

Abstract
Cyclic GMP-selective phosphodiesterase type 5 (PDE5) has been traditionally thought to play little role in cardiac myocytes, yet recent studies using selective inhibitors such as sildenafil suggest it can potently modulate acute and chronic cardiac stress responses. To date, evidence for myocyte PDE5 expression and regulation has relied on small-molecule inhibitors and anti-sera, leaving open concerns regarding non-specific immune-reactivity, and off-target drug effects. To directly address both issues, we engineered a robust PDE5-gene silencing shRNA (inserted into miRNA-155 cassette) and DsRed-PDE5 fusion protein, both coupled to a CMV promoter and incorporated into adenoviral vectors. PDE5 mRNA and protein knockdown eliminated anti-sera positivity on immunoblots and fluorescent immuno-histochemistry in neonatal and adult cardiomyocytes, and suppressed PDE5 enzyme activity. Stimulation of myocyte hypertrophy by phenylephrine was blunted by PDE5 gene silencing in a protein kinase G dependent manner, and this effect was similar to that from sildenafil with no additive response by both combined. DsRed-PDE5 fusion protein expression showed normal z-band localization in adult myocytes but was diffuse in eNOS-/- myocytes; echoing reported findings with anti-sera. PDE5 over-expression increased enzyme activity and amplified natriuretic peptide gene expression from phenylephrine stimulation. These data confirm PDE5 expression, activity, and targeted inhibition by sildenafil in cardiomyocytes, as well as the role of this PDE in cardiomyocyte hypertrophy modulation.
Keywords: phosphodiesterase 5A, sildenafil, protein kinase G
Introduction
Intracellular cGMP is an important second messenger regulating a broad range of acute and chronic cellular functions. In the vascular system, cGMP and its primary effecter kinase cGK play a pivotal role in regulating vascular tone, platelets, and endothelial function [1]. In the heart, cGMP/cGK signaling appears to function more like a brake system, having less impact on rest function but the capacity to counter acute and chronic stress responses and remodeling. An example of acute modulation is its suppression of β-adrenergic/cAMP stimulation by altering phosphodiesterase type 2 activity [2] or cGK phosphorylation of calcium handling and sarcomeric proteins (e.g. L-type calcium channel [3] and troponin I [4]). Chronic activation of cGMP synthesis suppresses cardiac remodeling in response to pressure-overload [5], whereas genetic deletion of its synthetic enzymes exacerbates the response [6].
Cyclic GMP is generated by guanylate cyclase(GC) coupled to either nitric oxide(NO) or natriuretic peptide(NP) stimulation, and is hydrolyzed by members of the phosphodiesterase (PDE) super-family (reviewed in [7]). In the cardiovascular system, the latter includes cGMP-specific phosphodiesterase type 5 (PDE5) and possibly PDE9 [8], and dual substrate enzymes -PDE1, activated by calcium-calmodulin [9], and PDE2, activated by cGMP binding to regulatory domains that enhance its hydrolysis of cAMP [2]. Of these, PDE5 was discovered first and remains the best characterized [10]. It is the only PDE currently targeted for treating clinical disease (erectile dysfunction and pulmonary hypertension) [11]. PDE5 was first found in platelets, and later in vascular smooth muscle and endothelial cells, with high expression levels reported in the corpus cavernosum and lung [12]. It has three isoforms, PDE5A1, 5A2 and 5A3, that differ only in the initial portion of exon 1 in the N-terminus, and there are no known functional differences between them [10]. PDE5A1 and A2 mRNAs are detected in a wide variety of tissues including heart [13], whereas PDE5A3 is only expressed in vascular smooth muscle.
Despite its expression, the importance of PDE5 for the heart and even more specifically for cardiac muscle cells had long been considered minimal [12,14,15]. For one thing, PDE5 RNA, protein expression and enzyme activity in heart are 2 orders of magnitude lower than in lung [16]. Basal effects of small molecule PDE5 inhibitors such as sildenafil appear negligible in both normal and failing myocardium and hearts [17-20], nor do they impact β-adrenergic stimulated function in human or experimental heart failure [17,18]. However, more recent studies revealed potent cardiac effects from selective PDE5 inhibitors in hearts subjected to acute or chronic stress, suppressing β-adrenergic stimulated contractility in normal mammalian hearts including human [16,17,21], ameliorating ischemia/reperfusion [22] and anthracycline [23] induced myocardial damage, and blunting and reversing pressure-overload induced hypertrophy, fibrosis, and chamber remodeling [24]. Adult and neonatal myocyte studies have reported low level gene and protein expression as well as physiologic effects from PDE5 inhibitors [16,17,22,25]. Based on these and other data, the National Institutes of Health is to commence a multi-center trial (RELAX) to test the utility of sildenafil for treating heart failure with a preserved ejection fraction (diastolic heart failure).
While these results have renewed interest in PDE5 cardiac regulation, debate regarding its myocyte expression and physiologic role in these cells persists [10,12], as all prior data have depended upon the use of pharmacologic inhibitors and anti-sera. This has left open concerns regarding tissue and antibody specificity, and possible off-target effects [10,12] such as sildenafil-inhibition of PDE1 [11]. Though PDE5 has been known for >20 years, there are remarkably no genetic studies yet reported involving gain or loss of expression. To address these questions, we developed and tested genetic gain and deletion strategies to test whether PDE5 is indeed expressed, active, and relevant to hypertrophic signaling in isolated cardiac myocytes.
Methods
Plasmids
Short-hairpin RNAs were designed based on mouse PDE5A sequence (shRNAPDE5-1899: FORWARD 5′-TGCTGTTTCAGAGCAGCAAACATGCAGTTTTGGCCACTGACTGACTGCATG TTCTGCTCTGAAA-3′ and REVERSE 5′-CCTGTTTCAGAGCAGAACATGCAGTCAGTCAGTGGCCAAAACTGCATGTTTGCTGCTCTGAAAC-3′; shRNAPDE5-2066 FORWARD 5′-TGCTGAAATGATGGTGTTCCATGATGGTTTTGGCCACTGACTGACCATCATGGCACCATCATTT-3′ and REVERSE 5′-CCTGAAATGATGGTGCCATGATGGTCAGTCAGTGGCCAAAACCATCATGGAA-3′)and inserted into pcDNA 6.2-GW/EmGFP-miR-155 vector (Invitrogen) [26] retaining miRNA regulatory sequences required for efficient shRNA processing (Fig 1A). This was transferred by recombinase cloning into pAd/CMV/V5-DEST™ vector (Invitrogen) to generate AdV-gfp-shRNAPDE5a. AdV were CsCl purified and titered at 1.5-3.0•1010 pfu /ml. Green fluorescent protein (gfp) enabled transfection to be confirmed (AdV-gfp virus was the control), and the miRNA construct allowed use of the CMV promotor enhancing gene knock-down. An adeno-associated virus version AAV2/9 DsRed/shRNAPDE5a (3.47×1012 GC/ml) was also made (assisted by the AAV-core of University of Pennsylvania). A DsRed monomer-PDE5a fusion protein vector was constructed by cloning PDE5A1 using mouse lung cDNA (FORWARD : 5′ACGAACTCGAGTTATGGAACGAGCG GGCCCCAACT-3′, REVERSE: 5′-ATATTCCCGGGTCAGTCAGTCCCGCTTGC CCT-3′) inserted into a shuttle vector (Invitrogen), subcloned into a pDsRed-C1 vector (Clontech), and inserted into AdV type-V driven by the CMV promoter (AdV/DsRed-PDE5a).
Figure 1.
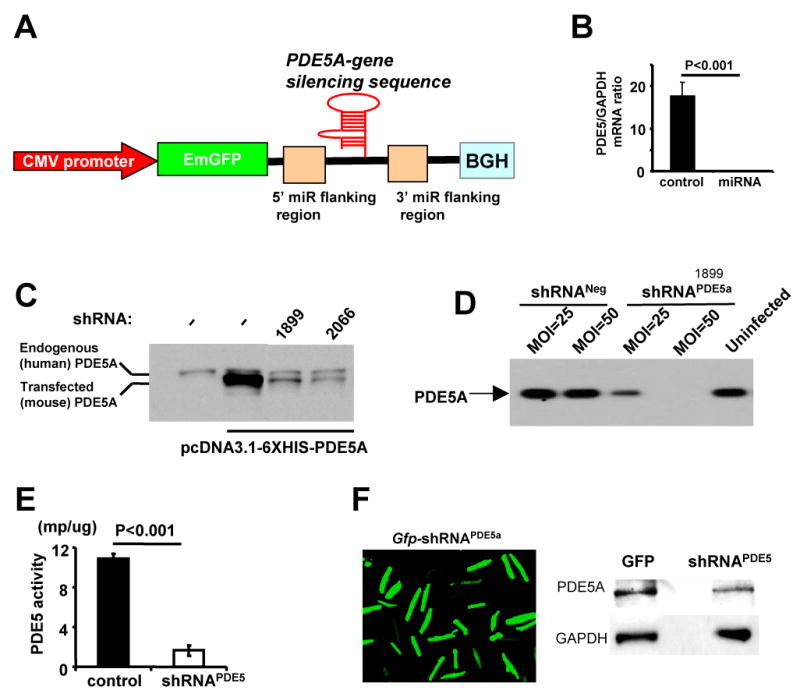
A) Schematic of shRNAmiR construct. See method for details. B) AdV-gfp-shRNAPDE5a suppresses PDE5a mRNA in RNCM. C) Mouse PDE5a overexpression in HEK293 cells is blocked by two different shRNAPDE5a ; D) Endogenous PDE5 expression in RNCM are inhibited AdV-gfp-shRNAPDE5a but not control AdV-gfp transfection E) Basal PDE5a enzyme activity is suppressed by AdV-gfp-shRNAPDE5a ; F) Adult mouse myocytes infected with AdV-gfp-shRNAPDE5a show reduced protein expression.
RNA and Protein Assays
RNA was extracted from rat neonatal myocyte using Trizol Reagent (Invitrogen) and then cDNA was generated using Superscript First-Strand Synthesis System for RT-PCR (Invitrogen). Real-time PCR reactions were carried out using SyberGreen Master Mix according to the manufacturer's protocol (Applied Biosystems). Data were captured and analyzed on Gene-Amp 5700 Sequence detection system (Applied Biosystems). The relative concentration of BNP, ANP mRNA was calculated by dividing their concentration by that of the GAPDH control gene. Primers are listed as following: rat ANP (Forward 5′-ATACAGTGCGGTGTCCAACACAGA-3′, Reverse 5′-TGACCTCATCTTCTACCGGCATCT-3′), rat BNP (Forward 5′-ATGCAGAAGCTGCTGGAG CTGATA-3′, Reverse 5′-CTTCTGCCCAAAGCAGCTTGAACT-3′), Rat GAPDH (Forward 5′-GACATGCCGCCTGGAGAAAC-3′, Reverse 5′-AGCCCAGGATGCCCTTTAGT-3′), rat PDE5A (Forward 5′-AACTCGTGGCAGCCGAATTCTTTG-3′, Reverse 5′-TGTTCATTAGATCAGCGGGCTCCA-3′). Protein was analyzed from frozen cell lysates using PDE5a antibody (Cell Signaling).
In vivo gene transfer and confocal staining
Adult myocyte gene transfer was performed in intact hearts (n=2-3) by direct LV injection (1.2×109 Pfu for AdV/DsRed-PDE5A1, 1011 GC for AAV2/9 DsRed/shRNAmiRPDE5a, followed by primary myocyte isolation 48 hrs (AdV) or 6 weeks (AAV2/9) later. Myocytes (n=5-10/heart) were incubated overnight with anti-sera for α-actinin (1:500 dilution; Sigma-Aldlich) or PDE5a (1:250 dilution; Cell Signaling), followed by incubation with secondary anti-mouse and anti-rabbit Alexa 488(green) or Alexa 546(red) antibodies(Molecular Probes)respectively for 1 hour. Cells were imaged on a Zeiss inverted epifluorescence microscope with argon-krypton laser confocal scanning system (UltraVIEW; PerkinElmer Life Sciences).
Cell culture studies
Rat neonatal cardiac myocytes (RNCM) were isolated from 1-2-day-old Sprague-Dawley rats [24], and cultured cells stimulated with 20μM phenylephrine or 0.01-0.1μM Endothelin-1, with or without 1μM sildenafil. Protein synthesis was assessed by [3H]leucine incorporation [9]. Total low-Km cGMP-PDE activity was assayed using a fluorescence polarization assay (Molecular Devices) under linear conditions with and without PDE5A inhibitor (sildenafil; 0.1-1μM).
Statistical Analysis
Quantitative assays were performed in triplicate and results expressed as means±SD. The Student's t test and one way ANOVA was used to test for significance of differences between groups.
Results
PDE5a gene silencing and Enzyme Activity and Anti-Sera Detection
Infection of RNCMs with gfp-shRNAPDE5a led to complete suppression of PDE5a mRNA expression (Fig 1B). Over-expression of mouse PDE5a in HEK293 cells was markedly and specifically suppressed by two different shRNAPDE5a (2066-mouse only; 1899- mouse and rat), while intrinsic human PDE5a was unaffected (Fig 1C). Similar knock-down of endogenous PDE5a protein was achieved in RNCMs (Fig 1D) and accompanied by reduced enzyme activity (Fig. 1E). Adult myocyte transfection was also achieved, though with somewhat less robust gene knock-down efficiency (-50% after 4-days, Fig 1F). Both shRNAPDE5a were designed close to C-terminal of PDE5, they silence all three PDE5A variants expression.
To test the specificity of immunohistochemistry for PDE5a staining, RNCMs were transfected with shRNAPDE5a and then probed with PDE5a anti-sera showed negative staining, unlike controls transfected with AdV-gfp (Fig 2A). Adult myocytes were isolated from hearts transfected in vivo with DsRed-shRNAPDE5a via an AAV vector. Infected cells identified by DsRed fluorescence lacked PDE5a anti-sera staining (green), whereas non-transfected cells remained positive for PDE5a (Fig 2B). Thus, the PDE5a immunohistochemistry signal is specific to the protein.
Figure 2.
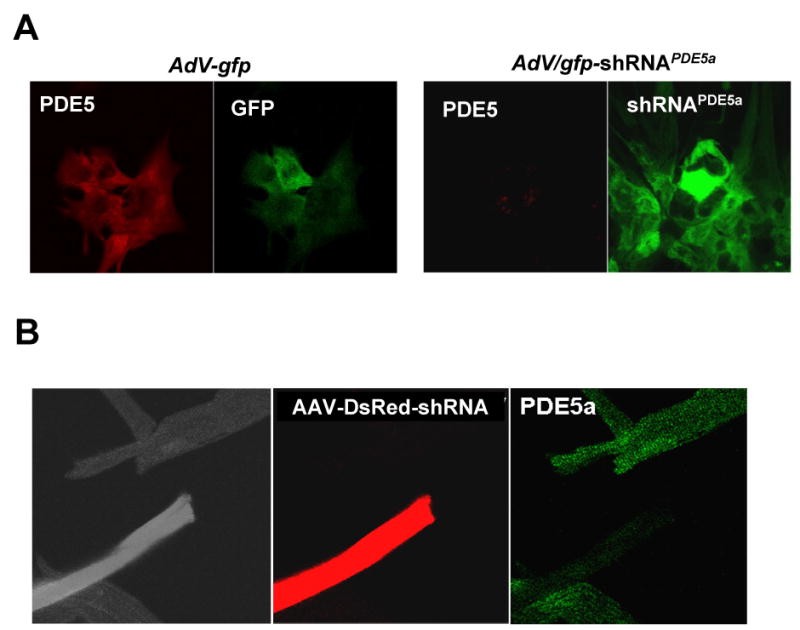
A) fluorescent immunohistochemistry of RNCM infected with AdV containing gfp or gfp- shRNAPDE5a . The latter shows suppression of anti-sera PDE5a immunostaining; B) Adult myocytes infected in vivo with AAV encoding DsRed-shRNAPDE5a were isolated and stained for PDE5a. Transfected cells (red) no longer co-stained for PDE5a (green).
PDE5a gene silencing and myocyte hypertrophic signaling
To test the role of PDE5a in cardiomyocyte hypertrophic responses, shRNAPDE5a was expressed in RNCM and cells stimulated by phenylephrine (PE, 20μM×24hr). The induction of hypertrophy signaling was indexed by A- and B-type natriuretic peptide gene expression, and cell growth by 3H-leucine incorporation. PE increased BNP ∼2.5 fold (Fig 3A), and this was suppressed to a similar extent by PDE5a gene silencing or sildenafil. Combining both yielded a similar level of suppression, indicating the specificity of sildenafil for PDE5a. The reduced BNP expression was prevented by co-incubation with the PKG antagonist rp-8Br-PET-cGMPs (30 μM) supporting the importance of PKG stimulation to the PDE5a silencing effect. Similar results were obtained in ANP expression (Fig 3B). In additional studies, we assessed 3H-leucine incorporation in cells exposed to endothelin-1, and found the increase was also blocked by transfection with shRNAPDE5a (Fig 4).
Figure 3.

Cardiomyocyte hypertrophic signaling reflected by natriuretic peptide gene expression induced by phenylephrine (PE) is suppressed similarly by PDE5a gene silencing and by sildenafil. The effect of gene silencing is blocked by co-incubation with a PKG inhibitor; A) Results for BNP stimulation, B) Results for ANP gene expression.
Figure 4.

Myocyte 3H-leucine incorporation from endothelin-1 stimulation is blocked by PDE5a gene silencing.
Myocyte PDE5a overexpression stimulates hypertrophy
As recently reported [27], DsRed-PDE5A1 fusion protein localized to z-bands in adult myocytes, while it was diffuse in eNOS-/- myocytes(Fig 5A), consistent with prior evidence based on immuno-fluorescence and immune-electron microscopy [16,28] The converse of anti-hypertrophic effects from blocking PDE5a is pro-hypertrophic effects from an increase in the enzyme. To test this, RNCM were transfected with DsRed-PDE5a. The effect was a 6-fold increase in enzyme activity (Fig. 5B). Stimulation by PE increased ANP and BNP gene expression, but both rose much more in cells overexpressing PDE5a compared to cells transfected with AdV-DsRed alone (Fig 5C).
Figure 5.
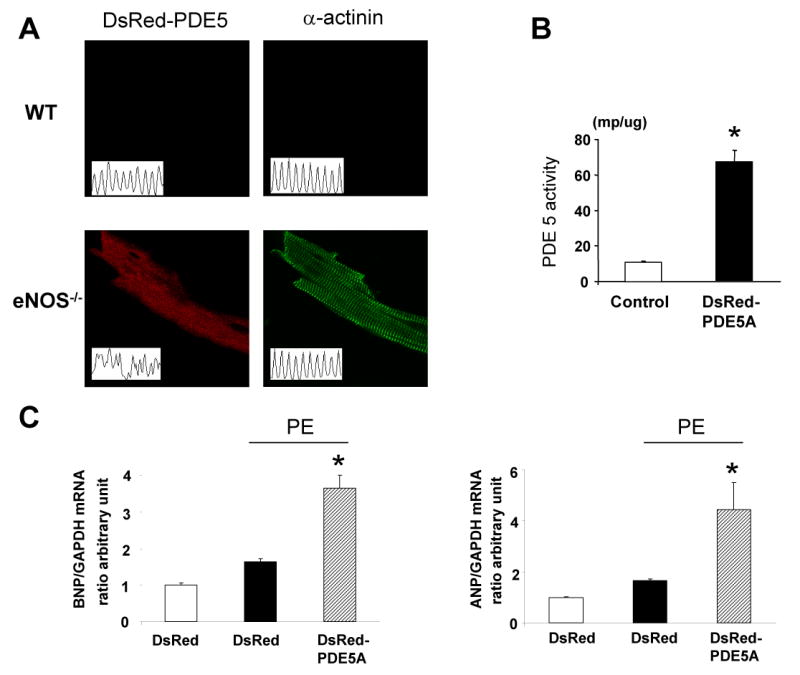
A) Localization of DsRed-PDE5a is at the z-band of the cardiac myocyte, similar to results reported using anti-sera, while it became diffuse in eNOS-/- cardiomyocytes. B) Over-expression of DsRed-PDE5a in neonatal myocytes increases enzyme activity. C) Enhanced PDE5a expression augments PE-induced stimulation of ANP and BNP gene expression.
Discussion
Growing evidence for a cardiac regulatory role of PDE5A in cardiac disease has countered earlier presumptions that it was not expressed at sufficient levels and un-important in the heart. However, controversy has persisted given the reliance on anti-sera and small molecule inhibitors for all prior studies. The present study aimed to address both concerns. Using a novel and robust gene silencing vector that greatly suppressed PDE5A gene transcription (all splice variants), we provide strong evidence for the presence of native and active enzyme in cardiac myocytes, and its relevance to stress modulation. The latter studies further support selective targeting of sildenafil to PDE5A for these effects.
While the sources of cGMP have long been well characterized, the specific PDE species responsible for its hydrolytic regulation are only now being revealed with development of new selective antagonists. Currently, several PDEs are proposed to contribute to this regulation in the heart. One is the dual substrate Ca2+-calmodulin dependent PDE1. It was first thought not to be expressed in myocytes [29], but recently found (PDE1c) in human myocytes where it appeared to contribute a large component of cAMP and cGMP hydrolytic activity in vitro [9]. Its physiologic role in the intact cell and/or heart remains unknown. PDE2, another dual substrate enzyme, is also expressed in heart and myocytes and can hydrolyze a cGMP pool localized at the outer cell membrane in vitro [30]. It is also stimulated by cGMP to hydrolyze cAMP, which appears to co-regulate myocytes responses to β-agonists [2].
Despite its cGMP selectivity, PDE5A was long considered a minor contributor in the heart given low expression and activity in myocytes under rest conditions. Yet, evidence that inhibitors such as sildenafil conferred protective effects against hypertrophy [24], ischemia [31], and apoptosis led to re-emerging interest [32]. For ischemia/apoptosis, this effect appears to involve NO signaling and mitochondrial protection [33], while anti-hypertrophic effects involve suppression of transcriptional regulators such as NFAT (nuclear factor of activated T-cells), and enzymes such as calcineurin, Rho-kinase, and Akt. Activation of cGK is though to play an important role in all three conditions. The upregulation of PDE5A expression and activity in hearts under stress may help explain an enhanced role in disease conditions [24,25].
Almost all the cardiac work to date has been performed using sildenafil. While this is also the most widely used agent clinically, it is not the most selective among small molecule inhibitors. The most potent is vardenafil (IC50 <1 nM, compared to 7 nM for sildenafil), but tadalafil is most selective for PDE5A. All three also inhibit PDE1, with about a 2-order magnitude higher IC50 over that for PDE5A in the case of sildenafil and vardenafil, and >103 fold higher concentration for tadalafil [34]. Given the wide expression of PDE1 in the cardiovascular system and new evidence of its presence in myocytes, this potential cross-reactivity is relevant. The sildenafil concentration most often used in cellular studies is 0.1-1 mM, above the IC50 for both PDE5A and near or higher that for PDE1, though intracellular concentrations maybe lower. The present data showing sildenafil identically suppressed acute myocyte hypertrophic stimulation (NP gene expression) as did PDE5A gene silencing, and that their combination had no additive effect indicates this regulation does not involve PDE1. The ability of PDE5A expression to itself enhance hypertrophic stimulation is also consistent with a primary role.
PDE5A protein expression in the cardiac myocyte has been documented by several laboratories using poly-clonal anti-sera [16,25,35,36] – though immunoblot protein levels have always been low, somewhat out of proportion to that seen by immuno-fluorescent confocal imaging. Part of the disparity may be due to sub-cellular localization of PDE5A in particulate fractions, specifically myocyte z-bands [16,28]. We have shown this localization requires NOS activity [16] and more specifically, cGMP generated by NO-activated soluble guanylate cyclase [37], and in their absence, the protein reversibly becomes more diffuse in the myocyte. This change has functional consequences, for one reducing the effectiveness of PDE5A inhibition for blunting acute b-adrenergic stimulation [16,17,37]. The present data provide key support to the specificity of the polyclonal anti-sera that has been employed for both confocal imaging and immunoblot analyses. Furthermore, migration of the dsRed-PDE5A fusion protein to z-bands in wild-type cells, but diffuse localization in myocytes lacking NOS3 (eNOS) is consistent with prior anti-sera derived findings.
The present study did not aim at elucidating new mechanisms underlying PDE5A cardiac regulation; such analysis is the subject of other ongoing investigations in ours and other laboratories. It also leaves open the question of whether intact cardiac effects of PDE5A inhibitors principally involve myocyte targeting or reflect activity on other cell types such as fibroblasts, smooth muscle cells, and endothelial cells. Clarification of this question will require cell-specific gene targeting models in vivo, efforts that are also ongoing. However, the findings do lend strong support to relevant myocyte PDE5A expression and regulatory activity, justifying such genetic models, and expanding our understanding of potential PDE5A inhibitor targets. They should help shift the prior paradigm, where PDE5A was considered only relevant to the vasculature, to one that encompasses myocyte involvement as well.
Acknowledgments
Supported by National Health Institute (Heart Lung and Blood Institute) Grants PO1 HL-77180 (DAK), HL-59408 (DAK), P50 HL084946 (DAK, MTC), HL-73935 (MTC), and T32 HL-007227 (DAK), SDG award (HCH, ET), and the Belfer Laboratory Fund, and the Abraham and Virginia Weiss Professorship (DAK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hofmann F, Feil R, Kleppisch T, Schlossmann J. Physiol Rev. 2006;86:1–23. doi: 10.1152/physrev.00015.2005. [DOI] [PubMed] [Google Scholar]
- 2.Mongillo M, Tocchetti CG, Terrin A, Lissandron V, Cheung YF, Dostmann WR, Pozzan T, Kass DA, Paolocci N, Houslay MD, Zaccolo M. Circ Res. 2006;98:226–234. doi: 10.1161/01.RES.0000200178.34179.93. [DOI] [PubMed] [Google Scholar]
- 3.Yang L, Liu G, Zakharov SI, Bellinger AM, Mongillo M, Marx SO. Circ Res. 2007;101:465–474. doi: 10.1161/CIRCRESAHA.107.156976. [DOI] [PubMed] [Google Scholar]
- 4.Layland J, Li JM, Shah AM. J Physiol. 2002;540:457–467. doi: 10.1113/jphysiol.2001.014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zahabi A, Picard S, Fortin N, Reudelhuber TL, Deschepper CF. J Biol Chem. 2003;278:47694–47699. doi: 10.1074/jbc.M309661200. [DOI] [PubMed] [Google Scholar]
- 6.Holtwick R, van Eickels M, Skryabin BV, Baba HA, Bubikat A, Begrow F, Schneider MD, Garbers DL, Kuhn M. J Clin Invest. 2003;111:1399–1407. doi: 10.1172/JCI17061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bender AT, Beavo JA. Pharmacol Rev. 2006;58:488–520. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- 8.Wang P, Wu P, Egan RW, Billah MM. Gene. 2003;314:15–27. doi: 10.1016/s0378-1119(03)00733-9. [DOI] [PubMed] [Google Scholar]
- 9.Vandeput F, Wolda SL, Krall J, Hambleton R, Uher L, McCaw KN, Radwanski PB, Florio V, Movsesian MA. J Biol Chem. 2007;282:32749–32757. doi: 10.1074/jbc.M703173200. [DOI] [PubMed] [Google Scholar]
- 10.Kass DA, Champion HC, Beavo JA. Circ Res. 2007;101:1084–1095. doi: 10.1161/CIRCRESAHA.107.162511. [DOI] [PubMed] [Google Scholar]
- 11.Ghofrani HA, Osterloh IH, Grimminger F. Nat Rev Drug Discov. 2006;5:689–702. doi: 10.1038/nrd2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin CS, Lin G, Xin ZC, Lue TF. Curr Pharm Des. 2006;12:3439–3457. doi: 10.2174/138161206778343064. [DOI] [PubMed] [Google Scholar]
- 13.Kotera J, Fujishige K, Imai Y, Kawai E, Michibata H, Akatsuka H, Yanaka N, Omori K. Eur J Biochem. 1999;262:866–873. doi: 10.1046/j.1432-1327.1999.00450.x. [DOI] [PubMed] [Google Scholar]
- 14.Reffelmann T, Kloner RA. Circulation. 2003;108:239–244. doi: 10.1161/01.CIR.0000081166.87607.E2. [DOI] [PubMed] [Google Scholar]
- 15.Zusman RM, Morales A, Glasser DB, Osterloh IH. Am J Cardiol. 1999;83:35C–44C. doi: 10.1016/s0002-9149(99)00046-6. [DOI] [PubMed] [Google Scholar]
- 16.Takimoto E, Champion HC, Belardi D, Moslehi J, Mongillo M, Mergia E, Montrose DC, Isoda T, Aufiero K, Zaccolo M, Dostmann WR, Smith CJ, Kass DA. Circ Res. 2005;96:100–109. doi: 10.1161/01.RES.0000152262.22968.72. [DOI] [PubMed] [Google Scholar]
- 17.Senzaki H, Smith CJ, Juang GJ, Isoda T, Mayer SP, Ohler A, Paolocci N, Tomaselli GF, Hare JM, Kass DA. FASEB J. 2001;15:1718–1726. doi: 10.1096/fj.00-0538com. [DOI] [PubMed] [Google Scholar]
- 18.Cremers B, Scheler M, Maack C, Wendler O, Schafers HJ, Sudkamp M, Bohm M. J Cardiovasc Pharmacol. 2003;41:734–743. doi: 10.1097/00005344-200305000-00010. [DOI] [PubMed] [Google Scholar]
- 19.Corbin J, Rannels S, Neal D, Chang P, Grimes K, Beasley A, Francis S. Curr Med Res Opin. 2003;19:747–752. doi: 10.1185/030079903125002522. [DOI] [PubMed] [Google Scholar]
- 20.Takimoto E, Champion HC, Belardi D, Moslehi J, Mongillo M, Mergia E, Montrose DC, Isoda T, Aufiero K, Zaccolo M, Dostmann WR, Smith CJ, Kass DA. Circ Res. 2005;96:100–109. doi: 10.1161/01.RES.0000152262.22968.72. [DOI] [PubMed] [Google Scholar]
- 21.Borlaug BA, Melenovsky V, Marhin T, Fitzgerald P, Kass DA. Circulation. 2005;112:2642–2649. doi: 10.1161/CIRCULATIONAHA.105.540500. [DOI] [PubMed] [Google Scholar]
- 22.Das A, Xi L, Kukreja RC. J Biol Chem. 2005 doi: 10.1074/jbc.M404706200. [DOI] [PubMed] [Google Scholar]
- 23.Fisher PW, Salloum F, Das A, Hyder H, Kukreja RC. Circulation. 2005;111:1601–1610. doi: 10.1161/01.CIR.0000160359.49478.C2. [DOI] [PubMed] [Google Scholar]
- 24.Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, Bedja D, Gabrielson KL, Wang Y, Kass DA. Nat Med. 2005;11:214–222. doi: 10.1038/nm1175. [DOI] [PubMed] [Google Scholar]
- 25.Nagendran J, Archer SL, Soliman D, Gurtu V, Moudgil R, Haromy A, Aubin C, Webster L, Rebeyka IM, Ross DB, Light PE, Michelakis ED. Circulation. 2007;116:238–248. doi: 10.1161/CIRCULATIONAHA.106.655266. [DOI] [PubMed] [Google Scholar]
- 26.Chung KH, Hart CC, Al-Bassam S, Avery A, Taylor J, Patel PD, Vojtek AB, Turner DL. Nucleic Acids Res. 2006;34:e53. doi: 10.1093/nar/gkl143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagayama T, zhang M, Hsu S, Takimoto E, Kass DA. J Pharmacol Exp Ther. 2008 doi: 10.1124/jpet.108.137422. [DOI] [PubMed] [Google Scholar]
- 28.Kass DA, Takimoto E, Nagayama T, Champion HC. Cardiovasc Res. 2007;75:303–314. doi: 10.1016/j.cardiores.2007.02.031. [DOI] [PubMed] [Google Scholar]
- 29.Sonnenburg WK, Rybalkin SD, Bornfeldt KE, Kwak KS, Rybalkina IG, Beavo JA. Methods. 1998;14:3–19. doi: 10.1006/meth.1997.0561. [DOI] [PubMed] [Google Scholar]
- 30.Castro LR, Verde I, Cooper DM, Fischmeister R. Circulation. 2006;113:2221–2228. doi: 10.1161/CIRCULATIONAHA.105.599241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salloum F, Yin C, Xi L, Kukreja RC. Circ Res. 2003;92:595–597. doi: 10.1161/01.RES.0000066853.09821.98. [DOI] [PubMed] [Google Scholar]
- 32.Fisher PW, Salloum F, Das A, Hyder H, Kukreja RC. Circulation. 2005;111:1601–1610. doi: 10.1161/01.CIR.0000160359.49478.C2. [DOI] [PubMed] [Google Scholar]
- 33.Kukreja RC, Salloum F, Das A, Ockaili R, Yin C, Bremer YA, Fisher PW, Wittkamp M, Hawkins J, Chou E, Kukreja AK, Wang X, Marwaha VR, Xi L. Vascul Pharmacol. 2005;42:219–232. doi: 10.1016/j.vph.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 34.Bischoff E. Int J Impot Res. 2004;16 1:S11–S14. doi: 10.1038/sj.ijir.3901208. [DOI] [PubMed] [Google Scholar]
- 35.Giordano D, De Stefano ME, Citro G, Modica A, Giorgi M. Biochim Biophys Acta. 2001;1539:16–27. doi: 10.1016/s0167-4889(01)00086-6. [DOI] [PubMed] [Google Scholar]
- 36.Das A, Xi L, Kukreja RC. J Biol Chem. 2005;280:12944–12955. doi: 10.1074/jbc.M404706200. [DOI] [PubMed] [Google Scholar]
- 37.Nagayama T, zhang M, Hsu S, Takimoto E, Kass DA. J Pharmacol Exp Ther. 2008;326:380–387. doi: 10.1124/jpet.108.137422. [DOI] [PubMed] [Google Scholar]