

Abstract
Phenylalkylamines that possess conformationally rigidified furanyl moieties in place of alkoxy arene ring substituents have been shown previously to possess the highest affinities and agonist functional potencies at the serotonin 5-HT2A receptor among this chemical class. Further, affinity declines when both furanyl rings are expanded to the larger dipyranyl ring system. The present paper reports the synthesis and pharmacological evaluation of a series of “hybrid” benzofuranyl–benzopyranyl phenylalkylamines to probe further the sizes of the binding pockets within the serotonin 5-HT2A agonist binding site. Thus, 4(a-b), 5(a-b), and 6 were prepared as homologs of the parent compound, 8-bromo-1-(2,3,6,7-tetrahydrobenzo[1,2-b:4,5-b’]difuran-4-yl)-2-aminopropane 2, and their affinity, functional potency, and intrinsic activity were assessed using cells stably expressing the rat 5-HT2A receptor. The behavioral pharmacology of these new analogs was also evaluated in the two-lever drug discrimination paradigm. Although all of the hybrid isomers had similar, nanomolar range receptor affinities, those with the smaller furanyl ring at the arene 2-position (4a-b) displayed a 4- to 15-fold greater functional potency than those with the larger pyranyl ring at that position (5a-b). When the furan ring of the more potent agonist 4b was aromatized to give 6, a receptor affinity similar to the parent difuranyl compound 2 was attained, along with a functional potency equivalent to 2, 4a, and 4b. In drug discrimination experiments using rats trained to discriminate LSD from saline, 4b was more than two times more potent than 5b, the latter having a potency similar to the classic hallucinogenic amphetamine 1 (DOB).
Keywords: phenethylamines, hallucinogen, 5-HT receptor agonist, structure-activity relationship
1. Introduction
Agonist activity at the serotonin 5-HT2A receptor appears to be essential for the observed psychopharmacology of psychedelic agents such as LSD, mescaline, psilocin, and 5-MeO-DMT, compounds that exert unique and dramatic effects on certain aspects of consciousness.1 A key aspect to understanding the unusual effects of psychedelics is study of the receptor-ligand interaction at the molecular level and how it modulates second messenger generation subsequent to receptor activation.
Recently, we have been particularly interested in experimental validation of our h5-HT2A receptor homology model.2 This model has proven to have predictive power in designing simple conformationally constrained phenethylamine agonists for this receptor,3,4 and we have been interested in extending it to provide a more comprehensive description of the key amino acid residues involved in receptor binding and activation.5 As a member of the type A G-protein coupled receptor (GPCR) family, we reasoned that understanding this receptor would provide general concepts that might extend across the GPCR field.
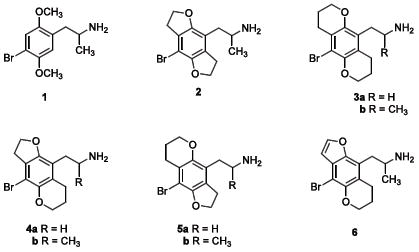
We previously discovered that constraining the 2- and 5-methoxy groups of prototypical substituted phenethylamine ligands such as 2,5-dimethoxy-4-bromoamphetamine (DOB) 1 into rigid heterocycles gave highly potent compounds such as 2.6 These findings clearly demonstrated that the methoxy groups adopted particular orientations upon binding, presumably to direct their unshared electron pairs toward polar complementary residues within the agonist binding site, most likely serine residues S239 and S159.2,5 We also had found that expanding the two heterocyclic rings to two, six-membered dihydropyran moieties, as in 3, led to an almost ten-fold decrease in activity, both in vitro and in vivo.7 The most reasonable explanation for these findings was that the receptor space around one, or both, of the oxygen atoms was sterically constricted.
This present study extends that earlier work, where now the activity of the two isomeric pyrano/furano “hybrid” analogues (4a-b versus 5a-b) is examined to determine which, if either, of the oxygen atoms lies within a more constrained steric area. Our virtual docking experiments place the 2-oxygen atom downward in the binding cavity, whereas the 5-oxygen atom is projected upward toward the extracellular face of the receptor.3,5,8 These observations suggest that the oxygen atom that is more deeply embedded in the receptor might be the one that has greater steric restriction around it. The present experiments tested that hypothesis, and compared compounds 4a and 4b with 5a and 5b, respectively, for affinity and functional potency at the 5-HT2A receptor. We also examined 4b and 5b for an LSD-like behavioral effect in rats. Because the lipophilic properties of these latter two compounds are virtually identical, any difference in activity would be related to pharmacodynamic, rather than pharmacokinetic effects. In addition, because it was known that aromatization of the furan rings in 2 greatly enhanced activity,9 we aromatized the furan ring of 4b to afford 6 and examined the effect on receptor affinity and potency.
2. Results
2.1. Chemistry
It was envisioned that all target molecules 4-6 could be prepared readily from their common tricyclic intermediate 12 (Scheme 1). This asymmetric pyrano/furano hybrid ring system required that the precursors of each oxygen heterocycle (the respective bromoethyl and bromopropyl ether moieties) be constructed independently, as outlined in the scheme. Inititally, para-methoxy phenol 7 was alkylated with dibromoethane to provide the diether 8. (Our early attempts to build the larger pyran ring first, by alkylating with 1,3-dibromopropane, proved to be considerably less efficient.) Following dibromination of 8, the aromatic methyl ether of 9 was selectively cleaved using B-bromo-9-BBN.10,11 This mild demethylating agent was found to be highly effective, whereas the more reactive traditional reagents, HBr, BBr3 and BCl3, were all found to cleave both of the ether moieties of 9. Due to the moderately unstable nature of phenol 10, after isolation it was immediately alkylated with 1,3-dibromopropane to afford the desired diether 11. Finally, the simultaneous tandem ring-closure of 11 to yield key tricyclic intermediate 12 was readily achieved using n-butyllithium.
Scheme 1.
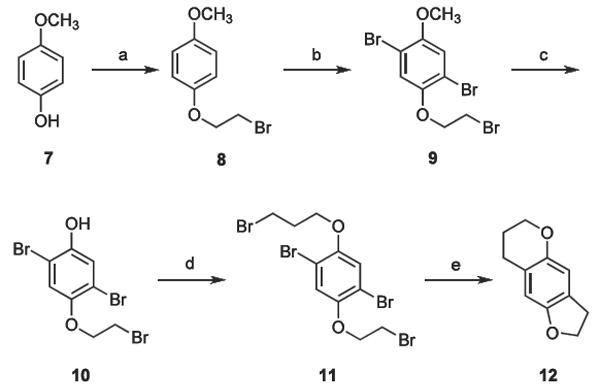
(a) 1,2-dibromoethane, K2CO3, acetone, reflux; (b) Br2, CH2Cl2, Fe; (c) B-bromo-9-BBN, CH2Cl2, reflux 14 days; (d) 1,3-dribromopropane, K2CO3, acetone, reflux; (e) n-butyllithium, THF, 0 °C.
With 12 in hand, formylation using dichloromethyl methyl ether in the presence of a tin (IV) catalyst produced regioisomers 13 and 14 (Scheme 2). Separation of these isomers by selective recrystallization and flash column chromatography12 gave 13 and 14 in a 5:1 ratio, respectively. Both aldehydes were then carried through the parallel sequence of reactions shown in Scheme 2. Thus, following condensation of the aldehydes with nitromethane or nitroethane, the resulting nitroalkenes 15 and 16 were fully reduced in the presence of LiAlH4 to afford primary amines 17 and 18. Target compounds 4a-b and 5a-b were then produced in good yield by brominating the remaining aromatic positions of 17 and 18, respectively.
Scheme 2.
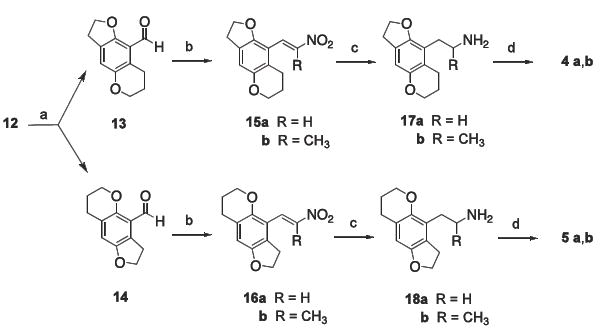
(a) dichloromethyl methyl ether, SnCl4, CH2Cl2, 0 °C; (b) RCH2NO2, ammonium acetate, 80 °C; (c) LiAlH4, THF, reflux; (d) Br2, AcOH.
Aromatization of the furan ring of 4b was accomplished using a method analogous to that employed by Parker, et al.9 As shown in Scheme 3, dihydrofuran 17b was protected as its N-trifluoroacetamide 19, brominated to provide 20, and then oxidized using 2,3-dichloro-5,6-dicyano-l,4-benzoquinone (DDQ) to afford the protected intermediate 21. Deprotection in strong base gave 6, the final target molecule. The relatively minor amounts of 14 obtained from the formylation reaction in Scheme 2 precluded a parallel synthesis of aromatized 5b; fortunately, 4b proved to be the more potent isomer, and its oxidized analog 6 sufficiently demonstrated the effects of aromatization of the furan ring.
Scheme 3.
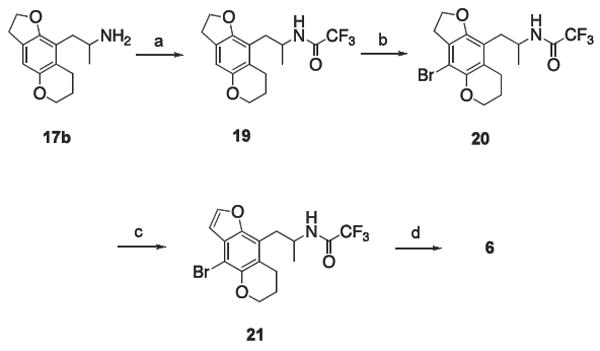
(a) ethyl trifluoroacetate, Et3N, THF, reflux; (b) AcOH, Br2, dioxane; (c) DDQ, toluene, reflux; (d) 5 M KOH, MeOH.
2.2 Pharmacology
All new target compounds were assessed for their affinity at the rat 5-HT2A receptor, using displacement of [3H]-ketanserin. In addition, functional potency and intrinsic activity were determined using the inositol phosphate accumulation assay.13 The affinities and potencies of the new compounds were compared with the prototype dimethoxy compound, DOB 1, and its rigidified difurano and dipyrano analogs, 2 and 3, respectively. These data are presented in Table 1, along with the clog P values of all compounds. Behavioral pharmacology for new compounds 4b and 5b was assessed using the two-lever drug discrimination paradigm,14-8 and those results are presented in Table 2, along with the comparative data for 1.
Table 1.
Analysis of biological activity at the rat 5-HT2A receptor. Ki values for [3H]-ketanserin competition displacement assays in NIH-3T3 cells expressing the rat 5-HT2A receptor and functional potency for stimulating inositol phosphate accumulation in the same cells.
| Drug | clog P | 5-HT2AKi, nM (SEM) | n | EC50, nM (SEM) | Intrinsic Activity % 5-HT (SEM) | n |
|---|---|---|---|---|---|---|
| 1 | 2.45 | 13 (6.0) | 2 | 22 (3.8) | 73 (5) | 3 |
| 2 | 2.65 | 1.0 (0.12) | 5 | 13 (1.9) | 88 (4) | 3 |
| 3a | 3.46 | 4.5 (0.76) | 3 | 241 (46) | 61 (9) | 3 |
| 3b | 3.77 | 3.1 (0.55) | 4 | 258 (45) | 72 (2) | 3 |
| 4a | 2.90 | 11 (1.4) | 3 | 9.4 (1.2) | 61 (6) | 3 |
| 4b | 3.21 | 3.6 (0.73) | 6 | 19 (1.9) | 86 (7) | 5 |
| 5a | 2.90 | 8.4 (0.35) | 3 | 146 (30) | 64 (12) | 3 |
| 5b | 3.21 | 5.3 (0.57) | 7 | 78 (11) | 74 (8) | 4 |
| 6 | 3.77 | 1.0 (0.12) | 5 | 13 (1.9) | 102 (6) | 3 |
Table 2.
Results from substitution tests in LSD-trained rats. All drugs were injected 30 min before tests. N = number of rats used for tests. % D = the percentage of rats with disrupted behavior. % SDL = the percentage of rats that selected the drug-appropriate lever. If the drug was one that completely substituted for the training drug, the method of Litchfield and Wilcoxon20 was used to determine the ED50 and 95% confidence interval (95% CL).
| Drug | Dose
|
N | % D | % SDL | ED50 (95% CL)
|
||
|---|---|---|---|---|---|---|---|
| mg/kg | μmole/kg | mg/kg | μmole/kg | ||||
| 1 | 0.25 | 0.80 | 13 | 46 | 29 | 0.35 | 1.06 |
| 0.38 | 1.21 | 10 | 40 | 50 | (0.27-0.46) | (0.82-1.36) | |
| 0.50 | 1.61 | 14 | 36 | 89 | |||
|
| |||||||
| 4b | 0.09 | 0.25 | 12 | 0 | 33 | 0.14 | 0.38 |
| 0.17 | 0.50 | 11 | 0 | 54 | (0.08-0.23) | (0.24-0.60) | |
| 0.35 | 1.00 | 12 | 0 | 83 | |||
|
| |||||||
| 5b | 0.17 | 0.50 | 12 | 17 | 10 | 0.40 | 1.16 |
| 0.35 | 1.00 | 12 | 0 | 41 | (0.26-0.61) | (0.77-1.75) | |
| 0.69 | 2.00 | 10 | 0 | 80 | |||
As seen in Table 1, the two new hybrid congeners, 4 and 5, as well as the dipyrano compounds 3,7 had approximately 4-5 fold lower affinity than the difurano analog 2, suggesting that the smaller furan rings are better tolerated by the receptor than the larger pyran rings. There was no significant difference in receptor affinity between 4 and 5. As we had previously observed with the aromatization of 2,9,19 oxidation of the dihydrofuran ring of 4b to give 6 increased receptor affinity nearly four-fold (compare Ki values of 4b and 6).
When functional potency was examined (see EC50 values in Table 1), 4b had about four-fold higher potency in stimulating inositol phosphate accumulation, compared to 5b, and was nearly as potent as the difuranyl compound 2. Surprisingly, although the receptor affinities were nearly the same for the dipyranyl compound 3b and the smaller hybrid compound 4b, the latter was functionally more than an order of magnitude more potent than 3b in stimulating inositol phosphate accumulation. Further, aromatization of the furan ring of 4b to give 6 had virtually no effect on potency, although receptor affinity was enhanced. Comparison of 3a with 3b, 4a with 4b, and 5a with 5b also shows that the addition of the alpha methyl slightly enhances intrinsic activity in this series, confirming the earlier conclusions drawn by Parrish, et al.8
The drug discrimination (DD) paradigm routinely has been used in our laboratory as a screen for evaluating the behavioral activity and hallucinogenic potential of newly synthesized molecules.14-16 This assay system models human hallucinogenic effects by examining the LSD-like discriminative stimulus properties produced in animals.17-18 Thus, compounds 1, 4b, and 5b were evaluated in the two-lever DD assay using a group of rats trained to discriminate the effects of ip injections of saline from those of d-lysergic acid N,N-diethylamide (LSD) tartrate (0.08 mg/kg; 186 nmol/kg) (Table 2). As can be seen in Table 2, 4b has nearly three times the in vivo potency of 5b, whereas the ED50s for 1 and 5b are not significantly different from one another.
3. Discussion
Previously, we demonstrated that when the two methoxy groups of 1 are constrained into dihydrofuran rings, as in 2, both receptor affinity and functional potency are significantly increased.9 In a prior study, we also had shown that expansion of both furan rings to the larger, six-membered pyrans led to a nearly ten-fold loss of affinity and in vivo behavioral activity, although compounds 3 were still significantly more potent than the flexible prototype 1.7 In this report, new compounds 4-6 further support those earlier observations and confirm the proper orientation of the arene oxygen lone pair electrons for optimal activity at the 5-HT2A receptor.
On the basis of virtual docking studies, we have hypothesized that the 5-HT2A receptor has less steric tolerance around the oxygen moiety that projects downward into the receptor than the one projected toward the extracellular face of the receptor.2 Those virtual docking studies are supported by our recent mutagenesis experiments and indicate that the 5-oxygen of 1 projects upward, toward the extracellular face of the receptor, whereas the 2-oxygen projects downward into the receptor binding cavity.5 In the present study, a comparison of the activities of 4 and 5 is consistent with this hypothesis. Although this conclusion is not as evident in the affinity data, it is clearly apparent when functional potency is assessed. Indeed, because affinity simply measures how tightly the ligand binds to the receptor, measures of functional potency probably better reflect general complementarity between the agonist and the receptor.
Intrinsic activity also is increased slightly by addition of the alpha-methyl to the side chain (compare 3a with 3b, 4a with 4b, and 5a with 5b). We previously reported a similar result for other substituted hallucinogenic phenylalkylamines.8 Although there is no definitive explanation for this observation, our virtual docking studies8 prompt us to speculate that the alpha-methyl may have favorable van der Waals interactions with a hydrophobic residue in trans-membrane helix 6. Although aromatization of the furan rings of 2 leads to the most potent simple phenethylamine type agonists presently known,9,19 aromatization of the furan ring of 4b to 6 had only a slight, and non-significant, effect on intrinsic activity. It should be noted, however, that this aromatization did increase receptor affinity nearly four-fold.
Of the new compounds reported here, those with the smaller O-heterocycle (furan) at the 2-position of the aryl ring (4a, 4b, and 6) clearly have higher affinities and functional potencies than those with the smaller O-heterocycle (furan) at the 5-position (5a and 5b). These findings support our current hypothesis that the 5-HT2A receptor is more tolerant of steric bulk in the region that interacts with the 5-oxygen function.
Data from the in vivo behavioral studies using the drug discrimination paradigm, where rats are trained to discriminate LSD from saline, also are consistent with these conclusions. Thus, analog 4b, which had a four-fold higher functional potency than 5b, proved to be nearly three times more potent than 5b in this in vivo assay. Because 4 and 5 have essentially identical pharmacokinetic properties (compare clog P values in Table 1), as well as similar affinities and intrinsic activities, the in vivo potency seems to be primarily attributable to the functional potency of the ligand, and thus to a highly complementary agonist–receptor interaction. Nonetheless, 5b, where the 2-oxygen is incorporated into a pyran ring and the 5-oxygen into a furan ring, still produced a discriminative stimulus effect similar to that of the more flexible prototype 1. It should be noted that the loss of potency of 5b, when compared with 4b, was not as dramatic as was observed earlier for compounds 3, where both oxygen atoms are incorporated into larger pyran rings.7
The high potency and intrinsic activity of compounds with an extended aromatic system, i.e.6, suggests that they are also closely complementary to the agonist binding site of the receptor. Because the endogenous agonist ligand, serotonin, is based on a bicyclic aromatic indole nucleus, we speculate that the aromatic furan rings better mimic the indole system and provide a more extensive pi-pi stacking partner for an aromatic residue within the receptor. Furthermore, the differences in hydrophobicity between 4b, 5b, and 6 are most likely so small as to not contribute significantly to the different in vivo activities of these agents; rather, the primary factors influencing activity and potency in this class appear to be the steric-electronic effects of the heterocyclic moieties of the 2 and 5 positions.
In conclusion, we have demonstrated that the different regioisomeric O-heterocycles in this homologous series of phenethylamines are not equally well accommodated by the receptor. In particular, we have shown that the steric space around the 2-oxygen appears to be more constrained than around the 5-position, as seen when comparing the potencies of 4b with 5b. Future SAR work probing the extent of these different spatial size constraints is warranted and is currently underway in our laboratories.
4. Experimental
4.1. Chemistry
All reactions were carried out under an inert atmosphere unless otherwise indicated. Melting points were determined using a Thomas-Hoover capillary melting point apparatus and are uncorrected. Thin-layer chromatography (TLC) was performed using Baker-flex silica gel plastic-backed plates with fluorescent indicator, developing with CH2Cl2, and visualizing under UV light. 1H-NMR spectra were obtained using a 300 MHz Bruker AC-300 or a 300 MHz Bruker ARX-300 NMR spectrometer with TMS as the internal standard. Abbreviations used in NMR analyses are as follows: b = broad, s = singlet, d = doublet, t = triplet, q= quartet, p = pentet, m = multiplet, Ar = aromatic. GC-MS analyses were obtained using a Varian 3800 GC coupled to a Varian Saturn 2000 ion trap MS. Infrared spectra were recorded on a Midac Prospect FT-IR spectrometer. Elemental analyses were performed by Quantitative Technologies, Inc. (QTI) of Whitehouse, NJ, USA, and reported values are within ± 0.4% of the calculated values. High-resolution mass spectrometry was performed with a Perkin-Elmer Voyager matrix-assisted laser desorption ionization time-of-flight (MALDI–TOF) mass spectrometer located in the Biophysics Instrumentation Facility at the University of Wisconsin–Madison Biophysics Instrumentation Facility. All solvents (acetone, diethyl ether, CH2Cl2, THF, ethyl acetate, hexanes, acetic acid, toluene) were obtained from Fisher Scientific and used as received. Most inorganic reagents, along with 4-methoxyphenol, 1,3-dibromopropane, 1,2-dibromoethane, 1.0 M B-bromo-9-BBN in CH2Cl2, dichloromethyl methyl ether, DDQ, ethyl trifluoroacetate, nitromethane, and nitroethane were used as received from Sigma-Aldrich.
4.2. Synthesis of 13
4.2.1. 1-(2-bromoethoxy)-4-methoxybenzene (8).15
A solution of 7 (10.1 g, 81.3 mmol) dissolved in acetone (100 mL) was added over a 12 h period to a mixture of 1,2-dibromoethane (35 mL, 0.41 mol), finely powdered K2CO3 (35 g, 0.25 mol), and acetone (300 mL) at reflux. The reaction was held at reflux for 125 h before cooling and removing the K2CO3 by filtration. After solvent removal in vacuo, the remaining residue was combined with CH2Cl2, washed with 2.5 M NaOH, 1 M HCl, and brine, and dried over anhydrous MgSO4. Evaporation of the volatiles in vacuo yielded an orange solid that was treated with a 10% CH2Cl2-90% hexanes solution (100 mL) to precipitate the dimeric byproduct. Following filtration, the filtrate was evaporated in vacuo and recrystallized from EtOAc/hexanes to yield 8.42 g (44.8%) of 8 as translucent sheet-like crystals (mp 50-51 °C) with a 1H-NMR spectrum identical to that reported by Pomilio and Tettamanzi.15 Low-resolution ESIMS, m/z (rel intensity) 136, 151, 178.
4.2.2. 1,4-dibromo-2-(2-bromoethoxy)-5-methoxybenzene (9)
Bromine (21.8 g, 0.137 mol) in CH2Cl2 (50 mL) was added dropwise to a mixture of 8 (14.9 g, 64.6 mmol) dissolved in CH2Cl2 (500 mL) with a catalytic amount of Fe filings. The reaction was allowed to continue for 24 h before the mixture was washed with 3M NaOH, 3M HCl, and brine. The organic phase was dried with MgSO4 and the solvent was removed in vacuo to provide an orange solid that was recrystallized from EtOAc/hexanes to yield 21.5 g (85.3%) of 9 as off-white crystals: mp 64-65 °C. 1H NMR (CDCl3) δ 3.66 (t, 2, ArOCH2CH2Br, J = 6.4 Hz), 3.87 (s, 3, ArOCH3), 4.30 (t, 2, ArOCH2CH2Br, J = 6.4 Hz), 7.11 (s, 1, ArH), 7.17 (s, 1, ArH). Low-resolution EIMS, m/z 386/388/390 (M+), 309, 281. Anal calcd for C9H9Br3O2: C, 27.80; H, 2.31. Found: C, 27.69; H, 2.08.
4.2.3. 2,5-Dibromo-4-(2-bromoethoxy)phenol (10)
Under an inert atmosphere, 1.0 M B-Br-9-BBN (522 mL, 0.522 mol) was added dropwise to a solution of 9 (45.0 g, 0.116 mol) in CH2Cl2 (1 L). The reaction was stirred for 14 days before the volatiles were removed in vacuo to give a yellow colored oil (that emitted white fumes). The oil was stirred under N2 for 2 h until the white fumes subsided, and was then taken up in Et2O (500 mL) and cooled to 0 °C. Ethanolamine (37.9 mL, 1.2 equivalents relative to B-Br-9-BBN) was added to precipitate the B-Br-9-BBN adduct, and this solid was removed by filtration, washing thoroughly on the filter with Et2O. Solvent was removed from the filtrate in vacuo to yield a yellow oil that was taken up into CH2Cl2 and washed with 3M HCl. The organic solution was then extracted several times with 3M NaOH. The combined basic extracts were acidified with 12 M HCl, and the solution was extracted with CH2Cl2. The organic fractions were combined, washed once with brine, dried with MgSO4, and the solvent removed in vacuo to give a yellow oil. The crude oil was purified by flash column chromatography on silica gel (eluting with CH2Cl2) to yield 29.5 g (73.7%) of 10 as a fine white powder: mp 106-107 °C. IR (NaCl, cm-1) 3437, 3054, 2986, 1266. 1H NMR (CDCl3) δ 3.65 (t, 2, ArOCH2CH2Br, J = 6.4 Hz), 4.27 (t, 2, ArOCH2CH2Br, J = 6.4 Hz), 5.29 (s, 1, ArOH), 7.07 (s, 1, ArH), 7.28 (s, 1, ArH). Low-resolution EIMS, m/z 372/374/376 (M+), 295, 267. Anal calcd for C8H7Br3O2: C, 25.63; H, 1.86; found: C, 25.80; H, 1.64.
4.2.4. 1,4-dibromo-2-(2-bromoethoxy)-5-(3-bromo-pro-poxy)benzene (11)
In a manner analogous to the preparation of 8, 10 (17.5 g, 46.6 mmol) was alkylated with 1,3-dibromopropane to give 18.3 g (79.0%) of 11 as white needle-like crystals: mp 67-68 °C. 1H NMR (CDCl3) δ 2.34 (p, 2, ArOCH2CH2CH2Br), 3.64 (t, 2, ArOCH2CH2CH2Br), superimposed upon 3.67 (t, 2, ArOCH2CH2Br), 4.10 (t, 2, ArOCH2CH2CH2Br, J = 5.7 Hz), 4.27 (t, 2, ArOCH2CH2Br, J = 6.3 Hz), 7.17 (s, 1, ArH), 7.28 (s, 1, ArH). Low-resolution EIMS, m/z 492/494/496 (M+), 107. Anal calcd for C11H12Br4O2: C, 26.65; H, 2.42; found: C, 26.85; H, 2.20.
4.2.5. 3,6,7,8-tetrahydro-2H-furo[2,3-g]chromene (12)
Diether 11 (17.6 g, 35.5 mmol) was dissolved in anhydrous THF (75 mL) and cooled to 0 °C, followed by the dropwise addition of 10.9 mL (109 mmol) of 10 M n-butyllithium in hexanes. The reaction was held at 0 °C for 4 h before quenching with H2O (10 mL). Solvent removal in vacuo gave a red oil that was taken up into Et2O and washed with H2O and brine. The organic phase was dried with MgSO4 and the solvent was removed in vacuo to afford a red oil that was recrystallized from EtOAc/hexanes to yield 4.3 g (69%) of 12 as orange crystals: mp 75-76 °C. 1H NMR (CDCl3) δ 1.96 (p, 2, ArOCH2CH2CH2, J = 5.2, 6.3 Hz), 2.72 (t, 2, ArOCH2CH2CH2, J = 6.3 Hz), 3.12 (t, 2, ArOCH2CH2, J = 8.5 Hz), 4.12 (t, 2, ArOCH2CH2CH2, J = 5.2 Hz), 4.56 (t, 2, ArOCH2CH2, J = 8.5 Hz), 6.46 (s, 1, ArH), 6.65 (s, 1, ArH). Low-resolution ESIMS, m/z (rel intensity) 176/178 (M+, 94/100). Anal calcd for C11H12O2: C, 74.98; H, 6.86; found: C, 74.65; H, 6.96.
4.2.6. 3,6,7,8-tetrahydro-2H-furo[2,3-g]chromene-9-carbaldehyde (13) and 3,6,7,8-tetrahydro-2H-furo[2,3-g]-chromene-4-carbaldehyde (14)
A solution of 12 (8.29 g, 47.1 mmol) dissolved in CH2Cl2 (200 mL) was stirred and cooled to 0 °C. The solution was charged with SnCl4 (6.07 mL, 51.8 mmol) and the reaction was held at 0 °C for 15 min before the slow addition of dichloromethyl methyl ether (5.9 mL, 52 mmol). The reaction was halted after 4 h by the addition of H2O (50 mL). The reaction mixture was partitioned and the organic phase was set aside. The aqueous layer was extracted with CH2Cl2, and the organic fractions were combined and washed several times with 3M HCl and once with brine. The organic phase was dried with MgSO4 and the solvent was removed under reduced pressure to give a dark red oil (10.29 g) that contained the two regioisomers 13 and 14. The oil was recrystallized from EtOAc/hexanes to afford 3.91 g (40.6%) of pure 13. The remaining oil, containing both 13 and 14, was subjected to silica gel flash chromatography (eluting with 95% CH2Cl2/5% hexane) to afford 14, the first to elute, as a yellow solid: mp 72-73 °C. IR (NaCl, cm -1) 3053, 2986, 1265. 1H NMR (CDCl3) δ 1.96 (p, 2, ArOCH2CH2CH2), 2.79 (t, 2, ArOCH2CH2CH2, J = 6.5 Hz), 3.45 (t, 2, ArOCH2CH2, J = 8.8 Hz), 4.22 (t, 2, ArOCH2CH2CH2, J = 5.2 Hz), 4.55 (t, 2, ArOCH2CH2, J = 8.8 Hz), 6.70 (s, 1, ArH), 10.47 (s, 1, ArCHO). Low-resolution EIMS, m/z (rel intensity) 204 (M+, 100). Anal calcd for C12H12O3: C, 70.57; H, 5.92; found: C, 70.36; H, 6.08.
The second to elute, 13, was isolated as a bright yellow solid: mp 71-72°C. IR (NaCl, cm -1) 3054, 2986, 1264. 1H NMR (CDCl3) δ 1.96 (p, 2, ArOCH2CH2CH2), 3.11 (t, 2, ArOCH2CH2CH2), superimposed upon 3.17 (t, 2, ArOCH2CH2, J = 8.7 Hz), 4.11 (t, 2, ArOCH2CH2CH2, J = 5.1 Hz), 4.65 (t, 2, ArOCH2CH2, J = 8.4 Hz), 6.92 (s, 1, ArH), 10.37 (s, 1, ArCHO). Low-resolution EIMS, m/z (rel intensity) 204 (M+, 100). Anal calcd for C12H12O3: C, 70.57; H, 5.92; found: C, 70.47; H, 5.98.
4.3. Synthesis of 4a and 5a
4.3.1. 9-[(E)-2-nitroethenyl]-3,6,7,8-tetrahydro-2H-furo-[2,3-g]chromene (15a)
Aldehyde 13 (1.37 g, 6.69 mmol) and ammonium acetate (0.539 g, 7.00 mmol) were dissolved in nitromethane (12 mL) and heated at 90 °C for 2.5 h. Volatiles were removed in vacuo to give an orange solid that was dissolved in CH2Cl2 and washed with 3 M HCl, H2O, and once with brine. The organic phase was dried using MgSO4 and the solvent was removed in vacuo, leaving 1.61 g (97.3%) of a dark orange solid that was recrystallized from MeOH to afford 1.36 g (82.6%) of 15a as an orange crystalline solid: mp 130-131 °C. 1H NMR (CDCl3) δ 2.04 (p, 2, ArOCH2CH2CH2), 2.87 (t, 2, ArOCH2CH2CH2, J = 6.9 Hz), 3.18 (t, 2, ArOCH2CH2, J = 8.4 Hz), 4.09 (t, 2, ArOCH2CH2CH2, J = 5.1 Hz), 4.67 (t, 2, ArOCH2CH2, J = 8.7 Hz), 6.80 (s, 1, ArH), 8.11 (s, 2, ArCH=CH). Low-resolution EIMS, m/z (rel intensity) 247 (M+, 100). Anal calcd for C13H13NO4: C, 63.15; H, 5.30; N, 5.66; found: C, 62.88; H, 5.40; N, 5.57.
4.3.2. 4-[(E)-2-nitroethenyl]-3,6,7,8-tetrahydro-2H-furo-[2,3-g ]chromene (16a)
Similar to the preparation of 15a, aldehyde 14 (0.127 g, 1.65 mmol) was condensed with nitromethane to give 0.301 g (73.8%) of 16a as fluffy bright-orange colored crystals: mp 139-140 °C. 1H NMR (CDCl3) δ 2.01 (p, 2, ArOCH2CH2CH2), 2.77 (t, 2, ArOCH2CH2CH2), 3.31 (t, 2, ArOCH2CH2), 4.26 (t, 2, ArOCH2CH2CH2), 4.58 (t, 2, ArOCH2CH2), 6.61 (s, 1, ArH), 7.94 (d, 1, ArCH=CH), 8.01 (d, 1, ArCH=CH). Low-resolution EIMS, m/z (rel intensity) 247 (M+, 100). Anal calcd for C13H13NO4: C, 63.15; H, 5.30; N, 5.66; found: C, 63.08; H, 5.22; N, 5.51.
4.3.3. 2-(3,6,7,8-tetrahydro-2H-furo[2,3-g]chromen-9-yl)-ethanamine (17a)
A dry reaction flask was purged with N2, charged with anhydrous THF (100 mL), and cooled to 0 °C. A 1M solution of LiAlH4 in THF (25.0 mL, 25.0 mmol) was then added dropwise. Once the addition was complete, 15a (2.41 g, 9.76 mmol) dissolved in anhydrous THF (25 mL) was added to the suspension over a 30 min period. The flask was held at reflux for 4 h before the reaction was quenched by the addition of 2.5 M NaOH (15 mL). Celite was added to the flask and the mixture was filtered, washing the flask and filtered solids thoroughly with CH2Cl2. The yellow filtrate was concentrated under vacuum, dissolved in CH2Cl2, and washed with 3M HCl. The aqueous extracts were combined, cooled to 0 °C, and basified by addition of 5M NaOH. The basic solution was extracted with CH2Cl2, and the organic extract was washed with brine and dried over MgSO4. The volatiles were removed in vacuo to yield 17a as a yellow oil. This free amine was converted to its hydrochloride salt by dissolving in Et2O and adding 1.1 equiv of 1M HCl in anhyd EtOH. The salt was recrystallized from EtOH/Et2O to afford 0.934 g (37.4%) of 17a•HCl as a white crystalline solid: mp 279-280 °C. 1H NMR (free base in CDCl3) δ 1.11 (bs, 2, NH2), 1.96 (p, 2, ArOCH2CH2CH2), 2.69 (t, 2, ArCH2CH2, J = 6.6 Hz), superimposed onto 2.69 (t, 2, ArOCH2CH2CH2, J = 7.2 Hz), 2.85 (t, 2, ArCH2CH2, J = 6.6 Hz), 3.12 (t, 2, ArOCH2CH2, J = 8.4 Hz), 4.06 (t, 2, ArOCH2CH2CH2, J = 5.1 Hz), 4.45 (t, 2, ArOCH2CH2, J = 8.4 Hz), 6.56 (s, 1, ArH). Low-resolution ESIMS, m/z (rel intensity) 220 (M + H, 67), 203 ((M + H) − NH3, 100). Anal calcd for C13H17NO2•HCl: C, 61.06; H, 7.09; N, 5.47; found: C, 60.91; H, 7.05; N, 5.44.
4.3.4. 2-(3,6,7,8-tetrahydro-2H-furo[2,3-g]chromen-4-yl)-ethanamine (18a)
Similar to the preparation of 17a, 16a (0.572 g, 2.31 mmol) was reduced to give 0.408 g (68.9%) of 18a as pale yellow oil that was converted to its hydrochloride salt 18a•HCl with the addition of 1M HCl in anhyd EtOH: mp 272 °C. 1H NMR (free base in CDCl3) δ 1.27 (bs, 2, NH2), 1.93 (p, 2, ArOCH2CH2CH2), 2.65 (t, 2, ArCH2CH2) superimposed onto 2.70 (t, 2, ArOCH2CH2CH2), 2.87 (t, 2, ArCH2CH2), 3.11 (t, 2, ArOCH2CH2), 4.10 (t, 2, ArOCH2CH2CH2), 4.49 (t, 2, ArOCH2CH2), 6.34 (s, 1, ArH). Low-resolution ESIMS, m/z (rel intensity) 220 (M + H, 100), 203 ((M + H) − NH3, 49), 175. High resolution MALDI–TOF m/z calculated for C13H18NO2 (M + H) 220.1346, found 220.1332.
4.3.5. 2-(4-bromo-3,6,7,8-tetrahydro-2H-furo[2,3-g]-chromen-9-yl)ethanamine (4a)
A 0.776 M solution of Br2 in HOAc (0.75 mL, 0.58 mmol) was added dropwise to a solution of 17a•HCl (0.333 g, 0.520 mmol) dissolved in glacial AcOH (10 mL). The reaction was stirred for 3.5 h before the volatiles were removed in vacuo. The white solid that remained was taken up into 3M HCl, this solution was washed with Et2O,then made basic by the addition of 5M NaOH and extracted several times with CH2Cl2. The organic extracts were combined, washed once with brine, dried over MgSO4, filtered, and the filtrate reduced in vacuo to give 0.144 g (93.0%) of 4a as a pale yellow oil. The free amine was converted to its hydrochloride salt with the addition of 1.1 equivalents of 1M HCl in anhyd EtOH, and recrystallization from EtOH/Et2O to afford 4a•HCl as a white, crystalline solid: mp >300 °C. 1H NMR (free base in CDCl3) δ 1.09 (bs, 2, NH2), 1.98 (p, 2, ArOCH2CH2CH2), 2.63 (t, 2, ArCH2CH2, J = 7.2 Hz), 2.73 (t, 2, ArOCH2CH2CH2, J = 6.3 Hz), 2.83 (q, 2, ArCH2CH2), 3.18 (t, 2, ArOCH2CH2, J = 8.1 Hz), 4.17 (t, 2, ArOCH2CH2CH2, J = 5.1 Hz), 4.51 (t, 2, ArOCH2CH2, J = 8.4 Hz). Low-resolution ESIMS, m/z (rel intensity) 298/300 (M + H, 71/66), 281/283 ((M + H) − NH3, 100/96). Anal calcd for C13H16BrNO2•HCl: C, 46.66; H, 5.12; N, 4.18; found: C, 46.48; H, 4.96; N, 4.07.
4.3.6. 2-(9-bromo-3,6,7,8-tetrahydro-2H-furo[2,3-g]-chromen-4-yl)ethanamine (5a)
In a manner analogous to that reported for the preparation of 4a, 18a•HCl (0.051 g, 0.20 mmol) was brominated to yield 0.051 g (86%) of 5a as a pale yellow oil that was taken up in Et2O and converted to its hydrochloride salt 5a•HCl with the addition of 1M HCl in anhydrous EtOH: mp >300 °C. 1H NMR (free base in CDCl3) δ 1.24 (bs, 2, NH2), 1.98 (p, 2, ArOCH2CH2CH2), 2.65 (t, 2, ArCH2CH2) superimposed onto 2.71 (t, 2, ArOCH2CH2CH2), 2.86 (t, 2, ArCH2CH2), 3.24 (t, 2, ArOCH2CH2), 4.06 (t, 2, ArOCH2CH2CH2), 4.60 (t, 2, ArOCH2CH2). Low-resolution ESIMS, m/z (rel intensity) 298/300 (M + H, 100/96), 281/283 ((M + H) − NH3, 69/67). Anal calcd for C13H16BrNO2•HCl: C, 46.66; H, 5.12; N, 4.18; found: C, 46.73; H, 5.07; N, 4.02.
4.4. Synthesis of 4b and 5b
4.4.1. 9-[(1E)-2-nitroprop-1-en-1-yl]-3,6,7,8-tetrahydro-2H-furo[2,3-g]chromene (15b)
In a manner analogous to that used in the preparation of 15a, aldehyde 13 (3.50 g, 17.1 mmol) was condensed with nitroethane to give 2.90 g (64.7%) of 15b as orange needle-like crystals: mp 88-89 °C. IR (NaCl, cm -1) 3054, 2986, 1422, 1264. 1H NMR (CDCl3) δ 1.96 (p, 2, ArOCH2CH2CH2), 2.16 (s, 3, CH3), 2.63 (t, 2, ArOCH2CH2CH2, J = 6.4 Hz), 3.19 (t, 2, ArOCH2CH2, J = 8.4 Hz), 4.11 (t, 2, ArOCH2CH2CH2, J = 5.2 Hz), 4.55 (t, 2, ArOCH2CH2, J = 8.5 Hz), 6.76 (s, 1, ArH), 7.84 (s, 1, ArCH=C). Low-resolution EIMS, m/z 262 (M+), 244 (M − OH). Anal calcd for C14H15NO4: C, 64.35; H, 5.78; N, 5.36; found: C, 64.07; H, 5.64; N, 5.29.
4.4.1. 4-[(1E)-2-nitroprop-1-en-1-yl]-3,6,7,8-tetrahydro-2H-furo[2,3-g]chromene (16b)
Aldehyde 14 (0.623 g, 3.05 mmol) and benzylamine (0.399 mL, 3.66 mmol) were dissolved in benzene (50 mL) and heated at reflux for 2h with a Dean-Stark trap for H2O removal. After cooling to room temperature, the solvent was removed in vacuo to give a yellow oil that was combined with nitroethane (40 mL) and AcOH (0.55 mL, 9.2 mmol) and stirred for 48 h. The volatiles were removed in vacuo, leaving behind a yellow oil that was taken up in Et2O and washed with 1M HCl, H2O, and once with brine. The organic phase was dried with MgSO4 and the solvent was removed under reduced pressure to give a yellow oil that was recrystallized from MeOH to afford 0.40 g (50%) of 16b as an orange crystalline solid: mp 103-104 °C. IR (NaCl, cm -1) 3054, 2986, 1422, 1264. 1H NMR (CDCl3) δ 1.98 (p, 2, ArOCH2CH2CH2), 2.16 (s, 3, CH3), 2.77 (t, 2, ArOCH2CH2CH2, J = 6.6 Hz), 3.01 (t, 2, ArOCH2CH2, J = 8.7 Hz), 4.15 (t, 2, ArOCH2CH2CH2, J = 5.1 Hz), 4.53 (t, 2, ArOCH2CH2, J = 8.4 Hz), 6.55 (s, 1, ArH), 7.92 (s, 1, ArCH=C). Low-resolution EIMS, m/z 261 (M+), 244 (M − OH). Anal calcd for C14H15NO4: C, 64.35; H, 5.78; N, 5.36; found: C, 64.23; H, 6.03; N, 5.25.
4.4.2. 1-(3,6,7,8-tetrahydro-2H-furo[2,3-g]chromen-9-yl)-propan-2-amine (17b)
In a manner analogous to that used in the preparation of 17a and 18a, 15b (2.01 g, 7.69 mmol) was reduced to afford 1.70 g (94.9%) of 17b as the free amine. The hydrochloride salt 17b•HCl was formed by the addition 1M HCl in anhydrous EtOH, and the solid was recrystallized from MeOH/EtOAc: mp 255-256 °C. 1H NMR (free base in CDCl3) δ 1.15 (d, 3, ArCH2CHCH3, J = 6.3 Hz), 1.62 (bs, 2, NH2), 1.98 (p, 2, ArOCH2CH2CH2), 2.58 (d, 2, ArCH2CHCH3, J = 6.9 Hz), superimposed upon 2.59 (t, 2, ArOCH2CH2CH2), 2.71 (m, 1, ArCH2CHCH3, J = 6.6 Hz), 3.14 (t, 2, ArOCH2CH2), 4.08 (t, 2, ArOCH2CH2CH2, J = 5.1 Hz), 4.47 (t, 2, ArOCH2CH2, J = 8.7 Hz), 6.58 (s, 1, ArH). Low-resolution ESIMS, m/z (rel intensity) 234 (M + H, 64), 217 ((M + H) − NH3, 100). Anal calcd for C14H19NO2•HCl: C, 62.33; H, 7.47; N, 5.19. Found: C, 62.10; H, 7.34; N, 5.11.
4.4.3. 1-(3,6,7,8-tetrahydro-2H-furo[2,3-g]chromen-4-yl)-propan-2-amine (18b)
In a method analogous to that used for the preparation of 17a, 17b, and 18a, 16b (0.200 g, 0.765 mmol) was reduced to yield 0.142 g (79.7%) of 18b as the free amine that was converted to its hydrochloride salt by the addition 1M HCl in anhydrous EtOH: mp 270-271 °C. 1H NMR (free base in CDCl3) δ 1.21 (d, 3, ArCH2CHCH3, J = 6.3 Hz), 1.95 (p, 2, ArOCH2CH2CH2, J = 5.1, 6.3 Hz), 2.55 (bs, 2, NH2), superimposed upon 2.65 (d, 2, ArCH2CHCH3, J = 6.9 Hz), 2.74 (t, 2, ArOCH2CH2CH2, J = 6.6 Hz), 3.11 (m, 2, ArOCH2CH2), 3.40 (m, 1, ArCH2CHCH3), 4.12 (t, 2, ArOCH2CH2CH2, J = 5.1 Hz), 4.49 (t, 2, ArOCH2CH2, J = 8.4 Hz), 6.37 (s, 1, ArH). Low-resolution ESIMS, m/z (rel intensity) 234 (M + H, 100), 217 ((M + H) - NH3, 77). Anal calcd for C14H19NO2•HCl: C, 62.33; H, 7.47; N, 5.19; found: C, 61.98; H, 7.66; N, 5.11.
4.4.4. 1-(4-bromo-3,6,7,8-tetrahydro-2H-furo[2,3-g]-chromen-9-yl)propan-2-amine (4b)
In a manner similar to the preparation of 4a and 5a, amine 17b (0.818 g, 3.51 mmol) was brominated to give 0.441 g (63.9%) of 4b as a yellow oil that was converted to its hydrochloride salt by the addition of 1M HCl in anhydrous EtOH: mp 269-270 °C. 1H NMR (free base in CDCl3) δ 1.13 (d, 3, ArCH2CHCH3), 1.48 (bs, 2, NH2), 1.99 (p, 2, ArOCH2CH2CH2), 2.52 (t, 2, ArOCH2CH2CH2), superimposed upon 2.53 (d, 2, ArCH2CHCH3), 2.74 (m, 1, ArCH2CHCH3), 3.18 (t, 2, ArOCH2CH2, J = 8.4 Hz), 4.18 (t, 2, ArOCH2CH2CH2, J = 5.1 Hz), 4.51 (t, 2, ArOCH2CH2, J = 9.3 Hz). Low-resolution ESIMS, m/z (rel intensity) 312/314 (M + H, 91/84). Anal calcd for C14H18BrNO2•HCl: C, 48.23; H, 5.49; N, 4.02; found: C, 47.85; H, 5.21; N, 4.10.
4.4.5. 1-(9-bromo-3,6,7,8-tetrahydro-2H-furo[2,3-g]-chromen-4-yl)propan-2-amine (5b)
In a manner analogous to that used for the preparation of 4a, 4b, and 5a, amine 18b (0.183 g, 7.84 mmol) was brominated to give 0.156 g (63.8%) of 5b that was converted to its hydrochloride salt by the addition of 1 M HCl in anhydrous EtOH: mp 277-278 °C. 1H NMR (free base in CDCl3) δ 1.21 (d, 3, ArCH2CHCH3), 1.95 (p, 2, ArOCH2CH2CH2), 2.55 (bs, 2, NH2), superimposed upon 2.65 (d, 2, ArCH2CHCH3), 2.73 (t, 2, ArOCH2CH2CH2), 3.11 (m, 2, ArOCH2CH2), 3.40 (m, 1, ArCH2CHCH3), 4.12 (t, 2, ArOCH2CH2CH2), 4.48 (m, 2, ArOCH2CH2). Low-resolution ESIMS, m/z (rel intensity) 312/314 (M + H, 100/95). Anal calcd for C14H18BrNO2•HCl: C, 48.23; H, 5.49; N, 4.02; found: C, 48.37; H, 5.47; N, 3.92.
4.5. Synthesis of 6
4.5.1. 2,2,2-trifluoro-N-[1-methyl-2-(3,6,7,8-tetrahydro-2H-furo[2,3-g]chromen-9-yl)ethyl]acetamide (19)
Compound 17b (1.52 g, 6.51 mmol) was dissolved in anhydrous THF (250 mL) and cooled to 0 °C, followed by the addition of ethyl trifluoroacetate (1.71 mL, 14.3 mmol) and triethylamine (0.953 mL, 6.84 mmol). The reaction was stirred at 0 °C for 30 min before being brought to reflux for 24 h. The volatiles were removed in vacuo, leaving behind a light orange solid that was taken up in CH2Cl2, washed with 3M HCl, and once with brine. The organic phase was dried with MgSO4, filtered, and the solvent was removed in vacuo to afford 1.58 g (73.9%) of a pale orange solid that was recrystallized from EtOAc to give 1.27 g (59.3%) of 19 as fluffy white crystals: mp 156-157 °C. 1H NMR (CDCl3) δ 1.32 (d, 3, ArCH2CHCH3, J = 6.6 Hz), 2.01 (p, 2, ArOCH2CH2CH2), 2.67 (m, 2, ArCH2CHCH3), superimposed upon 2.71 (m, 2, ArOCH2CH2CH2), 3.71 (t, 2, ArOCH2CH2, J = 8.1 Hz), 4.02 (m, 1, ArCH2CHCH3), superimposed upon 4.05 (m, 2, ArOCH2CH2CH2), 4.50 (t, 2, ArOCH2CH2, J = 8.7 Hz), 6.61 (s, 1, ArH), 7.64 (bs, 1, NH). Low-resolution ESIMS, m/z (rel intensity) 328 (M − H, 100). High resolution MALDI–TOF m/z calculated for C16H19F3NO3 (M + H) 330.1323, found 330.1312.
4.5.2. N-[2-(4-bromo-3,6,7,8-tetrahydro-2H-furo[2,3-g]-chromen-9-yl)-1-methylethyl]-2,2,2-trifluoroacetamide (20)
Amide 19 (0.538 g, 1.63 mmol) was dissolved (with heating) in AcOH (15 mL) and dioxane (5 mL). The solution was cooled to 15 °C and covered with foil, followed by the dropwise addition of a bromine-dioxane solution (8.10 mL: 0.10 mL bromine in 8.00 mL of dioxane) over a 10 min period. The reaction was stirred at 25 °C for 6 h before being quenched with H2O and a satd sodium bisulfite solution. The mixture was extracted with CH2Cl2, the organic fractions were combined and washed once with brine, dried with MgSO4, and the volatiles were removed in vacuo. The off-white solid residue was recrystallized from EtOAc/hexanes to yield 0.425 g (63.9%) of 20 as white crystals: mp 156 °C. 1H NMR (CDCl3) δ 1.31 (d, 3, ArCH2CHCH3, J = 6.6 Hz), 2.02 (p, 2, ArOCH2CH2CH2), 2.68 (m, 2, ArCH2CHCH3), superimposed upon 2.72 (m, 2, ArOCH2CH2CH2), 3.22 (t, 2, ArOCH2CH2, J = 8.7 Hz), 4.17 (m, 1, ArCH2CHCH3), superimposed upon 4.21 (m, 2, ArOCH2CH2CH2), 4.55 (t, 2, ArOCH2CH2, J = 8.4 Hz), 7.41 (bs, 1, NH). Low-resolution CIMS, m/z 408/410 (M + H), 329 ((M + H) − Br). Anal calcd for C16H17BrF3NO3: C, 47.08; H, 4.20; N, 3.43; found: C, 47.36; H, 4.14; N, 3.40.
4.5.3. N-[2-(4-bromo-7,8-dihydro-6H-furo[2,3-g]chromen-9-yl)-1-methylethyl]-2,2,2-trifluoroacetamide (21)
A solution of 20 (0.408 g, 0.999 mmol) and 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) (0.590 g, 2.60 mmol) in toluene (50 mL) was heated at reflux for 3.5 h and was cooled to room temperature. The volatiles were removed in vacuo to afford a brown oil that was purified by silica gel flash chromatography (eluting with CH2Cl2) to afford a light yellow solid that was recrystallized from EtOAc/hexanes to afford 0.152 g (37.4%) of 21 as white crystals. 1H NMR (free base in CDCl3) δ 1.31 (d, 3, ArCH2CHCH3, J = 6.6 Hz), 2.08 (p, 2, ArOCH2CH2CH2), 2.92 (qd, 2, ArOCH2CH2CH2), 3.07 (qd, 2, ArCH2CHCH3), 4.27 (m, 1, ArCH2CHCH3), superimposed upon 4.28 (m, 2, ArOCH2CH2CH2), 6.57 (bs, 1, NH), 6.75 (d, 1, ArOCH=CH, J = 2.1 Hz), 7.56 (d, 1, ArOCH=CH, J = 2.1 Hz). Low-resolution EIMS, m/z 404/406 (M - H), 69 (CF3+). High resolution MALDI–TOF m/z calculated for C16H15BrF3NO3 (M + H) 406.0263, found 406.0260.
4.5.4. 1-(4-bromo-7,8-dihydro-6H-furo[2,3-g]chromen-9-yl)propan-2-amine (6)
A solution of 21 (0.146 g, 0.359 mmol) in MeOH (20 mL) was cooled to 0 °C, 5M KOH (10 mL) was added, and the stirred reaction mixture was allowed to warm to room temperature over 5h. The mixture was diluted with Et2O and extracted several times with 3M HCl. The aqueous fractions were combined, cooled to 0 °C, made basic by the addition of 5M KOH, and extracted several times with CH2Cl2. The organic fractions were combined, washed with brine, and dried with MgSO4. After filtration, the solvent was removed under reduced pressure to afford 0.109 g (98.2%) of 6 as yellow oil. The hydrochloride salt was formed by adding 1.1 equivalents of 1M HCl in anhydrous EtOH to the free amine dissolved in Et2O. 1H NMR (free base in CDCl3) δ 1.23 (d, 3, ArCH2CHCH3), 2.06 (p, 2, ArOCH2CH2CH2), superimposed upon 2.07 (m, 2, ArCH2CHCH3), 2.49 (bs, 2, NH2), 2.92 (m, 2, ArOCH2CH2CH2), 3.02 (d, 2, ArCH2CHCH3), 4.27 (t, 2, ArOCH2CH2CH2), 6.78 (d, 1, ArOCH=CH, J = 2.1 Hz), 7.58 (d, 1, ArOCH=CH, J = 2.1 Hz). Low-resolution ESIMS, m/z (rel intensity) 310/312 (M + H, 72/79), 293/295 ((M + H) − NH3, 92/100). High resolution MALDI–TOF m/z calculated for C14H18BrNO2 (M + H) 310.0428, found 310.0437.
4.6. Pharmacology
4.6.1. Materials
[3H]-ketanserin was obtained from PerkinElmer Life and Analytical Sciences (Boston, MA). Serotonin (5-HT) was obtained from Sigma-Aldrich (St. Louis, MO).
4.6.2. Cell Culture Methods
NIH-3T3 cells stably expressing the rat 5-HT2A receptor (GF-6; 5500 fmol/mg) were the kind gift of Dr. David Julius (University of California, San Francisco, CA). Cells were maintained as described previously by Braden et al.23 All DMEM media contained 300 μg/ml G-418 (Sigma-Aldrich).
4.6.3. Radioligand Binding Assays
Membrane preparations and competition binding assays were performed as previously described.7,24 Competition binding assays were carried out using 0.5nM [3H]-ketanserin in the presence of increasing concentrations of test compound (ca. 10 pM - 10 μM). Kd values and competition curves were calculated using Prism (Graphpad Software, San Diego, CA).
4.6.4. Inositol Phosphate Accumulation Assays
The ability of test compounds to stimulate radiolabeled inositol phosphate accumulation in NIH-3T3 cells stably expressing the rat 5-HT2A receptor was performed as previously described.13 Each assay plate was normalized to wells stimulated with water (0%) and 10 μM serotonin (100%).
4.6.5. Behavioral Pharmacology Methods
Animals
Four male Sprague-Dawley rats (Harlan Laboratories, Indianapolis, IN) weighing 200-220 g at the beginning of the drug discrimination study were trained to discriminate LSD (186 nmol/kg; 0.08 mg/kg) from saline. The animal protocols used in the present experiments were consistent with current NIH principles of good laboratory animal care and were approved by the Purdue University Animal Care and Use Committee.
Discrimination Training and Testing
A fixed ratio (FR) 50 schedule of food reinforcement in a two-lever paradigm was used. This method is described in detail elsewhere.25 Briefly, training sessions lasted 15 min and were conducted at the same time each day. Training continued until an accuracy of at least 85% was attained for eight of ten consecutive sessions. Once criterion performance was attained, test sessions were interspersed between training sessions, either one or two times per week. At least one drug and one saline session separated each test session. Rats were required to maintain the 85% correct responding criterion on training days in order to be tested. In addition, test data were discarded when the accuracy criterion of 85% was not achieved on the two training sessions following a test session. Test drugs were administered i.p. 30 min prior to sessions; test sessions were run under conditions of extinction, with rats removed from the operant chamber when 50 presses were emitted on one lever. If 50 presses on one lever were not completed within 5 min, the session was ended and scored as a disruption. In combination tests, antagonists were administered 30 min before the training drug. The percentage of rats selecting the drug lever (% SDL) for each dose of test compound was determined. Treatments were randomized at the beginning of the study.
Acknowledgments
This work was supported by NIH grant DA02189 from NIDA (DEN), the UW-La Crosse Undergraduate Research Program, and a Purdue MCMP summer research fellowship awarded to DMS. The authors would like to thank Stewart Frescas at Purdue for his technical assistance with the syntheses, and Amy Harms at UW-Madison for her assistance with the HRMS analyses.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nichols DE. Hallucinogens. Pharmacol Ther. 2004;101:131–181. doi: 10.1016/j.pharmthera.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Chambers JJ, Nichols DE. A homology-based model of the human 5-HT2A receptor derived from an in silico activated G-protein coupled receptor. J Comput Aided Mol Des. 2002;16:511–520. doi: 10.1023/a:1021275430021. [DOI] [PubMed] [Google Scholar]
- 3.McLean TH, Parrish JC, Braden MR, Marona-Lewicka D, Gallardo-Godoy A, Nichols DE. 1-aminomethylbenzo-cycloalkanes: conformationally restricted hallucinogenic phenethylamine analogues as functionally selective 5-HT2A receptor agonists. J Med Chem. 2006;49:5794–5803. doi: 10.1021/jm060656o. [DOI] [PubMed] [Google Scholar]
- 4.McLean TH, Chambers JJ, Parrish JC, Braden MR, Marona-Lewicka D, Kurrasch-Orbaugh D, Nichols DE. C-(4,5,6-trimethoxyindan-1-yl)methanamine: a mescaline analogue designed using a homology model of the 5-HT2A receptor. J Med Chem. 2006;49:4269–4274. doi: 10.1021/jm060272y. [DOI] [PubMed] [Google Scholar]
- 5.Braden MR, Nichols DE. Assessment of the roles of serines 5.43(239) and 5.46(242) for binding and potency of agonist ligands at the human serotonin 5-HT2A receptor. Mol Pharmacol. 2007;72(5):1200–1209. doi: 10.1124/mol.107.039255. [DOI] [PubMed] [Google Scholar]
- 6.Monte AP, Marona-Lewicka D, Parker MA, Wainscott DB, Nelson DL, Nichols DE. Dihydrobenzofuran analogues of hallucinogens. 3. Models of 4-substituted (2,5-dimethoxyphenyl)alkylamine derivatives with rigidified methoxy groups. J Med Chem. 1996;39:2953–2961. doi: 10.1021/jm960199j. [DOI] [PubMed] [Google Scholar]
- 7.Whiteside MS, Kurrasch-Orbaugh D, Marona-Lewicka D, Nichols DE, Monte A. Substituted hexahydrobenzodipyrans as 5-HT2A/2C receptor probes. Bioorg Med Chem. 2002;10:3301–3306. doi: 10.1016/s0968-0896(02)00209-2. [DOI] [PubMed] [Google Scholar]
- 8.Parrish JC, Braden MR, Gundy E, Nichols DE. Differential phospholipase Cactivation by phenylalkylamine serotonin 5-HT2A receptor agonists. J Neurochem. 2005;95:1575–1584. doi: 10.1111/j.1471-4159.2005.03477.x. [DOI] [PubMed] [Google Scholar]
- 9.Parker MA, Marona-Lewicka D, Lucaites VL, Nelson DL, Nichols DE. A novel (benzodifuranyl)-aminoalkane with extremely potent activity at the 5-HT2A receptor. J Med Chem. 1998;41:5148–5149. doi: 10.1021/jm9803525. [DOI] [PubMed] [Google Scholar]
- 10.Brown HC, Krishnamurthy S, Yoon NM. 9-Bora-bicyclo[3.3.1]nonane in tetrahydrofuran as a new selective reducing agent in organic synthesis. J Org Chem. 1976;41(10):1778–1791. [Google Scholar]
- 11.Bhatt MV. B-bromo-9-borabicyclo[3.3.1]nonane: a convenient and selective reagent for ether cleavage. J Organometallic Chem. 1978;156:221–226. [Google Scholar]
- 12.Still WC, Kahn M, Mitra A. Rapid chromatographic technique for preparative separations with moderate resolution. J Org Chem. 1978;43:2923–2925. [Google Scholar]
- 13.Kurrasch-Orbaugh DM, Watts VJ, Barker EL, Nichols DE. Serotonin 5-hydroxytryptamine-2A receptor-coupled phosholipase C and phospholipase A2 signaling pathways have different receptor reserves. J Pharmacol Exp Ther. 2003;304:229–237. doi: 10.1124/jpet.102.042184. [DOI] [PubMed] [Google Scholar]
- 14.Appel JB, Cunningham KA. The use of drug discrimination procedures to characterize hallucinogenic drug actions. Psychopharmacol Bull. 1986;22:959–967. [PubMed] [Google Scholar]
- 15.Arnt J. Characterization of the discriminative stimulus properties induced by 5-HT1 and 5-HT2 agonists in rats. Pharmacol Toxicol. 1989;64:165–172. doi: 10.1111/j.1600-0773.1989.tb00623.x. [DOI] [PubMed] [Google Scholar]
- 16.Glennon RA, Rosecrans JA, Young R. Drug-induced discrimination: a description of the paradigm and a review of its specific application to the study of hallucinogenic agents. Medicinal Res Rev. 1983;3:289–340. doi: 10.1002/med.2610030305. [DOI] [PubMed] [Google Scholar]
- 17.Cunningham KA, Appel JB. Neuropharmacological reassessment of the discriminative stimulus properties of d-lysergic acid diethylamide (LSD) Psychopharmacology. 1987;91:67–73. doi: 10.1007/BF00690929. [DOI] [PubMed] [Google Scholar]
- 18.Nielsen EJ. Discriminative stimulus properties of lysergic acid diethylamide in the monkey. J Pharmacol Exp Ther. 1985;234:244–249. [PubMed] [Google Scholar]
- 19.Chambers JJ, Kurrasch-Orbaugh DM, Parker MA, Nichols DE. Enantiospecific synthesis and pharmacological evaluation of a series of super-potent, conformationally restricted 5-HT2A/2C receptor agonists. J Med Chem. 2001;44:1003–1010. doi: 10.1021/jm000491y. [DOI] [PubMed] [Google Scholar]
- 20.Litchfield JT, Wilcoxon FA. Simplified method of evaluating dose-effect experiments. J Pharmacol Exp Ther. 1949;96:99–112. [PubMed] [Google Scholar]
- 21.Pomilio AB, Tettamanzi MC. NMR study of substituted 1-bromo-2-aryloxyethanes and monosubstituted xanthones. Magn Reson Chem. 1996;34(2):165–171. [Google Scholar]
- 22.Harwood LM. “Dry column” flash chromatography. Aldrichimica Acta. 1985;18(1):25. [Google Scholar]
- 23.Braden MR, Parrish JC, Naylor JC, Nichols DE. Molecular interactions of serotonin 5-HT2A receptor residues Phe339 (6.51) and Phe340 (6.52) with superpotent N-benzyl phenethylamine agonists. Mol Pharmacol. 2006;70:1956–1964. doi: 10.1124/mol.106.028720. [DOI] [PubMed] [Google Scholar]
- 24.Chambers JJ, Kurrasch-Orbaugh DM, Nichols DE. Translocation of the 5-alkoxy substituent of 2,5-dialkoxyarylalkylamines to the 6-position: effects on 5-HT(2A/2C) receptor affinity. Bioorg Med Chem Lett. 2002;12:1997–1999. doi: 10.1016/s0960-894x(02)00306-2. [DOI] [PubMed] [Google Scholar]
- 25.Marona-Lewicka D, Thisted RA, Nichols DE. Distinct temporal phases in the behavioral pharmacology of LSD: dopamine D2 receptor-mediated effects in the rat and implications for psychosis. Psychopharmacology (Berl) 2005;180:427–435. doi: 10.1007/s00213-005-2183-9. [DOI] [PubMed] [Google Scholar]