

Abstract
Background
We reviewed our experience with micronodular adrenal hyperplasia (MAH), its pigmented variant primary pigmented nodular adrenocortical disease (PPNAD), and the association with Carney’s Complex (CNC) in order to better characterize the disorders.
Methods
This study is a retrospective analysis of clinical data and operative reports of 34 patients identified with MAH and/or PPNAD who underwent resection between 1969 and 2006 at the Clinical Research Center, an inpatient research hospital, at the National Institutes of Health. Symptoms and anthropometric and biochemical data were used to evaluate effect of resection.
Results
Fifteen patients (44%) presented as adults and 19 (56%) as children. Twenty five patients (74%) presented with non-cyclic Cushing syndrome and nine patients (26%) presented with cyclic Cushing Syndrome. Thirty one patients underwent bilateral resection; this was curative biochemically in 30 patients. Fourteen operations were performed laparoscopically (41%), and 20 were perfomed as open resections (59%). There was one post-operative complication in the laparoscopic group (7%) and 6 complications in the open group (30%) (p=0.20). Follow-up was available for 25 patients (74%). Statistically significant improvements in anthropometrics were observed for both adults and children. The most frequent manifestation of CNC requiring additional operation was cardiac myxoma which was associated strongly with an atypical (cyclic) presentation of Cushing Syndrome (p=0.009).
Conclusion
Cushing Syndrome due to MAH and PPNAD may be cured by bilateral adrenal resection. All patients should be screened for manifestations of CNC at the time of adrenal diagnosis with particular attention to cardiac disease.
INTRODUCTION
Cushing syndrome is caused by chronic glucocorticoid excess that may be adrenocorticotropic hormone (ACTH)-dependent or independent. ACTH-independent processes account for approximately 15–20% of cases in adults [1,2] and 15% in children over age seven [3]. Of these ACTH-independent causes, almost all are due to adrenal adenoma or carcinoma [1, 2, 3]. Micronodular adrenal hyperplasia (MAH), and its pigmented variant, primary pigmented nodular adrenocortical disease (PPNAD), are rare, ACTH-independent processes; their true incidence is unknown but is thought currently to be more frequent than estimated previously [4]. Macronodular adrenocortical disease (or ACTH-independent macronodular adrenocortical hyperplasia – AIMAH) is another rare condition that is most frequently seen in older patients [2].
An account of seven patients with PPNAD who underwent bilateral adrenalectomy at the Mayo Clinic between 1920 and 1986 was presented at the 1986 meeting of the American Association of Endocrine Surgeons [5]. In the late 90s, it was recognized that the majority of the PPNAD cases were part of a multiple neoplasia syndrome known as Carney Complex (CNC) [6].
Since that time, CNC has been characterized further. A 2007 review identified more than 500 patients with CNC who had been registered by the NIH-Mayo clinic and the Cochin Hospital international registry in Paris, France [7]. The constellation of manifestations of CNC include characteristic skin pigmentation, cardiac and cutaneous myxomas, PPNAD, large-cell calcifying Sertoli cell neoplasms, thyroid nodules or cancer, acromegaly, psammomatous melanotic schwannomas (PMS), breast ductal adenomas, and other lesions [7]. Cardiac myxomas were found at presentation in 53 % of 338 patients with CNC and represent a significant risk to the patient [6]. A mutation of PRKAR1A, a tumor suppressor gene, has been identified in more than half of CNC kindreds [6]. Families that do not carry the PRKAR1A mutation are thought to map to the CNC2 locus and possibly other loci [7]. Genetic defects in another gene, PDE11A, were identified recently in a few kindreds with MAH [8] and, rarely, in the general population [9].
MAH and PPNAD represent a diagnostic challenge given the rarity of the disorder. Although they are bilateral processes, the diagnosis can be difficult to make and may be mistaken for more commonly seen adrenal disorders. It is important to establish the diagnosis pre-operatively in order to avoid the need for re-operation after unilateral resection. Once the diagnosis of MAH is suspected, it is also important to screen for CNC and its potentially lethal manifestations. In the present investigation, we reviewed retrospectively the clinical presentation and treatment of PPNAD and other forms of bilateral MAH. This is a timely study given the absence of related references for more than a decade, and the recent recognition of the possible higher prevalence of these disorders.
METHODS
Thirty four patients with MAH who were operated on at the National Institutes of Health from 1969 to 2006 were identified. These patients were admitted to the NIH under clinical protocols 95CH0059 and 00CH0160; they all gave informed consent, and the studies were approved by the NICHD IRB. Demographic information, family history, signs and symptoms at presentation, duration of symptoms, anthropomorphic measures, and other findings on examination were collected from each chart. Based on guidelines of the Center for Disease Control, patients were recorded as children and adolescents if they were ≤ 19 years of age at presentation.
Patients were also classified by the presence or absence of cyclic Cushing Syndrome. Pre-operative symptoms and examination findings were recorded separately for the patients with non-cyclic Cushing Syndrome. Most of the patients with cyclic Cushing Syndrome were seen multiple times, with varying complaints and findings, until the diagnosis of an adrenal source of Cushing Syndrome was able to be made definitively.
The biochemical diagnosis of Cushing Syndrome was made using standard laboratory assays. Due to the long history of the study, patients were tested with a variety of assays. In recent years, a more standardized protocol was followed: Cushing Syndrome was diagnosed by 24-hr assay for urinary free (unbound) cortisol (UFC) and corrected for body surface area in children [1,10]. ACTH levels were measured to confirm ACTH-independence. A challenge with ovine corticotropin releasing hormone (oCRH) for ACTH and cortisol was also administered to exclude pituitary and ectopic ACTH-secreting lesions [1,10,11]. Liddle’s test, the administration of low-dose and high-dose dexamethasone, was used to differentiate among adrenal sources of Cushing Syndrome and to establish the diagnosis of PPNAD [12].
All patients underwent adrenal imaging in order to localize the process and for pre-operative planning. In recent years, imaging has been done using computed tomography (CT) [13]. In past years, patients underwent adrenal vein sampling and iodocholesterol scan, but these studies are not done currently for MAH patients. All CTs were reviewed again for this study collectively by members of the endocrine, radiology, and surgery teams.
All peri-operative data and operative notes were reviewed, and data were collected regarding pre-operative preparation, operative approach, complications, and outcome. Follow-up data were reviewed for all patients for whom it was available. Manifestations of CNC both at presentation and follow-up were recorded. Mutation status was also reported. Patients with cyclic Cushing Syndrome were analyzed both within the full cohort and separately in order to identify any trends associated with this unique presentation of ACTH-independent Cushing Syndrome.
Anthropometric data were compared in a paired fashion within the adult and pediatric cohorts at presentation and in follow-up using the Wilcoxon signed rank test. Comparisons of binary parameters between patients with cyclic and non-cyclic Cushing Syndrome were made using Fisher’s exact test. Mehta’s modification to Fisher’s exact test was used to compare pathology (in three groups) between the patients with isolated adrenal disease and those with CNC. [14] Comparisons of continuous, numeric data between patients with cyclic and non-cyclic Cushing Syndrome were done using an exact Wilcoxon rank sum test. The categorized ACTH level distributions were compared between groups using an exact Cochran-Armitage test for trend. All p-values are two-tailed and were not adjusted for multiple comparisons. [15]
RESULTS
Demographic information is presented for all patients in Table 1. The majority of patients in the full cohort were female, and a slight majority presented as children. Nine patients (26%) presented with cyclic Cushing Syndrome. There was a family history of CNC, PPNAD, or both in 9 of the 34 patients. Two of these 9 patients had cyclic and 7 had non-cyclic Cushing Syndrome.
Table 1.
Patient Characteristics (n=34)
| Adults | Children | Noncyclic CS* | Cyclic CS* | |
|---|---|---|---|---|
| Total n (%) | 15 (44) | 19 (56) | 25 (74) | 9 (26) |
| Males n (%) | 2 (13) | 6 (32) | 6 (24) | 2 (22) |
| Females n (%) | 13 (87) | 13 (68) | 19 (76) | 7 (78) |
| Mean age of presentation (yrs) ± SEM | 32 ± 3 | 10 ± 1 | 20 ± 3 | 19 ± 4 |
| Range of presentation (yrs) | 20–57 | 2.7–19 | 2.7–57 | 3.9–35 |
| Mean symptom duration (mos) ± SEM | 51 ± 11 | 22 ± 4 | 28 ± 7 | 54 ± 12 |
| Range of duration (mos) | 6–168 | 3–47 | 3–168 | 15–132 |
CS: Cushing Syndrome
Symptoms and signs of the 25 patients with non-cyclic Cushing Syndrome are listed in Table 2. Available anthropometric data are presented in Table 3.
Table 2.
Symptoms and Exam Findings in Non-cyclical Patients (n=25)
| Symptom | n (%) | Exam Finding | n (%) |
|---|---|---|---|
| Weight gain | 23 (92) | Cushing fat distribution | 21 (84) |
| Excessive hair | 11 (44) | Hirsutism | 14 (56) |
| Growth arrest* | 10 (40) | Striae | 13 (52) |
| Fatigue | 10 (40) | Hyperpigmentation | 11 (44) |
| Weakness | 8 (32) | Lentigines | 9 (36) |
| Psychiatric symptoms | 5 (20) | Acne | 7 (28) |
| History of hypertension | 6 (24) | Muscle weakness | 6 (24) |
| Problems associated with CNC+ | 4 (16) | Acanthosis nigricans | 3 (12) |
| Fractures | 4 (16) | Cardiac murmur | 3 (12) |
| Sleep disturbance | 3 (12) | Skin myxoma | 2 (8) |
growth arrest was reported in 9/19 pediatric patients and 1 adult patient whose symptoms had been present since age 18
CNC: Carney’s Complex
Table 3.
Anthropometric Data
| Adults | Presentation | n | 1 year follow up | n | change | p* |
|---|---|---|---|---|---|---|
| Mean BMI ± SEM | 30 ± 1.6 | 15 | 27 ± 1.4 | 8 | −2 ± 0.5 | 0.016 |
| Children | Presentation | n | 1 year follow up | n | change | p* |
| Mean Height for age % ± SEM | 26 ± 6.3 | 17 | 34 ± 8.2 | 12 | 13 ± 3.7 | 0.003 |
| Mean Weight for age % ± SEM | 77 ± 6.7 | 19 | 56 ± 33 | 13 | −20 ± 5.1 | 0.002 |
p value calculated using Wilcoxon signed rank test
Biochemical assays that established the diagnosis of Cushing Syndrome are presented in Figure 1. The standard Liddle’s test did not become part of the diagnostic investigation of MAH until 1998 [12]; only patients seen at the NIH after the mid-90s had this investigation.
Figure 1. Laboratory data.

Comparisons of continuous, numerical data between patients with cyclic and non-cyclic Cushing Syndrome were done using an exact Wilcoxon rank sum test. The categorized ACTH level distributions were compared between groups using an exact Cochran-Armitage test for trend.
Adrenal CTs were obtained at presentation in all patients except for one patient who first presented in 1969 (33/34). Of 33 CTs, 24 were read as consistent with micronodular and/or hyperplastic disease, and 9 were read as normal. Representative images are shown in Figure 2. Seven patients underwent an iodocholesterol scan; one patient had adrenal venous sampling.
Figure 2. Adrenal CT images.
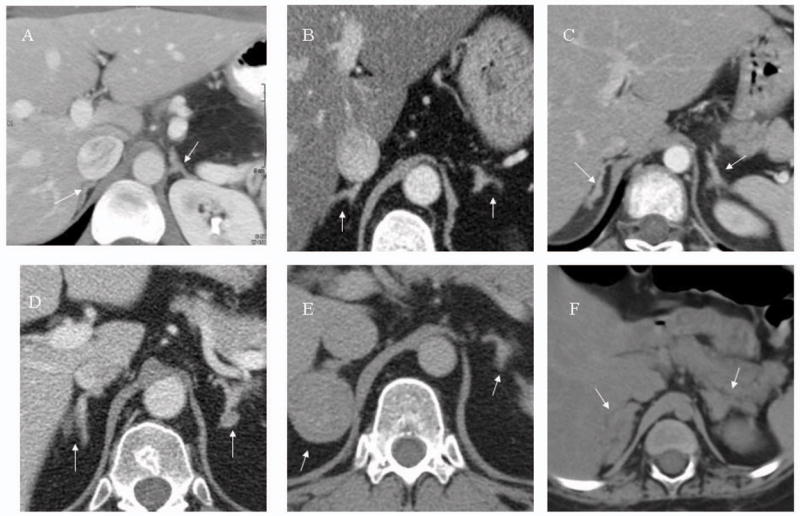
Digitally processed CT images reproduced at 200% magnification. Arrows point to adrenal glands.
- Normal adrenal glands in a patient without adrenal disease
- Essentially normal appearing adrenal glands in a patient with PPNAD.
- Bilateral micronodular hyperplasia
- Classic “beads-on-a-string” appearance of adrenal glands in a patient with PPNAD
- Right adrenal adenoma in the setting of bilateral micronodular hyperplasia
- Bilateral micronodular hyperplasia with more than one macronodule.
Five patients had severe hypercortisolemia pre-operatively and received adrenolytic medications for up to 8 weeks prior to operation [16]. Medications included ketoconazole, aminoglutethimide, and mitotane. One pediatric patient required four anti-hypertensive medications for blood pressure control pre-operatively. Fourteen operations (41%) were performed laparoscopically, while 18 (53%) were performed via an open approach. Two laparoscopic operations were attempted and converted to open (6%) for technical reasons.
Thirty-one patients underwent bilateral resection which was curative biochemically in 30 (97%) of these patients. The remaining patient underwent re-exploration of the adrenal beds, no gross adrenal tissue was seen, and multiple frozen sections were negative; following this operation, however, she was cured of disease. Three patients underwent unilateral resection. Two of these resections were terminated after unilateral resection; neither patient was cured biochemically, but symptoms were more manageable and both patients elected not to undergo resection of the contralateral side. The final patient had been referred to the NIH after an incomplete resection and was cured after the remaining adrenal gland was removed.
There was one post-operative complication among the 14 laparoscopic patients (7%) which consisted of a brief episode of transient unexplained acute renal failure postoperatively. There were six post-operative complications among the 20 open adrenal resections (30%), consisting of a staphylococcus bacteremia, two wound infections, bleeding requiring re-operation, and pneumonia. The final complication was a case of pancreatitis which resulted in a mortality.
All patients received glucocorticoid supplementation during and after adrenalectomy. All patients were discharged on hydrocortisone and fludrocortisone. The mean discharge dose of hydrocortisone was 12.8 mg per meter squared (± 0.4) for the 31 patients who were cured at first operation. The standard discharge dose of fludrocortisone was 100 mcg (four patients required more). Six patients presented with hypertension and were taking one or often multiple anti-hypertensive medications pre-operatively. Post-operatively, hypertension in these patients was cured or improved. Five patients were able to stop all anti-hypertensive therapy while the remaining patient regimen required just one medication.
Long-term follow-up was available for 25 of the 34 patients (74%). Mean follow-up was 6.7 years (± 1.2) with a range of 1 to 27 years. Measurements for height and weight are shown in Table 3. All 25 patients reported improved or resolved symptoms of Cushing Syndrome. Complete resolution of Cushing Syndrome symptoms in these patients took place by 9–12 months post-operatively. Post-operative UFCs were reported undetectable in all patients who were cured of their disease. There were no long term complications in any of the 33 surviving patients. Patients that had unilateral adrenalectomy did not require any glucocorticoid replacement but all patients who had bilateral adrenalectomy continued physiologic replacement for life. Three patients experienced episodes of acute gluco- or mineralocorticoid deficiency almost always in the context of a concurrent illness such as viral syndrome, dehydration or other disease.
Most PPNAD patients also had CNC (18, 53%). Patients with isolated MAH were the second largest group with micronodular adrenal disease (7, 21%). There were six patients that had isolated MAH with pigmentation (21%). There was only one patient with PPNAD and no CNC signs (3%), and one patient with MAH and PPNAD and CNC signs (3%). The difference between patients with PPNAD and CNC (19) and those with other forms of MAH and no CNC (isolated, 14) was statistically significant (p<0.001). Representative pathologic images are shown in Figure 3. The CNC patient who had MAH/PPNAD did not have the PRKAR1A mutation, making the clinical diagnosis of CNC uncertain. Of note, one of the isolated MAH patients was identified with a mutation in the PDE11A gene [8].
Figure 3. Pathology: PPNAD.

Digitally processed photomicrographs of adrenal glands with hematoxylin and eosin stain. Left panel: PPNAD with discrete nodules within the section. Entire tissue section imaged with Mosaix© software, 10X. Right panel: Pigmented Nodule. 40X magnification of area surrounded by box in left panel.
The results of the Liddle’s test were correlated with the histopathology. An increase of 100% or greater in UFC on day 6 was observed in 5 of 6 non-cyclic PPNAD patients and both cyclic PPNAD patients, as well as MAH/PPNAD patient in each cohort. An increase of 50% was observed in two patients with MAH, one with PPNAD, and one with MAH/PPNAD in the patients with non-cyclic Cushing Syndrome. The Liddle’s test was negative in one MAH/PPNAD patient with non-cyclic Cushing Syndrome and one MAH patient with cyclic Cushing Syndrome.
All patients were screened for CNC at initial evaluation (except for the few patients who presented before CNC was fully described) and in follow-up. The most frequent manifestations of CNC in the 19 patients with both adrenal disease and CNC are shown in Table 4. There was a difference in the incidence of cardiac myxomas between patients with cyclic versus those with non-cyclic Cushing Syndrome (6/6 vs. 4/13, p = 0.009).
Table 4.
| Manifestation | All Patients | Cyclical CS§ with CNC | Non-cyclical CS with CNC | p[] |
|---|---|---|---|---|
| n=19 | n=6 | n=13 | ||
| PRKAR1A mutation | 18 (95%) | 6 | 12 | 1.00 |
| PPNAD¶ | 18 (95%) | 6 | 12 | 1.00 |
| Pigmentation | 12 (63%) | 5 | 7 | 0.33 |
| Cardiac Myxoma | 10 (53%) | 6 | 4 | 0.009 |
| LCCST# (males) n=5 | 4 (80%) | 2 | 2 | 1.00 |
| Thyroid cancer or nodule | 12 (63%) | 5** | 7 | 0.22 |
| PMS++ | 3 (21%) | 1 | 2 | 1.00 |
CNC: Carney’s Complex
MAH: Micronodular Adrenal Hyperplasia
One patient’s CNC status is still pending.
CS: Cushing syndrome
calculated using Fisher’s exact test.
PPNAD: Primary Pigmented Nodular Adrenocortical Disease
LCCST: large-cell calcifying Sertoli cell tumors
2 patients with cyclical CS had cancer; one died of follicular cancer.
psammomatous melanotic schwannomas
Of the 10 patients with cardiac myxomas, 6 had undergone resection prior to coming to attention for adrenal disease. Two of these patients were diagnosed at the time of evaluation for adrenal disease at NIH, and adrenalectomy was delayed in order to proceed with resection of the myxoma first. This delay also occurred at an outside hospital, delaying a planned unilateral adrenalectomy for one patient, and she was referred to the NIH for CNC and PPNAD evaluation after resection of the myxoma. Of note, two patients suffered strokes from their myxomas prior to resection. Finally, three patients were diagnosed with cardiac myxomas in follow-up after adrenalectomy.
Of the 12 patients with thyroid cancer or nodules, 2 patients were diagnosed with cancer. One patient was cured of papillary cancer after resection, but the second patient died of metastatic follicular cancer. Four of five males in the series were found to have testicular calcifications suggestive of Sertoli cell neoplasms; these patients were all followed by ultrasonographic surveillance. Although only three of 19 patients (16%) had proven schwannomas, all of these patients required resection of these lesions due to symptoms.
DISCUSSION
Patients with MAH and PPNAD in our series had classic findings of suppressed ACTH, loss of diurnal cortisol variation, and lack of cortisol and ACTH responses to CRH. The Liddle’s test can be a final arbiter of the diagnosis; PPNAD is diagnosed when a 50% increase of UFC is seen over baseline on day 6 of the test [12]. The cause of this increase has been shown to be increased expression of glucocorticoid receptor (GR) in PPNAD nodules [17, 18], but the mechanism of GR stimulation is unknown. The present study shows that not all patients with the non-pigmented variant of bilateral micronodular hyperplasia, MAH, respond to dexamethasone as patients with PPNAD.
The patients who were not cured after unilateral resection illustrate the importance of bilateral resection. The patient who continued to have biochemical evidence of Cushing Syndrome after bilateral resection but who was cured after complete excision of residual tissue within the operative fields also demonstrates how even small-to-microscopic nests of cells affected by micronodular forms of adrenocortical hyperplasia can continue to cause glucocorticoid excess.
There was no difference in cure rate between the laparoscopic and open approaches. Our preferred approach is laparoscopic if possible in order to minimize morbidity. Improvement in patient status was immediate after resection in those patients who were hypertensive pre-operatively. Anthropometrics reveal statistically significant decreases in BMI for the adult cohort and significant increases and decreases in height for age and weight for age percentiles respectively for the pediatric cohort. Patients were maintained safely on glucocorticoid and mineralocorticoid replacement. As in other series of patients with ACTH-independent Cushing Syndrome, none of our patients developed Nelson’s syndrome [13,19].
Hitherto, the micronodular adrenocortical hyperplasias have not been subdivided into different diseases. Over the last decade it has become clear that there are at least two different diseases, MAH and its pigmented variant PPNAD [4]; patients with intermediate histologic findings also exist (MAH/PPNAD). The main difference between the two forms is the presence of lipofuscin pigmentation (PPNAD) or its absence (MAH); mutations of the PRKAR1A gene are also seen almost exclusively in PPNAD [20]. Representative pathologic examples of PPNAD and MAH in this series are shown in Figures 3 and 4 respectively. It should be noted that although PPNAD is a widely accepted term, MAH was only proposed recently by Stratakis and colleagues [4,8] and awaits final classification and terminology.
Figure 4. Pathology: MAH.

Digitally processed photomicrographs of adrenal glands with hematoxylin and eosin stain. Left panel: MAH with multiple nodules and hyperplasia throughout the section. Entire tissue section imaged with Mosaix© software, 10X. Right panel: Non-pigmented nodule. 40X magnification of area surrounded by box in left panel.
The frequency of CNC manifestations other than adrenal disease in our series are similar to previous reports of patients with CNC [6]. Cardiac myxomas, thyroid cancer, and psammomatous melanotic schwannoma were the most frequent causes of related operations in our patients. Cardiac myxomas, occurring in 53% of our patients, were the most serious, and have been reported previously to cause more than 50% of mortalities in these patients [7]. Three adrenal operations were delayed in order to first address the cardiac myxomas and two patients suffered strokes from these lesions prior to resection. Three patients were diagnosed with cardiac myxomas in follow-up, and four patients had multiple myxomas requiring multiple operations. These data highlight the importance of screening patients with suspected PPNAD for CNC both at the time of presentation, in order to avoid a catastrophic intra-operative event during adrenalectomy and in follow-up.
It is interesting that in our series, cyclic presentation of Cushing Syndrome was significantly associated with cardiac myxoma. Cyclic symptomatology is a rare presentation of Cushing Syndrome and has been described primarily in the pediatric literature and often associated with PPNAD [21]. Cyclic (or periodic) Cushing Syndrome is a cause of atypical Cushing Syndrome which is difficult to diagnose because of incomplete suppression of the hypothalamic-pituitary-adrenal axis and, therefore, the inability to use dexamethasone suppression testing toward that purpose. The lack of proper testing coupled with the periodicity of symptomatology makes the diagnosis of cyclic Cushing Syndrome extremely hard. The etiology of the association of cyclic Cushing Syndrome and cardiac myxoma is unclear. Possible pathophysiologic explanations for this association include hemodynamic changes affecting adrenocortical function due to the presence of a heart neoplasm or the known production of the inflammatory cytokine, interleukin-6 (IL-6), by heart myxomas [22]. IL-6 is an important regulator of adrenocortical function [23].
Our data as well as previously published reports [5] demonstrate that MAH and PPNAD are curable disorders by adrenalectomy. The diagnosis should be considered whenever an adrenal source of Cushing Syndrome is established and should be excluded whenever imaging is equivocal or family history is suggestive. Liddle’s test should be used for the diagnosis. Laparoscopic bilateral resection is curative with careful technique and carries minimal morbidity. Gluco- and mineralocorticoid replacement should be started immediately after stress doses are tapered. Patients with PPNAD should be followed closely for other manifestations of CNC, especially cardiac myxomas.
Acknowledgments
This study was supported in part by the intramural program of the National Institute of Child Health and Human Development through a grant, Z01HD000642-04 (PI: CS Stratakis).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Arnaldi G, Angeli A, Atkinson AB, et al. Diagnosis and complications of Cushing’s syndrome: A consensus statement. J Clin Endocrinol Metab. 2003;88(12):5593–5602. doi: 10.1210/jc.2003-030871. [DOI] [PubMed] [Google Scholar]
- 2.Lacroix A, Bordeau I. Bilateral adrenal Cushing syndrome: Macronodular adrenal hyperplasia and primary pigmented nodular adrenocortical disease. Endocrinol Metab Clin N Am. 2005;34:441–458. doi: 10.1016/j.ecl.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 3.Magiakou MA, Chrousos GP. Cushing syndrome in children and adolescents: Current diagnosis and therapeutic strategies. J Endocrinol Invest. 2002;25:181–194. doi: 10.1007/BF03343985. [DOI] [PubMed] [Google Scholar]
- 4.Stratakis CA. Adrenocortical tumors, primary pigmented adrenocortical disease (PPNAD)/Carney Complex, and other bilateral hyperplasias: The NIH studies. Horm Metab Res. 2007;39(6):467–473. doi: 10.1055/s-2007-981477. [DOI] [PubMed] [Google Scholar]
- 5.Grant C, Carney JA, Carpenter PC, van Heerden JA. Primary pigmented nodular adrenocortical disease: Diagnosis and management. Surgery. 1986;100:1178–1183. [PubMed] [Google Scholar]
- 6.Stratakis CA, Kirschner LS, Carney JA. Clinical and Molecular Features of the Carney Complex: Diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab. 2001;86(9):4041–4046. doi: 10.1210/jcem.86.9.7903. [DOI] [PubMed] [Google Scholar]
- 7.Boikos SA, Stratakis CA. Carney complex: the first 20 years. Curr Opin Oncol. 2007;19:24–29. doi: 10.1097/CCO.0b013e32801195eb. [DOI] [PubMed] [Google Scholar]
- 8.Horvath A, Boikos S, Giatzakis C, Robinson-White A, Groussin L, Griffin KJ, Stein E, Levine E, Delimpasi G, Hsiao HP, Keil M, Heyerdahl S, Matyakhina L, Libe R, Fratticci A, Kirschner LS, Cramer K, Gaillard RC, Bertagna X, Carney JA, Bertherat J, Bossis I, Stratakis CA. A genome-wide scan identifies mutations in the gene encoding phosphodiesterase 11A4 (PDE11A) in individuals with adrenocortical hyperplasia. Nat Genet. 2006;38(7):794–800. doi: 10.1038/ng1809. [DOI] [PubMed] [Google Scholar]
- 9.Horvath A, Giatzakis C, Robinson-White A, Boikos S, Levine E, Griffin K, Stein E, Kamvissi V, Soni P, Bossis I, de Herder W, Carney JA, Bertherat J, Gregersen PK, Remmers EF, Stratakis CA. Adrenal hyperplasia and adenomas are associated with inhibition of phosphodiesterase 11A in carriers of PDE11A sequence variants that are frequent in the population. Cancer Res. 2006;66(24):11571–5. doi: 10.1158/0008-5472.CAN-06-2914. [DOI] [PubMed] [Google Scholar]
- 10.Batista DL, Riar J, Keil M, Stratakis CA. Diagnostic tests for children who are referred for the investigation of Cushing syndrome. Pediatrics. 2007;120(3):575–86. doi: 10.1542/peds.2006-2402. [DOI] [PubMed] [Google Scholar]
- 11.Nieman LK, Oldfield EH, Wesley R, Chrousos GP, Loriaux DL, Cutler GB. A simplified morning ovine corticotrophin-releasing hormone stimulation test for the differentiation of adrenocorticotropin-dependent Cushing’s Syndrome. J Clin Endocrinol Metab. 1993;77 (5):1308–1312. doi: 10.1210/jcem.77.5.8077325. [DOI] [PubMed] [Google Scholar]
- 12.Stratakis CA, Sarlis N, Kirschner LS, Carney JA, Doppman JL, Nieman LK, Chrousos GP, Papanicolaou DA. Paradoxical response to dexamethasone in the diagnosis of primary pigmented nodular adrenocortical disease. Ann Intern Med. 1999;131:585–591. doi: 10.7326/0003-4819-131-8-199910190-00006. [DOI] [PubMed] [Google Scholar]
- 13.Doppman JL, Travis WD, Nieman L, Miller DL, Chrousos GP, Gomez MT, Cutler GB, Loriaux DL, Norton JA. Cushing syndrome due to primary pigmented nodular adrenocortical disease: findings at CT and MR Imaging. Radiology. 1989;172:415–420. doi: 10.1148/radiology.172.2.2748822. [DOI] [PubMed] [Google Scholar]
- 14.Mehta CR, Patel NR. A network algorithm for performing Fisher’s exact test in r x c contingency tables. J Am Stat Assoc. 1983;78:427–434. [Google Scholar]
- 15.Agresti A. Categorical Data Analysis. New York: John Wiley and Sons, Inc.; 1990. pp. 79–129. [Google Scholar]
- 16.Morris D, Grossman A. The medical management of Cushing’s syndrome. Ann NY Acad Sci. 2002;970:119–133. doi: 10.1111/j.1749-6632.2002.tb04418.x. [DOI] [PubMed] [Google Scholar]
- 17.Bourdeau I, Lacroix A, Schurch W, Caron P, Antakly T, Stratakis CA. Primary pigmented nodular adrenocortical disease: paradoxical responses of cortisol secretion to dexamethasone occur in vitro and are associated with increased expression of the glucocorticoid receptor. J Clin Endocrinol Metab. 2003;88:3931–3937. doi: 10.1210/jc.2002-022001. [DOI] [PubMed] [Google Scholar]
- 18.Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, Cho-Chung YS, Stratakis CA. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet. 2000;26(1):89–92. doi: 10.1038/79238. [DOI] [PubMed] [Google Scholar]
- 19.Travis WD, Tsokos M, Doppman JL, Nieman L, Chrousos GP, Cutler GB, Loriaux DL, Norton JA. Primary pigmented nodular adrenocortical disease. Am J Surg Path. 1989;13(11):921–930. [PubMed] [Google Scholar]
- 20.Bertherat J, Groussin L, Sandrini F, Matyakhina L, Bei T, Stergiopoulus S, Papageorgiou T, Bourdea I, Kirschner LS, Vincent-Dejean C, Perlemoine K, Gicquel C, Bertagna X, Stratakis CA. Molecular and functional analysis of PRKARIA and its locus (17q22–24) in sporadic adrenocortical tumors: 17q losses, somatic mutations, and protein kinase A expression and activity. J Cancer Res. 2003;63:5308–5319. [PubMed] [Google Scholar]
- 21.Gunther DF, Bourdeau I, Matyakhina L, Cassarino D, Kleiner DE, Griffin K, Courkoutsakis N, Abu-Asab M, Tsokos M, Keil M, Carney JA, Stratakis CA. Cyclical Cushing syndrome presenting in infancy: An early form of primary pigmented nodular adrenocortical disease, or a new entitity? J Clin Endocrinol Metab. 2004;89(7):3173–3182. doi: 10.1210/jc.2003-032247. [DOI] [PubMed] [Google Scholar]
- 22.Mochizuki Y, Okamura Y, Iida H, Mori H, Shimada K. Interleukin-6 and “complex” cardiac myxoma. Ann Thorac Surg. 1998;66:931–3. doi: 10.1016/s0003-4975(98)00569-4. [DOI] [PubMed] [Google Scholar]
- 23.Mastorakos G, Chrousos GP, Weber JS. Recombinant interleukin-6 activates the hypothalamic-pituitary-adrenal axis in humans. J Clin Endocrinol Metab. 1993;77(6):1690–4. doi: 10.1210/jcem.77.6.8263159. [DOI] [PubMed] [Google Scholar]