

Abstract
Transcription factor NF-κB controls the expression of multiple genes involved in immunity and inflammation. The initial activation and duration of NF-κB signaling is regulated by posttranslational modifications to IκB kinase, which earmarks inhibitors of NF-κB for degradation. Prior studies suggest that K63-linked ubiquitination of NEMO (NF-κB essential modulator), an IκB kinase regulatory subunit, is critical for NF-κB and MAPK signaling following engagement of Ag receptors. We now demonstrate that NF-κB and MAPK pathways are largely unaffected in primary cells from mice harboring a ubiquitination-defective form of NEMO, NEMO-KR. TLR- but not Ag receptor-induced cellular responses are impaired in NEMO-KR mice, which are more resistant to LPS-induced endotoxic shock than wild-type animals. Thus, one function of NEMO ubiquitination is to fine tune innate immune responses under TLR control.
Nuclear factor-κB signaling is biochemically coupled to multiple receptors that mediate innate and adaptive immunity (1, 2). These include Ag receptors (AgRs)3 on lymphocytes, pattern recognition receptors on innate immune cells (TLRs), and receptors for proinflammatory cytokines. NF-κB is regulated from the cytoplasmic compartment by protein inhibitors, including IκBα, IκBβ, and IκBε (1). Proinflammatory agonists such as TNF, IL-1, or LPS induce phosphorylation of IκB proteins, which are targeted for K48-linked ubiquitination and degradation (3). The signal-dependent enzyme that phosphorylates IκB proteins is a multicomponent complex composed of two IκB kinases (IKKα and IKKβ) and a noncatalytic subunit called NF-κB essential modulator (NEMO or IKKγ) (4). NEMO contains a C-terminal zinc finger motif that is required for NF-κB activation by certain agonists. Mutations within this domain can cause skin inflammation or humoral immunodeficiencies in humans (5).
In vitro studies suggest that K63-linked ubiquitination within the NEMO zinc finger domain (Lys392 in mice, Lys399 in humans) plays a key role in TCR signaling (6, 7). TCR ligation drives the assembly of a signaling complex that includes IKK and the protein-ubiquitin (Ub) ligase TRAF6. In collaboration with the Ub-conjugating enzyme Ubc13, TRAF6 mediates K63-linked ubiquitination of NEMO (3). Targeted disruption of the Ubc13 gene impairs T cell activation as well as TLR signaling in macrophages and B cells (6, 8). Ubc13 deficiency in T cells causes defects in NF-κB and MAPK activation, suggesting that NEMO ubiquitination at Lys392 impinges on both pathways (6). However, Ubc13 targets multiple proteins, raising the possibility that ubiquitination of a substrate other than NEMO regulates NF-κB and MAPK signaling.
To address this issue, we introduced a point mutation (Lys392→Arg392 or K392R) into the mouse germline that specifically disrupts K63-linked ubiquitination of NEMO at Lys392 while sparing other Ubc13 substrates (NEMO-KR mice). NEMO-KR mice develop normal numbers of lymphocytes with no apparent defect in AgR signaling. However, primary macrophages and dendritic cells (DCs) from NEMO-KR mice exhibit blunted cytokine responses to TLR agonists. Consistent with this, survival rates among NEMO-KR mice are improved relative to those of control animals when challenged with doses of LPS that elicit lethal endotoxic shock. Surprisingly, NF-κB and MAPK signaling are largely unaffected in NEMO-KR cells, suggesting that Ubc13 impinges on these pathways via a mechanism independent of NEMO ubiquitination. We conclude that K63-linked ubiquitination of NEMO at Lys-392 functions as a rheostat for TLR-dependent responses in innate immunity.
Materials and Methods
NEMO-KR mice
The NEMO-targeting vector contained a point mutation in the Lys392 codon (AAG) that converted it to an Arg codon (CGG). A floxed phosphoglycerate kinase (PGK)-neomycin (Neo) cassette was inserted into a StuI site between exons 9 and 10. The short and long homology arms were generated from the NcoI-StuI fragment encompassing exon 9 (1.6 kb) and the StuI-KpnI fragment encompassing exon 10 (4.0 kb). Generation of NEMO-KR mice (Mouse Genome Informatics no.3775942; Ikbkgtm1dwb) was performed as described (9). All experiments were conducted according to guidelines of the Animal Care and Use Committee at Vanderbilt University (Nashville, TN).
Immunizations
Keyhole limpet hemocyanin (KLH; Sigma-Aldrich) in CFA (Difco) was injected i.p. (100 μg/50 μl/mouse). Serum collection and ELISA for KLH-specific Abs were performed as described (10).
Cell purification
Splenic B and T lymphocytes were purified using CD43-negative selection and pan-T cell isolation kits, respectively (Miltenyi Biotec). For macrophages, mice were injected i.p. with 3% thioglycolate (2 ml; Sigma-Aldrich). Four days postinjection, peritoneal exudate cells were collected and cultured overnight in complete RPMI 1640 medium. Adherent cells were used as peritoneal macrophages. Bone marrow DC (BMDC) and macrophages were generated as described (11, 12).
Cell-based assays
For cytokine responses, macrophages or BMDC were treated with agonists for 24h(5 × 104 cells/well). Levels of TNF, IFN-γ, and IL-6 in supernatants were measured using a BD CBA6 kit (BD Biosciences). Mouse IL-12/p40 was detected by ELISA (BD OptEIA Set) following cellular stimulation with LPS (100 ng/ml) and IFN-γ (30 ng/ml).
Biochemical analyses
Macrophage lysates were prepared and probed on Western blots for NF-κB and MAPK signaling components as described (8).
Results
Generation of NEMO-KR mice
To assess the physiologic role of NEMO ubiquitination, we introduced a point mutation into the mouse germline that prevents Ub attachment to Lys392. The X-linked NEMO allele in embryonic stem (ES) cells was targeted as shown in Fig. 1A. Homologous recombinants were confirmed by Southern blotting (Fig. 1B). The floxed PGK-Neo cassette was deleted from the KR-Neo allele following transfection of ES cells with a Cre expression vector, yielding the NEMO-KR allele (Fig. 1B). These cells were used to generate NEMO-KR mice, from which the mutation was verified by sequence analysis. Male NEMO-KR mice were born in the expected Mendelian ratio and exhibited no overt phenotypes.
FIGURE 1.
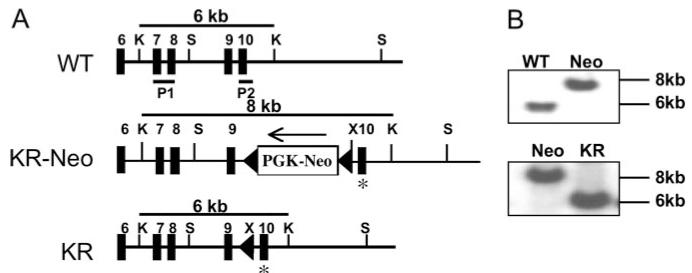
Characterization of NEMO-KR mice. A, Structure of the mouse NEMO gene and knock-in alleles. Filled boxes represent NEMO coding exons. The asterisk denotes the Lys→Arg mutation at codon 392. P1 and P2 correspond to probes used for Southern blotting. Restriction enzyme sites: K, KpnI; S, SacI; X, XhoI. Cre-mediated deletion of PGK-Neo from KR-Neo ES cells generated the NEMO-KR allele. B, Southern blot analysis. Probes and restriction fragments are shown in A. The NEMO gene is X-linked and generates only a single band in male ES cells.
Role of NEMO ubiquitination in adaptive immunity
Disruption of the gene encoding Ubc13 inhibits signal-dependent ubiquitination of NEMO and causes a profound deficiency in mature T cells (6, 8). Because Ubc13 modifies multiple target proteins, NEMO-KR mice provide a tractable model to define the in vivo contribution of NEMO ubiquitination to this phenotype. Unlike Ubc13 knockout mice, NEMO-KR animals had normal levels of mature T lymphocytes as well as B cells, thymocytes, macrophages, DCs, and specialized lymphocyte subsets (B1, invariant NKT, and regulatory T cells) (Fig. 2A; data not shown). Steady-state levels of Ig isotypes were comparable in NEMO-KR and control sera (data not shown).
FIGURE 2.
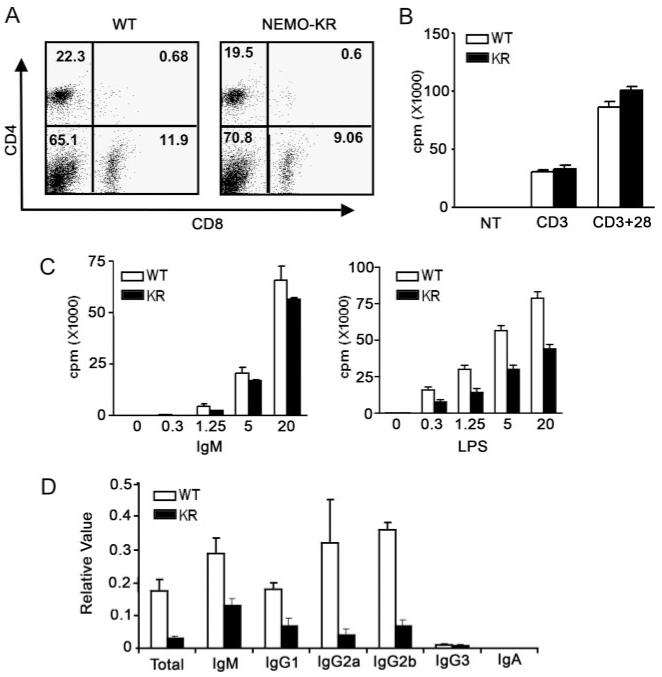
Impaired humoral immunity in NEMO-KR mice. A, Normal T cell populations in NEMO-KR mice. Splenocytes were stained with Abs for CD4/CD8 and analyzed by FACS. B, T cell proliferation. Purified T cells (106 cells/ml) were cultured with plate-bound anti-CD3 (10 μg/ml), anti-CD3 plus CD28 (10 plus 2 μg/ml, respectively), or not treated (NT). New DNA synthesis was measured by [3H]thymidine uptake. Data in B and C are presented as mean values (±SEM) for three mice (representative of at least two independent experiments). C, B cell proliferation. Purified B cells were cultured with escalating doses of anti-IgM or LPS as indicated (μg/ml) and analyzed as in B. D, Humoral immunity. Mice were immunized with KLH in CFA and sera were collected 2 wk postimmunization. Relative levels of KLH-specific Abs were measure by ELISA. Values represent the mean (±SEM) for four pairs of mice (representative of three independent experiments).
Studies of transformed T cells and Ubc13-deficient lymphocytes suggested an important role for NEMO ubiquitination in TCR/NF-κB signaling (6, 7). In contrast, the proliferative response of mature T cells and thymocytes from NEMO-KR mice was normal following TCR cross-linking or treatment with PMA/ionomycin (Fig. 2B; data not shown). Splenic B cells from NEMO-KR mice displayed normal proliferative responses to BCR or CD40 ligation, whereas TLR4-induced growth was modestly inhibited following treatment with LPS (Fig. 2C; data not shown). We conclude that K63-linked ubiquitination of NEMO at Lys-392 plays no discernible role in AgR-mediated proliferation of primary lymphocytes ex vivo.
To determine whether the NEMO-KR mutation affects humoral immunity, we injected mice with KLH, a T-dependent immunogen. KLH elicited a robust Ab response in control animals (Fig. 2D); however, this response was consistently attenuated in NEMO-KR mice. These findings indicate that NEMO ubiquitination modulates intracellular signaling pathways involved in humoral immunity, perhaps including those that emanate from TLR4. Although defective AgR signaling may contribute to the observed in vivo phenotype, our ex vivo data suggest otherwise (Figs. 2, B and C).
Impaired cytokine expression by NEMO-KR cells
In addition to its role in B cell activation, TLR4 is important for pathogen recognition by innate immune cells, including macrophages and DC. To test whether the TLR4 signaling defect in B cells is lineage specific, we stimulated peritoneal-derived macrophages with LPS and measured cytokine secretion. LPS-induced production of IL-6 and IL-12/p40 by NEMO-KR macrophages was significantly impaired, whereas TNF and MCP-1 secretion was only modestly reduced relative to wild-type (WT) controls (Fig. 3A). Moreover, agonists specific for TLR2 (heat-killed Listeria monocytogenes), TLR1/TLR2 (tripalmitoylated lipopeptide), and TLR2/TLR6 (N terminus of lipoprotein 44 from Mycoplasma salivarium) induced lower levels of IL-6 secretion by NEMO-KR vs WT macrophages (Fig. 3B). Secretion of IL-12/p40 was also diminished, albeit modestly, in NEMO-KR macrophages (data not shown).
FIGURE 3.
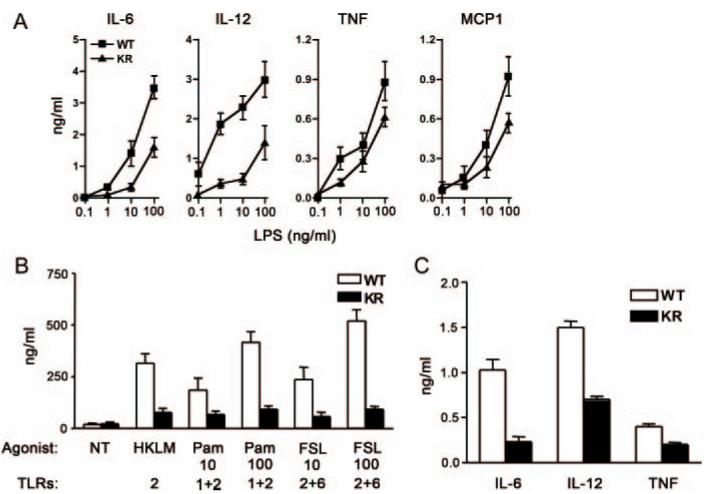
Blunted cytokine responses in NEMO-KR cells. A, Cytokine expression. Peritoneal macrophages were cultured with the indicated concentrations of LPS in the presence (IL-12/p40 assays) or absence (IL-6, TNF, and MCP-1 assays) of 30 ng/ml IFN-γ (24 h). Data represent mean cytokine concentrations (±SEM) in culture supernatants from five mice (representative of three independent experiments). B, Peritoneal macrophages were cultured with ligands for TLR2 (heat-killed L. monocytogenes (HKLM); 108 cells/ml), TLR1/2 (tripalmitoylated lipopeptide (Pam); 10 and 100 ng/ml), and TLR 2/6 (N terminus of lipoprotein 44 from M. salivarium (FSL); 10 and 100 ng/ml). After 24 h, IL-6 was measured in culture supernatants as in A. NT, Not treated. C, BMDC were stimulated with LPS (1 ng/ml; 24 h) and cytokines were measured as in A. Data are presented as mean cytokine concentrations from triplicate cultures (±SEM; representative of three independent experiments).
In complementary studies with BMDC cultures (Fig. 3C)we found that the NEMO-KR mutation attenuated secretion of IL-6, IL-12/p40, and TNF following LPS treatment (1 ng/ml). Higher doses of LPS rescued the defect in cytokine secretion by BMDC from NEMO-KR mice (>10 ng/ml, data not shown). This defect could not be attributed to impaired activation or Ag presentation by BMDC because LPS induced similar levels of costimulatory molecules (MHC II and CD40) and proliferation of allogeneic lymphocytes in both genetic backgrounds (data not shown). Thus, K63-linked ubiquitination of NEMO impinges on multiple TLR signaling pathways to regulate cytokine production by macrophages and DC.
NF-κB and MAPK signaling in NEMO-KR cells
Ubc13 deficiency leads to profound defects in MAPK but not NF-κB signaling in macrophages (8), whereas both of these pathways are impaired in Ubc13-deficient thymocytes (6). To determine whether NEMO ubiquitination contributes to these lineage-specific phenotypes, we performed biochemical studies with macrophages and thymocytes. Initial experiments confirmed the following: 1) NEMO-KR protein is expressed at normal levels; 2) NEMO-KR associates efficiently with IKKβ; and 3) the point mutation inhibits LPS-induced ubiquitination of NEMO but not TRAF6 in macrophages (Fig. 4A; data not shown). Blockade of NEMO ubiquitination had no major effect on LPS-induced degradation of IκB proteins in macrophages, although the kinetics of IκBβ and IκBε breakdown were delayed slightly (Fig. 4B). Consistent with this, LPS-induced activation of IKKβ and nuclear NF-κB was unaffected in NEMO-KR macrophages over a 10-h time course (data not shown). Similar levels of MAPK phosphorylation (p38, JNK, and ERK) were observed in NEMO-KR and WT macrophages treated with LPS (Fig. 4C). NF-κB and MAPK signaling was also normal in thymocytes treated with PMA/ionomycin or in cytokine-stimulated mouse embryonic fibroblasts (IL-1β and TNF) despite a defect in NEMO ubiquitination (data not shown). Thus, the NF-κB and MAPK signaling defects reported for Ubc13-deficient cells are not likely attributed to disruption of NEMO ubiquitination at Lys392.
FIGURE 4.
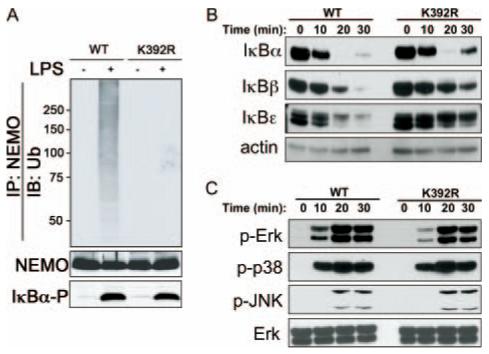
NF-κB and MAPK activation in NEMO-KR macrophages. A, Bone marrow macrophages were stimulated with LPS (100 ng/ml for 10 min). Whole cell lysates were boiled in 1% SDS and diluted to 0.1% SDS before NEMO immunoprecipitation (IP). Western blots prepared from NEMO immunocomplexes were probed with anti-Ub Abs (top). A portion of each lysate (5%) was directly blotted (IB) and probed with Abs for NEMO (loading control; middle) or phospho-IκBα (IκBα-P) (activation control; bottom). B and C, Peritoneal macrophages (5 × 106 cells/ml) were stimulated with LPS (100 ng/ml) for the indicated times. Whole cell lysates were analyzed on immunoblots using Abs specific for IκB proteins (B) or phosphorylated (p-) forms of MAPKs (C). Relative levels of β-actin and total Erk provide loading controls.
Resistance of NEMO-KR mice to LPS-induced shock
Acute exposure of the innate immune system to LPS leads to persistent NF-κB expression, excessive production of macrophage-derived cytokines (e.g., TNF), and a lethal inflammatory response called endotoxic shock (13). To test whether NEMO ubiquitination regulates this response, we measured serum cytokine levels and mortality rates among mice injected with lethal doses of LPS. As shown in Fig. 5A, 100% of the WT mice succumbed within 3 days after LPS injection, whereas 60% of NEMO-KR animals survived over the 5-day period. In keeping with these results, serum cytokine levels of TNF, IL-6, and IFN-γ in NEMO-KR mice treated with LPS were significantly reduced relative to WT animals (Fig. 5B). We conclude that K63-linked ubiquitination of NEMO at Lys-392 contributes significantly to TLR-dependent inflammatory responses in vivo.
FIGURE 5.
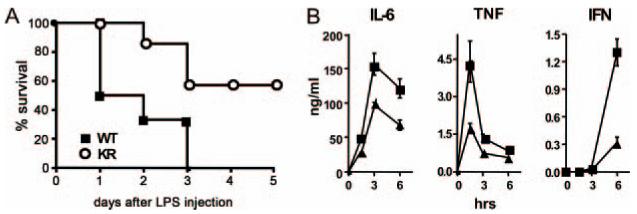
NEMO-KR mice are resistant to LPS-induced endotoxic shock. A, Age-matched WT (n = 6) and NEMO-KR (n = 7) mice were injected with LPS (2 mg per 20 g of body weight). Mortality was assessed daily. Three independent survival studies were performed with similar results (p < 0.05). B, Sera from mice in A were collected at the indicated times. TNF, IL-6, and IFN-γ levels were measured as in Fig. 3 (closed squares, WT; closed triangles, NEMO-KR). Data are presented as mean values (±SEM, representative of two independent experiments).
Discussion
We show that K63-linked ubiquitination of NEMO at Lys392 modulates cytokine responses under TLR control in innate immune cells. Prior studies with transformed T cells and Ubc13-deficient thymocytes suggested that this NEMO modification regulates TCR signaling to NF-κB and MAPKs (6, 7). Remarkably, we found that NEMO ubiquitination is largely dispensable for AgR-induced lymphocyte responses as well as TLR-induced activation of NF-κB and MAPK signaling ex vivo. Thus, many of the biochemical and cellular defects associated with Ubc13 deficiency must be mechanistically linked to modification of Ubc13 substrates other than NEMO. Of note, naturally occurring mutations in NEMO can cause skin disorders and profound immunodeficiencies. Neither of these pathologic phenotypes was manifest in NEMO-KR mice, suggesting that K63-linked ubiquitination impinges on only a subset of NEMO functions.
NEMO-KR mice display increased resistance to endotoxic shock and reduced levels of proinflammatory cytokines in their serum following LPS injection. Impaired cytokine production by NEMO-KR macrophages likely contributes to this phenotype. Paradoxically, LPS-triggered activation of the NF-κB and MAPK pathways was largely unaffected in NEMO-KR macrophages, suggesting that NEMO ubiquitination may regulate a distinct signaling pathway. Alternatively, minor quantitative changes in NF-κB and/or MAPK signaling are translated into physiologically significant shifts in cytokine production. Notwithstanding, our findings highlight K63-linked ubiquitination of NEMO as an important regulatory step in the development of a systemic inflammatory response.
Acknowledgments
We thank Drs. J. Hawiger, X.Y. Lui, and D. Lui for guidance with in vivo LPS administration.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
This work is supported by National Institutes of Health Grants AI052379, CA082556, CA081065, AI070305, AI042284, AI061721, and HL068744 and by a grant from the Leukemia and Lymphoma Society (to Z.-H.W.).
Disclosures
The authors have no financial conflict of interest.
- AgR
- Ag receptor
- DC
- dendritic cell
- BMDC
- bone marrow DC
- ES
- embryonic stem
- IKK
- IκB kinase
- KLH
- keyhole limpet hemocyanin
- NEMO
- NF-κB essential modulator
- Neo
- neomycin
- PGK
- phosphoglycerate kinase
- TRAF6
- TNFR-associated factor 6
- Ub
- ubiquitin
- WT
- wild type.
References
- 1.Li Q, Verma IM. NF-κB regulation in the immune system. Nat. Rev. Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 2.Schulze-Luehrmann J, Ghosh S. Antigen-receptor signaling to nuclear factor κB. Immunity. 2006;25:701–715. doi: 10.1016/j.immuni.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 3.Chen ZJ. Ubiquitin signalling in the NF-κB pathway. Nat. Cell Biol. 2005;7:758–765. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perkins ND. Integrating cell-signalling pathways with NF-κB and IKK function. Nat. Rev. Mol. Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 5.Courtois G, Gilmore TD. Mutations in the NF-κB signaling pathway: implications for human disease. Oncogene. 2006;25:6831–6843. doi: 10.1038/sj.onc.1209939. [DOI] [PubMed] [Google Scholar]
- 6.Yamamoto M, Sato S, Saitoh T, Sakurai H, Uematsu S, Kawai T, Ishii KJ, Takeuchi O, Akira S. Cutting edge: pivotal function of Ubc13 in thymocyte TCR signaling. J. Immunol. 177:7520–7524. doi: 10.4049/jimmunol.177.11.7520. [DOI] [PubMed] [Google Scholar]
- 7.Zhou H, Wertz I, O’Rourke K, Ultsch M, Seshagiri S, Eby M, Xiao W, Dixit VM. Bcl10 activates the NF-κB pathway through ubiquitination of NEMO. Nature. 2004;427:167–171. doi: 10.1038/nature02273. [DOI] [PubMed] [Google Scholar]
- 8.Yamamoto M, Okamoto T, Takeda K, Sato S, Sanjo H, Uematsu S, Saitoh T, Yamamoto N, Sakurai H, Ishii KJ. Key function for the Ubc13 E2 ubiquitin-conjugating enzyme in immune receptor signaling. Nat. Immunol. 2006;7:962–970. doi: 10.1038/ni1367. [DOI] [PubMed] [Google Scholar]
- 9.Afshar R, Pierce S, Bolland DJ, Corcoran A, Oltz EM. Regulation of IgH gene assembly: role of the intronic enhancer and 5′DQ52 region in targeting DHJH recombination. J. Immunol. 2006;176:2439–2447. doi: 10.4049/jimmunol.176.4.2439. [DOI] [PubMed] [Google Scholar]
- 10.Naim JO, Satoh M, Buehner NA, Ippolito KM, Yoshida H, Nusz D, Kurtelawicz L, Cramer SF, Reeves WH. Induction of hypergammaglobulinemia and macrophage activation by silicone gels and oils in female A. SW mice. Clin. Diagn. Lab. Immunol. 2000;7:366–370. doi: 10.1128/cdli.7.3.366-370.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447:972–978. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 12.Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 13.Kawai T, Akira S. TLR signaling. Cell Death Differ. 2006;13:816–825. doi: 10.1038/sj.cdd.4401850. [DOI] [PubMed] [Google Scholar]