

Summary
The framework for modern studies of speciation was established as part of the Neo-Darwinian synthesis of the early twentieth century. Here we evaluate this framework in the light of recent empirical and theoretical studies. Evidence from experimental studies of selection, quantitative genetic studies of species’ differences, and the molecular evolution of ‘isolation’ genes, all agree that directional selection is the primary cause of speciation, as initially proposed by Darwin. Likewise, as suggested by Dobzhansky and Mayr, gene flow does hold species together, but probably more by facilitating the spread of beneficial mutants and associated hitchhiking events than by homogenizing neutral loci. Reproductive barriers are important as well in that they preserve adaptations, but as has been stressed by botanists for close to a century, they rarely protect the entire genome from gene flow in recently diverged species. Contrary to early views, it is now clear that speciation can occur in the presence of gene flow. However, recent theory does support the long-held view that population structure and small population size may increase speciation rates, but only under special conditions and not because of the increased efficacy of drift as suggested by earlier authors. Rather, low levels of migration among small populations facilitates the rapid accumulation of beneficial mutations that indirectly cause hybrid incompatibilities.
Keywords: gene flow, introgression, population size, population subdivision, reproductive isolation, selection, selective sweep, speciation
Introduction
The study of speciation was formally initiated by the publication of Darwin’s famous monograph, On the Origin of Species by Natural Selection, which posited that species differences are caused by natural selection (Darwin, 1859). However, Darwin did not fully explain how species differed from locally adapted populations or how conspecific populations were able to evolve as a unit. These issues were clarified by the biological species concept (Dobzhansky, 1937; Mayr, 1942), which emphasized the importance of gene flow for holding species together and reproductive barriers for keeping them apart. Mayr (1942, 1954) also argued forcefully that geographic isolation of populations was critical to species formation and that speciation was stimulated by small population size. However, note that Mayr’s (1954) paper on founder effect speciation was preceded by Lewis (1953), who argued that speciation in Clarkia was facilitated by founder events and the fixation of chromosomal rearrangements in peripheral populations.
Although these ideas provided a framework for speciation studies over the past 60 yr, they have not been universally accepted. Indeed, there is inherent conflict between Darwin’s focus on selection, which operates most efficiently in large populations, and Mayr’s emphasis on the creative role of small population size. Likewise, some authors have argued that there is too little gene flow to hold species together (Ehrlich & Raven, 1969), and others have shown that species boundaries often are semipermeable to introgression (Rieseberg & Wendel, 1993; Arnold, 1997). It also has become increasingly apparent that geographic isolation is not required for speciation (McNeilly & Antonovics, 1968), and that the effects of population size and structure on speciation rates are complex (Rice & Hostert, 1993; Orr & Orr, 1996; Church & Taylor, 2002).
In this paper, we briefly review evidence from recent theoretical and empirical studies that bear on these problems. We focus mostly on aspects that remain controversial or where we personally have made contributions toward their resolution. When possible, we have tried to emphasize botanical contributions.
Selection and speciation
Many kinds of evidence have been used to assess the role of selection in the evolution of species differences and reproductive isolation. These include: analyses of patterns of selection in contemporary hybrid zones or experimental hybrids; the application of Orr’s (1998) quantitative trait locus (QTL) sign test to species differences, which allows the history of selection on a given trait to be determined; analyses of the molecular evolution of genes that contribute to reproductive isolation; experimental population genetic studies; and comparative analyses. Because of space constraints, we focus on the first three approaches. Note, however, that detailed reviews of results from experimental population genetic studies and comparative analyses have been published elsewhere (Rice & Hostert, 1993; Barraclough & Nee, 2001; Schilthuizen, 2001). Also, it should be evident that different methods may apply best to different questions or traits. For example, neither phenotypic selection analysis nor the QTL sign test are useful for studying the role of selection in the origin of postzygotic barriers such as hybrid sterility or inviability.
Contemporary patterns of selection
The simplest and most direct way to test for a role for selection in speciation is to ask whether species differences are currently being maintained by selection. If so, this would imply that the differences arose by selection as well. Such inferences are not robust, however, because of temporal and spatial variation in the strength and direction of selection (Schemske & Bierzychudek, 2001).
Most selection studies at the species level employ experimental or natural hybrids that have been placed in the habitat of one or both parental species. A recent survey of the literature yielded 47 such studies, of which 31 involved plants and 16 involved animals (Lexer et al., 2003). Although evidence of selection for or against the hybrids was detected in all but one of the studies, only eight experiments reported the strength of selection on individual phenotypic traits. Fortunately, all eight were from plants. The eight studies generated 149 estimates of directional selection gradients, of which 56% were significant, with a mean selection gradient of 0.12 ± 0.01. By contrast, only 25% of trait polymorphisms within populations were found to be under significant selection in a review of selection in natural populations (Kingsolver et al., 2001). These results imply that the majority of trait differences between species (at least for the taxa studied here) are indeed maintained by selection and that selection is quite strong. Note, however, that results from selection studies are sensitive to sample size (Kingsolver et al., 2001). With larger samples, the proportion of traits under significant selection is likely to increase, although estimates of the strength of selection may decrease.
Historical patterns of selection: the QTL sign test
As mentioned earlier, the fact that species differences are currently maintained by selection doesn’t necessarily prove that the differences were caused by selection. Thus, methods that can estimate the history of selection on a trait or gene are needed to fully understand the speciation process. One such method is based on the direction of effects of QTLs that contribute to phenotypic differences (Orr, 1998). If a trait has a continuous history of directional selection, then QTL effects should be in the same direction within a line. By contrast, if a trait has diverged under neutrality, QTLs with opposing effects should be common. Orr (1998) has formalized these predictions with the creation of a QTL sign test that compares observed numbers of ± QTLs in a given line with those expected under neutral conditions.
A recent application of Orr’s (1998) sign test to the QTL literature in both wild and domesticated plants (Rieseberg et al., 2002) revealed that, as predicted by theory, domestication traits had a much lower proportion of opposing QTLs (0.06 ± 0.03, least square mean ± standard error) than did other kinds of traits that were segregating in the same wild domesticated mapping populations (0.18 ± 0.03). Analyses of crosses involving taxonomically diverse wild populations also revealed widespread selection; all categories of traits had QTL proportions that deviated significantly from neutral expectations (Rieseberg et al., 2002). Thus, directional selection appears to be a major contributor to phenotypic differentiation in essentially all kinds of traits and organisms.
There are caveats associated with this conclusion, however. In particular, QTL magnitudes were not included in this analysis, and Orr (1998) has pointed out that major QTLs fixed during an initial bout of natural selection may overshoot the phenotypic optimum, and minor QTLs in the opposite direction may evolve later to bring the trait back to the phenotypic optimum.
Although the signature of directional selection is widespread, there was variation among trait categories, suggesting that certain kinds of traits were exposed to stronger and more consistent selection than other trait categories. For the purposes of this review, the most interesting result was that intraspecific traits had almost twice the proportion of opposing QTLs as interspecific traits (0.25 vs 0.14; P = 0.02), suggesting that species differences are more likely to be a product of divergent selection than intraspecific differences.
We have extended this analysis further in the present paper by asking whether traits that contribute directly to assortative mating (e.g. flowering time; flower color, pollination syndrome, etc.) are more or less strongly selected than ordinary species differences. This was accomplished by adding the category ‘assortative mating trait vs other’ to the ANOVA model described by Rieseberg et al. (2002). As expected, the proportion of opposing QTLs (0.12 ± 0.15, least square mean ± tandard error) for assortative mating traits was significantly lower than that predicted under a model of neutral divergence. Indeed, this is the lowest proportion of opposing QTLs observed for any trait category in comparisons involving wild populations. Nonetheless, QTL proportions did not differ significantly between assortative mating traits and ordinary species differences. Thus, although most assortative mating traits appear to have diverged as a consequence of divergent natural selection, there is no evidence that they have been more strongly or consistently selected than other kinds of species differences.
Historical patterns of selection: molecular evolution of isolation genes
Another method for assessing historical patterns of selection during the speciation process is to examine the molecular evolution of genes that contribute to reproductive isolation. This approach tests for selection by asking whether there is an excess of nonsynonymous substitutions (dN) relative to neutral expectations (i.e. dN/dS > 1), significant variation in substitution rates among codons within a gene (Nielsen & Yang, 1998), or reduced variability in the gene and flanking sequences caused by a recent selective sweep (Wang et al., 1999).
Only a handful of genes have been identified that are known to contribute to reproductive isolation, and most of these are from animals. These ‘isolation genes’ can be divided into two groups, based on whether they contribute to pre or postzygotic isolation. Those involved in prezygotic isolation include: period, a clock gene which modifies song rhythm in Drosophila (Wheeler et al., 1991) and timing of mating behavior in both Drosophila (Tauber et al., 2003) and melon fly (Miyatake et al., 2002); bindin, a gamete recognition protein in sea urchins that mediates species-specific attachment to an egg-surface receptor during fertilization (Metz & Palumbi, 1996); lysin, a sperm protein that species-specifically creates a hole in the egg envelope during abalone fertilization (Lee et al., 1995); VERL, the egg vitelline envelope receptor for lysin (Galindo et al., 2003); desat2, which is responsible for a cuticular hydrocarbon pheromone polymorphism that contributes to behavioral isolation between geographic races of Drosophila melanogaster (Takahashi et al., 2001); and S-RNase-based self-incompatibility (SI) that causes unilateral interspecific incompatibility in Nicotiana (Hancock et al., 2003). Other plant genes that are likely to contribute to reproductive isolation, but for which definitive proof is lacking, include rapidly evolving pollen coat proteins that may mediate species-specific pollen recognition (Mayfield et al., 2001), flowering time genes such as FRIGIDA (Johanson et al., 2000) and Hd1 (Yano et al., 2000), and flower color genes such as the anthocyanin2 (an2) locus that is the main determinant of floral color differences between Petunia integrifolia and P. axillaris (Quattrocchio et al., 1999).
The six genes proven to contribute to prezygotic isolation all show the signature of positive selection. For bindin, lysin, VERL, and SI, the evidence of selection is in the form of an excess of nonsynonymous substitutions across all or part of the studied gene. The rapid rate of protein evolution exhibited by these genes is likely a result of continuous coevolution of the gamete recognition system, driven by sexual selection or sexual conflict for the animal reproductive proteins (bindin, lysin, and VERL), and frequency dependent selection for SI. By contrast, period and desat2 are not unusually variable and evidence for positive selection comes from evidence of selective sweeps (i.e. reduced variation) associated with the gene or selected mutation (Ford & Aquadro, 1996; Wang & Hey, 1996; Takahashi et al., 2001). Note that in all cases, selection is not for reproductive isolation. Rather, isolation appears to have evolved as a byproduct of positive selection for some other function.
We are aware of four genes that have been shown to contribute to postzygotic isolation, including Odysseus (Ods), which induces hybrid male sterility in crosses between Drosophila mauritiana and D. simulans (Ting et al., 1998); Hybrid male rescue (Hmr), which causes lethality and female sterility in hybrids among D. melanogaster and its sibling species (Barbash et al., 2003); Nup96, a nuclear pore protein that causes hybrid lethality in crosses between D. melanogaster and D. simulans (Presgraves et al., 2003); and xmrk-2, a growth factor receptor that is overexpressed in hybrids between Xiphophorus (platyfish) species, causing tumor development (Froschauer et al., 2001). Three of these (Ods, Hmr and Nup96) evolve rapidly and display an excess of replacement substitutions, indicative of positive Darwinian selection. Unlike the gamete recognition proteins described above, little is known about the selective forces causing the high rates of protein evolution at these loci, although Ting et al. (1998) speculate that sexual selection may drive rapid sequence evolution in Ods because it is expressed in Drosophila testis. As far as we are aware, no evidence for positive selection has been reported for xmrk-2, and variation (small chromosomal rearrangements) in regulatory rather than coding sequence appears to be responsible for the hybrid inviability associated with this gene (Froschauer et al., 2001).
In sum, 9 of the 10 genes currently known to contribute to reproductive isolation appear to have diverged as a consequence of Darwinian natural selection, a result that agrees nicely with the prominent role for selection implied by the experimental selection studies and the QTL sign test. There is a major caveat, however. The identified ‘isolation’ genes are biased toward animals and toward reproductive proteins, which may diverge more rapidly than other kinds of characters because of sexual selection or sexual conflict (Wyckoff et al., 2000). Although reproductive proteins in plants also appear to evolve rapidly (Mayfield et al., 2001), other kinds of traits that contribute to reproductive isolation (e.g. differences in morphological, life history, and physiological adaptations to different habitats) seem less likely to be involved in the coevolutionary ‘chases’ that lead to rapid protein divergence. These ‘ordinary’ species differences probably are caused by natural selection as well, but it may be that the selective sweeps will sometimes be too ancient to detect them.
A prominent role for natural selection does not necessarily mean that stochastic forces are unimportant. One means by which genetic drift might contribute, for example, is through the fixation of neutral or underdominant chromosomal rearrangements (White, 1978). Unless strongly underdominant (and thus unlikely to be fixed in the first place), chromosomal rearrangements will act mostly to reduce effective gene flow rates in regions close to chromosomal breakpoints (Rieseberg, 2001). As a consequence, selected differences are predicted to accumulate most quickly on rearranged chromosomes (Navarro & Barton, 2003a), a prediction which has been confirmed for divergence among species of Drosophila (Noor et al., 2001) and between humans and chimpanzees (Navarro & Barton, 2003b). Rearrangements may be particularly important during the early stages of speciation when effective gene flow rates may otherwise be high enough to prevent differentiation at most loci.
Of course, it may be that many rearrangements become established as a consequence of selection favoring a particular combination of alleles (Charlesworth & Charlesworth, 1980) or position effects, rather than drift. Unfortunately, little is known about the evolutionary forces underlying the establishment of chromosomal rearrangements, although it does appear that transposable elements often contribute to their origin (Gray, 2000).
Stochastic forces may also play a significant role in the divergence of duplicate genes following polyploidy or segmental duplication, and models have been developed for the evolution of hybrid incompatibilities as a result of reciprocal silencing of duplicated genes (Werth & Windham, 1991) or divergent resolution of regulatory sequences (Lynch & Force, 2000). Although both phenomena are well-documented in the literature, neither has yet been shown to cause hybrid incompatibilities. Plant genomes tend to be more redundant than those of animals, however, and may therefore provide a more fertile substrate for evolutionary changes that involve duplicate genes.
Gene flow and speciation
Although battered and scarred from constant criticism over the past 70 yr, the biological species concept remains the most widely employed species concept in evolutionary biology, and its focus on gene flow and reproductive barriers continues to provide the motivation for most empirical and theoretical studies of speciation (Schilthuizen, 2001). It is not possible to list the many criticisms of the biological species concept in the space provided here, let alone respond to them. A frequent objection by phylogeneticists is that the ability to interbreed is symplesiomorphic. However, this criticism only makes sense if the phenotypic clusters recognized by naturalists represent monophyletic lineages, and this is demonstrably not the case. Not only are species often of recurrent origin (Levin, 2001), but newly derived sister species follow a common time course of change subsequent to speciation of polyphyly → paraphyly → monophyly, with 4 N generations required to achieve reciprocal monophyly for organellar genes and even longer times for nuclear genes (Avise, 2000). More serious criticisms relate to the observation that there appears to be too little gene flow among populations of some species to hold them together (Ehrlich & Raven, 1969) and too much gene flow among other species to keep them apart. Among Eukaryotes, these problems are perhaps most pronounced in plants, resulting in the widespread rejection of the biological species concept (and species reality) by botanists (Mishler, 1999). Below, we show that the problem of too little gene flow is ameliorated by recognition that even very low levels of gene flow are sufficient for the spread of advantageous alleles and that the problem of too much flow is mitigated by recognition that whole genome isolation is not required for species divergence.
Too little gene flow
Students of speciation have primarily focused on the conservative role of gene flow, in which high levels of gene flow (Nem > 4, where Nem is the effective number of migrants per generation) serve to homogenize populations at neutral loci (Hartl & Clark, 1997). It was recognized more than three decades ago, however, that levels of gene flow in many species are not nearly this high (Ehrlich & Raven, 1969). Indeed, for many plant and animal species, estimates of Nem fall well below one (Fig. 1), the level of gene flow required to prevent divergence at neutral loci (Wright, 1931).
Fig. 1.
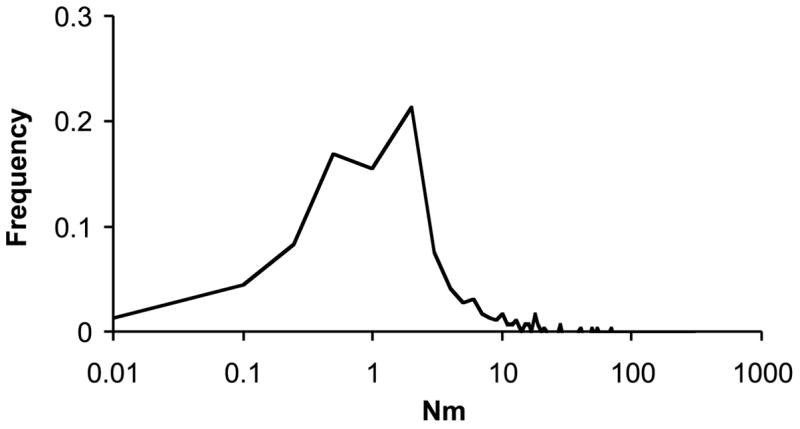
Frequency histogram of migration rate (Nem) for total nuclear data from 290 plant studies. Data are derived from 130 studies reporting Fst, Gst Nem, and Fst values published in Molecular Ecology from May 1992 until December 2002, and Gst values from 160 allozyme studies (Ward et al., 1992). For studies not reporting Nem values, Nem was calculated from Fst, Gst, or analogous statistics for nuclear markers as Nem = 1 − Fst/4 * Fst (Wright, 1931). Data are displayed on a logarithmic scale, binned for Nem = 0.01, 0.1, 0.25, 0.5, 1, and binned into 1-unit intervals thereafter.
Consideration of the creative role of gene flow as a mechanism for the spread of advantageous alleles offers a potential solution to this problem (Rieseberg & Burke, 2001). Only very low levels of gene flow are required for the spread of advantageous alleles and fixation times are much less than for their neutral counterparts (Slatkin, 1976). Thus, it is conceivable that species’ populations could remain connected through repeated selective sweeps of favored mutations and associated hitchhiking events or ‘genetic draft’ (Gillespie, 2001).
Is this scenario likely? In low gene flow species, population subdivision greatly reduces the rate of allelic spread, particularly for weakly selected or neutral alleles (Slatkin, 1976; Whitlock, 2003). Thus, one concern is whether a favored allele will spread to fixation before it goes extinct. A second concern is whether selective sweeps are frequent enough to produce cohesion. If they are rare or restricted to a handful of loci, the level of connectedness might not be sufficient to account for the apparent cohesiveness observed for many species in nature.
We are aware of only two studies that have modeled the effects of population subdivision on the probability of and/or time to fixation of beneficial alleles (Slatkin, 1976; Whitlock, 2003). Slatkin (1976) employed a one-dimensional stepping stone model to estimate the time it would take for a favored allele to spread across the range of a species that comprised 20 demes. He showed that the rate of spread of a beneficial mutation depended strongly on the strength of selection and the long-distance migration rate. We have extended Slatkin’s model by computer simulation to include a second dimension and a leptokurtic dispersal function (Fig. 2). Our results confirm that moderately selected alleles will spread rapidly across a species range despite low levels of migration, although the time to fixation is somewhat reduced relative to that of Slatkin’s calculations, presumably as a result of the use of the leptokurtic dispersal function.
Fig. 2.
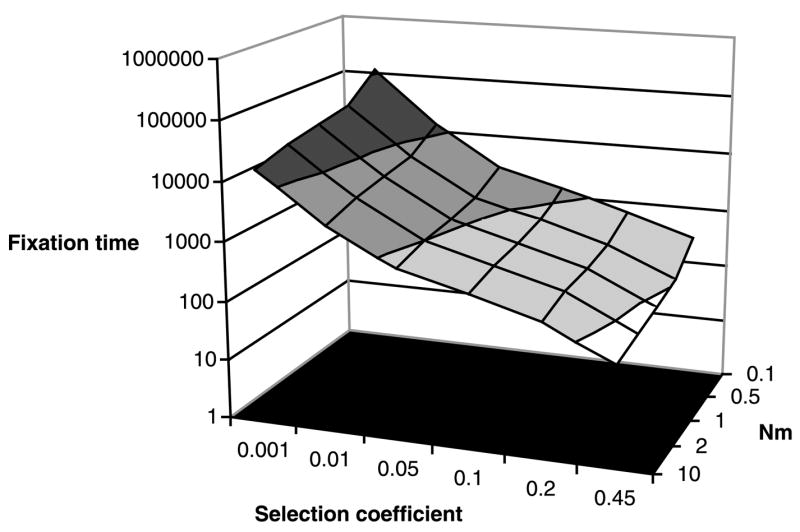
Fixation time (in generations) by selection coefficient and Nem for a subdivided population of 106 diploid individuals with nonoverlapping generations. Subpopulations, each consisting of 2500 individuals, are arranged in a 20 × 20 grid. Migration occurs among subpopulations according to a leptokurtic dispersal function, with a mean dispersal distance of one subpopulation.
Whitlock (2003) provides estimates of both the time to and probability of fixation for beneficial alleles in island, stepping stone, and extinction-recolonization models. Unfortunately, only a subset of parameter space is illustrated by his figures. Nonetheless, his results appear to be fully compatible with Slatkin’s work. As expected, the strength of selection has a huge impact on both the probability of and time to fixation. Weakly selected alleles spread at a glacial pace and almost always go extinct, whereas strongly beneficial alleles spread much faster and have a much higher probability of fixation.
In sum, the effects of population subdivision are to greatly increase fixation times relative to panmictic populations with a slight positive effect on fixation probabilities. More importantly, however, population subdivision magnifies the differences in time to fixation for strongly and weakly selected alleles. Thus, weakly selected alleles that spread to fixation in panmictic populations are less likely to do so in subdivided populations, possibly biasing fixed interspecific differences toward major genes.
So are selection coefficients for mutations underlying species differences large enough to validate a model in which species are held together by the spread of beneficial alleles? Rieseberg & Burke (2001) generated crude estimates of what selection coefficients might be by calculating the average percentage variance explained by QTLs that contribute to species differences in plants, multiplying by selection differentials for phenotypic traits in wild plant populations and then halving to account for diploidy. However, their calculations were based on a very small number of studies. We have updated this analysis with the inclusion of data from Kingsolver et al. (2001), who report 993 linear selection gradients and 753 selection differentials for phenotypic traits in 62 studies of natural populations; Lexer et al. (2003), who provide 149 estimates of directional selection gradients and 27 selection differentials from 8 studies of experimental hybrids in natural populations; and C. L. Morjan & L. H. Rieseberg (unpublished), in which 133 selection gradients and 96 selection differentials from experimentally manipulated or disturbed populations were compiled from 26 studies. The expanded analysis corroborates the general conclusions of Rieseberg & Burke (2001). Selection coefficients associated with moderate to large QTLs are likely to be large enough to ensure rapid spread and fixation, whereas very minor QTLs (PVE < 0.01) may be trapped in local populations and are less likely to contribute to fixed differences between species (Fig. 3). Of course, if most phenotypic differentiation were to occur in a local population, followed by range expansion, then even small QTLs might contribute to species differences in such a scenario.
Fig. 3.
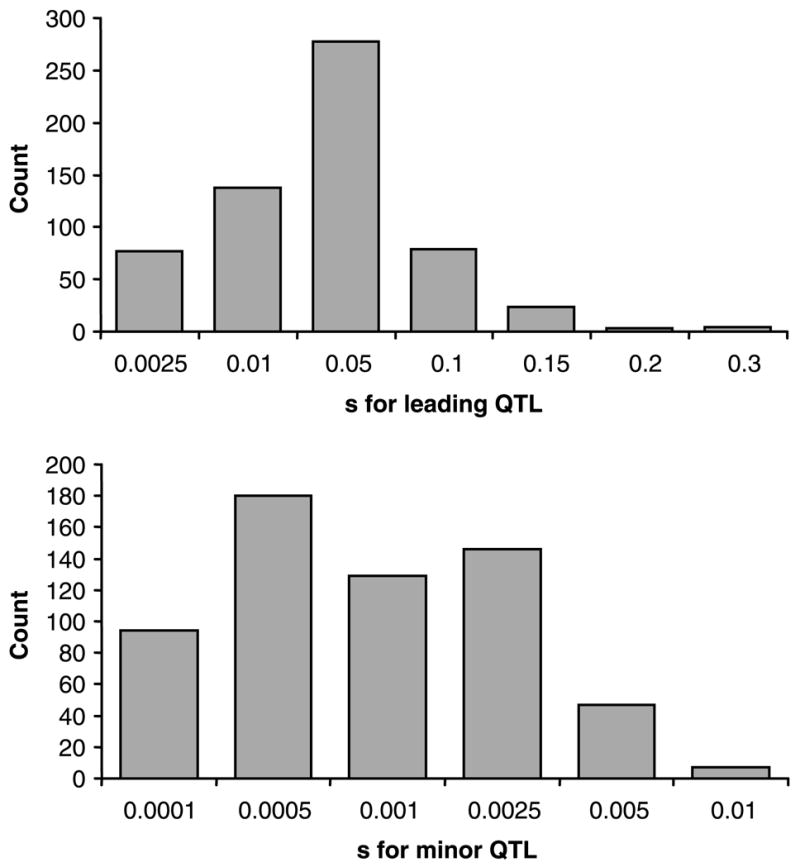
Distributions for the estimated strength of selection (s) for leading quantitative trait locis (QTLs) (top panel) and minor QTLs (bottom panel) underlying phenotypic traits in plants. s was calculated by multiplying the average percent variance explained (PVE) for leading QTLs for 50 traits (31.1%) by 604 selection gradients for phenotypic traits from a literature review and halving for diploidy. The bottom panel was calculated by multiplying the 604 selection gradients by expected PVE values for minor QTLs (1%) and halving for diploidy.
Another issue alluded to earlier concerns the frequency of selective sweeps. If they are frequent and involve genes scattered across the genome, it is plausible that, in concert with hitchhiking effects, the whole genome could remain connected. On the other hand, if cohesion is limited to a small number of major genes, then we could have a situation in which a species is evolving collectively at a handful of genes while simultaneously diverging at other loci through drift or local adaptation (Rieseberg & Burke, 2001).
Comparative sequencing studies are beginning to shed light on this question. For example, Smith & Eyre-Walker (2002) have recently shown that 45% of all amino-acid substitutions between Drosophila species appear to have been fixed by selection. We do not yet have comparable data from plants, although Barrier et al. (2003) suggest that c. 5% of genes differentiating Arabidopsis thaliana and A. lyrata have diverged as a consequence of positive selection. Comparative EST sequencing of wild sunflowers (S. Church et al., unpublished) suggest a similar percentage of sunflower genes are under selection. These values, if reliable, are consistent with the maintenance of genome-wide species cohesion through repeated selective sweeps and genetic hitchhiking. Of course this doesn’t rule out the possibility that some loci are simultaneously diverging through local selection. Indeed, fairly weak selection can overcome the effects of migration at a given locus, so this is expected.
Too much gene flow
The fact that many otherwise ‘good’ species continue to exchange genetic material long after they have embarked on independent and irreversible evolutionary trajectories has been known and accepted by botanists for close to a century (Ostenfeld, 1927; Grant, 1981). Thus, there seems little reason to exhaustively review the literature on the permeability of species barriers other than to comment that the evidence for introgression is becoming increasingly widespread and robust (Grant, 1981; Rieseberg & Wendel, 1993; Arnold, 1997; Wendel & Doyle, 1998) and now extends beyond plants to animal groups such as Drosophila in which introgression was once thought to be rare or absent (Wang et al., 1997).
Coincident with more rigorous evidence of introgression has been the development of theory, which indicates that divergence in the presence of gene flow is possible as long as the strength of selection at a given locus exceeds the migration rate (Maynard Smith, 1966). This is not an unlikely condition, resulting in widespread acceptance of models for sympatric and parapatric speciation. The same basic theory applies to zones of secondary contact (i.e. hybrid zones) between species that have diverged in allopatry, but with one main difference. In hybrid zones there often are genome–wide associations or linkage disequilibrium between the traits and genes characteristic of a given taxon (Barton & Hewitt, 1985). That is, multilocus parental genotypes are over-represented relative to recombinant genotypes. Although some linkage disequilibrium among selected traits is likely in zones of primary contact, genome wide disequilibrium is unlikely until the very latest stages of sympatric and parapatric speciation.
The primary consequence of linkage disequilibrium is to retard the movement of genes across larger chromosomal segments, or across the entire genome if many genes contribute to reproductive isolation. However, in most hybrid zones, the number of genes contributing to reproductive isolation is not sufficient to create genome-wide reproductive isolation (Barton & Hewitt, 1985; Rieseberg et al., 1999). As a consequence neutral or universally favorable alleles move easily across the species barrier, unless they are tightly linked to negatively selected alleles. This contrasts with the restricted movement of alleles that contribute to reproductive isolation and genes or markers tightly linked to them, whose movement across the zone will decline in proportion to the selection:recombination ratio (Barton, 1979). That is, strongly selected alleles will have a greater impact on linked loci than will weakly selected alleles, and tightly linked loci will be more affected than unlinked or loosely linked loci. For example, Ting et al. (2000) has shown that the Drosophila sterility locus, Odysseus, protects just 2 kb of the genome from introgression. These findings have led to a renewed interest in the role of chromosomal rearrangements as reproductive barriers, because rearrangements reduce effective recombination rates, thereby extending the effects of linked isolation genes (Rieseberg, 2001).
In sum, both empirical and theoretical data indicate that reproductive barriers are likely to be semipermeable to gene flow in recently diverged species or taxa in which hybrid incompatibilities evolve slowly. Because the term ‘reproductive isolation’ implies whole-genome isolation (Wu, 2001), evidence of widespread introgression in both plants and animals does appear to conflict with a strict interpretation of the biological species concept.
How should evolutionary biologists respond to this conflict? One possibility would be to apply the biological concept very strictly, with absolute reproductive isolation required for species status. This approach is logically attractive, but stumbles in its application to real organisms, primarily because biological barriers to successful reproduction evolve at hugely different rates in different organismal groups (Rieseberg, 2001), and such barriers are not a requirement for divergent evolution. Strict implementation would lead to such absurdities as according similar taxonomic rank to cryptic species of Drosophila, genera of plants and birds, and phyla of bacteria.
Another possibility would be to employ alternative species concepts that do not rely on reproductive isolation. A variety of evolutionary forces are assumed to contribute to species cohesion (e.g. common descent, stabilizing and parallel selection, genetic constraints, gene flow/reproductive isolation), and species concepts have been devised that emphasize one or all these mechanisms (Templeton, 1989). However, gene flow/reproductive isolation is unique among these cohesive mechanisms in that it acts primarily at the population and species level. Selection, by contrast, acts most strongly on individuals, and common descent and genetic constraint contribute equally to cohesion at all taxonomic levels. Thus, there is considerable justification for focusing on the only distinguishing property of species (gene flow/reproductive isolation), while at the same time recognizing its imperfections as a species diagnostic.
If complete reproductive isolation is not a requirement for species status, then how much is required? One answer to this question relates to the key difference between locally adapted populations/geographic races and species: Divergence among locally adapted populations and even geographic races is almost always ephemeral, whereas evolutionary divergence among species is preserved by reproductive isolation (Futuyma, 1989). Thus, the amount of reproductive isolation required for species status is simply that necessary to preserve the evolutionary changes associated with population systems following changes in the location of suitable habitat (e.g. following climatic shifts) or in the proximity of other population systems or species (i.e. changes in levels of gene flow).
This may not seem helpful, but because reproductive barriers themselves are sensitive to shifts in selection or gene flow, the permanence of species may be equated with the permanence of the reproductive barriers that protect them. Premating barriers may be erased by shifts in patterns of selection, and two-locus hybrid incompatibilities are sensitive to changes in gene flow. By contrast, chromosomal barriers and complex Dobzhansky–Muller (Dobzhansky, 1937; Muller, 1942) incompatibilities (epistatic incompatibilities that appear in hybrids as a result of interaction between alleles that did not lower fitness when they were individually substituted within the diverging lineages) tend to be robust to either kind of challenge and attest to the likely longevity or permanence of the population system associated with them. Thus, while speciation often is initiated by the development of premating barriers, the evolution of complex postzygotic barriers is required to set these differences in ‘concrete’. In this context, note that plant species seemingly isolated by prezygotic barriers alone often show considerable transmission ratio distortion in hybrids (Whitkus, 1998; Fishman et al., 2001), implying that postzygotic barriers are present as well.
Population structure and speciation
As alluded to in the introduction, early models of speciation emphasized the importance of geographic isolation, because gene flow was thought to prevent geographically proximal populations from diverging through drift and selection (Mayr, 1942). However, studies over the past 40 yr have shown time and again that speciation may occur despite gene flow between populations (McNeilly & Antonovics, 1968; Caisse & Antonovics, 1978; Church & Taylor, 2002). These studies have inspired the development of spatially explicit models to explore more complex and realistic predictions of the effects of selection, drift, and gene flow on the likelihood, rate, and tempo of speciation.
One of the earliest studies to incorporate spatially explicit models of population structure and gene flow showed that speciation could occur between connected populations, regardless of gene flow (Caisse & Antonovics, 1978). The authors used a computer simulation of 10 linearly arranged populations that shared genes through stepping stone or exponential migration patterns to investigate the evolution of reproductive isolation along a cline. Their model confirmed the earlier study of Maynard Smith (1966), demonstrating that divergence will occur when the strength of selection at a given locus (s) is greater than the migration rate (m). Furthermore, if the gene under selection also caused assortative mating (or was linked to a gene causing assortative mating) the development of reproductive isolation was straightforward (Caisse & Antonovics, 1978). Reproductive isolation also occurred in the absence of linkage or pleiotropy, but selection against hybrids had to be strong. These early models, and others like them (Karlin & McGregor, 1972), have shown repeatedly that reproductive barriers may arise and be maintained between hybridizing populations provided that selection against hybrids is sufficiently strong relative to migration (Dieckmann & Doebeli, 1999; Kondrashov & Kondrashov, 1999; Doebeli & Dieckmann, 2003).
More recently, speciation models have investigated the role of spatial structure and gene flow on the evolution of Dobzhansky–Muller type incompatibilities, which may be the most common cause of postzygotic reproductive isolation. In these models, all populations are initially identical (genotype AAbb). Mutations occur independently across populations, with one population becoming AABB and a second population becoming aabb. There is no reduced fitness within either population as a result of these mutations; however, the hybrid genotype, AaBb, has drastically reduced fitness or is inviable. These populations are now reproductively isolated as a consequence of random mutation and genetic drift or natural selection acting to fix alternate mutations. However, reproductive isolation between the populations was not the target of selection. Once the barrier is established though, selection against hybrids maintains the newly diverged species. Empirical studies have confirmed that hybrid sterility or inviability can result from such multi–locus interactions (Orr, 1997). These incompatibilities can accumulate rapidly in geographically isolated populations, in a ‘snowball effect’, which is the square of the time since the separation of the populations (Orr, 1995). A similar effect occurs in spatially structured populations, even with low levels of migration, where the number of pairwise incompatibilities between populations can eventually increase quadratically (Kondrashov, 2003).
If the accumulation of Dobzhansky–Muller incompatibilities represents a plausible scenario for speciation, what are the consequences of population subdivision on the patterns and timing of speciation events? Recent theoretical models have focused on this issue, with conflicting results. Early students of speciation postulated that speciation was most likely to occur when species were subdivided into small populations (Wright, 1931; Lewis, 1953; Mayr, 1954) because of the increased efficiency of genetic drift. However, Orr & Orr (1996) showed that, if selection acts to fix alternate alleles in different populations, the time to speciation increases as population size decreases. This results from the scattering of new mutations across many small populations in a subdivided species. As a consequence, more mutations must arise in a subdivided species before any two populations accumulate a sufficient number of mutations to be incompatible.
However, more recent models show that the relationship between population size and speciation rate breaks down when migration is introduced (Gavrilets et al., 2000b; Church & Taylor, 2002; Gavrilets & Gibson, 2002). One such model considers a population subdivided into demes of equal size and arranged spatially in a circular stepping stone model (Church & Taylor, 2002). Initially, all demes were identical genetically and diverged through the accumulation of beneficial mutations. Low levels of migration between neighboring demes was allowed, with all compatible, positively selected genes entering and going to fixation in a deme. The results of this model agreed with Orr & Orr (1996) in that small deme size inhibited speciation in a strictly allopatric model. However, this result was reversed when low levels of migration were introduced; speciation rates accelerate with nonzero rates of migration (Fig. 4). Simply put, migration increased the rate at which a given deme accumulated mutations, with subsets of demes within the population evolving in concert.
Fig. 4.
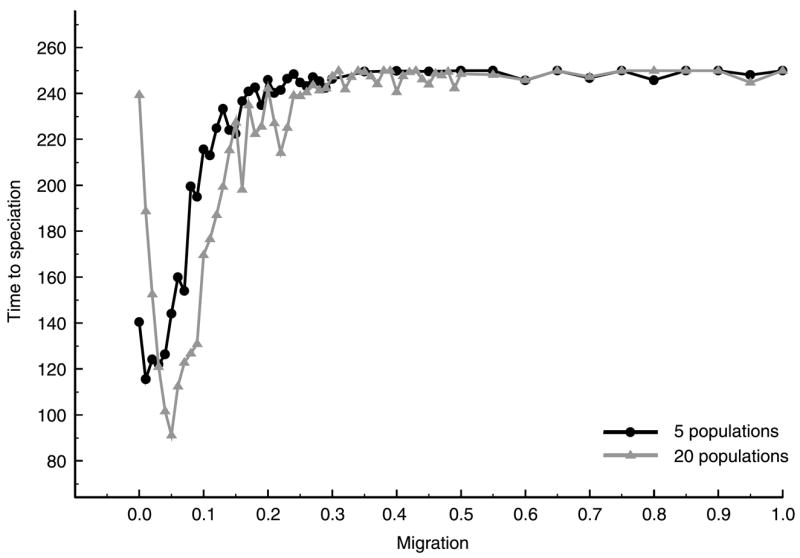
The effect of population fragmentation and migration on the time to speciation in a stepping stone model with 5 or 20 subpopulations. Time to speciation was the number of mutational events until a pair of populations accumulated an arbitrarily defined number of incompatibilities required for speciation. Migration was the average number of migration events occurring across the entire population. The simulation was stopped when speciation was not completed before mutations occurred at all 250 loci therefore speciation times of 250 signify that the time to speciation was at least 250 mutational events.
Other models of parapatric speciation have elucidated geographic patterns of speciation in spatially structured populations (Gavrilets et al., 2000b). It is thought that, in nature, most migration occurs between neighboring populations, resulting in geographic differentiation of populations (Endler, 1977). The extreme cases of this are found in so-called ‘ring species’, which exchange migrants only with their neighbors, allowing high levels of genetic differentiation among geographically isolated populations (Wake, 1997). In these situations, new species may form as a result of extensive geographic differentiation, despite low levels of gene flow. Theoretical studies suggest that speciation in such circumstances can occur as rapidly as a few hundred to a few thousand generations in spite of the exchange of several migrants per generation between neighboring populations (Gavrilets et al., 2000b).
Building on parapatric models, recent studies have also incorporated metapopulation dynamics, the phenomenon of local extinction and colonization of populations that is common among natural populations (Hanski & Gilpin, 1997). In these models, divergence among populations is erased by extinction and recolonization rather than migration (Gavrilets et al., 2000a). It follows that increased rates of population turnover (extinction and colonization) decreases the rate of speciation (Gavrilets et al., 2000a). More generally, the model develops an excellent framework for modeling speciation in a metapopulation, but more realistic spatial structures and migration models need to be incorporated to make this model more biologically relevant.
Analyses of Dobzhansky–Muller incompatibilities in clinal populations generate similar results (Bengtsson, 1985; Barton & Bengtsson, 1986; Gavrilets, 1997). Although persistent migration can break down barriers to gene exchange between populations (Barton & Bengtsson, 1986), speciation can result from the accumulation of Dobzhansky–Muller incompatibilities if migration rates are low and selection against intermediates is strong (Gavrilets, 1997). Moreover, genotypes of intermediate populations in a cline tend to be quite different from those that would be formed by hybridization between the more geographically distant populations at either end of the cline (Gavrilets, 1997).
Empirical studies of spatially structured populations or species often reveal patterns of ecological and genetic divergence that are strongly correlated with geographic structure. These include parallel adaptation to the same range of habitats (Turesson, 1922, 1925, 1930; Clausen et al., 1940, 1948), as well as parallel geographic patterns in the distribution of alleles among populations and species (Soltis et al., 1997; Avise, 2000; Barraclough & Vogler, 2000; Church et al., 2003). The theoretical models discussed above indicate that this geographic variation could in fact contribute to speciation. They also provide a sounder basis for interpreting geographic patterns seen in nature. The primary caveat associated with many of these studies relates to their focus on Dobzhansky–Muller interactions, which, for reasons described earlier, seem unlikely to initiate speciation. More complex models that include various kinds of isolating mechanisms and empirically validated assumptions regarding mutation rate, migration rate, and population size and structure will be needed before confident conclusions can be made regarding the role of geographic structure in speciation.
Acknowledgments
The authors’ research in these areas is funded by the U.S. National Institutes of Health (GM059065 to L.H. Rieseberg), and the U.S. National Science Foundation (Postdoctoral Research Grants in Bioinformatics to S.A. Church and C.L. Morjan). We thank Troy Wood for constructive comments on the manuscript.
References
- Arnold ML. Natural hybridization and evolution. New York, USA: Oxford University Press; 1997. [Google Scholar]
- Avise JC. Phylogeography. Cambridge, MA, USA: Harvard University Press; 2000. [Google Scholar]
- Barbash DA, Siino DF, Tarone AM, Roote J. A rapidly evolving MYB-related protein causes species isolation in Drosophila. Proceedings of the National Academy of Sciences, USA. 2003;100:5302–5307. doi: 10.1073/pnas.0836927100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barraclough TG, Nee S. Phylogenetics and speciation. Trends in Ecology and Evolution. 2001;16:391–399. doi: 10.1016/s0169-5347(01)02161-9. [DOI] [PubMed] [Google Scholar]
- Barraclough TG, Vogler AP. Detecting the geographic pattern of speciation from species-level phylogenies. American Naturalist. 2000;155:419–434. doi: 10.1086/303332. [DOI] [PubMed] [Google Scholar]
- Barrier M, Bustamante CD, Yu JY, Purugganan MD. Selection on rapidly evolving proteins in the Arabidopsis genome. Genetics. 2003;163:723–733. doi: 10.1093/genetics/163.2.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton NH. Gene flow past a cline. Heredity. 1979;43:333–339. [Google Scholar]
- Barton NH, Bengtsson BO. The barrier to genetic exchange between hybridizing populations. Heredity. 1986;56:357–376. doi: 10.1038/hdy.1986.135. [DOI] [PubMed] [Google Scholar]
- Barton NH, Hewitt GM. Analysis of hybrid zones. Annual Review of Ecology and Systematics. 1985;16:113–148. [Google Scholar]
- Bengtsson BO. The flow of genes through a genetic barrier. In: Greenwood JJ, Harvey PH, Slatkin M, editors. Evolution essays in honor of John Maynard Smith. Cambridge, UK: Cambridge University Press; 1985. pp. 31–42. [Google Scholar]
- Caisse M, Antonovics J. Evolution in closely adjacent plant populations. IX. Evolution of reproductive isolation in clinal populations. Heredity. 1978;40:371–384. [Google Scholar]
- Charlesworth D, Charlesworth B. Sex differences in fitness and selection for centric fusions between sex chromosomes and autosomes. Genetical Research. 1980;35:205–214. doi: 10.1017/s0016672300014051. [DOI] [PubMed] [Google Scholar]
- Church SA, Kraus J, Mitchell JC, Church DR, Taylor DR. Evidence for multiple Pleistocene refugia in the postglacial expansion of the eastern tiger salamander, Ambystoma tigrinum tigrinum. Evolution. 2003;57:372–383. doi: 10.1554/0014-3820(2003)057[0372:EFMPRI]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Church SA, Taylor DR. The evolution of reproductive isolation in spatially structured populations. Evolution. 2002;56:1859–1862. doi: 10.1111/j.0014-3820.2002.tb00200.x. [DOI] [PubMed] [Google Scholar]
- Clausen JD, Keck DD, Hiesey WM. Experimental studies of the nature of species. I. Effect of varied environments on western North American plants. Stanford, CA, USA: Carnegie Institute of Washington Publishers; 1940. p. 520. [Google Scholar]
- Clausen JD, Keck DD, Hiesey WM. Experimental studies of the nature of species. III. Environmental responses of climatic races of Achillea. Stanford, CA, USA: Carnegie Institute of Washington Publishers; 1948. p. 581. [Google Scholar]
- Darwin D. On the origin of species. London, UK: John Murray; 1859. [Google Scholar]
- Dieckmann U, Doebeli M. On the origin of species by sympatric speciation. Nature. 1999;400:354–357. doi: 10.1038/22521. [DOI] [PubMed] [Google Scholar]
- Dobzhansky TH. Genetics and the origin of species. New York, USA: Columbia University Press; 1937. [Google Scholar]
- Doebeli M, Dieckmann U. Speciation along environmental gradients. Nature. 2003;421:259–264. doi: 10.1038/nature01274. [DOI] [PubMed] [Google Scholar]
- Ehrlich PR, Raven PH. Differentiation of populations. Science. 1969;165:1228–1232. doi: 10.1126/science.165.3899.1228. [DOI] [PubMed] [Google Scholar]
- Endler JA. Geographic variation, speciation, and clines. Princeton, NJ, USA: Princeton University Press; 1977. [PubMed] [Google Scholar]
- Fishman L, Kelly AJ, Morgan E, Willis JH. A genetic map in the Mimulus guttatus species complex reveals transmission ratio distortion due to heterospecific interactions. Genetics. 2001;159:1701–1716. doi: 10.1093/genetics/159.4.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford MJ, Aquadro CF. Selection on X-linked genes during speciation in the Drosophila athabasca complex. Genetics. 1996;144:689–703. doi: 10.1093/genetics/144.2.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froschauer A, Korting C, Bernhardt W, Nanda I, Schmid M, Schartl M, Volff JN. Genomic plasticity and melanoma formation in the fish Xiphophorus. Marine Biotechnology. 2001;3:S72–S80. doi: 10.1007/s10126-001-0049-7. [DOI] [PubMed] [Google Scholar]
- Futuyma DJ. Macroevolutionary consequences of speciation: Inferences from phytophagous insects. In: Otte D, Endler JA, editors. Speciation and its Consequences. Sunderland, MA USA: Sinauer Associates; 1989. pp. 557–578. [Google Scholar]
- Galindo E, Vacquier VD, Swanson WJ. Positive selection in the egg receptor for abalone sperm lysine. Proceedings of the National Academy of Sciences, USA. 2003;100:4639–4643. doi: 10.1073/pnas.0830022100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrilets S. Hybrid zones with Dobzhansky-type epistatic selection. Evolution. 1997;51:1027–1035. doi: 10.1111/j.1558-5646.1997.tb03949.x. [DOI] [PubMed] [Google Scholar]
- Gavrilets S, Acton R, Gravner J. Dynamics of speciation and diversification in a metapopulation. Evolution. 2000a;54:1493–1501. doi: 10.1111/j.0014-3820.2000.tb00695.x. [DOI] [PubMed] [Google Scholar]
- Gavrilets S, Gibson N. Fixation probabilities in a spatially heterogeous environment. Population Ecology. 2002;44:51–58. [Google Scholar]
- Gavrilets S, Li H, Vose MD. Patterns of parapatric speciation. Evolution. 2000b;54:1126–1134. doi: 10.1111/j.0014-3820.2000.tb00548.x. [DOI] [PubMed] [Google Scholar]
- Gillespie JH. Is the population size of a species relevant to its evolution? Evolution. 2001;55:2161–2169. doi: 10.1111/j.0014-3820.2001.tb00732.x. [DOI] [PubMed] [Google Scholar]
- Grant V. Plant speciation. New York, USA: Columbia University Press; 1981. [Google Scholar]
- Gray YHM. It takes two transposons to tango – transposable-mediated chromosomal rearrangements. Trends in Genetics. 2000;16:461–468. doi: 10.1016/s0168-9525(00)02104-1. [DOI] [PubMed] [Google Scholar]
- Hancock CN, Kondo K, Beecher B, McClure B. The S-locus and unilateral incompatibility. Philosophical Transactions of The Royal Society of London Series B-Biological Sciences. 2003;358:1133–1140. doi: 10.1098/rstb.2003.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanski I, Gilpin ME, editors. Metapopulation biology: ecology, genetics, and evolution. San Diego, CA, USA: Academic Press; 1997. [Google Scholar]
- Hartl DL, Clark AG. Principles of population genetics. underland, MA, USA: Sinauer Associates; 1997. [Google Scholar]
- Johanson U, West J, Lister C, Michaels S, Amasino R, Dean C. Molecular analysis of FRIGIDA, a major determinant of natural variation in Arabidopsis flowering time. Science. 2000;290:344–347. doi: 10.1126/science.290.5490.344. [DOI] [PubMed] [Google Scholar]
- Karlin S, McGregor J. Application of method of small parameters in multiniche population genetics models. Theoretical Population Biology. 1972;3:180–209. doi: 10.1016/0040-5809(72)90026-3. [DOI] [PubMed] [Google Scholar]
- Kingsolver JG, Hoekstra HE, Hoekstra JM, Berrigan D, Vignieri SN, Hill CE, Hoang A, Gibert P, Beerli P. The strength of phenotypic selection in natural populations. American Naturalist. 2001;157:245–261. doi: 10.1086/319193. [DOI] [PubMed] [Google Scholar]
- Kondrashov AS. Accumulation of Dobzhansky–Muller incompatibilities within a spatially structured population. Evolution. 2003;57:151–153. doi: 10.1111/j.0014-3820.2003.tb00223.x. [DOI] [PubMed] [Google Scholar]
- Kondrashov AS, Kondrashov FA. Interactions among quantitative traits in the course of sympatric speciation. Nature. 1999;400:351–354. doi: 10.1038/22514. [DOI] [PubMed] [Google Scholar]
- Lee YH, Ota T, Vacquier VD. Positive selection is a general phenomenon in the evolution of abalone sperm lysin. Molecular Biology and Evolution. 1995;12:231–238. doi: 10.1093/oxfordjournals.molbev.a040200. [DOI] [PubMed] [Google Scholar]
- Levin DA. The recurrent origin of plant races and species. Systematic Botany. 2001;26:197–204. [Google Scholar]
- Lewis H. The mechanism of evolution in the genus Clarkia. Evolution. 1953;7:1–20. [Google Scholar]
- Lexer C, Randell RA, Rieseberg LH. Experimental hybridization as a tool for studying selection in the wild. Ecology. 2003;84:1688–1699. [Google Scholar]
- Lynch M, Force AG. The origin of interspecific genomic incompatibility via gene duplication. American Naturalist. 2000;156:590–605. doi: 10.1086/316992. [DOI] [PubMed] [Google Scholar]
- Mayfield JA, Fiebig A, Johnstone SE, Preuss D. Gene families from the Arabidopsis thaliana pollen coat proteome. Science. 2001;292:2482–2485. doi: 10.1126/science.1060972. [DOI] [PubMed] [Google Scholar]
- Maynard Smith J. Sympatric speciation. American Naturalist. 1966;100:637–650. [Google Scholar]
- Mayr E. Systematics and the origin of species. New York, USA: Columbia University Press; 1942. [Google Scholar]
- Mayr E. Change of genetic environment and evolution. In: Huxley J, editor. Evolution as a process. London, UK: Allen & Unwin; 1954. pp. 156–180. [Google Scholar]
- McNeilly T, Antonovics J. Evolution in closely adjacent plant populations. IV. Barriers to gene flow. Heredity. 1968;23:205–218. [Google Scholar]
- Metz EC, Palumbi SR. Positive selection and sequence rearrangements generate extensive polymorphism in the gamete recognition protein bindin. Molecular Biology and Evolution. 1996;13:397–406. doi: 10.1093/oxfordjournals.molbev.a025598. [DOI] [PubMed] [Google Scholar]
- Mishler BD. Getting rid of species. In: Wilson RA, editor. Species: new interdisciplinary essays. Cambridge, USA: Harvard University Press; 1999. pp. 307–316. [Google Scholar]
- Miyatake T, Matsumoto A, Matsuyama T, Ueda HR, Toyosato T, Tanimura T. The period gene and allochronic reproductive isolation in Bactrocera cucurbitae. Proceedings of The Royal Society of London Series B-Biological Sciences. 2002;269:2467–2472. doi: 10.1098/rspb.2002.2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HJ. Isolating mechanisms, evolution, and temperature. Biological Symposia. 1942;6:71–125. [Google Scholar]
- Navarro A, Barton NH. Accumulating postzygotic isolation genes in parapatry: a new twist on chromosomal speciation. Evolution. 2003a;57:447–459. doi: 10.1111/j.0014-3820.2003.tb01537.x. [DOI] [PubMed] [Google Scholar]
- Navarro A, Barton NH. Chromosomal speciation and molecular divergence – Accelerated evolution in rearranged chromosomes. Science. 2003b;300:321–324. doi: 10.1126/science.1080600. [DOI] [PubMed] [Google Scholar]
- Nielsen R, Yang ZH. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics. 1998;148:929–936. doi: 10.1093/genetics/148.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noor MAF, Grams KL, Bertucci LA, Reiland J. Chromosomal inversions and the reproductive isolation of species. Proceedings of the National Academy of Sciences, USA. 2001;98:12084–12088. doi: 10.1073/pnas.221274498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HA. The population genetics of speciation: the evolution of hybrid incompatibilities. Genetics. 1995;139:1805–1813. doi: 10.1093/genetics/139.4.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HA. Haldane’s rule. Annual Review of Ecology and Systematics. 1997;28:195–218. doi: 10.1146/annurev.ecolsys.28.1.85. [DOI] [PubMed] [Google Scholar]
- Orr HA. Testing natural selection vs. genetic drift in phenotypic evolution using quantitative trait locus data. Genetics. 1998;149:2099–2104. doi: 10.1093/genetics/149.4.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HA, Orr LH. Waiting for speciation – the effect of population subdivision on the time to speciation. Evolution. 1996;50:1742–1749. doi: 10.1111/j.1558-5646.1996.tb03561.x. [DOI] [PubMed] [Google Scholar]
- Ostenfeld CH. The present state of knowledge on hybrids between species of flowering plants. Journal of the Royal Horticultural Society. 1927;53:31–44. [Google Scholar]
- Presgraves DC, Balagopalan L, Abmayr SM, Orr HA. Adaptive evolution drives divergence of a hybrid inviability gene between two species of Drosophila. Nature. 2003;423:715–719. doi: 10.1038/nature01679. [DOI] [PubMed] [Google Scholar]
- Quattrocchio F, Wing J, van der Woude K, Souer E, de Vetten N, Mol J, Koes R. Molecular analysis of the anthocyanin2 gene of petunia and its role in the evolution of flower color. Plant Cell. 1999;11:1433–1444. doi: 10.1105/tpc.11.8.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice WR, Hostert EE. Laboratory experiments on speciation – what have we learned in 40 years. Evolution. 1993;47:1637–1653. doi: 10.1111/j.1558-5646.1993.tb01257.x. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH. Chromosomal rearrangements and speciation. Trends in Ecology and Evolution. 2001;16:351–358. doi: 10.1016/s0169-5347(01)02187-5. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH, Burke JM. The biological reality of species: gene flow, selection, and collective evolution. Taxon. 2001;50:47–67. [Google Scholar]
- Rieseberg LH, Wendel J. Introgression and its consequences in plants. In: Harrison R, editor. Hybrid zones and the evolutionary process. New York, USA: Oxford University Press; 1993. pp. 70–109. [Google Scholar]
- Rieseberg LH, Whitton J, Gardner K. Hybrid zones and the genetic architecture of a barrier to gene flow between two wild sunflower species. Genetics. 1999;152:713–727. doi: 10.1093/genetics/152.2.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieseberg LH, Widmer A, Arntz AM, Burke JM. Directional selection is the primary cause of phenotypic diversification. Proceedings of the National Academy of Sciences, USA. 2002;99:12242–12245. doi: 10.1073/pnas.192360899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schemske DW, Bierzychudek P. Evolution of flower color in the desert annual Linanthus parryae: Wright revisited. Evolution. 2001;55:1269–1282. doi: 10.1111/j.0014-3820.2001.tb00650.x. [DOI] [PubMed] [Google Scholar]
- Schilthuizen M. Frogs, flies, and dandelions. Oxford, UK: Oxford University Press; 2001. [Google Scholar]
- Slatkin M. The rate of spread of an advantageous allele in a subdivided population. In: Karlin S, Nevo E, editors. Population genetics and ecology. New York, USA: Academic Press, Inc; 1976. pp. 767–780. [Google Scholar]
- Smith NGC, Eyre-Walker A. Adaptive protein evolution in Drosophila. Nature. 2002;415:1022–1024. doi: 10.1038/4151022a. [DOI] [PubMed] [Google Scholar]
- Soltis DE, Gitzendanner MA, Strenge DD, Soltis PS. Chloroplast DNA intraspecific phylogeography of plants from the Pacific Northwest of North America. Plant Systematics and Evolution. 1997;206:353–373. [Google Scholar]
- Takahashi A, Tsaur SC, Coyne JA, Wu CI. The nucleotide changes governing cuticular hydrocarbon variation and their evolution in Drosophila melanogaster. Proceedings of the National Academy of Sciences, USA. 2001;98:3920–3925. doi: 10.1073/pnas.061465098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauber E, Roe H, Costa R, Hennessy JM, Kyriacou CP. Temporal mating isolation driven by a behavioral gene in Drosophila. Current Biology. 2003;13:140–145. doi: 10.1016/s0960-9822(03)00004-6. [DOI] [PubMed] [Google Scholar]
- Templeton AR. The meaning of species and speciation: a genetic perspective. In: Otte D, Endler JA, editors. Speciation and its consequences. Sunderland, MA USA: Sinauer Associates; 1989. pp. 3–27. [Google Scholar]
- Ting CT, Tsaur SC, Wu CI. The phylogeny of closely related species as revealed by the genealogy of a speciation gene Odysseus. Proceedings of the National Academy of Sciences, USA. 2000;97:5313–5316. doi: 10.1073/pnas.090541597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting CT, Tsaur SC, Wu ML, Wu CI. A rapidly evolving homeobox at the site of a hybrid sterility gene. Science. 1998;282:1501–1504. doi: 10.1126/science.282.5393.1501. [DOI] [PubMed] [Google Scholar]
- Turesson G. The genotypical response of the plant species to the habitat. Hereditas. 1922;3:211–350. [Google Scholar]
- Turesson G. The plant species in relation to habitat and climate. Hereditas. 1925;6:147–236. [Google Scholar]
- Turesson G. The selective effect of climate upon the plant species. Hereditas. 1930;14:99–152. [Google Scholar]
- Wake DB. Incipient species formation in salamanders of the Ensatina complex. Proceedings of the National Academy of Sciences, USA. 1997;94:7761–7767. doi: 10.1073/pnas.94.15.7761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RL, Hey J. The speciation history of Drosophila pseudoobscura and close relatives: Inferences from DNA sequence variation at the period locus. Genetics. 1996;144:1113–1126. doi: 10.1093/genetics/144.3.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RL, Stec A, Hey J, Lukens L, Doebley J. The limits of selection during maize domestication. Nature. 1999;398:236–239. doi: 10.1038/18435. [DOI] [PubMed] [Google Scholar]
- Wang RL, Wakeley J, Hey J. Gene flow and natural selection in the origin of Drosophila pseudoobscura and close relatives. Genetics. 1997;147:1091–1106. doi: 10.1093/genetics/147.3.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward RD, Skibinski DO, Woodwark M. Protein heterozygosity, protein structure, and taxonomic differentiation. Evolutionary Biology. 1992;26:73–159. [Google Scholar]
- Wendel JF, Doyle JJ. Phylogenetic incongruence: window into genome history and molecular evolution. In: Soltis DE, Soltis PS, Doyle JJ, editors. Molecular systematics of plants II: DNA sequencing. Boston, MA, USA: Kluwer Academic Publishers; 1998. pp. 256–296. [Google Scholar]
- Werth CR, Windham MD. A model for divergent, allopatric speciation of polyploid pteridophytes resulting from silencing of duplicate-gene expression. American Naturalist. 1991;137:515–526. [Google Scholar]
- Wheeler DA, Kyriacou CP, Greenacre ML, Yu Q, Rutila JE, Rosbash M, Hall JC. Molecular transfer of a species-specific behavior from Drosophila-simulans to. Drosophila-Melanogaster. Science. 1991;251:1082–1085. doi: 10.1126/science.1900131. [DOI] [PubMed] [Google Scholar]
- White MJD. Modes of speciation. San Francisco, CA, USA: W.H. Freeman Co; 1978. [Google Scholar]
- Whitkus R. Genetics of adaptive radiation in Hawaiian and Cook Islands species of Tetramolopium (Asteraceae). II. Genetic linkage map and its implications for interspecific breeding barriers. Genetics. 1998;150:1209–1216. doi: 10.1093/genetics/150.3.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock MC. Fixation probability and time in subdivided populations. Genetics. 2003;164:767–779. doi: 10.1093/genetics/164.2.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S. Evolution in Mendelian populations. Genetics. 1931;16:97–159. doi: 10.1093/genetics/16.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CI. The genic view of the process of speciation. Journal of Evolutionary Biology. 2001;14:851–865. [Google Scholar]
- Wyckoff GJ, Wang W, Wu CI. Rapid evolution of male reproductive genes in the descent of man. Nature. 2000;403:304–309. doi: 10.1038/35002070. [DOI] [PubMed] [Google Scholar]
- Yano M, Katayose Y, Ashikari M, Yamanouchi U, Monna L, Fuse T, Baba T, Yamamoto K, Umehara Y, Nagamura Y, Sasaki T. Hd1, a major photoperiod sensitivity quantitative trait locus in rice, is closely related to the Arabidopsis flowering time gene CONSTANS. Plant Cell. 2000;12:2473–2483. doi: 10.1105/tpc.12.12.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]