity. Consistent with this, the addition of exogenous ATP to stimulate 26S proteasome activity had no significant effect on CTL-A (<10%). In these experiments, we find that total proteasome function is increased in heart failure, consistent with similar findings in a model of desmin-related cardiomyopathy,35 suggesting increased flux through proteasomal degradation pathways. Our data demonstrate that proteasomal and autophagic activities increase in parallel.
α-1-Antitrypsin deficiency is another proteinopathy in which autophagy is activated as a clearance mechanism. In this disease, proteolytic destruction of elastic connection tissue matrix occurs in lung because of unrestrained proteolysis. In liver, however, toxicity resulting from the accumulation of misfolded mutant α-1-antitrypsin leads to chronic liver inflammation and hepatocellular carcinoma. Recent studies have revealed dramatic increases in autophagic activity in liver, which serves to prevent toxic accumulation of mutant α-1-antitrypsin by selectively targeting insoluble aggregates.36
Thus, a growing body of evidence implicates autophagy as a protective response in genetic diseases associated with cytoplasmic aggregation-prone proteins. Here, we extend this to disease triggered by environmental stress. Importantly, recent studies demonstrating that pharmacological upregulation of autophagy is protective in a wide variety of disease models associated with intracellular protein aggregation raise the exciting prospect of autophagic activation as a novel therapeutic strategy.37 Our findings here extend this prospect to the myocardium.
Role in Heart Failure Progression
The presence of increased autophagic activity in animal models of heart failure and in samples from human failing hearts provides no insight into whether cardiomyocyte autophagy is beneficial or pathological. Indeed, it is possible that autophagic activity carries out different functions, depending on disease stage and severity or the nature of the components being degraded. For example, we have shown previously that genetic amplification of the autophagic response to acute-onset pressure stress leads to increased hypertrophic growth of the heart.3 On the other hand, if the protein aggregates themselves are toxic, increased autophagic removal could be beneficial. Furthermore, recent evidence strengthens the assertion that dysregulated autophagic activity induces a caspase-independent programmed cell death program (type II programmed cell death38-40). Nakai et al41 reported that cardiac-specific inactivation of atg5 early in cardiogenesis was not associated with an abnormal cardiac phenotype. However, subjecting these animals to pressure stress during adulthood triggered rapid and dramatic declines in cardiac function. Intriguingly, inactivation of atg5 in the heart after the mice had reached maturity led rapidly to cardiac hypertrophy, LV dilation, contractile dysfunction, and a syndrome consistent with heart failure.41 Clearly, to exploit autophagy as a therapeutic target, it is essential to identify when it is protective and when it contributes to the pathogenesis of disease.4
Evidence presented here indicates that autophagy is activated to eliminate damaged proteins that accumulate within the cardiomyocyte. Several reports point to functional defects in the ubiquitin-proteasome system in heart failure, resulting in accumulation of misfolded proteins.27 Consistent with this, we have observed increased ubiquitin staining in pressure-stressed ventricle, which is most prevalent in the basal septum. Therefore, activation of autophagy at early stages of the disease may be a protective mechanism to scavenge and eliminate misfolded, polyubiquitinated protein aggregates that have overwhelmed the degradative capacity of the proteasomal system.
Perspective
Cardiac hypertrophy is a major predictor of heart failure, a prevalent disorder with high mortality, and there is urgent need for novel therapies to prevent or reverse pathological cardiac remodeling. Our data point to a progression of protein damage, aggregation, and coalescence into aggresomes as a previously unrecognized mechanism of disease. Furthermore, our findings suggest that autophagic activity in cardiomyocytes is a general response to diverse stressors and contributes to disease pathogenesis in multiple contexts. As new advances in deciphering molecular regulation of autophagy emerge, it may soon be possible to enhance or inhibit autophagic activity selectively to affect heart disease meaningfully. In addition, given that some drugs already in clinical use alter the process of autophagy and organ systems in which increased autophagic activity is associated with disease, advances in this field are all the more urgent.
Acknowledgment
We gratefully acknowledge Dr Beth Levine (UT Southwestern) for helpful advice.
Sources of Funding
This work was supported by grants from the National Institutes of Health (HL-075173, HL-006296, HL-080144 to Dr Hill; HL-072016 to Dr Rothermel) and American Heart Association (0640084N to Dr Hill, 0655202Y to Dr Rothermel).
Footnotes
Disclosures
None.
The online Data Supplement can be found with this article at http://circ.ahajournals.org/cgi/content/full/CIRCULATIONAHA.107.763870/DC1.
References
- 1.Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–1380. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 2.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 3.Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA. Cardiac autophagy is a mal-adaptive response to hemodynamic stress. J Clin Invest. 2007;117:1782–1793. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rothermel BA, Hill JA. Myocyte autophagy in heart disease: friend or foe? Autophagy. 2007;3:632–634. doi: 10.4161/auto.4913. [DOI] [PubMed] [Google Scholar]
- 5.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 6.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–937. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 7.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ross CA, Poirier MA. Opinion: what is the role of protein aggregation in neurodegeneration? Nat Rev Mol Cell Biol. 2005;6:891–898. doi: 10.1038/nrm1742. [DOI] [PubMed] [Google Scholar]
- 9.Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- 10.Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107–1117. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 11.Fortun J, Dunn WA, Jr, Joy S, Li J, Notterpek L. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J Neurosci. 2003;23:10672–10680. doi: 10.1523/JNEUROSCI.23-33-10672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 13.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 14.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 15.Rothermel BA, Berenji K, Tannous P, Kutschke W, Dey A, Nolan B, Yoo KD, Demetroulis E, Gimbel M, Cabuay B, Karimi M, Hill JA. Differential activation of stress-response signaling in load-induced cardiac hypertrophy and failure. Physiol Genomics. 2005;23:18–27. doi: 10.1152/physiolgenomics.00061.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simpson P, McGrath A, Savion S. Myocyte hypertrophy in neonatal rat heart cultures and its regulation by serum and by catecholamines. Circ Res. 1982;51:787–801. doi: 10.1161/01.res.51.6.787. [DOI] [PubMed] [Google Scholar]
- 17.Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 18.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A. 1982;79:1889–1892. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kihara A, Noda T, Ishihara N, Ohsumi Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol. 2001;152:519–530. doi: 10.1083/jcb.152.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, Ohsumi Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001;20:5971–5981. doi: 10.1093/emboj/20.21.5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 25.Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–40292. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 26.Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science. 2002;296:1991–1995. doi: 10.1126/science.1067122. [DOI] [PubMed] [Google Scholar]
- 27.Wang X, Robbins J. Heart failure and protein quality control. Circ Res. 2006;99:1315–1328. doi: 10.1161/01.RES.0000252342.61447.a2. [DOI] [PubMed] [Google Scholar]
- 28.Weekes J, Morrison K, Mullen A, Wait R, Barton P, Dunn MJ. Hyperubiquitination of proteins in dilated cardiomyopathy. Proteomics. 2003;3:208–216. doi: 10.1002/pmic.200390029. [DOI] [PubMed] [Google Scholar]
- 29.Kostin S, Pool L, Elsasser A, Hein S, Drexler HC, Arnon E, Hayakawa Y, Zimmermann R, Bauer E, Klovekorn WP, Schaper J. Myocytes die by multiple mechanisms in failing human hearts. Circ Res. 2003;92:715–724. doi: 10.1161/01.RES.0000067471.95890.5C. [DOI] [PubMed] [Google Scholar]
- 30.French BA, van Leeuwen F, Riley NE, Yuan QX, Bardag-Gorce F, Gaal K, Lue YH, Marceau N, French SW. Aggresome formation in liver cells in response to different toxic mechanisms: role of the ubiquitin-proteasome pathway and the frameshift mutant of ubiquitin. Exp Mol Pathol. 2001;71:241–246. doi: 10.1006/exmp.2001.2401. [DOI] [PubMed] [Google Scholar]
- 31.Sanbe A, Osinska H, Saffitz JE, Glabe CG, Kayed R, Maloyan A, Robbins J. Desmin-related cardiomyopathy in transgenic mice: a cardiac amyloidosis. Proc Natl Acad Sci U S A. 2004;101:10132–10136. doi: 10.1073/pnas.0401900101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 34.Luker GD, Pica CM, Song J, Luker KE, Piwnica-Worms D. Imaging 26S proteasome activity and inhibition in living mice. Nat Med. 2003;9:969–973. doi: 10.1038/nm894. [DOI] [PubMed] [Google Scholar]
- 35.Chen Q, Liu JB, Horak KM, Zheng H, Kumarapeli AR, Li J, Li F, Gerdes AM, Wawrousek EF, Wang X. Intrasarcoplasmic amyloidosis impairs proteolytic function of proteasomes in cardiomyocytes by compromising substrate uptake. Circ Res. 2005;97:1018–1026. doi: 10.1161/01.RES.0000189262.92896.0b. [DOI] [PubMed] [Google Scholar]
- 36.Perlmutter DH. The role of autophagy in alpha-1-antitrypsin deficiency: a specific cellular response in genetic diseases associated with aggregation-prone proteins. Autophagy. 2006;2:258–263. doi: 10.4161/auto.2882. [DOI] [PubMed] [Google Scholar]
- 37.Berger Z, Ravikumar B, Menzies FM, Oroz LG, Underwood BR, Pangalos MN, Schmitt I, Wullner U, Evert BO, O'Kane CJ, Rubinsztein DC. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet. 2006;15:433–442. doi: 10.1093/hmg/ddi458. [DOI] [PubMed] [Google Scholar]
- 38.Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 39.Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–1228. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 40.Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, Baehrecke EH, Lenardo M. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci U S A. 2006;103:4952–4957. doi: 10.1073/pnas.0511288103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]