

Abstract
A cell line in which RD-HGA16 cells were stably transfected with the hTAAR 1 receptor was created and utilized to carry out a systematic evaluation of a series of β-phenethylamines. Fair agreement was observed with data obtained for aryl and ethylene chain substituted analogs in an AV12-664 cell line in which hemagglutinin-tagged hTAAR 1 was stably co-expressed with rat Gαs. Analogs with multiple substituents as well as analogs with bulky groups were found to be partial agonists. Analogs in which the primary amino group was converted to a secondary or a tertiary amino group by N-methylation were also partial agonists. Comparative Molecular Field Analysis (CoMFA) using the potency data yielded a regression coefficient r2 of 0.824. The steric field contribution to the model was 61% with the balance (39%) contributed by the electrostatic field. The collective results suggest that increasing steric bulk at both the amino nitrogen, particularly by N-dimethylation, and at the 4-position of the aromatic ring, leads to low efficacy ligands.
Keywords: β-PEA, trace amine, hTAAR 1, CoMFA
Introduction
Whether amino acid metabolites such as β-phenethylamine, p- and m- tyramine, octopamine, and tryptamine, which are present at potentially physiologically relevant concentration in the CNS, have a specific role in brain function has long been a matter of speculation. These so called “trace amines (TAs)” are known to have important functions in invertebrates, particularly in insects. The invertebrate receptors for trace amines, also referred to as octopamine receptors, are known to belong to the family of G-protein coupled receptors (GPCRs).1 Four of them have been distinguished using pharmacological tools and they have been found to show different coupling to second messenger systems, including activation and inhibition of adenylyl cyclase, activation of phospholipase C, and coupling to a chloride channel. Recently, octopamine receptors from mollusks and insects have been cloned. In humans and other mammals, the existence of receptors for TAs had, until recently, only been hypothesized, although it has been suggested that TAs may be involved in diverse conditions including migraine, depression, Parkinson’s disease, and schizophrenia, as reviewed recently.2
The identification of a new family of mammalian GPCRs, two of which are activated by TAs3 and their expression in amygdala, cerebellum, dorsal root ganglia, hippocampus, hypothalamus, medulla, and pituitary has rekindled speculation regarding the function of TAs in brain. Thus, it has been suggested that, in addition to the above-mentioned conditions, TAs may be implicated in psychosis, ADHD, substance abuse, eating disorders, and epilepsy.4 Complete identification of all members of this novel GPCR family5 indicated the existence of three distinct subfamilies, with the first subfamily containing four genes (or pseudogenes), the second containing one gene, and the third containing 11 and 14 genes in mouse and rat, respectively, but only five genes in chimpanzee and human. Because not all of these receptors were activated by TAs, the term TAAR (Trace Amine Associated Receptor) was proposed for this family of receptors.5
The low TAAR densities in native tissue required the development of expression systems for screening purposes. Rat (r), mouse (m), rhesus monkey (rh) and human (h) TAAR 1 have been cloned;3,5–7 rTAAR 1 was stably expressed in HEK 293 cells,5,6 as were mTAAR 15 and rhTAAR 1.8 Transient transfection has been reported for hTAAR 13 but attempted stable in vitro expression was frustrated by the lack of pharmacologic response to trace amines.6 This was attributed to intracellular sequestration of TAAR 1 in HEK 293 cells.9 A stable cell line with pharmacological properties resembling those of mTAAR 1 was achieved by transfection of HEK 293 cells with a human-rat chimera in which the coding sequence was modified by the addition of an influenza hemagluthinin viral leader sequence and the N-terminus, C-terminus, and third intracellular loop were replaced with the corresponding rTAAR 1 sequences.5 A stable cell line expressing hTAAR 1 in rGαsAV-12-664 cells has also been reported.10 We had stably expressed unmodified hTAAR 1 in both CHO-K1 and RD-HGA16 cells. The latter cell line was used to successfully create and validate a functional high throughput assay based on the coupling of the unmodified hTAAR 1 to Gq and the mobilization of internal calcium.11 This assay was used to evaluate a series of β-phenethylamine (β-PEA) analogs at hTAAR 1. The resulting potency data were utilized in CoMFA 3D-QSAR studies aimed at the development of a pharmacophore model of hTAAR 1.
Results
The effect of substitution on the aromatic moiety of β-PEA on both potency and efficacy at hTAAR 1 are shown in Table 1 and Table 2. The results are consistent with the data reported by others.5,10
Table 1.
Effect of Single Substituents on the Aromatic Moiety of β-PEA on Potency and Efficacy at hTAAR 1
| Substituent | Position | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 2 | 3 | 4 | |||||||
| No. | EC50 (nM) | Emax | No. | EC50 (nM) | Emax | No. | EC50 (nM) | Emax | |
| H | 1 | 129±13 | 100±9 | 1 | 129±13 | 100±9 | 1 | 129±13 | 100±9 |
| F | 2 | 65±2 | 91±2 | 3 | 150±13 | 93±2 | 4 | 740±210 | 81±3 |
| Cl | 5 | 77±5 | 72±1 | 6 | 189±20 | 105±17 | 7 | 2,900±1,000 | 104±7 |
| Br | 8 | 57±15 | 89±9 | 9 | 398±114 | 75±5 | 10 | 1,540±400 | 98±7 |
| I | 11 | 103a | 109a | ||||||
| CH3 | 12 | 355±76 | 97±7 | 13 | 638±160 | 93±12 | 14 | 4,380±300 | 96±1 |
| Et | 15 | 9,100a | 86a | ||||||
| t-Bu | 16 | >50,000b | ND | ||||||
| OCH3 | 17 | 144±13 | 95±1 | 18 | 1,444±25 | 73±4 | 19 | 5,980±580 | 106±2 |
| OH | 20 | 614±150 | 90±10 | 21 | 1,740±50 | 78±9 | 22 | 731±220 | 91±20 |
| CF3 | 23 | 3,950a | 69a | 24 | 11,600a | 78a | |||
| NO2 | 25 | >50,000c | NDd | ||||||
single determination
5,112,000 nM was the experimentally determined value used in the CoMFA
167,386 nM was the experimentally determined value used in the CoMFA
ND. Not determined
Table 2.
Effect of Multiple Substituents on the Aromatic Moiety of β-PEA on Potency and Efficacy at hTAAR 1
| Compound No. | Substituent/position | ||||||
|---|---|---|---|---|---|---|---|
| 2 | 3 | 4 | 5 | 6 | EC50 (nM) | Emax (%) | |
| 1 | H | H | H | H | H | 129±13 | 100±9 |
| 26 | Cl | H | H | H | Cl | 51±9 | 94±9 |
| 27 | Cl | H | H | H | F | 312±68 | 94±11 |
| 28 | F | H | OCH3 | H | F | 1,400±680 | 90±12 |
| 29 | F | H | OH | H | F | 767±180 | 103±14 |
| 30 | Cl | Cl | H | H | H | 1,160±210 | 84±8 |
| 31 | H | Cl | Cl | H | H | 3,430a | 78a |
| 32 | CH3 | H | H | CH3 | H | 377a | 84a |
| 33 | OCH3 | H | H | OCH3 | H | 2,010±120 | 69±3 |
| 34 | OCH3 | H | Cl | OCH3 | H | 3,750±460 | 30±6 |
| 35 | OCH3 | H | Br | OCH3 | H | 7,190±880 | 22±5 |
| 36 | OCH3 | H | CH3 | OCH3 | H | 6,478a | 36a |
| 37 | OCH3 | H | Et | OCH3 | H | 6,410±1,970 | 32±9 |
| 38 | OCH3 | H | OCH3 | OCH3 | H | Inactive | |
| 39 | H | H | CH2O | H | 5,030±4,000 | 63±1 | |
Single determination
Nearly all of the analogs were partial or full agonists. Substitution on the ethylene chain of β-PEA indicates that the addition of a single small substituent such as a methyl group to the β-carbon (40, 41) is well tolerated, but greater bulk, e.g. two methyl groups (42) or a phenyl substituent, (43) greatly reduces potency (Table 3). Similar, but somewhat larger, detrimental effects on potency result from introducing an α-methyl group, to give amphetamine (47, 48); even greater bulk, e.g. two α-methyl groups (to give phentermine, 49), sharply reduces potency. The α-substituted compounds have reduced efficacy at hTAAR 1 compared to the parent compound β-PEA (1). Converting the primary amino group in β-PEA and in some analogs to a secondary amino group by N-methylation leads to minor (~3-fold) reduction in potency, but further N-methylation, to afford the tertiary amines, reduces potency by a factor of ~30 (Table 4). As shown in the steric CoMFA field map (Figure 1) decreased potency is associated with increased steric bulk essentially all around the molecular perimeter (yellow colored regions) with the exception of an ortho position, α-substitution, and N-substitution (green colored regions). The electrostatic CoMFA filed map (Figure 2) indicates that negative charge along the ethylene chain is associated with increased potency (red regions) while positive charge in the 3- and 4- positions of the phenyl ring is associated with increased potency (blue regions).
Table 3.
Effect of Substitution on the Ethylene Chain of β-PEA on Potency and Efficacy at hTAAR 1
| Compound No. | substituent | position | EC50 (nM) | Emax (%) |
|---|---|---|---|---|
| 1 | H | 129±13 | 100±9 | |
| 40 | (S)- CH3 | β | 129±33 | 104±10 |
| 41 | (R)- CH3 | β | 361±60 | 97±8 |
| 42 | (CH3)2 | β | 7,880±3,600 | 70±14 |
| 43 | c(CH2)2 | β | 5,050±100 | 79±22 |
| 44 | i-Pr | β | 9,950±2,040 | 46±6 |
| 45 | Ph | β | 3,400±1,000 | 70±11 |
| 46 | CH2 | α/β | 1510±120 | 72±2 |
| 47 | (S)- CH3 | α | 935±260 | 74±7 |
| 48 | (R)- CH3 | α | 920±270 | 64±10 |
| 49 | (CH3)2 | α | 5,470±1,900 | 68±4 |
Table 4.
Effect of N-Methyl Substituents of β—PEA on Potency and Efficacy at hTAAR 1
| Compound | EC50 (nM) | Emax (%) | ||||
|---|---|---|---|---|---|---|
| No. | R=H R’=H |
R=H, R’= CH3 |
R= CH3 R’= CH3 |
R=H R’=H |
R=H, R’= CH3 |
R=CH3 R’= CH3 |
| Structure | ||||||
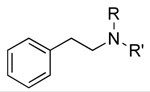 |
1 129±13 |
50 387±106ab |
51 100,000±50,000c |
1 100±9 |
50 69±4ab |
51 64±7c |
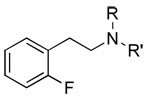 |
2 65±2 |
52 117±23 |
53 1,290±400 |
2 91±2 |
52 75±2 |
53 75±3 |
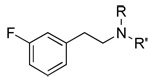 |
3 150±13 |
54 530±128 |
55 4,320±1,800 |
3 93±2 |
54 88±15 |
55 78±9 |
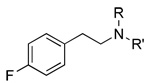 |
4 740±213 |
56 1,040±180 |
57 19,100±3,000 |
4 81±3 |
56 63±6 |
57 71±5 |
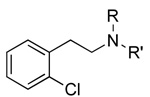 |
5 77±5 |
58 160±38 |
59 687±210 |
5 72±1 |
58 70±6 |
59 79±4 |
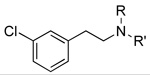 |
6 189±19 |
60 501±70 |
61 5,980±1,280 |
6 105±17 |
60 77±5 |
61 62±20 |
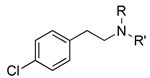 |
7 2,860±1,000 |
62 7,430±2,600 |
63 630,000±300,000 |
7 104±7 |
62 67±6 |
63 43±7 |
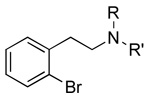 |
8 57±15 |
64 205±66 |
65 3,460±1,200 |
8 89±9 |
64 93±9 |
65 78±7 |
 |
9 398±114 |
66 1,070±300 |
67 8,300±2,000 |
9 89 |
66 71±9 |
67 88±10 |
 |
10 1,540±400 |
68 6,540±2,300 |
69 24,400±6,000 |
10 98±7 |
68 60±5 |
69 40±8 |
Figure 1. Contour plot of the CoMFA model steric fields (StdDev*Coeff).

The region of favorable steric field interaction is indicated by green contours, while the region of unfavorable steric interaction is indicated by yellow contours. β-PEA is shown for reference.
Figure 2. Contour plot of the CoMFA model electrostatic fields (StdDev* Coeff).

The region of favorable electrostatic interaction with external positive charge is indicated by a blue contour, while regions of favorable interaction with external negative charge are indicated by red contours. β-PEA is shown for reference.
Discussion
The good correspondence between our results, obtained by determining calcium flux in CHO cells stably expressing Ga16 (RD-HGA16 cells; Molecular Devices Corporation, Sunnyvale, CA) and hTAAR 1,11 and the literature data10 determined in rGαsAV12-664 cells, serves to validate both datasets. As observed by evaluating cAMP accumulation in rGαsAV12-664 cells stably expressing hTAAR 110 our data also show the positional aryl substituent effects on potency, i.e. 2>3>4 for each substituent with the exception of hydroxyl (−OH), where the potencies are essentially the same at all positions. In addition, any substituent at either the 4- or the 3-position of β-PEA serves to reduce potency at hTAAR 1. Some substituents at the 2-position, in particular 2-halo (F(2), Cl(5), Br(8) and I(11)), afford analogs that more potently activate hTAAR 1 than the endogenous ligand β-PEA, while analogs 2-substituted with a methyl (12) group show lower potency than β-PEA. Analogs of β-PEA with multiple aromatic substituents exhibit potencies that essentially represent the sum total of the effects of individual substituents on potency to activate hTAAR 1. Thus, for example, the EC50 value for 2,5-dimethoxy-β-PEA (33) (2,010±120 nM, see Table 2) is close to the sum of the EC50 values of 2-methoxy-β-PEA 17 (144±13 nM, Table 1) and 3-methoxy-β-PEA 18 (1,444±259 nM, Table 1) and addition of a 4-chloro- (7) or 4-methyl (14) substituent (EC50 = 2,900±1,000, 4380±300 nM, respectively, see Table 1) increases the EC50 by 2158 and 4890, respectively. Addition of a 4-methoxy substituent (19), known to exert a large negative effect on potency (EC50 = 5,980±580 nM, Table 1), leads to an inactive compound (38) (see Table 2). Conversely, the negative effect of a 4-methoxy (19) substituent on potency to activate hTAAR 1 is modulated by the enhancing effect of a 2-fluoro (2) substituent (EC50 = 65±2 nM, Table 1) to afford an EC50 = 1,400±680 nM for 2,6-difluoro-4-methoxy-β-PEA (28) (Table 2).
The most striking outcome of the CoMFA evaluation of this set of analogs is that the steric constraints associated with hTAAR 1 binding are very severe, suggesting that analogs with large N-substituents may align differently from analogs with large substituents in the meta and/or para position. This may account for the reduced potencies and efficacies (partial agonists) observed for these analogs at hTAAR 1. These observations are also consistent with the literature data10 determined for cAMP accumulation in rGασAV12-664 cells.
The effect of β-substitution on potency to activate hTAAR 1 (Table 3) is somewhat deleterious, but, interestingly, appears to be stereospecific. Thus, in both cell lines (CHO- Gα16 and rGαsAV12-664) the S-enantiomer 40 is essentially equally as potent as β-PEA (1) while the R-enantiomer 41 is some three-fold less potent; both enantiomers are full agonists. By contrast, Wainscott et al10 claim stereospecificity for the effect of α-substitution by a methyl group on potency to activate hTAAR 1 by evaluating cAMP accumulation in rGαsAV12-664 cells, with (R)-amphetamine (48) being the less potent isomer, whereas our data, obtained by determining calcium flux in CHO cells stably expressing Gα16 and hTAAR 1, do not show this effect. Considering the magnitude of the error bars associated with the two sets of data, there may in fact not be a significant difference between them. At the same time, three-fold greater potency was reported for (S)-amphetamine (47) relative to (R)-amphetamine (48) in HEK-293 cells heterologously and stably expressing h-rChTAAR 1 (human-rat chimera).12 However, the significantly larger difference in potency observed between β-PEA and p-tyramine (22) in the h-rChTAAR 1 cell line12 suggests that, as has been noted,10 these effects may be due to the distinct species differences observed for TAAR 1.
While stereospecific effects of α-substituents on potency are questionable, the effects on efficacy to activate hTAAR 1 may be stereospecific. Our data are not strong in that regard, but this conclusion is supported by the agreement between our data and the literature data10 for amphetamine (47, 48). Data obtained in the h-rChTAAR 1 cell line12 are also consistent with this conclusion, but may be confounded by the fact that even larger differences in potency between (S)- and (R)-amphetamine are observed in HEK-293 cells heterologously and stably expressing rTAAR 1. Tranylcypromine, (49) a hybrid of α and β substitution that is some 10-fold less potent than β-PEA, also appears to be a partial agonist in our calcium flux assay, but not in the cAMP assay in rGαsAV12-664 cells.10
The effects of N-methylation (Table 4), taken together with effects of aromatic substitution (Table 1 and Table 2) are consistent with the idea that β-PEA analogs with bulky substituents are poorly accommodated due to steric constraints, as shown in the CoMFA maps (Figure 1 and Figure 2). This results in decreased potency and efficacy. Thus, β-PEA analogs with large single substituents, particularly in the 4- position of the aromatic ring, as in 4-t-butyl β-PEA (16) and 4-nitro-β–PEA (25) (Table 1), and with multiple aromatic substituents, as in the series of 2,5-dimethoxy-β–PEA analogs (33, 38) (Table 2), have reduced efficacy. Increasing bulk at the amino end of the molecule by N-methylation and N-dimethylation (Table 4) has a profound effect on the potency of β–PEA with minimal effect on efficacy. Increasing steric bulk at both the amino nitrogen, particularly by N-dimethylation, and at the para position of the aromatic ring, profoundly affects both potency and efficacy.
Experimental
Chemicals
Derivatives and analogs of β-PEA were obtained from commercial sources, from the NIDA Research Technology Branch (Rockville, MD), or by synthesis. Specifically, compounds that were purchased from Sigma Aldrich (St. Louis, MO) were as follows: 2-phenylethylamine (1), 2-(2-fluorophenyl)ethylamine (2), 2-(3-fluorophenyl)ethylamine (3), 2-(4-fluorophenyl)ethylamine (4), 2-(2-chlorophenyl)ethylamine (5), 2-(3-clorophenyl)ethylamine (6) hydrochloride, 2-(4-clorophenyl)ethylamine (7), 2-(2-bromopheneyl)ethylamine (8), 2-(3-bromophenyl)ethylamine (9), 2-(4-bromophenyl)ethylamine (10), 2-(2-methylphenyl)ethylamine (12), 2-(3-methylphenyl)ethylamine (13), 2-(4-methylphenyl)ethylamine (14), 2-(4-ethylphenyl)ethylamine (15) hydrochloride, 2-(2-methoxyphenyl)ethylamine (17), 2-(3-methoxyphenyl)ethylamine (18), 2-(4-methoxyphenyl)ethylamine (19) hydrochloride, 2-(3-hydroxyphenyl)ethylamine (21) hydrochloride, 2-(4-hydroxyphenyl)ethylamine (22), 2-(3-trifluoromethylphenyl)ethylamine (23), 2-(4-trifluoromethyphenyl)ethylamine (24), 2-(4-nitrophenyl)ethylamine (25), 2-(2,6-dichlorophenyl)ethylamine (26), 2-(2,6-difluoro-4-methoxyphenyl)ethylamine (28) hydrochloride, 2-(2,3,-dichlorophenyl)ethylamine (30) hydrochloride, 2-(3,4,-dichlorophenyl)ethylamine (31) hydrochloride, (S)-(−)-2-phenylpropylamine (40), (R)-(+)-2-phenylpropylamine (41), 2,2-diphenylethylamine (45), trans-2-phenylcyclopropylamine (46), N-methyl-2-phenylelthylamine (50), N,N-diimethyl-2-phenylethylamine (51). The following 2 compounds were purchased from Acros Organics (Gee, Belgium): 2-(2-chloro-6-fluorophenyl)ethylamine (27) hydrochloride and 2-(2,5-dimethylphenyl)ethylamine (32) hydrochloride; 2-(4-t-Bu-phenyl)ethylamine (16) hydrochloride was purchased from ABCR (Karlsruhe, Germany). Compounds obtained from the NIDA Research Technology Branch (Rockville, MD) are: 2-(2,5-dimethoxyphenyl)ethylamine (33), 2-(4-chloro-2,5-dimethoxyphenyl)ethylamine (34), 2-(4-bromo-2,5-dimethoxyphenyl)ethylamine (35), 2-(2,5-dimethoxy-4-methylphenyl)ethylamine (36), 2-(2,5-dimethoxy-4-ethylphenyl)ethylamine (37), 2-(2,5-dimethoxy-4-methoxylphenyl)ethylamine (38) hydrochloride, (S)-(+)-amphetamine (47) hydrochloride, (R)-(−)-amphetamine (47) hydrochloride, phentermine (49) hydrochloride. The remaining compounds were synthesized and characterized as described below:
Synthesis
Melting points were determined on either a Thomas-Hoover capillary tube apparatus or on a Fisher-Johns Model 12–144 melting point apparatus. Thin layer chromatography was carried out using Merck silica gel 60F254 TLC plates or Baker-flex silica gel 1B-F flexible sheets; visualization was under UV or in an iodine chamber, as appropriate. Flash chromatography was conducted on silica gel 60 (40 µM). Nuclear magnetic resonance (NMR) spectra were determined on a Bruker Avance 300 MHz NMR spectrometer. Mass spectra were obtained using a Perkin-Elmer Sciex API 150EX mass spectrometer outfitted with an APCI source. Microanalyses were carried out by Atlantic Microlab, Inc.
2-(2-iodophenyl)ethylamine (11) Hydrochloride
To a solution of (2-iodophenyl)acetonitrile (2.0 g, 0.008 mol) in anhydrous THF (10 mL), BH3 • THF (1.0 M, 20 mL, 20 mmol) was slowly added. After refluxing for 16 h, the reaction mixture was chilled to 0 °C and CH3OH (10 mL) was added dropwise. The resulting mixture was concentrated, CH3OH (15 mL) was added, and the volatiles were removed in vacuo. The residue was treated with HCl (0.5N, 30 mL) and the solution was refluxed 1 h. After cooling to room temperature the mixture was washed with diethyl ether. The aqueous phase was basified to pH 14 with 30% NaOH then extracted with ether (3×10 mL). The combined extract was dried over Na2SO4 and concentrated. The HCl salt was prepared by adding 6% ethanolic HCl until no further precipitation occurred. The precipitated solid was collected by filtration and recrystallized from CH3OH/ether to yield 1.68 g (72%). Mp: 213 °C [lit.13 251–255 °C]; MS (ESI): m/z calcd for C8H11IN: 246.99, found (M+1): 248.10. Anal. calcd C8H10ClIN: C, 33.89; H, 3.91; N, 4.94. Found: C, 34.01; H, 3.93; N 4.86. 1H-NMR (DMSOd6, 300 MHz) δ (ppm): 2.99 (s, 4H, CH2CH2), 7.8-7.3 (4H,ArH). 13C-NMR (DMSOd6, 75.5 MHz) δ (ppm): 37.36, 37.67, 100.87, 128.84, 139.03, 129.37, 129.57, 129.97, 139.35, 139.74, 140.15.
2-(2-Hydroxyphenyl)ethylamine (20) Hydrochloride
A solution of 2-(2-methoxyphenyl)ethylamine (17) (484 mg, 3.30 mmol) in HCl (12N, 0.7 mL) was placed in a thick walled microwave test tube, covered with the test tube lid, lowered into the microwave apparatus (CEM Explorer Microwave) and microwaved for 10 minutes (83 psi, 160 °C). Reaction progress was followed by TLC (90/10/1); CHCl3:CH3OH:NH4OH. The reaction was complete after a total of 1 mL HCl was added and the heat increased to 170 °C. The resulting mixture was chilled to 0 °C, basified with NaOH (pH ~10-1), and extracted with EtOAc (3×20 mL). The combined organic extract was washed with H2O (30 mL) and saturated NaCl (30 mL) then dried over Na2SO4. The solid remaining after evaporation of the volatiles was taken up in EtOAc (5 mL) and treated with 6 % ethanolic HCl (10 mL). After freezing overnight (0 °C), the precipitated pink solid was collected by filtration (133 mg, 23%). Mp: 154 °C [lit.14 152–154 °C]; MS (ESI): m/z calcd for C8H11NO: 137.08, found (M+1): 138.4. Anal. calcd for C8H12ClNO: C, 55.34; H, 6.97; N, 8.07. Found: C, 55.12; H, 6.90; N, 7.97. 1H-NMR (DMSOd6, 300 MHz) δ (ppm): 2.80–2.83 (m, 2H, CH2), 2.85–2.92 (m, 2H, CH2), 6.71–6.76 (m, 1H, ArH), 6.85–6.88 (m, 1H, ArH), 7.04–7.09 (m, 2H, ArH). 13C-NMR (DMSOd6, 75.5 MHz) δ (ppm): 28.53, 38.84, 115.49, 119.33, 123.78, 128.15, 130.45, 155.80.
2-(4-Trifluoromethylphenyl)ethylamine (24) Hydrochloride
To a solution of 4-(trifluromethylphenyl)acetonirtile (423 mg, 2.28 mmol), in methanolic ammonia (2N, 10 mL), in a 250 mL Parr flask was added a scoop of Raney Nickel (in water). The resulting mixture was shaken overnight under hydrogen (50 psi), then filtered through Celite, rinsed with CH3OH, and then concentrated to a white solid. The HCl salt was generated using ethereal HCl (~10 mL) to give a white solid (51 mg, 12%). Mp: 187 °C; MS (ESI): m/z calcd for C9H11F3N: 189.18, found (M+1): 190.4. Anal. calcd for C9H11ClF3N: C, 47.91; H, 4.91; N, 6.21. Found: C, 47.7; H, 4.83; N, 6.15. 1H-NMR (DMSOd6, 300 MHz) δ (ppm): 3.01 (s, 4H, CH2CH2), 7.51 (d, J=7.8 Hz, 2H. ArH), 7.70 (d, J=7.5 Hz, 2H, ArH). 13C-NMR (DMSOd6, 75.5 MHz) δ (ppm): 30.81, 38.10, 122.3 (q, JCF= 271.8 Hz, CF3), 123.6 (q, JFCCC = 3.77 Hz, ArC-3), 125.7 (q, JFCC=31.98 Hz, ArC-4), 127.9, (ArC-2), 140.8 (ArC-1).
2-(2,6-Difluoro-4-hydroxyphenyl)ethylamine (29) Hydrochloride
A suspension of 2-(2,6-difluoro-4-methoxyphenyl)ethylamine (28) HCl (132 mg, 0.630 mmol) in 48% HBr (2 mL) was heated at 145 °C for 1.5 h and then concentrated in vacuo to a pale orange residue that was recrystallized twice from EtOH/ether. Mp: 254 °C; MS (ESI): m/z calcd for C8H9F2NO: 173.0, found (M+1): 174.0. Anal. calcd for C8H10ClF2NO: C, 37.82; H, 3.97; N, 5.51. Found: C, 37.87; H, 3.88; N, 5.43. 1H-NMR (DMSOd6, 300 MHz) δ (ppm): 2.89–2.9 (m, 2H, CH2), 2.82–2.84 m, 2H, CH2), 6.48 (d, JHF=21 Hz, 2H, ArH); 13C-NMR (DMSOd6, 75.5 MHz) δ (ppm): 19.2, 37.7, 98.6 (d, JCCF=27.6 Hz, ArC-3,5,) 101.8 (t, JCF=21.1 Hz, ArC-1), 157.8 (t, JFCCC=15.1 Hz, ArC-4) 160.8 (dd, JCF=241.6 Hz, JCCF= 12.5 Hz, ArC-2,6).
2-Methyl-2-phenylpropylamine (42) Hydrochloride
To a chilled solution of 1,1-dimethylbenzonittrile (145.13 mg, 1 mmol) and cobalt (II) chloride (237.9 mg, 1 mmol) in EtOH (10 mL) was added NaBH4 (75.66 mg, 2 mmol) in portions. A precipitate formed. After stirring the suspension 3 h under N2, it was poured into chilled 3N HCl (5 mL) and then washed with diethyl ether (2×20 mL). The aqueous phase was basified with 20% NaOH (pH 14) and extracted with diethyl ether (3×30 mL). The combined extract was dried over Na2SO4, filtered, and concentrated to an oil. The HCl salt was made using 6% methanolic HCl (10 mL). The recovered solid was recrystallized from CH3OH/ether to yield 28.5 mg (15%) of a white solid. Mp: 204 °C; MS (ESI): calcd for C10H15N: 149.1, found (M+1): 150.5. Anal. calcd for C10H16ClN: C, 64.68; H, 8.68; N, 7.54. Found: C, 63.91; H, 8.58; N, 7.61. 1H-NMR (CDCl3, 300 MHz) δ (ppm):1.3 (s, 3H, CH3), 2.7 (s, 2H, CH2), 7.3 (s, 5H, ArH). 13C-NMR (CDCl3, 75.5 MHz) δ (ppm): 26.3, 37.3, 51.1, 125.9, 127.1, 128.9, 144.1.
1-Phenylcyclopropanemethylamine (43) Hydrochloride
A solution of 1-phenyl-1-cyclopropylacetonitrile (1 g, 0.007 mol) in anhydrous THF (10 mL) was added to a chilled solution of LAH (1.0 M in THF, 6.98 mL, 6.98 mmol). A precipitate formed. After stirring overnight under N2 at room temperature the reaction mixture was poured over ice and 3N HCl and extracted with ether (2×10 mL). The aqueous phase was basified to pH 14 with 30% NaOH, then extracted with ether (3×10 mL), dried over Na2SO4 and concentrated. The residual oil was treated with 6% methanolic HCl. Evaporation of the solvent afforded a solid that was recrystallized twice from CH3OH/ether (336 mg, 26%). Mp: 176–177 °C [lit.15 175 °C]; MS (ESI): m/z calcd for C10H13N: 147.2, found (M+1): 148.4. Anal. calcd C10H14ClN: C, 65.39; H, 7.68; N, 7.63. Found: C, 65.39; H, 7.84; N, 7.58. 1H-NMR (CDCl3, 300 MHz) δ (ppm): 1.03–1.12 (m, 4H, CH2CH2) 3.07 (s, 2H, CH2N), 7.2–7.4 (s, 5H, ArH). 13C-NMR (CDCl3, 75.5 MHz) δ (ppm): 13.0, 24.6, 49.4, 128.1, 129.3, 129.9, 140.4.
3-Methyl-2-phenylbutylamine (44) Hydrochloride
A solution of 3-methyl-2-phenylbutyronitrile (500 mg, 3.14 mmol) in anhydrous THF (6 mL) was added dropwise to a chilled solution of LAH in THF (1.0 M, 3.46 mL, 3.46 mmol). After heating at 50 °C for 16 h the reaction mixture was slowly added to chilled 3N HCl, then washed with diethyl ether (3×30 mL). The aqueous phase was basified with chilled NaOH (pH 14) and extracted with diethyl ether (3×30 mL). The combined organic extract was dried over Na2SO4, filtered, then concentrated. The HCl salt was generated by stirring the residue in methanolic HCl, then concentrating the solution to a white solid that was recrystallized from CH3OH/ether (202 mg, 32%). Mp: 190–191 °C; MS (ESI): m/z calcd for C11H17N: 163.1, 165.5, found (M+1): 164.4, 166.5. Anal. calcd for C11H18ClN (+.13 mol H2O): C, 65.39; H, 9.11; N 6.93, Found: C, 65.39; H, 9.11; N, 6.95. 1H-NMR (300 mHz, DMSOd6) δ (ppm): 0.6 (d, 3H CH3), 0.88 (d, 3H, CH3), 1.9 (m, 1H, CHCH (CH3)2), 2.79 (d, 1H, CHCH CH2) 3.1–3.2 (m, 2H, CH2), 7.25–7.45 (m, 5H, ArH). 13C-NMR (DMSOd6) δ (ppm): 17.6, 19.7, 21.0, 30.7, 41.5, 49.9, 126.8, 128.3, 129.1, 139.6.
N-Methyl-2-(2-fluorophenyl)ethylamine (52) Hydrochloride (Method A)
A mixture of 2-(2-fluorophenyl)ethylamine (2) (1.066 g, 0.0077 mol), ethyl formate (24 mL, large excess), and formic acid (3 drops) was refluxed under N2 for 5 h. The reaction mixture was concentrated in vacuo, leaving an oil (1.339 g, 0.008 mol, 104%). The bulk of the formamide intermediate (1.323 g, 0.0079 mol) was dissolved in THF (anhydrous, 5 mL), cooled to 0 °C, mixed with boran-tetrahydrofuran complex (1M, 30 mL, 30 mmol). The mixture was refluxed under N2 for 2 h. The reaction solution was cooled to 0 °C, mixed slowly with 10% methanolic HCl (6 mL), and refluxed under N2 for 2 h. Removal of the volatiles afforded a brown solid (3.386 g) that was mixed with H2O (20 mL) and 3N HCl (8 mL), washed wih EtOAc (3×10 mL), basified to pH 14 with NaOH (10N, 3 mL) and extracted with EtOAc (3×15 mL). The combined extract was washed with brine (3×10 mL), dried over Na2SO4, concentrated in vacuo, leaving a colorless oil (518 mg, 3.38 mmol, 43%). The crude product (510 mg, 3.33 mmol) was dissolved in 10% methanolic HCl (6 mL) and the solution was refluxed under N2 for 2 hours, then concentrated in vacuo to give a light yellow solid (682 mg). Recrystallization from MeOH/EtOAc (30/1) gave light yellow crystals (308 mg). A solution of these crystals (300 mg) in 10% methanolic HCl (5 mL) was filtered through celite then concentrated in vacuo to give 52•HCl as a white solid (290 mg, 22%). Mp: 152–153 °C; MS (ESI): m/z calcd for C9H12FN: 153.2, found (M + 1): 154.1. Anal. calcd for C9H12FN•HCl: C, 57.00; H, 6.91; Cl, 18.69; F, 10.02; N, 7.39. Found: C, 57.01; H, 6.89; Cl, 18.91; F, 10.01; N, 7.49. 1H-NMR (CD3OD, 300 MHz) δ (ppm): 2.76 (s, 3H, CH3), 3.07–3.19 (m, 2H, CH2), 3.25–3.34 (m, 2H, CH2), 7.11–7.19 (m, 2H, ArH), 7.33–7.39 (m, 2H, ArH). 13C-NMR (CD3OD, 75.5 MHz) δ (ppm): 27.2, 27.3, 34.2, 50.5, 116.8, 117.1, 124.7, 124.9, 126.2, 126.3, 130.9, 131.0, 132.6, 132.7, 161.4, 164.6.
N,N-dimethyl-2-(2-fluorophenyl)ethylamine (53) (Method B)
To a cooled solution of 2-(2-fluorophenyl)ethylamine (2) (1.066 g, 0.0077 mol) in formic acid (1.4 mL, 96%, 35 mmol) was added formaldehyde (1.6 mL, 37%, 21.3 mmol) and the solution was refluxed under N2 for 16 h. After cooling to 0 °C, HCl (3N, 15 mL) was added and the mixture was washed with EtOAc (3×10 mL). The aqueous phase was basified to pH 14 with NaOH (10N, 5 mL) then extracted with EtOAc (3×15 mL). The combined extract was washed with brine (3×15 mL), dried over Na2SO4, and concentrated in vacuo, leaving a colorless oil (814 mg, 63%); MS (ESI): m/z calcd for C10H14FN: 167.2, found (M + 1): 168.3. Anal. calcd for C10H14FN: C, 71.82; H, 8.44; F, 11.36; N, 8.38. Found: C, 71.59; H, 8.50; F, 11.09; N, 8.32. 1H-NMR (CDCl3, 300 MHz) δ (ppm): 2.30 (s, 6H, (CH3)2), 2.50–2.61 (m, 2H, CH2), 2.79–2.84 (m, 2H, CH2), 7.00–7.08 (m, 2H, ArH), 7.16–7.21 (m, 2H, ArH), 13C-NMR (CDCl3, 75.5 MHz) δ (ppm): 27.48, 27.50, 45.4, 59.8, 59.9, 115.1, 115.4, 123.9, 124.0, 127.1, 127.4, 127.7, 127.8, 130.8, 130.9, 159.6, 162.8.
N-Methyl-2-(3-fluorophenyl)ethylamine (54) Hydrochloride
The free base 54 was prepared from 2-(3-fluorophenyl)ethylamine (3) by a slight modification of Method A. Specifically, workup of the borohydride reaction was by treatment of the cold reaction mixture with H2O (15 mL), stirring at 0 °C for 5 min, then mixing with NaOH (10%, 3 mL), stirring at 0 °C for 30 min, and extracting with EtOAc (3×15 mL). The combined extract was washed with brine (3×10 mL), dried over Na2SO4, concentrated in vacuo, leaving a colorless oil (1.037 g, 94%). The crude product (906 mg, 5.91 mmol) was purified using flash silica gel chromatography, eluting with hexane/EtOAc, to give a colorless oil (256 mg, 28%). The hydrochloride salt was prepared as described in Method A. Recrystallization from CH3OH/EtOAc (1/16) gave pure 54•HCl. Mp: 158–159 °C; MS (ESI): m/z calcd for C9H12FN: 153.2, found: 154.3 (M + 1). Anal. calcd for C9H13ClFN: C, 57.00; H, 6.91; Cl, 18.69; F, 10.02; N, 7.39. Found: C, 56.77; H, 6.87; Cl, 18.94; F, 9.78; N, 7.34. 1H-NMR (CD3OD, 300 MHz) δ (ppm): 2.70 (s, 3H, CH3), 3.03-2.97 (m, 2H, CH2), 3.30-3.22 (m, 2H, CH2), 7.11-6.97 (m, 3H, ArH-4,5,6), 7.38-7.31 (m, 1H, ArH-2). 13C-NMR (CD3OD, 75.5 MHz) δ (ppm): 33.2, 33.3, 34.1, 51.4, 115.3, 115.6, 116.9, 117.1, 126.0, 126.1, 132.1, 132.3, 140.7, 140.8, 163.3, 166.5.
N,N-Dimethyl-2-(3-fluorophenyl)ethylamine (55) Hydrochloride
Prepared from 2-(3-fluorophenyl)ethylamine (3) following Method B. The HCl salt was prepared by dissolving a portion of the oil (379 mg, 2.27 mmol) in CH3OH (2 mL), followed by addition of 1M HCl in Et2O (3 mL). The solvents were evaporated and the product was obtained as white solid (409 mg, 89%). Mp: 162–163 °C; MS (ESI): m/z calcd for C10H14FN: 167.2, found (M + 1): 168.5. Anal. calcd for C10H15ClFN: C, 58.97; H, 7.42; Cl, 17.41; F, 9.33; N, 6.88. Found: C, 58.96; H, 7.35; Cl, 17.42; F. 9.23; N, 6.79. 1H-NMR (CD3OD, 300 MHz) δ (ppm): 2.97 (s, 6H, (CH3)2), 3.15-3.01 (m, 2H, CH2), 3.44-3.31 (m, 2H, CH2), 7.06-7.00 (m, 1H, ArH-6), 7.33 (m, 1H, ArH-2), 7.18-7.12 (m, 2H, ArH-4,5). 13C-NMR (CD3OD, 75.5 MHz) δ (ppm) 31.8, 44.1, 59.8, 115.3, 115.6, 117.0, 117.3, 126.21, 126.24, 132.1, 132.2, 140.5, 140.6, 163.3, 166.5.
N-Methyl-2-(4-fluorophenyl)ethylamine (56) Hydrochloride
Prepared from 2-(4-fluorophenyl)ethylamine (4) using a slight modification of Method A. Specifically, for the workup the reaction mixture was cooled to 0 °C then mixed slowly with 10% hydrogen chloride methanol solution (12 mL). The mixture was refluxed under N2 for 2 h, and concentrated in vacuo. Pure HCl salt (662 mg, white solid, 46%) was obtained by recrystallization from CH3OH/EtOAc (15/1). Mp: 163–164 °C; MS (ESI): m/z calcd for C9H12FN: 153.2, found (M + 1): 153.7. Anal. calcd for C9H13ClFN: C, 57.00; H, 6.91; Cl, 18.69; F, 10.02; N, 7.39. Found: C, 57.02; H, 6.91; Cl, 18.59; F, 10.10; N, 7.37. 1H-NMR (CD3OD, 300 MHz) δ (ppm): 2.73 (s, 3H, CH3), 2.95-3.03 (m, 2H, CH2), 3.22-3.33 (m, 2H, CH2), 7.06-7.13 (m, 2H, ArH-2,6), 7.31–7.36 (m, 2H, rH-3,5), 13C-NMR (CD3OD, 75.5 MHz) δ (ppm): 32.8, 34.1, 51.8, 116.9, 117.2, 131.9, 132.1, 133.9, 134.0, 162.3, 165.5.
N,N-Dimethyl-2-(4-fluorophenyl)ethyl)amine (58) Hydrochloride
Prepared from 2-(4-fluorophenyl)ethylamine (4) using Method B. The pure HCl salt (331 mg, pale yellow solid, 46%) was obtained by recrystallization from CH3OH/EtOAc/Et2O (1/10/30). Mp: 146–147 °C; MS (ESI): m/z calcd for C10H14FN: 167.2, found (M + 1): 168.2. Anal. calcd for C10H14FN•HCl: C, 58.97; H, 7.42; Cl, 17.41; F, 9.33; N, 6.88. Found: C, 58.76; H, 7.38; Cl, 17.62; F, 9.51; N, 6.83. 1H-NMR (CD3OD, 300 MHz) δ (ppm): 2.97 (s, 6H, (CH3)2), 3.11–3.12 (m, 2H, CH2), 3.32–3.42 (m, 2H, CH2), 7.06–7.12 (m, 2H, ArH-2,6), 7.35–7.40 (m, 2H, ArH-3,5), 13C-NMR (CD3OD, 75.5 MHz) δ (ppm): 31.4, 44.0, 60.1, 116.9, 117.2, 132.1, 132.2, 133.7, 133.8, 162.3, 165.5.
N-Methyl-2-(2-chlorophenyl)ethylamine (58) Hydrochloride
The free base 58 was prepared from 5 following a slight modification of Method A. Specifically, the workup involved mixing the cooled reaction mixture with H2O (15 mL) stirring at 0 °C for 5 min, then mixing with NaOH (10%, 3 mL), stirring at 0 °C for 30 min, and extracting with EtOAc (3×15 mL). The combined extract was washed with brine (3×10 mL), dried over Na2SO4, concentrated in vacuo, leaving a colorless oil (1.265 g, 97%). The HCl salt was prepared by refluxing a solution of 58 in 10% methanolic hydrogen chloride (10 mL) under N2 for 3 h and then concentrating in vacuo. The residual light yellow solid was mixed with CH3OH (3 mL) and filtered through a Celite pad. Pure HCl salt (209 mg, white solid, 30%) was obtained by recrystallization from CH3OH/EtOAc (1/20). Mp: 137–138 °C; MS (ESI): m/z calcd for C9H12ClN: 169.7, found (M + 1): 170.3. Anal. calcd for C9H13Cl2N: C, 52.45; H, 6.36; Cl, 34.40; N, 6.80. Found: C, 52.43; H, 6.41; Cl, 34.15; N, 6.86. 1H-NMR (CD3OD, 300 MHz) δ (ppm): 2.64 (s, 3H, CH3), 3.03–3.20 (m, 2H, CH2CH2), 7.17–7.23 (m, 2H, ArH), 7.29–7.34 (m, 2H, ArH). 13C-NMR (CD3OD, 75.5 MHz) δ (ppm): 27.2, 27.3, 34.2, 50.5, 116.8, 117.1, 124.7, 124.9, 126.2, 126.3, 130.9, 131.0, 132.6, 132.7, 161.4, 164.6.
N,N-Dimethyl-2-(2-chlorophenyl)ethylamine (59) Hydrochloride
The free base 59 was prepared in 74% from 4 following Method B. The mixture obtained by dissolving 59 (970 mg, 5.28 mmol) in Et2O (5 mL), and adding ethereal HCl (1M, 8 mL) was centrifuged, and the precipitated solid was collected, washed with EtOAc (2×10 mL) and dried in vacuo to give the HCl salt (1.142 g, white solid, 98%). Mp: 165–167 °C; MS (ESI): m/z calcd for C10H14ClN: 183.4, found (M + 1): 184.3. Anal. calcd for C10H15Cl2N: C, 54.56; H, 6.87; Cl, 32.21; N, 6.36. Found: C, 54.43; H, 6.82; Cl, 32.01; N, 6.35. 1H-NMR (CD3OD, 300 MHz) δ (ppm): 3.01 (s, 6H, (CH3)2), 3.23–3.51 (m, 4H, CH2 CH2), 7.29–7.36 (m, 2H, ArH), 7.43–7.46 (m, 2H, ArH), 13C-NMR (CD3OD, 75.5 MHz) δ (ppm): 30.1, 44.2, 58.5, 129.2, 130.7, 131.3, 132.8, 135.42, 135.44.
N-Methyl-2-(3-chlorophenyl)ethylamine (60) Hydrochloride
A mixture of 2-(3-chlorophenyl)ethylamine (6) (1.119 g, 0.007 mol), ethyl formate (24 mL), and formic acid (3 drops) was refluxed under N2 for 5 h. The reaction mixture was concentrated in vacuo to give crude N-3-chlorophenethylformamide intermediate (light orange oil, 2.17 g, 0.012 mol). A solution of this intermediate (856 mg, 4.66 mmol) in THF (25 mL, anhydrous) was added dropwise to a suspension of LiAlH4 (756 mg, 19.9 mmol, pellet) in THF (10 mL, anhydrous) cooled to 0 °C under N2. The mixture was refluxed under N2 for 4 h, then cooled to 0 °C, mixed slowly with H2O (0.8 mL), NaOH (15%, 0.8 mL), and H2O (0.8 mL). The mixture was stirred at RT for 10 min, filtered through a Celite pad, and the solvent in the filtrate was evaporated. The residual colorless oil was dissolved with HCl (3N, 10 mL), washed with EtOAc (3×10 mL), basified to pH 14 with NaOH (10N, 4 mL), and extracted with EtOAc (3×10 mL). The combined extract was washed with brine (3×10 mL), dried over Na2SO4, and concentrated in vacuo, leaving a colorless oil (588 mg, 29%). The free amine was converted to HCl salt by dissolving the oil (576 mg, 3.40 mmol) in Et2O (15 mL), and mixing with HCl in Et2O (1M, 6 mL). The pure HCl salt (234 mg, 1.14 mmol, white solid, 33%) was obtained by recrystallization from CH3OH/EtOAc (1/30). Mp: 137–138 °C; MS (ESI): m/z calcd for C9H12ClN: 169.7, found (M + 1): 170.3. Anal. calcd for C9H13Cl2N: C, 52.45; H, 6.36; Cl, 34.40; N, 6.80. Found: C, 53.22; H, 6.48; Cl, 33.17; N, 6.85. 1H-NMR (CD3OD, 300 MHz) δ (ppm): 2.76 (s, 3H, CH3), 3.07–3.19 (m, 2H, CH2), 3.25–3.34 (m, 2H, CH2), 7.11–7.19 (m, 2H, ArH), 7.33–7.39 (m, 2H, ArH). 13C-NMR (CD3OD, 75.5 MHz) δ 27.2, 27.3, 34.2, 50.5, 116.8, 117.1, 124.7, 124.9, 126.2, 126.3, 130.9, 131.0, 132.6, 132.7, 161.4, 164.6.
N,N-Dimethyl-2-(3-chlorophenyl)ethylamine (61) Hydrochloride
The free base 61 (726 mg, 3.95 mmol, 55%) was prepared from 5 following Method B. Pure HCl salt (487 mg, pale yellow solid) was obtained by recrystallization from CH3OH/EtOAc (1/20). Mp: 147–148 °C; MS (ESI): m/z: calcd for C10H14ClN: 183.4, found (M + 1): 184.3. Anal. calcd for C10H15Cl2N: C, 54.56; H, 6.87; Cl, 32.21; N, 6.36. Found: C, 54.74; H, 6.96; Cl, 32.17; N, 6.45. 1H-NMR (CD3OD, 300 MHz) δ (ppm): 2.97 (s, 6H, (CH3)2), 3.11–3.12 (m, 2H, CH2), 3.32–3.42 (m, 2H, CH2), 7.06–7.12 (m, 2H, ArH), 7.35–7.40 (m, 2H, ArH), 13C-NMR (CD3OD, 75.5 MHz) δ (ppm): 31.4, 44.0, 60.1, 116.9, 117.2, 132.1, 132.2, 133.7, 133.8, 162.3, 165.5.
N-Methyl-2-(4-chlorophenyl)ethylamine (62) Hydrochloride
The free base 62 was prepared from 6 as described in Method A using a modified workup. Specifically, the workup involved mixing the cooled reaction mixture with H2O (15 mL) stirring at 0 °C for 5 min, then mixing with NaOH (10%, 3 mL), stirring at 0 °C for 30 min, and extracting with EtOAc (3×15 mL). The combined extract was washed with brine (3×10 mL), dried over Na2SO4, concentrated in vacuo, leaving a colorless oil (1.273 g, 99%). The oil (1.260 g) was dissolved in 10% methanolic hydrogen chloride (10 mL). The solution was refluxed under N2 for 3 h, then concentrated in vacuo to a light yellow solid (1.919 g). This oil was mixed with CH3OH (3 mL) and filtered through a Celite pad. Pure HCl salt (348 mg, white solid) was obtained by recrystallization from CH3OH/EtOAc (1/20). Mp: 181–182 °C; MS (ESI): m/z calcd for C9H12ClN: 169.7, found (M + 1): 170.3. Anal. calcd for C9H13Cl2N: C, 52.45; H, 6.36; Cl, 34.40; N, 6.80. Found: C, 52.39; H, 6.41; Cl, 34.23; N, 6.83. 1H-NMR (CD3OD, 300 MHz) δ (ppm): 2.57 (s, 3H, CH3), 2.83–2.89 (m, 2H, CH2), 3.12–3.19 (m, 2H, CH2), 7.08–7.20 (m, 4H, ArH), 13C-NMR (CD3OD, 75.5 MHz) δ (ppm): 33.2, 34.2, 51.5, 128.75, 128.85, 130.3, 131.9, 136.1, 140.4.
N,N-Dimethyl-2-(4-chlorophenyl)ethylamine (63) Hydrochloride
The free base 63 was prepared from 7 following Method B. The mixture obtained by dissolving the oil (946 mg, 5.15 mmol) in Et2O (5 mL) and adding ethereal HCl (1M, 8 mL) was centrifuged, and the precipitated solid was collected, washed with EtOAc (2×10 mL) and dried in vacuo (1.047 g, white solid, 92%). Mp: 162–163 °C; MS (ESI): m/z calcd for C10H14ClN: 183.4, found (M + 1): 184.3. Anal. calcd for C10H15Cl2N: C, 54.56; H, 6.87; Cl, 32.21; N, 6.36. Found: C, 54.49; H, 6.94; Cl, 32.36; N, 6.38. 1H-NMR (CD3OD, 300 MHz) δ (ppm): 2.97 (s, 6H, (CH3)2), 3.11–3.12 (m, 2H, CH2), 3.32–3.42 (m, 2H, CH2), 7.06–7.12 (m, 2H, ArH), 7.35–7.40 (m, 2H, ArH), 13C-NMR (CD3OD, 75.5 MHz) δ (ppm): 31.4, 44.0, 60.1, 116.9, 117.2, 132.1, 132.2, 133.7, 133.8, 162.3, 165.5.
N-Methyl-2-(2-bromophenyl)ethylamine (64) Hydrochloride
The free base 64 was prepared from 2-(2-bromophenyl)ethylamine (8) using a slight modification of Method A. Specifically, the chilled borohydride reaction mixture was treated with H2O (10 mL) followed by 10% NaOH (until basic), stirred for 30 min at 0° under N2 and then extracted with EtOAc (3×30 mL). The combined organic extract was washed with H2O (20 mL), sat’d NaCl (20 mL), then dried over Na2SO4 and concentrated to a solid. The solid was taken up in diethyl ether (5 mL) and 6% ethanolic HCl (10 mL) was added. Concentration afforded a solid that was recrystallized from CH3OH/ether (69 mg, 8% yield). Mp: 109 °C; MS (ESI): m/z calcd for C9H11BrN: 213.02, 215.01., found (M+1): 214.1, 21.1. Anal. calcd for C9H12BrClN: C, 43.14; H, 5.23; N, 5.59. Found: C, 43.07; H, 5.10; N, 5.53. 1H-NMR (DMSOd6, 300 MHz) δ (ppm): 2.58 (s, 4H, CH2CH2), 3.1 (s, 3H, CH3), 7.25–7.27 (m, 1H, ArH), 7.3–7.4 (m, 2H, ArH), 7.62–7.65 (d 1H, ArH). 13C-NMR (DMSOd6, 75.5 MHz) δ (ppm): 32.02, 32.63, 47.74, 124.08, 128.5, 129.45, 131.29, 133.07, 136.79.
N,N-Dimethyl-2-(2-bromophenyl)ethylamine (65) Hydrochloride
The free base 65 was prepared from 8 following method B. Treatment of the oil with 6% ethanolic HCl (10 mL) afforded the hydrochloride salt (363 mg, 40%). Mp: 205 °C; MS (ESI): m/z calcd for C10H14BrN: 227.03, 229.03, found: 228.4, 230.4. Anal. Calcd for C10H15BrClN: C, 45.39; H, 5.71; N, 5.29. Found: C, 45.43; H, 5.69; N, 5.37. 1H-MNR (DMSOd6, 300 MHz) δ (ppm): 2.81 (s, 6H, (CH3)2), 3.2 (m, 4H, CH2CH2), 7.37–7.39 (m, 1H, ArH), 7.39–7.41 m, (2H, ArH), 7.62–7.65 (d, 1H, ArH). 13C-NMR (DMSOd6, 75.5 MHz) δ (ppm): 30.54, 42.63, 56.18, 124.0, 125.77, 128.8, 129.71, 131.55, 133.55, 135.92.
N-Methyl-2-(3-bromophenyl)ethylamine (66) Hydrochloride
The free base 66 was prepared from 2-(3-bromophenyl)ethylamine (9) using a slight modification of Method A. Specifically, the chilled borohydride reaction mixture was treated with H2O (10 mL) followed by 10% NaOH (until basic), stirred for 30 min. at 0 °C under N2 and then extracted with EtOAc (3×30 mL). The combined organic extract was washed with H2O (20 mL), sat’d NaCl (20 mL), then dried over Na2SO4 and concentrated to a solid. The solid was taken up in diethyl ether (5 mL) and 6% ethanolic HCl (10 mL) was added. Concentration afforded a solid that was recrystallized from CH3OH/ether (600 mg, 50%). Mp: 124 °C; MS (ESI): m/z calcd for C9H11BrN: 213.02, 215.2, found (M+1): 214.1, 216.2. Anal. calcd for C9H13BrClN: C, 43.14; H, 5.23; N, 5.59. Found: C, 43.04; H, 5.21; N, 5.59. 1H-NMR (DMSOd6, 300 MHz) δ (ppm): 2.56 (s, 2H, CH3), 2.9 (m, 2H, CH2), 3.12 (m, 2H, CH2), 7.2–7.3 (m, 3H, ArH), 7.3–7.45 (m, 1H, ArH). 13C-NMR (DMSOd6, 75.5 MHz) δ (ppm): 31.18, 32.69, 47.74, 122.17, 128.19, 129.96, 131.06, 131.71, 140.68.
N,N-Dimethyl-2-(3-bromophenyl)ethylamine (67) Hydrochloride
The free base 67 was prepared from 9 following method B and the hydrochloride salt (395 mg, 60 %) was obtained by treatment of 67 with 6% ethanolic HCl (10 mL). Mp: 163 °C; MS (ESI): m/z calcd for C10H14BrN: 227.03, 229.03, found: 228.4, 230.5. Anal. Calcd for C10H15BrClN: C, 45.39; H, 5.71; N, 2.29. Found: C, 45.39; H, 5.73; N, 5.29. 1H-NMR (DMSOd6, 300 MHz) δ (ppm): 2.77 (s, 6H, (CH3)2), 3.05 (m, 2H, CH2), 3.2 (m, 2H, CH2), 7.32–7.34 (m, 2H, ArH), 7.45–7.48 (m, 1H, ArH), 7.61 (s, 1H, ArH). 13C-NMR (DMSOd6, 75.5 MHz) δ (ppm): 29.55, 42.19, 54.04, 122.19, 128.24, 130.0, 131.09, 131.79, 140.31.
N-Methyl-2-(4-bromophenyl)ethylamine (68) Hydrochloride
The free base 68 was prepared from 2-(4-bromophenyl)ethylamine (10) using a slight modification of Method A. Specifically, the chilled borohydride reaction mixture was treated with H2O (10 mL) followed by 10% NaOH (until basic), stirred for 30 min at 0° under N2 and then extracted with EtOAc (3×30 mL). The combined organic extract was washed with H2O (20 mL), sat’d NaCl (20 mL), then dried over Na2SO4 and concentrated to a solid. The solid was taken up in diethyl ether (5 mL) and 6% ethanolic HCl (10 mL) was added. Concentration afforded a solid that was recrystallized from CH3OH/ether (660 mg, 65%). Mp: 196 °C; MS (ESI): m/z calcd for C9H11BrN: 213.02, 215.1, found (M+1): 214.0, 216.1. Anal. calcd for C9H12BrClN: C, 43.14; H, 5.23; N, 5.59; Found: C, 43.13; H, 5.22; N, 5.56. 1H-NMR (DMSOd6, 300 MHz) δ (ppm): 2.5 (s, 3H, CH3), 2.9 (m, 2H, CH2), 3.1 (m, 2H, CH2), 7.23 (d, 2H, ArH), 7.54 (d. 2H, ArH). 13C-NMR (DMSOd6, 75.5 MHz) δ (ppm): 31.03, 32.55, 49.04, 120.18, 131.28, 131.78, 137.0.
N,N-Dimethyl--2-(4-bromophenyl)ethylamine (69) Hydrochloride
The free base 69 was prepared from 10 following method B and the hydrochloride salt (428 mg, 65%) was obtained by treatment of 69 with 6% ethanolic HCl (10 mL). Mp: 196 °C; MS (ESI): m/z calcd for C10H14BrN: 227.03, 229.03, found: 228.4, 230.5. Anal. calcd for C10H15BrClN: C, 45.93; H, 5.71; N, 5.21. Found: C, 44.91; H, 5.68; N, 5.21. 1H-NMR (DMSOd6, 300 MHz) δ (ppm): 2.77 (s, 6H, (CH3)2), 3.10, (m, 2H, CH2), 3.25 (m, 2H, CH2) 7.20 (d, 2H, ArH), 7.54 (d, 2H, ArH). 13C-NMR (DMSOd6, 75.5 MHz) δ (ppm): 29.41, 42.19, 57.05, 120.22, 131.32, 131.81, 136.92.
hTAAR 1 Activation Assay
Receptor activation was assayed in CHO cells stably expressing Gα16 (RD-HGA16 cells; Molecular Devices Corporation, Sunnyvale, CA) and hTAAR 1 as previously described.11 Briefly, these cells were plated in HAM’s F-12 medium with 10% FBS, 400 ug/mL hygromycin and 400 µg/mL geneticin at 30,000 cells/well in 96-well black clear-bottom plates and incubated at 37 °C, 5% CO2, the night before the assay. Activation of hTAAR 1 via test compounds was assessed the next day using the Calcium 3 Assay Kit (Molecular Devices; Sunnyvale, CA) per manufacturer’s specifications except that the Calcium 3 dye was used at 1/3 the suggested concentration. The cells were incubated with the dye at 37 °C for 1 h. Test compounds were evaluated using 7–8 different concentrations run in duplicate and a six-point β-PEA curve was run on each assay plate to monitor assay performance. The effect of test compound on internal calcium mobilization was determined with a FlexStation set for bottom read with 485 nm excitation and 525 nm emission wavelengths with a 515 nm emission cutoff. Basal or unstimulated fluorescence intensity was recorded for 13 sec before the addition of test compound. The effect of test compound was determined by subtracting the minimum from the maximum fluorescence intensity during a 47-sec recording period following the addition of test compound. A three-parameter logistic equation was fit to the data to calculate the EC50 and Emax values (Prism version 4.0; GraphPad, Inc. San Diego CA). The data represent the mean ± S.E. from at least three different experiments except were noted.
Molecular Modeling
The potency data (EC50) for the 68 β-phenethylamines (Table 1–Table 4) were analyzed using Comparative Molecular Field Analysis (CoMFA). The starting conformation of each structure was set to the global energy minimum conformation of the parent structure (β-PEA) and then energy-optimized using the MMFF94 force field. In addition, MMFF94 charges were calculated for each atom. The CoMFA region was defined as extending 4 A beyond every molecule in all directions. Lower corner coordinates = (−10.393577, −8.368728, −6.689531), Top corner coordinates = (9.141720, 8.391055, 9.467717). The default grid spacing of 2 A was used. The default sp3 carbon probe atom was used with the default +1 charge. The structures were aligned by the common phenethylamine substructure ([N]CCC=CC=CC=C) using the Sybyl “Align Database” command. The hTAAR 1 EC50 values (Table 1–Table 4) were utilized to calculate “pEC50” (defined as –log(EC50)). The default CoMFA row weighting (“SAME_WEIGHTS_FOR_ALL”) and scaling (“COMFA_STANDARD”) were used. A validated PLS analysis was performed on the dependent variable “pEC50” on all 68 compounds using the leave-one-out (LOO) method starting with 8 components; no compounds were left out. The optimum number of components (5) was based on the cross validated standard error of prediction (0.66 for five components). Carrying out the final PLS analysis with five components yielded a regression coefficient r2 of 0.824 (predicted as actual pEC50). The steric field contribution to the model was 61% with the balance (39%) contributed by the electrostatic field. Actual values of StdDev*Coeff of the final five-component model were contoured at 0.01 and −0.01 for both steric and electrostatic fields; these are shown in Figure 1 (steric) and Figure 2 (electrostatic).
Acknowledgements
Support for this research from the NIDA (R01-DA-19327-01) is gratefully acknowledged. The authors thank Dr. Bruce Blough for compounds from his collection in support of NIDA grant R01-DA12970, and D. Zhong, L. D. Mayer, M. Porter, and K. Warner for their technical assistance. The authors would also like to express their appreciation to the manuscript reviewers for their constructive suggestions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Roeder T. Prog Neurobiol. 1999;59:533–561. doi: 10.1016/s0301-0082(99)00016-7. [DOI] [PubMed] [Google Scholar]
- 2.Berry MD. J Neurochem. 2004;90:257–271. doi: 10.1111/j.1471-4159.2004.02501.x. [DOI] [PubMed] [Google Scholar]
- 3.Borowsky B, Adham N, Jones KA, Raddatz R, Arymshym R, Ogozalek KL, Durkin MM, Laklani PP, Bonini JA, Pathirana S, Boyle N, Po X, Kouranova E, Lichtblau H, Ochoa FY, Branchek TA, Gerald C. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:8966–8971. doi: 10.1073/pnas.151105198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Branchek TA, Blackburn TP. Cur Opin Pharmacol. 2003;3:90–97. doi: 10.1016/s1471-4892(02)00028-0. [DOI] [PubMed] [Google Scholar]
- 5.Lindemann L, Ebeling M, Kratochwil NA, Bunzow JR, Grandy DK, Hoener MC. Genomics. 2005;85:372–385. doi: 10.1016/j.ygeno.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 6.Bunzow JR, Sonders MS, Arttmagangkul S, Harrison lM, Zhamg G, Quigley DI, Darland T, Suchland KL, Pasumamula S, Kennedy JL, Olson SB, Magenis RE, Amara SG, Grandy DK. Mol Pharmacol. 2001;60:1181–1188. doi: 10.1124/mol.60.6.1181. [DOI] [PubMed] [Google Scholar]
- 7.Miller GM, Verrico C, Jassen A, Konar M, Yang H, Panas H, Mary B, Johnson R, Madras BK. J Pharmacol Exp Ther. 2005;313:983–994. doi: 10.1124/jpet.105.084459. [DOI] [PubMed] [Google Scholar]
- 8.Xie Z, Westmoreland SV, Bahn ME, Chen GL, Yang H, Vallender EJ, Yao WD, Madras BK, Miller GM. J Pharmacol Exp Ther. 2007;321:116–127. doi: 10.1124/jpet.106.116863. [DOI] [PubMed] [Google Scholar]
- 9.Grandy DK. Pharmacol Ther. 2007;116:355–390. doi: 10.1016/j.pharmthera.2007.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wainscott DB, Little SP, Yin T, Tu Y, Rocco VP, He JX, Nelson DL. J Pharmacol Exp Ther. 2007;320:475–485. doi: 10.1124/jpet.106.112532. [DOI] [PubMed] [Google Scholar]
- 11.Navarro HA, Gilmour BP, Lewin AH. J Biomol Screen. 2006;11:688–693. doi: 10.1177/1087057106289891. [DOI] [PubMed] [Google Scholar]
- 12.Reese EA, Bunzow JR, Arttamangkul S, Sonders MS, Grandy DK. J Pharmacol Exp Ther. 2007;321:178–186. doi: 10.1124/jpet.106.115402. [DOI] [PubMed] [Google Scholar]
- 13.Hays SJ, Caprathe BW, Gilmore JL, Amin N, Emmerling MR, Michael W, Nadimpalli R, Nath R, Raser KJ, Stafford D, Watson D, Wang K, Jaen JC. J Med Chem. 1998;41:1060–1067. doi: 10.1021/jm970394d. [DOI] [PubMed] [Google Scholar]
- 14.Schwartz MA, Zoda M, Vishnuvajjala B, Mami I. J Org Chem. 1976;41:2502–2503. [Google Scholar]
- 15.Krohs W, Ther L, Vogel G. Germany: 1962. p. 9. [Google Scholar]