

Abstract
The intestinal hormones CCK and PYY3–36 inhibit gastric emptying and food intake via vagal afferent neurons. Here we report that CCK regulates the expression of Y2R, at which PYY3–36 acts. In nodose ganglia from rats fasted up to 48 h, there was a fivefold decrease of Y2R mRNA compared with rats fed ad libitum; Y2R mRNA in fasted rats was increased by administration of CCK, and by refeeding through a mechanism sensitive to the CCK1R antagonist lorglumide. Antibodies to Y2R revealed expression in both neurons and satellite cells; most of the former (89 ± 4%) also expressed CCK1R. With fasting there was loss of Y2R immunoreactivity in CCK1R-expressing neurons many of which projected to the stomach, but not in satellite cells or neurons projecting to the ileum or proximal colon. Expression of a Y2R promoter-luciferase reporter (Y2R-luc) in cultured vagal afferent neurons was increased in response to CCK by 12.3 ± 0.1-fold and by phorbol ester (16.2 ± 0.4-fold); the response to both was abolished by the protein kinase C inhibitor Ro-32,0432. PYY3–36 stimulated CREB phosphorylation in rat nodose neurons after priming with CCK; in wild-type mice PYY3–36 increased Fos labeling in brainstem neurons but in mice null for CCK1R this response was abolished. Thus Y2R is expressed by functionally distinct subsets of nodose ganglion neurons projecting to the stomach and ileum/colon; in the former expression is dependent on stimulation by CCK, and there is evidence that PYY3–36 effects on vagal afferent neurons are CCK dependent.
Keywords: satiety, Y2, PYY, CCK, ghrelin, vagus
Introduction
In recent years, increasing attention has been given to the peripheral mechanisms that influence food intake (Dockray, 2003; Murphy and Bloom, 2006). Signals from the gastrointestinal tract that are now recognized to limit meal size by terminating feeding include non-nutritive distension of the stomach (Gonzalez and Deutsch, 1981), and nutrient-dependent release of cholecystokinin (CCK) and the lipid mediator oleylethanolamide from the proximal small intestine (Gibbs et al., 1973; Rodríguez de Fonseca et al., 2001; Fu et al., 2003; Moran and Kinzig, 2004), and peptide YY (PYY)3–36 and GLP-1 from the ileum and colon (Murphy and Bloom, 2006). In the absence of food, other gastrointestinal signals stimulate appetite, notably the gastric hormone ghrelin (Cummings and Overduin, 2007), and the lipid amide anandamide (Gómez et al., 2002). In each case, there is evidence for signaling via afferent neurons of the vagus nerve (Smith et al., 1981; Rodríguez de Fonseca et al., 2001; Date et al., 2002; Gómez et al., 2002; Fu et al., 2003; Burdyga et al., 2004; Abbott et al., 2005; Burdyga et al., 2006a).
Extensive interactions between the various gastrointestinal stimuli acting on vagal afferent neurons are now emerging. For instance, CCK actions on vagal afferent neurons are potentiated by gastric distension and leptin (Schwartz et al., 1993; Wang et al., 1997; Peters et al., 2004), and inhibited by orexigenic factors such as orexin A and ghrelin (Burdyga et al., 2003; Date et al., 2005). In addition, it now appears that CCK regulates the expression of genes encoding certain G-protein coupled receptors and peptide transmitters in vagal afferent neurons. Thus, expression of receptors for endocannabinoids (CB1R), and melanin concentrating hormone (MCH1R), is increased in fasted rats and decreased by refeeding via endogenous CCK (Burdyga et al., 2004, 2006b). These effects do not reflect nonspecific changes in gene expression involving all receptors, because in the same circumstances there is little or no change in the expression of receptors for orexin (Ox1R), leptin (ObR), or the CCK1R itself, which is the target of endogenous CCK. Interestingly, expression of the neuropeptide transmitter MCH, which stimulates food intake, is increased in vagal afferent neurons in response to energy restriction and downregulated by CCK; conversely a peptide transmitter associated with inhibition of food intake, cocaine-, and amphetamine-regulated transcript (CART), is stimulated by CCK (Burdyga et al., 2006b; de Lartigue et al., 2007).
The changes in gene expression in vagal afferent neurons described above indicate a simple form of memory within these cells by which neurochemical phenotype reflects food intake over the previous 24–48 h. The data also suggest a previously unsuspected role for CCK in determining the capacity for orexigenic signaling at the level of vagal afferent neurons. In the present study we asked whether CCK might also act to enhance satiety signals. The intestinal peptide PYY3–36 induces satiety via vagal pathways (Abbott et al., 2005), and Y2R (at which PYY3–36 acts) is expressed in nodose ganglion neurons (Zhang et al., 1997; Koda et al., 2005). We sought, therefore, to test the hypothesis that CCK regulates expression of Y2R. We report here that in vagal afferent neurons projecting to the stomach, the expression of Y2R is indeed regulated by CCK acting via protein kinase C (PKC).
Materials and Methods
Animals.
Studies were made using adult male Wistar rats (250–350 g, ∼10 weeks old) housed at 22°C under a 12 h light/dark cycle with ad libitum access to food and water unless otherwise stated. Rats were killed by carbon dioxide inhalation and nodose ganglia either fixed in 4% paraformaldehyde (PFA) or immersed in RNA later (Ambion), or taken for culture. The experiments were conducted under appropriate UK Home Office personal and project licenses.
Experiments with mice were performed using CCK1R null mice (Mouse Biology Program, UC Davis) and their wild-type counterparts 129S6/SvEv (Taconic). Mice were of initial weight 18–20 g (8–10 weeks of age) and were maintained on regular laboratory chow (Purina Laboratory). Mice were fasted overnight before experiments but allowed ad libitum access to water. The institutional guidelines for care and use of laboratory animals were followed throughout the study in accordance with a protocol approved by the Institutional Animal Use and Care Committee, UC Davis.
Human tissue.
Human nodose ganglia were obtained from tissue removed during radical dissection of the neck for removal of a glomus tumor (Burdyga et al., 2003). After resection, the ganglia were rapidly divided into several segments and fixed in PFA for morphological studies. The work was approved by the Multi-Centre and Local Ethics Committee of Salford and Trafford Authority, and written consent was obtained from all patients.
Fasting–refeeding.
In some experiments rats were fasted up to 48 h with water ad libitum. Some animals were then refed for up to 2 h, and others received CCK8s (Bachem) by intraperitoneal injection (10 nmol), PYY3–36 (5 nmol; Tocris Bioscience), or saline, and were killed up to 15 h later. Before refeeding in these experiments, rats received either the CCK1R antagonist lorglumide (10 mg/kg, i.p.; gift from Rotta Pharmaceuticals) or saline.
RT-PCR.
Total RNA was extracted from pooled (10–12) nodose ganglia in TriReagent (Sigma). Briefly, samples were treated with DNase, reverse transcribed, and processed for RT-PCR using BIOTAQ DNA polymerase (Bioline) and the following primers: 5′-CAAGAGCATGCGCACAGTAACCAA-3′ (forward) and 5′-CTCCCCGGGCCATTTCTCAGT-3′ (reverse) to amplify a 397 bp region encoding nucleotides 233–630 of the published sequence (GenBank accession number NM_023968) of rat Y2R mRNA; 5′-GACCCCTTCATTGACCTCAACT-3′ (forward) and 5′-CTCAGTGTAGCCCAGGATGCC-3′ (reverse) to amplify rat GAPDH (GenBank accession number XO2231). The PCR product for Y2R was gel purified (MinElute gel extraction kit QIAGEN) and cloned into pGEM-T Easy (Promega), and its sequence was confirmed using an automated dideoxy method.
qPCR.
Quantitative analysis of Y2R transcripts was performed on total RNA extracted as described above with first-strand cDNA synthesis using avian myeloblastosis virus reverse transcriptase and random hexamer primers (Promega). Real-time PCR was performed using double-dye (FAM-TAMRA) oligonucleotide 5′-CCATCAGCGAAGGCACAAAACGACC-3′ and specific primers for rat Y2R: 5′-TTAGTCCTGGAGCTGCAAGTGA-3′-sense and 5′-ACCACGCACACGAGCATTT-3′-antisense. Results were expressed as Y2R mRNA relative to 18S (Eurogentec) using the comparative CT method (Livak and Schmittgen, 2001).
Immunohistochemistry.
Cryostat sections of nodose ganglia were rinsed in 0.1 m PBS, permeabilized in alcohol, and processed for single- or double-labeling immunofluorescence as described previously (Burdyga et al., 2002, 2003). For detection of Y2R in rat tissue we used a rabbit polyclonal Y2R antibody (1:200; Neuromics Antibodies) and for colocalization with CCK in human tissue we used a goat polyclonal Y2R antibody (1:80; Santa Cruz Biotechnology). CCK1R was localized using an affinity-purified goat polyclonal antibody (1:100; Santa Cruz Biotechnology) or rabbit polyclonal antibody raised against the N-terminal region of rat CCK1R (1:100; gift of the late John Walsh). Goat polyclonal antibody to glial fibrillary acidic protein (GFAP) (1:100; Santa Cruz Biotechnology) and mouse anti-glutamine synthetase monoclonal antibody (1:700; Chemicon) were used for identification of glial cells (Hanani, 2005). Vanilloid receptor (VR)-1 was detected by an affinity-purified goat polyclonal antibody (1:100; Santa Cruz Biotechnology) and phosphoCREB by mouse monoclonal antibody to phosphoCREB (Ser133) clone 634–2 (1:200; Millipore). Affinity-purified goat polyclonal antibodies to ObR (1:100; Research Diagnostics), OxR1, and GHS1R (1;100; Santa Cruz Biotechnology) were used in dual-labeling immunohistochemistry. Secondary antibodies were used as appropriate and included fluorescein (FITC)-conjugated AffiniPure donkey anti-rabbit, anti-goat, or anti-mouse IgG and Texas Red-conjugated AffiniPure donkey anti-rabbit, anti-goat, or anti-mouse IgG (1:100; Jackson ImmunoResearch Laboratories). Specificity of immunostaining was determined by omitting primary antibody and by preincubation with an excess of appropriate peptide. Sections were examined using an Axioplan Universal microscope and images were processed using the Axio Vision 3.0 Imaging system with deconvolution options (Carl Zeiss Vision). The quantification of neurons coexpressing Y2R, CCK1R, GHS1R, ObR, or Ox1R was made using nodose ganglia from 3–5 rats. Sections passing through the full-length of the ganglia were selected, and the appropriate double immunostaining and counting was performed on 5 sections per ganglion, with the sections being separated by 90 μm. Results are expressed as percentage of labeled cells expressing one or the other epitope, or both.
In situ hybridization.
Cryostat sections (10 μm) of frozen rat nodose ganglia were thaw mounted on UV-treated poly-l-lysine-coated slides, fixed in 4% PFA in 1× PBS, and acetylated in 0.25 m acetic anhydride–0.1 m triethanolamine (10 min) and processed for in situ hybridization as previously described (Burdyga et al., 2004) using oligonucleotide probes complementary to bases 448–488, 751–793, and 877–915 of the rat Y2R receptor (Eurogentec) 3′-end labeled with [35S]dATP (10 mCi/ml; PerkinElmer) (Zhang et al., 1997). Silver grains were visualized using an Axioplan Universal microscope and images processed using an AxioVision 3.0 Imaging system (Carl Zeiss Vision) with dark- and bright-field illumination. Control slides were hybridized with a 100-fold excess of unlabeled oligonucleotides.
Retrograde tracing.
Rats were anesthetized with diazepam (2.5 mg/kg i.m.) and hypnorm (0.3 ml/kg, i.m.). A suspension of True Blue (Sigma-Aldrich) in distilled water (5% w/v) was injected into the ventral wall of the stomach or proximal colon using a 10 μl Hamilton microsyringe. After each injection the needle was left in the place for up to 1 min to minimize leakage of dye along the needle tract, and the injection site was then swabbed with saline. Animals were given prophylactic antibiotic (Baytril, 0.2 ml/kg, i.m.) and after 7 d some were fasted for 24 h. True Blue staining was visualized before dual-labeling immunohistochemistry, and images were processed as describe above.
Cloning of the rat Y2R promoter.
An upstream sequence corresponding to 3034 bp of the wild-type human Y2R promoter, together with 94 bp of exon 1, was amplified by PCR from human genomic DNA and cloned into pGEM-T Easy vector. The promoter fragment was excised from pGEM-T Easy and cloned into the luciferase reporter vector pGL4.10 [luc2] between XhoI and Hind III sites of the multiple cloning region to give Y2R-luc. All PCRs were performed with Phusion High-Fidelity DNA polymerase (New England BioLabs) in a PerkinElmer Genamp 2400 thermal cycler. The integrity of all constructs was confirmed by automated dideoxy sequencing.
Cell culture.
Nodose ganglia were dissected under aseptic conditions and digested for 90 min at 37°C in 5 ml of Ca2+ and Mg2+-free HBSS containing 50 μl of 100 mm DTT and 1 mg/ml collagenase type Ia (Roche Diagnostics). Cells were dispersed by gentle trituration through siliconized Pasteur pipettes, washed twice with HEPES-buffered DMEM (HDMEM) containing 10% fetal calf serum (FCS) (Hyclone/Perbio Science) supplemented with 1% pen/strep solution and 2% antibiotic/antimycotic solution, and then plated onto 4 well chamber slides and maintained in HDMEM with 10% FCS at 37°C in a 5% CO2 atmosphere. Neurons were cultured for 4 d, and media changed every 48 h as previously described (de Lartigue et al., 2007).
Luciferase assay.
Medium was removed from cultured neurons and cells transfected with firefly-luciferase constructs and a Renilla luciferase construct as an internal control (Promega). Cells were transfected with 1.5 μg of DNA per well, 1 μl of vortexed combimag per μg DNA, 6 μl of Transfast per μg DNA, in 200 μl of HDMEM per well and incubated on a magnetic plate for 15 min at 22°C. Medium containing 10% FCS was then added and cells incubated at 37°C for 24 h before treatment with drugs as indicated below. Cells were then lysed using passive lysis buffer (Promega) and luciferase activity was determined with a dual-luciferase reporter assay system (Promega) using a LumiCount Platereader (Packard BioScience) according to the manufacturer's protocol. Results are presented, as fold increase over unstimulated control with a value of 1.0 signifying no change in luciferase activity.
Determination of fos immunoreactivity in brainstem in response to PYY3–36.
This method has been described in detail previously (Whited et al., 2007). Briefly, fasted mice were injected with vehicle (0.1 ml of 0.9% saline) or PYY3–36 (5 μg/kg IP; Bachem). After 2 h, mice were deeply anesthetized with sodium pentobarbital (100 mg/kg IP; Western Medical Supply) and transcardially perfused with 20 ml of heparinized saline followed by 25 ml of 4% paraformaldehyde and postperfusion fixed for 1 h. Sections (100 μm) were cut using a vibratome and processed for immunocytochemistry using an anti-fos protein antibody (1:2000; Santa Cruz Biotechnology). Immunoreactivity was visualized using the avidin–biotin complex reaction method using diaminobenzimide. Images were taken on a Provis (Olympus) microscope and analyzed using Corel Paint Shop Pro. Neurons in the mid-NTS (bregma −7.76 to −7.32) were determined to be immunopositive by color and size. The number of labeled neurons per section was counted (n = 3 sections) and summed for each mouse.
Statistics.
The results are expressed as means ± SE, and comparisons were made by ANOVA and Student's t test as appropriate.
Results
Y2R mRNA abundance
Initial studies using RT-PCR indicated that the expression of Y2R transcripts in nodose ganglia was decreased in fasted rats compared with rats fed ad libitum (Fig. 1A). Quantification by real-time PCR revealed that Y2R transcripts were decreased fivefold with fasting. Administration of CCK8s to fasted rats reversed the decrease in Y2R transcript abundance and so too did refeeding. Importantly, the effects of refeeding were inhibited by prior administration of the CCK1R antagonist lorglumide, indicating a role for endogenous CCK in regulating Y2R expression (Fig. 1B).
Figure 1.

RT-PCR and real-time PCR show changes in Y2R transcript abundance in nodose ganglia neurons of rats depending on energy intake. A, RT-PCR product of predicted size (397 bp) for Y2R in nodose ganglia is decreased in rats fasted for 48 h (lane 2) compared with those that fed ad libitum (lane 1). Note similar abundance of GAPDH products; lane 3, 100 bp calibration marker. B, Real-time PCR for Y2R mRNA reveals significantly decreased abundance in rats fasted for 48 h, compared with those that fed ad libitum; CCK8s (10 nmol, i.p.) administrated to fasted rats restores Y2R mRNA abundance, as does refeeding for 2 h through a mechanism sensitive to prior administration of the CCK1R antagonist lorglumide; n = 3; *p < 0.05.
Y2R localization
To determine whether the differences in Y2R mRNA abundance were reflected at the protein level, we used immunohistochemistry to study the distribution in fed and fasted rats. In the former, we identified Y2R immunoreactivity in many neurons of the mid and caudal parts of the nodose ganglia (Fig. 2A–O). In fasted rats, there were substantially fewer labeled neurons, although a subset still exhibited immunoreactivity that was as intense as that in ganglia from animals fed ad libitum. Examination of the time course of decrease in Y2R labeled neurons with fasting indicated 50% decrease after 12–13 h (Fig. 2). Administration of CCK to fasted rats increased the number of neurons expressing Y2R (Fig. 2C); the increase in labeled neurons after CCK was relatively rapid and peaked at 2–3 h after the injection (Fig. 2). Refeeding of fasted rats also increased Y2R immunoreactivity (Fig. 2), and prior administration of the CCK1R antagonist lorglumide blocked this response, consistent with a role for endogenous CCK in regulating Y2R expression (Fig. 2).
Figure 2.

CCK regulates expression of Y2R in vagal afferent neurons. A, B, Expression of Y2R in rats fasted for 48 h (A) is decreased in the mid-caudal regions of the nodose ganglion compared with rats fed ad libitum (B), although some neurons still express Y2R in fasted rats. C, After administration of CCK8s (10 nmol, i.p.) to fasted rats, there is a substantial increase of Y2R-positive neurons. D, For comparison, control samples show low abundance of Y2R 2 h after administration of vehicle. E, Refeeding of fasted rats for 2 h increases expression of Y2R in nodose neurons. F, In contrast, in rats treated with a CCK1R antagonist (lorglumide) 15 min before refeeding, the increase of Y2R after refeeding was inhibited. G–I, At higher power, dual staining with antibodies to Y2R (G, red) and glutamine synthetase (H, green) reveals that Y2R immunoreactivity is localized to satellite cells (open arrowheads) as well as neurons (filled arrows); I, overlay of panels G and H showing colocalization in satellite cells (yellow, filled arrowheads). J–L, In rats fed ad libitum, there is Y2R in both neurons (J, red, filled arrows) and satellite cells (K, green, open arrows; L, overlay with colocalization in satellite cells indicated by filled arrowheads). However, in fasted rats (M–O), there is decreased expression of Y2R in neuronal cell bodies (M, filled arrows) but not satellite cells (N, open arrows; O, overlay with colocalization in satellite cells indicated by filled arrowheads). P–T, Localization of Y2R in human nodose ganglia. P–R, Dual staining with antibodies to Y2R (P, red) and GFAP (Q, green) shows that Y2R is expressed in small and large neurons (red, filled arrows) as well as satellite cells (open arrows) (R, overlay with colocalization to satellite cells indicated by open arrowheads). S, Colocalization of Y2R (red) and CCK1R (green) in small and large neurons indicated by open arrows. T, Colocalization of Y2R (red) and VR1 (green) in small neurons (open arrows). U–W, Quantification of CCK-dependent changes in Y2R-positive neurons. U, Progressive decrease in the numbers of Y2R-immunoreactive cells in nodose ganglia after nutrient withdrawal. Exponential curve-fitting (OriginPro7.5) indicated 50% decrease in abundance of Y2R-immunoreactive neurons at 12.5 h (n = 4). V, Intraperitoneal administration of CCK8s (10 nmol) to rats fasted for 24 h increased the number of Y2R-immunoreactive neurons with a peak at 2 h (n = 4). W, Fasting for 24 h decreased the number of Y2R-immunoreactive neurons (***p < 0.001). Refeeding for 2 h increased the number of Y2R-immunoreactive neurons (**p < 0.01) and the CCK1R antagonist lorglumide administered 15 min before refeeding prevented the increase in Y2R-immunoreactive neurons (**p < 0.01) (n = 5–6). Scale bars: 100 μm in A–F, 25 μm in G–I and P–T, and 40 μm in J–O.
In addition to neurons expressing Y2R, there was expression in a subset of small cells that had a characteristic crescent-shaped morphology. These cells were identified as satellite cells on the basis of expression of glutamine synthetase (Fig. 2G–I) (Miller et al., 2002; Hanani, 2005); there was no apparent difference in Y2R immunoreactivity in fed and fasted rats in this cell population (Fig. 2J–O).
In situ hybridization indicated that in nodose ganglia from rats fed ad libitum, there was expression of Y2 mRNA in cells with the morphology of both neurons and satellite cells (Fig. 3). The latter were most readily seen where they surrounded neurons that do not express Y2 receptors. In fasted rats, the localization to neurons was very substantially depleted, but it was still possible to identify Y2R in cells surrounding neuronal profiles. Administration of CCK to fasted rats, stimulated neuronal Y2R expression, but had no detectable effect on the satellite cells (Fig. 3).
Figure 3.

Localization of Y2R by in situ hybridization in rat nodose ganglia. A, B, In ganglia from rats fed ad libitum (A), there is localization to neurons (white arrows), whereas in rats fasted for 48 h (B), neuronal profiles are typically not associated with Y2R. C, In fasted rats treated with CCK8s (10 nmol, i.p.), there is localization of Y2R to neurons. D–F, Crescent-shaped cells surrounding neuronal profiles and corresponding to satellite cells (black arrows) express Y2R in ganglia from rats fed ad libitum (D), in fasted rats (E), and in fasted rats treated with CCK (F) with no apparent differences. Scale bars, 25 μm.
Y2R expression in human nodose ganglia
Because previous studies had identified differences between rat and human nodose ganglia in receptor expression (Burdyga et al., 2003), we examined Y2R in human nodose ganglion recovered at surgery. As in the rat, Y2R immunoreactivity was found in both neurons and satellite cells (Fig. 2P–R). The latter are more prominent in human nodose ganglia (Burdyga et al., 2003) and are associated with Ox1R immunoreactivity. In the present study, Y2R was identified in satellite cells (positive for GFAP immunoreactivity) surrounding large neurons, which resembles the distribution of OxR1. Additionally, there were populations of large and small neurons that typically coexpressed Y2R and CCK1R (Fig. 2S). The small neurons (8–15 μm) expressing Y2R were also strongly positive for VR1 (Fig. 2T).
Neurochemical phenotype of Y2R-expressing nodose neurons
To further characterize the rat nodose ganglion neurons expressing Y2R, and to relate their phenotype to previous studies, we examined colocalization with other receptors. It is known that a high proportion of CCK1R-expressing neurons also express GHS1R, Ox1R, and ObR; in the present study we found that Y2R expression was also found in CCK1R-expressing neurons in rats fed ad libitum, and consistent with previous findings there was also coexpression with GHS1R, ObR, and Ox1R (Fig. 4A–H). When the pattern of colocalization was examined in fasted rats, there was in each case a lower proportion of labeled neurons exhibiting Y2R expression, consistent with food-dependent expression of Y2R but not of CCK1R, GHS1R, ObR, or Ox1R (Fig. 4I–L).
Figure 4.

Colocalization of Y2R with GHS1R, ObR, Ox1R, or CCK1R in rat nodose neurons. A–D, In nodose ganglion neurons of rats fed ab libitum, overlay (yellow, open arrows) reveals colocalization of Y2R (green) and GHS1R (red) (A); Y2R (green) and ObR (red) (B); Y2R (green) and Ox1R (red) (C); and Y2R (red) and CCK1R (green) (D). E–H, In rats fasted 48 h, there is a reduction of Y2R (green in E–G, red in H) but not GHS1R (E, red), ObR (F, red), Ox1R (G, red), or CCK1R (H, green). I, Percentage of labeled neurons containing Y2R only (black bars) or GHS1R only (open bars), or exhibiting colocalization of Y2R and GHS1R (gray bars) in fed and fasted rats. J–L, The same data for ObR, Ox1R, and CCK1R. Immunoreactive cells were counted in sections through mid and caudal regions of rat nodose ganglia after double-labeling immunocytochemistry with appropriate antibodies. All immunoreactive neurons were recorded, and double-labeled cells were counted once; the percentages assigned to each class are shown; n = 4–6 ganglia. Scale bars in A–H, 50 μm.
Retrograde tracing identifies functionally distinct subpopulations of Y2R nodose neurons
We reasoned that the presence of distinct subsets of Y2R-expressing neurons that differed on the basis of sensitivity to previous energy intake, might be related to functions in serving different regions of the gut. We therefore used retrograde labeling from stomach, ileum, and colon to identify the nodose ganglion neurons projecting to these regions. In animals fed ad libitum, ∼70% of neurons that projected to stomach, 50% of neurons from the colon, and 80% from the ileum expressed Y2R (Fig. 5). However in fasted rats, <10% of neurons from the stomach exhibited Y2R immunoreactivity, whereas the proportion of neurons from the colon and ileum was not significantly changed (Fig. 5U). As expected, a high proportion of neurons serving the stomach also expressed CCK1R, and this did not change with feeding, providing evidence that the changes in Y2R did not represent nonspecific alterations in receptor abundance (Fig. 5V).
Figure 5.
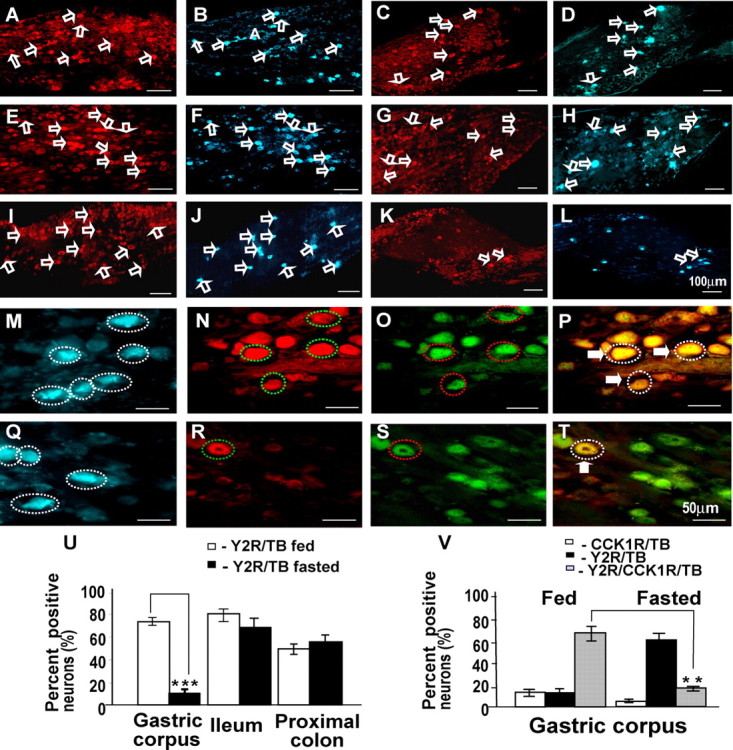
Nodose ganglion neurons projecting to the stomach, ileum, and colon retrogradely labeled with True Blue also express Y2R. A–D, Expression of Y2R in nodose neurons (open arrows) in fed rats projecting to the colon (A) indicated by True Blue fluorescence (B); in fasted rats, there is also Y2R (C) in neurons labeled with True Blue (D) from the colon. E–H, In fed rats, there is Y2R (E) in neurons labeled (open arrows) with True Blue from the ileum (F); in fasted rats, there is also Y2R (G) in neurons labeled with True Blue (H) from the ileum. I–L, In fed rats, there is Y2R in neurons labeled (open arrows) with True Blue from the stomach (I, J), whereas in fasted rats, there is decreased abundance of neurons expressing Y2R (K) and labeled with True Blue from the stomach (L). M–T, At higher power, many neurons (broken circles) retrogradely labeled from stomach in fed animals (M, blue) also contained Y2R (N, red) and CCK1R (O, green) as indicated by the overlay (P; triple-labeled neurons are enclosed in dotted lines and marked by filled arrows). With fasting, neurons projecting to the stomach (Q, blue) only rarely express Y2R (R, red) with CCK1R (S, green) as indicated by the overlay (T, yellow; circled neuron and filled arrow). U–V, Quantification of Y2R-positive vagal afferent neurons projecting to the stomach, ileum, and proximal colon in rats fed ad libitum or fasted. U, Fasting for 48 h decreased the number of neurons projecting to the gastric corpus and expressing Y2R, but expression in neurons projecting to the proximal colon and ileum was unchanged. V, Quantification of Y2R- and CCK1R-immunoreactive neurons projecting to gastric corpus. Compared with rats fed ad libitum in which most neurons serving the stomach expressed Y2R and CCK1R, in fasted rats most did not express Y2R; n = 5; **p < 0.01, ***p < 0.001. Scale bars: 100 μm in A–L and 50 μm in M–T.
Y2 receptor expression in vitro
To determine the cellular mechanisms by which CCK regulated Y2R expression, we cloned ∼3 kb of Y2R promoter into a luciferase reporter vector. The Y2R-luc vector, or a control empty vector, was then transfected into rat nodose ganglion cells in culture; CCK strongly stimulated expression of Y2R-luc, but had no effect on the empty vector (Fig. 6A). Because other transcriptional responses to CCK in these cells are mediated by PKC, we then examined the effect of phorbol ester on Y2R expression. The response to PMA was substantial and as expected was inhibited by the PKC inhibitor Ro-32,0432 (Fig. 6B,C). Consistent with the hypothesis that CCK activated PKC, the inhibitor also virtually abolished responses to CCK. Moreover, consistent with previous studies that demonstrated inhibition of transcriptional responses to CCK by ghrelin, we found that ghrelin also reversed the action of CCK on Y2R-luc expression (Fig. 6D).
Figure 6.
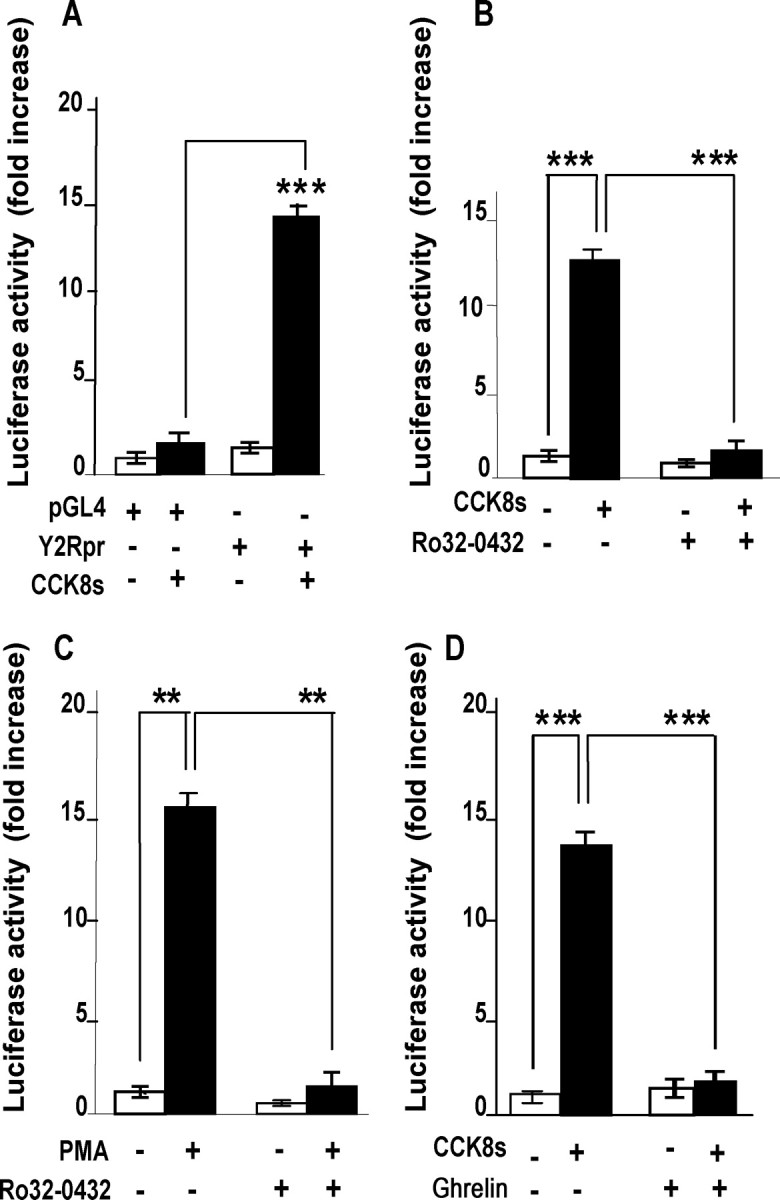
CCK regulates Y2R expression in luciferase promoter-reporter assays. A, In cultured vagal afferent neurons transfected with empty vector (pGL4), 10 nm CCK8s for 16 h had no effect on luciferase activity, whereas there was significant stimulation of luciferase activity in neurons transfected with Y2R-luc (Y2Rpr). B, In vagal afferent neurons transfected with Y2R-luc, the response to 10 nm CCK8s was inhibited by 1 μm Ro-32,0432. C, PMA (100 nm) also stimulated Y2R expression, and this was inhibited by Ro-32,0432. D, Ghrelin inhibited CCK8s stimulation of Y2R-luc expression in cultured vagal afferent neurons. Responses to CCK8s are expressed relative to the appropriate control; n = 5–15; **p < 0.005, ***p < 0.001.
Functional consequences of CCK-dependent control of Y2 abundance
To determine whether CCK-regulated Y2R expression influenced the capacity to respond to PYY3–36, we examined two experimental models. We made use of the fact that stimulation of vagal afferent neurons leads to rapid increases in nuclear phosphoCREB (Burdyga et al., 2004; de Lartigue et al., 2007). In fasted rats, ∼6% of neurons exhibited nuclear phosphoCREB staining; peripheral administration of PYY3–36 had no significant effect on the numbers of neurons exhibiting nuclear phosphoCREB (Fig. 7A). After administration of CCK8s, there was an increase in nuclear phosphoCREB, and the subsequent administration of PYY3–36 produced a further statistically significant increase in neurons expressing nuclear phosphoCREB compared with CCK alone and compatible with CCK-stimulation of Y2R expression (Fig. 7A). In a second experimental model of CCK-dependent vagal afferent signaling (Whited et al., 2007), we examined activation of brainstem neurons revealed by induction of fos. In wild-type mice, exogenous administration of PYY3–36 increased the number of fos-expressing neurons (Fig. 7B), but in CCK1R null mice induction of fos in response to peripheral administration of PYY3–36 was significantly reduced compared with wild-type mice again in keeping with a role for CCK in determining Y2R signaling.
Figure 7.

CCK-dependent effects of PYY3–36 on phosphoCREB in rat nodose ganglion and fos in mouse brainstem. A, In rats fasted for 24 h, intraperitoneal PYY3–36 (5 nmol) did not stimulate phosphoCREB compared with vehicle; administration of CCK (10 nmol) 3 h before vehicle increased phosphoCREB, and this was further increased 15 min after PYY3–36 (*p < 0.5) (n = 4). B, In nucleus tractus solitarius in wild-type mice, there was increased fos abundance after intraperitoneal PYY3–36, but in mice null for CCK1R this response was abolished (***p < 0.001; n = 4–5).
Discussion
The main finding of the present study is that CCK stimulates Y2R expression in vagal afferent neurons serving the stomach via activation of PKC. These findings are interesting because they provide the first example of CCK–stimulated expression in vagal afferent neurons of a receptor associated with satiety signaling and inhibition of gastric emptying. Cholecystokinin is released by dietary fat and protein and also inhibits food intake and gastric emptying; the present data therefore suggest that the actions of CCK in delaying nutrient delivery to the small intestine might be enhanced through stimulation of Y2R expression and enhanced capacity for PYY3–36 signaling. We show also that the action of CCK is exerted on vagal afferent neurons from the stomach but not ileum or colon, suggesting a previously unsuspected functional heterogeneity in Y2R signaling via vagal afferent neurons.
Immmunoreactive Y2R was identified in both neurons and satellite cells in nodose ganglia. This pattern of expression seems to be a feature of both human and rat nodose ganglia. Previous studies have noted that OxR1 receptors are expressed in both neurons and satellite cells in human nodose ganglia, whereas CCK1R, CB1R, and MCH1R appear to be expressed in neurons but not satellite cells (Burdyga et al., 2003, 2004, 2006b). The present report would appear to be the first example of expression in both nodose ganglion neurons and satellite cells of a receptor associated with anorexic signaling. These observations raise the question of whether the Y2R ligand PYY3–36 might exert indirect effects on vagal afferent signaling via satellite cells. Importantly, however, we found that whereas there was CCK-dependent expression of Y2R in neurons, there was no evidence of regulated expression in satellite cells. The satellite cells in rat nodose ganglion have not previously been studied in any detail, and the demonstration that they express Y2R will open the way to future functional studies.
Previous studies have indicated that nutrient withdrawal downregulates expression of receptors associated with orexigenic signaling in vagal afferent neurons; to the best of our knowledge the present study is the first to provide evidence of nutrient-dependent upregulation of the expression of receptors associated with anorexic signaling in these neurons (Burdyga et al., 2004, 2006b). Taken in the context of other work (de Lartigue et al., 2007), the present data indicate that CCK acts reciprocally to determine the capacity for both orexigenic and anorexic signaling via the vagus nerve. Thus, although the expression of certain receptor types in nodose ganglion neurons appears to be independent of energy intake and can be considered constitutive, others are expressed through CCK1R-regulated mechanisms. In the presence of low plasma CCK (after food withdrawal) there is an enhanced capacity for orexigenic signaling that is reversed by postprandial increases in CCK, whereas in animals fed ad libitum the present data indicate an enhanced capacity for signaling via anorexic receptors. Earlier work has reported that expression of Y2R in nodose ganglia is increased by axotomy (Zhang et al., 1997); in control animals the proportion of neurons expressing Y2R was similar to that in the present study (50–55%) but 7 d after axotomy this increased to ∼70%. The induction of Y2R in nodose ganglion neurons that do not normally express the receptor implies a different mechanism to the one reported here involving expression dependent on nutrient ingestion.
The main hormonal ligand of vagal Y2R is generally thought to be PYY3–36. This is generated from PYY released by enteroendocrine L-cells found mainly in the ileum and colon (Ali-Rachedi et al., 1984; Böttcher et al., 1984), through cleavage by dipeptidyl aminopeptidase IV. The peptide neurotransmitter NPY is also a putative ligand for Y2R. In the periphery, NPY is found in some sympathetic neurons, but on present evidence this is unlikely to provide the primary input to vagal afferent Y2R.
There has been considerable interest in the role of PYY3–36 in regulating food intake (Batterham et al., 2002, 2003). There have been some difficulties in reproducing the original findings, which have been attributed to failure to take account of the effect of stress on feeding responses to PYY3–36 (Halatchev et al., 2004; Abbott et al., 2006). There is expression of Y2R in CNS neurons, as well as vagal afferent neurons, and both central effects of peripheral PPY3–36 (Halatchev and Cone, 2005) on food intake, and effects mediated by vagal afferent neurons (Abbott et al., 2005; Koda et al., 2005) have been reported. The demonstration that vagal neurons projecting to the ileum and colon seem not to exhibit CCK-regulated expression of Y2R is interesting because the peripheral fibers of these neurons are presumably exposed to locally high concentrations of PYY3–36 acting via a paracrine mechanism. In contrast, the population of neurons exhibiting CCK-modulated Y2R expression that serve the stomach is presumably exposed to PYY3–36 acting as a hormone. Many studies of the effect of PYY on food intake have used protocols that involved prior fasting (Challis et al., 2003; Halatchev et al., 2004; Abbott et al., 2006) and these presumably select for responses of the colonic-projecting neurons. It is now clear that in the future it will be necessary to consider how different populations of Y2R-expressing neurons contribute to the satiety effects of endogenous PYY3–36.
The half-time for downregulation of Y2R after nutrient withdrawal was ∼12 h; after administration of CCK8s to fasted rats there was increased expression that peaked after 2–3 h. These relatively rapid changes in expression occur over periods similar to those of the postprandial release of CCK (Dockray, 2006). Several lines of evidence provide support for the idea that CCK-control of Y2R expression is functionally relevant. The action of PYY in delaying gastric emptying (Pappas et al., 1986) has recently been shown to be inhibited by the CCK1R antagonist devazepide (Whited et al., 2007). Moreover, the present data show that in mice null for CCK1R there is depressed fos stimulation in the brainstem (which is known to be a consequence of vagal afferent neuron excitation) in response to PYY3–36. Similarly, we found pretreatment of fasted rats with CCK to induce Y2R enhanced the capacity of PYY3–36 to stimulate nodose neurons as indicated by nuclear phosphoCREB abundance. It is worth noting that mice null for CCK1R are not obese whereas an obese phenotype is characteristic of the OLETF rat in which the CCK1R locus is deleted (Moran and Bi, 2006). In both cases, however, there is increased meal size. Previous work directed at understanding the different phenotypes in the two models has focused on hypothalamic mechanisms (Bi et al., 2004, 2007). The present data suggest it will now be fruitful to focus on mechanisms determining the neurochemical phenotype of vagal afferent neurons in seeking to understand in more detail the link between CCK signaling and obesity.
It is recognized that the action of CCK on vagal afferent nerve discharge is potentiated by gastric distension and leptin (Schwartz et al., 1991, 1993; Barrachina et al., 1997; Wang et al., 1997; Peters et al., 2004, 2005), and inhibited by ghrelin and orexin A (Burdyga et al., 2003; Date et al., 2005). The present data suggest that ghrelin inhibits the action of CCK on Y2R expression. Recent studies have shown that in the case of stimulation of CART expression by CCK in vagal afferent neurons, the inhibitory action of ghrelin is attributable to exclusion from the nucleus of the transcription factor CREB (de Lartigue et al., 2007). Although further work is required to define in detail the range of modulatory inputs acting on Y2R expression, it appears nevertheless that CCK occupies a central position in determining whether a variety of other gut-derived signals are able to influence food intake via vagal pathways. We suggest, therefore, that CCK can be considered to act as a gatekeeper that controls the capacity of vagal afferent pathways to respond to other signals influencing food intake and autonomic function.
Footnotes
This work was supported by the Biotechnology and Biological Sciences Research Council, the Wellcome Trust, the Medical Research Council, and National Institutes of Health Grant DK 41004 (H.E.R.). We are grateful to the late John Walsh for the supply of antibody to CCK-1 receptors and to Rotta Pharmaceuticals for a gift of lorglumide.
References
- Abbott CR, Monteiro M, Small CJ, Sajedi A, Smith KL, Parkinson JR, Ghatei MA, Bloom SR. The inhibitory effects of peripheral administration of peptide YY(3–36) and glucagon-like peptide-1 on food intake are attenuated by ablation of the vagal-brainstem-hypothalamic pathway. Brain Res. 2005;1044:127–131. doi: 10.1016/j.brainres.2005.03.011. [DOI] [PubMed] [Google Scholar]
- Abbott CR, Small CJ, Sajedi A, Smith KL, Parkinson JR, Broadhead LL, Ghatei MA, Bloom SR. The importance of acclimatisation and habituation to experimental conditions when investigating the anorectic effects of gastrointestinal hormones in the rat. Int J Obes (Lond) 2006;30:288–292. doi: 10.1038/sj.ijo.0803137. [DOI] [PubMed] [Google Scholar]
- Ali-Rachedi A, Varndell IM, Adrian TE, Gapp DA, Van Noorden S, Bloom SR, Polak JM. Peptide YY (PYY) immunoreactivity is co-stored with glucagon-related immunoreactants in endocrine cells of the gut and pancreas. Histochemistry. 1984;80:487–491. doi: 10.1007/BF00495439. [DOI] [PubMed] [Google Scholar]
- Barrachina MD, Martínez V, Wang L, Wei JY, Taché Y. Synergistic interaction between leptin and cholecystokinin to reduce short-term food intake in lean mice. Proc Natl Acad Sci U S A. 1997;94:10455–10460. doi: 10.1073/pnas.94.19.10455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batterham RL, Cowley MA, Small CJ, Herzog H, Cohen MA, Dakin CL, Wren AM, Brynes AE, Low MJ, Ghatei MA, Cone RD, Bloom SR. Gut hormone PYY(3–36) physiologically inhibits food intake. Nature. 2002;418:650–654. doi: 10.1038/nature00887. [DOI] [PubMed] [Google Scholar]
- Batterham RL, Cohen MA, Ellis SM, Le Roux CW, Withers DJ, Frost GS, Ghatei MA, Bloom SR. Inhibition of food intake in obese subjects by peptide YY3–36. N Engl J Med. 2003;349:941–948. doi: 10.1056/NEJMoa030204. [DOI] [PubMed] [Google Scholar]
- Bi S, Scott KA, Kopin AS, Moran TH. Differential roles for cholecystokinin a receptors in energy balance in rats and mice. Endocrinology. 2004;145:3873–3880. doi: 10.1210/en.2004-0284. [DOI] [PubMed] [Google Scholar]
- Bi S, Chen J, Behles RR, Hyun J, Kopin AS, Moran TH. Differential body weight and feeding responses to high-fat diets in rats and mice lacking cholecystokinin 1 receptors. Am J Physiol Regul Integr Comp Physiol. 2007;293:R55–R63. doi: 10.1152/ajpregu.00002.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böttcher G, Sjolund K, Ekblad E, Hakanson R, Schwartz TW, Sundler F. Coexistence of peptide YY and glicentin immunoreactivity in endocrine cells of the gut. Regul Pept. 1984;8:261–266. doi: 10.1016/0167-0115(84)90034-x. [DOI] [PubMed] [Google Scholar]
- Burdyga G, Spiller D, Morris R, Lal S, Thompson DG, Saeed S, Dimaline R, Varro A, Dockray GJ. Expression of the leptin receptor in rat and human nodose ganglion neurones. Neuroscience. 2002;109:339–347. doi: 10.1016/s0306-4522(01)00474-2. [DOI] [PubMed] [Google Scholar]
- Burdyga G, Lal S, Spiller D, Jiang W, Thompson D, Attwood S, Saeed S, Grundy D, Varro A, Dimaline R, Dockray GJ. Localization of orexin-1 receptors to vagal afferent neurons in the rat and humans. Gastroenterology. 2003;124:129–139. doi: 10.1053/gast.2003.50020. [DOI] [PubMed] [Google Scholar]
- Burdyga G, Lal S, Varro A, Dimaline R, Thompson DG, Dockray GJ. Expression of cannabinoid CB1 receptors by vagal afferent neurons is inhibited by cholecystokinin. J Neurosci. 2004;24:2708–2715. doi: 10.1523/JNEUROSCI.5404-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdyga G, Varro A, Dimaline R, Thompson DG, Dockray GJ. Ghrelin receptors in rat and human nodose ganglia: putative role in regulating CB-1 and MCH receptor abundance. Am J Physiol Gastrointest Liver Physiol. 2006a;290:G1289–G1297. doi: 10.1152/ajpgi.00543.2005. [DOI] [PubMed] [Google Scholar]
- Burdyga G, Varro A, Dimaline R, Thompson DG, Dockray GJ. Feeding-dependent depression of melanin-concentrating hormone and melanin-concentrating hormone receptor-1 expression in vagal afferent neurones. Neuroscience. 2006b;137:1405–1415. doi: 10.1016/j.neuroscience.2005.10.057. [DOI] [PubMed] [Google Scholar]
- Challis BG, Pinnock SB, Coll AP, Carter RN, Dickson SL, O'Rahilly S. Acute effects of PYY3–36 on food intake and hypothalamic neuropeptide expression in the mouse. Biochem Biophys Res Commun. 2003;311:915–919. doi: 10.1016/j.bbrc.2003.10.089. [DOI] [PubMed] [Google Scholar]
- Cummings DE, Overduin J. Gastrointestinal regulation of food intake. J Clin Invest. 2007;117:13–23. doi: 10.1172/JCI30227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Date Y, Murakami N, Toshinai K, Matsukura S, Niijima A, Matsuo H, Kangawa K, Nakazato M. The role of the gastric afferent vagal nerve in ghrelin-induced feeding and growth hormone secretion in rats. Gastroenterology. 2002;123:1120–1128. doi: 10.1053/gast.2002.35954. [DOI] [PubMed] [Google Scholar]
- Date Y, Toshinai K, Koda S, Miyazato M, Shimbara T, Tsuruta T, Niijima A, Kangawa K, Nakazato M. Peripheral interaction of ghrelin with cholecystokinin on feeding regulation. Endocrinology. 2005;146:3518–3525. doi: 10.1210/en.2004-1240. [DOI] [PubMed] [Google Scholar]
- de Lartigue G, Dimaline R, Varro A, Dockray GJ. Cocaine- and amphetamine-regulated transcript: stimulation of expression in rat vagal afferent neurons by cholecystokinin and suppression by ghrelin. J Neurosci. 2007;27:2876–2882. doi: 10.1523/JNEUROSCI.5508-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dockray GJ. The brain-gut axis. In: Yamada T, Alpers DH, Kaplowitz L, Laine L, Owyang C, Powell DW, editors. Textbook of gastroenterology. Philadelphia: Lippincott Williams and Wilkins; 2003. pp. 77–92. [Google Scholar]
- Dockray GJ. Gastrointestinal hormones: gastrin, cholecystokinin, somatostatin and ghrelin. In: Johnson LR, editor. Physiology of the gastrointestinal tract. Amsterdam: Elsevier Academic; 2006. pp. 91–120. [Google Scholar]
- Fu J, Gaetani S, Oveisi F, Lo Verme J, Serrano A, Rodríguez de Fonseca F, Rosengarth A, Luecke H, Di Giacomo B, Tarzia G, Piomelli D. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-alpha. Nature. 2003;425:90–93. doi: 10.1038/nature01921. [DOI] [PubMed] [Google Scholar]
- Gibbs J, Young RC, Smith GP. Cholecystokinin elicits satiety in rats with open gastric fistulas. Nature. 1973;245:323–325. doi: 10.1038/245323a0. [DOI] [PubMed] [Google Scholar]
- Gómez R, Navarro M, Ferrer B, Trigo JM, Bilbao A, Del Arco I, Cippitelli A, Nava F, Piomelli D, Rodríguez de Fonseca F. A peripheral mechanism for CB1 cannabinoid receptor-dependent modulation of feeding. J Neurosci. 2002;22:9612–9617. doi: 10.1523/JNEUROSCI.22-21-09612.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez MF, Deutsch JA. Vagotomy abolishes cues of satiety produced by gastric distension. Science. 1981;212:1283–1284. doi: 10.1126/science.7233218. [DOI] [PubMed] [Google Scholar]
- Halatchev IG, Cone RD. Peripheral administration of PYY(3–36) produces conditioned taste aversion in mice. Cell Metab. 2005;1:159–168. doi: 10.1016/j.cmet.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Halatchev IG, Ellacott KL, Fan W, Cone RD. Peptide YY3–36 inhibits food intake in mice through a melanocortin-4 receptor-independent mechanism. Endocrinology. 2004;145:2585–2590. doi: 10.1210/en.2003-1754. [DOI] [PubMed] [Google Scholar]
- Hanani M. Satellite glial cells in sensory ganglia: from form to function. Brain Res Brain Res Rev. 2005;48:457–476. doi: 10.1016/j.brainresrev.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Koda S, Date Y, Murakami N, Shimbara T, Hanada T, Toshinai K, Niijima A, Furuya M, Inomata N, Osuye K, Nakazato M. The role of the vagal nerve in peripheral PYY3–36-induced feeding reduction in rats. Endocrinology. 2005;146:2369–2375. doi: 10.1210/en.2004-1266. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Miller KE, Richards BA, Kriebel RM. Glutamine-, glutamine synthetase-, glutamate dehydrogenase- and pyruvate carboxylase-immunoreactivities in the rat dorsal root ganglion and peripheral nerve. Brain Res. 2002;945:202–211. doi: 10.1016/s0006-8993(02)02802-0. [DOI] [PubMed] [Google Scholar]
- Moran TH, Bi S. Hyperphagia and obesity in OLETF rats lacking CCK-1 receptors. Philos Trans R Soc Lond B Biol Sci. 2006;361:1211–1218. doi: 10.1098/rstb.2006.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran TH, Kinzig KP. Gastrointestinal satiety signals II. Cholecystokinin. Am J Physiol Gastrointest Liver Physiol. 2004;286:G183–G188. doi: 10.1152/ajpgi.00434.2003. [DOI] [PubMed] [Google Scholar]
- Murphy KG, Bloom SR. Gut hormones and the regulation of energy homeostasis. Nature. 2006;444:854–859. doi: 10.1038/nature05484. [DOI] [PubMed] [Google Scholar]
- Pappas TN, Debas HT, Chang AM, Taylor IL. Peptide YY release by fatty acids is sufficient to inhibit gastric emptying in dogs. Gastroenterology. 1986;91:1386–1389. doi: 10.1016/0016-5085(86)90191-5. [DOI] [PubMed] [Google Scholar]
- Peters JH, Karpiel AB, Ritter RC, Simasko SM. Cooperative activation of cultured vagal afferent neurons by leptin and cholecystokinin. Endocrinology. 2004;145:3652–3657. doi: 10.1210/en.2004-0221. [DOI] [PubMed] [Google Scholar]
- Peters JH, McKay BM, Simasko SM, Ritter RC. Leptin-induced satiation mediated by abdominal vagal afferents. Am J Physiol Regul Integr Comp Physiol. 2005;288:R879–R884. doi: 10.1152/ajpregu.00716.2004. [DOI] [PubMed] [Google Scholar]
- Rodríguez de Fonseca F, Navarro M, Gómez R, Escuredo L, Nava F, Fu J, Murillo-Rodríguez E, Giuffrida A, LoVerme J, Gaetani S, Kathuria S, Gall C, Piomelli D. An anorexic lipid mediator regulated by feeding. Nature. 2001;414:209–212. doi: 10.1038/35102582. [DOI] [PubMed] [Google Scholar]
- Schwartz GJ, McHugh PR, Moran TH. Integration of vagal afferent responses to gastric loads and cholecystokinin in rats. Am J Physiol. 1991;261:R64–R69. doi: 10.1152/ajpregu.1991.261.1.R64. [DOI] [PubMed] [Google Scholar]
- Schwartz GJ, McHugh PR, Moran TH. Gastric loads and cholecystokinin synergistically stimulate rat gastric vagal afferents. Am J Physiol. 1993;265:R872–R876. doi: 10.1152/ajpregu.1993.265.4.R872. [DOI] [PubMed] [Google Scholar]
- Smith GP, Jerome C, Cushin BJ, Eterno R, Simansky KJ. Abdominal vagotomy blocks the satiety effect of cholecystokinin in the rat. Science. 1981;213:1036–1037. doi: 10.1126/science.7268408. [DOI] [PubMed] [Google Scholar]
- Wang YH, Taché Y, Sheibel AB, Go VL, Wei JY. Two types of leptin-responsive gastric vagal afferent terminals: an in vitro single-unit study in rats. Am J Physiol. 1997;273:R833–R837. doi: 10.1152/ajpregu.1997.273.2.R833. [DOI] [PubMed] [Google Scholar]
- Whited KL, Tso P, Raybould HE. Involvement of apolipoprotein A-IV and cholecystokinin1 receptors in exogenous peptide YY3 36-induced stimulation of intestinal feedback. Endocrinology. 2007;148:4695–4703. doi: 10.1210/en.2006-1665. [DOI] [PubMed] [Google Scholar]
- Zhang X, Shi T, Holmberg K, Landry M, Huang W, Xiao H, Ju G, Hökfelt T. Expression and regulation of the neuropeptide Y Y2 receptor in sensory and autonomic ganglia. Proc Natl Acad Sci U S A. 1997;94:729–734. doi: 10.1073/pnas.94.2.729. [DOI] [PMC free article] [PubMed] [Google Scholar]