

Abstract
Background and Purpose
Previous reports indicate that compared with normoxia, 100% ventilatory O2 during early reperfusion after global cerebral ischemia decreases hippocampal pyruvate dehydrogenase activity and increases neuronal death. However, current standards of care after cardiac arrest encourage the use of 100% O2 during resuscitation and for an undefined period thereafter. Using a clinically relevant canine cardiac arrest model, in this study we tested the hypothesis that hyperoxic reperfusion decreases hippocampal glucose metabolism and glutamate synthesis.
Methods
After 10 minutes of cardiac arrest, animals were resuscitated and ventilated for 1 hour with 100% O2 (hyperoxic) or 21% to 30% O2 (normoxic). At 30 minutes reperfusion, [1-13C]glucose was infused, and at 2 hours, brains were rapidly removed and frozen. Extracted metabolites were analyzed by 13C nuclear magnetic resonance spectroscopy.
Results
Compared with nonischemic controls, the hippocampi from hyperoxic animals had elevated levels of unmetabolized 13C-glucose and decreased incorporation of 13C into all isotope isomers of glutamate. These findings indicate impaired neuronal metabolism via the pyruvate dehydrogenase pathway for carbon entry into the tricarboxylic acid cycle and impaired glucose metabolism via the astrocytic pyruvate carboxylase pathway. No differences were observed in the cortex, indicating that the hippocampus is more vulnerable to metabolic changes induced by hyperoxic reperfusion.
Conclusions
These results represent the first direct evidence that hyperoxia after cardiac arrest impairs hippocampal oxidative energy metabolism in the brain and challenge the rationale for using excessively high resuscitative ventilatory O2
Keywords: 13C-glucose, pyruvate dehydrogenase, mitochondria, cardiac arrest, glutamate, energy metabolism
Global cerebral ischemia/reperfusion induces a wide range of cellular alterations, including impaired activity of enzymes required for cerebral energy metabolism. Oxidative molecular modifications play a significant role in postischemic damage and may ultimately lead to inhibition of cellular ATP regeneration, thereby contributing to the pathophysiology of delayed neuronal cell death.1–3
Normal cerebral energy metabolism relies on glycolysis to form pyruvate, which undergoes oxidative decarboxylation catalyzed by the pyruvate dehydrogenase complex (PDHC) to form acetyl coenzyme A (CoA) and NADH.4 Although astrocytes and neurons take up similar amounts of glucose, neurons metabolize most of the acetyl CoA derived from glucose.4 Acetyl CoA enters the tricarboxylic acid (TCA) cycle and combines with oxaloacetate, producing intermediates including α-ketoglutarate, which is converted to glutamate and further metabolized to glutamine in astrocytes.4 During ischemia, anaerobic glycolysis produces excessive lactate, which falls during reperfusion but can remain higher when using hyperoxic compared with normoxic resuscitation.1,5
Compared with normoxic reperfusion, hyperoxia immediately after global cerebral ischemia increases lipid peroxidation, worsens neurologic outcome,1 elevates nitrotyrosine levels, and reduces PDHC immunoreactivity.6 In contrast, exposure to hyperbaric O2 a few hours after global ischemia improves outcome, suggesting that the brain is most vulnerable to oxidative stress early during reperfusion when energy metabolism is most abnormal.7 Reactive oxygen and nitrogen species inhibit key metabolic enzymes, including the PDHC2,3 and the α-ketoglutarate dehydrogenase complex.8 We therefore hypothesized that after global ischemia, animals resuscitated under hyperoxic conditions exhibit decreased hippocampal aerobic energy metabolism compared with sham-operated controls and animals resuscitated with a normoxic procedure. Because the hippocampus is selectively vulnerable to neuronal death after ischemia/reperfusion,9,10 we also hypothesized that fewer changes would be found in the cortex, which is more resistant to injury.
Materials and Methods
[1-13C]glucose was purchased from Cambridge Isotope Laboratories. All other chemicals and reagents were of the highest quality and purchased from Sigma Aldrich unless otherwise stated.
Canine Cardiac Arrest and Resuscitation Model
Animal experimentation was performed according to the guidelines of the Institutional Animal Use and Care Committee of the University of Maryland, Baltimore. The model used for these studies has been utilized extensively to study global cerebral ischemia/reperfusion,1,7,11,12 and the current protocol was described previously.6 In brief, adult purebred female beagles weighing 9 to 12 kg were anesthetized initially with an intravenous injection of pentobarbital (12.5 mg/kg). Prolonged anesthesia was then induced with an infusion of α-chloralose (75 mg/kg). Core temperature was monitored for the duration of the experiment and maintained between 37°C and 38.5°C with a heating blanket and heat lamps.
The experimental timeline is shown in Figure 1. After surgical preparation, cardiac arrest was induced with an electrical train of currents generated by a Grass stimulator applied directly to the epicardium.1,11 Ventilation was terminated on verification of ventricular fibrillation. Before the onset of 10 minutes of cardiac arrest, animals were randomly assigned to 1 of 3 resuscitative protocols. In the hyperoxic group, resuscitation was performed with 100% ventilatory O2 during the open-chest cardiopulmonary resuscitation and for the first hour of reperfusion. During the next hour, the ventilator settings were adjusted to maintain arterial pO2 at >80 and <120 mm Hg. Dogs in the normoxic group were resuscitated with 21% O2, and then inspired O2 was rapidly adjusted between 21% and 30% during the 2-hour reperfusion to maintain pO2 at >80 and <120 mm Hg. Sham-operated (nonischemic) control dogs were ventilated on room air and underwent all anesthetic and surgical procedures but did not undergo cardiac arrest or resuscitative drug delivery. Open-chest cardiopulmonary resuscitation was continued for 3 minutes, at which time internal defibrillation was performed at 5 J. pCO2 was maintained between 25 and 35 mm Hg in all animals for the duration of the experiment. Exclusion criteria included temperature <37°C, systolic arterial pressure <60 mm Hg at any time after resuscitation, or inability to maintain pO2 or pCO2 within stated limits. No animals were excluded from this study. At 30 minutes reperfusion, animals were infused in the femoral vein with 0.2 g/kg [1-13C]glucose, dissolved in sterile water, over a 30-minute period. Blood was drawn at 30-minute intervals relative to the initiation of reperfusion to determine glucose concentration.
Figure 1.
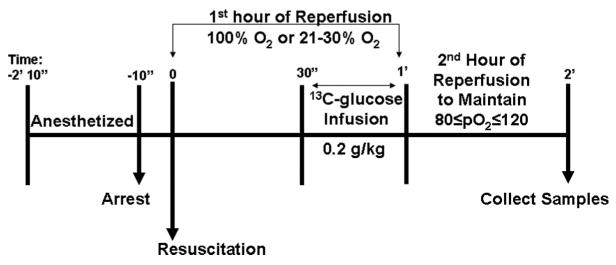
Experimental timeline. Chloralose-anesthetized animals underwent 10 minutes of cardiac arrest followed by 2 hours of reperfusion under hyperoxic (100% O2) or normoxic (21% to 30% O2) conditions for the first hour. [1-13C]glucose (0.2 mg/kg) was infused from 30 to 60 minutes of reperfusion. During the second hour of reperfusion, the ventilator settings were adjusted to maintain arterial pO2 at >80 and <120 mm Hg. At the end of the second hour, brains were removed and immediately immersed in liquid N2 (see Methods for details).
Postischemic Tissue Processing
After the 2-hour reperfusion, a craniotomy was performed on the anesthetized animal and the skull was resected to expose the brain. A 3-cm-long/2-cm-wide/1-cm-thick portion of the right frontal cortex was quickly removed and immersed in liquid N2 within 10 seconds. Each cerebral hemisphere was then rapidly removed and immersed in liquid N2, also within 10 seconds. Animals were euthanized with an intravenous injection of a pentobarbital-based euthanasia solution immediately thereafter. All samples were stored at −80°C until further processing. When removed from storage, each frozen hemisphere was placed in a temperature-controlled box (0°C), and the hippocampus was surgically dissected. The hippocampus from 1 normoxia-resuscitated animal was accidentally destroyed during this process, and therefore, both it and the cortex sample from the same animal were not assayed. The sample of cortex and the pooled hippocampi from both hemispheres were placed separately in dounce homogenizers (Fisher) containing 7% ice-cold perchloric acid (PCA). Samples were homogenized as they thawed and centrifuged for 5 minutes (Beckman J2-MC, Rotor 25.5) at 4°C and 7500 rpm. Two hundred microliters of the supernatant was stored at −80°C, and aliquots were later derivatized with o-phthaldialdehyde for high-performance liquid chromatography (HPLC) analysis (Gilson model 121) of amino acids13 and analyzed for total lactate.14 The pellets were reextracted with ice-cold deionized water and centrifuged. The PCA extracts (supernatants) were pooled and neutralized to pH 7.0, shell-frozen, lyophilized, and reconstituted in D2O with 0.4% dioxane added as an internal standard for quantification. Samples (0.6 mL) were transferred to a 5-mm nuclear magnetic resonance (NMR) tube (Wilmad) for analysis by NMR spectroscopy. Pellets were solubilized in 1N NaOH and analyzed for protein by the method of Lowry (Bio-Rad Laboratories).
NMR Spectroscopy
Proton-decoupled 125.5-MHz 13C-NMR spectra were acquired on a Varian Inova 500 MHz spectrometer with a broadband detection probe and a 30° pulse angle, 25 KHz spectral width, and 64K data points. An acquisition time of 1.3 seconds and a 4.3-second relaxation delay were used. The number of scans was 11 000 to 13 000 for cortical samples and 18 000 to 20 000 for hippocampal samples. All spectra were corrected for nuclear Overhauser effects by using correction factors obtained by comparing peak intensities from spectra collected with the decoupler on throughout the experiment to spectra decoupled only during acquisition.13 A line broadening of 2 Hz was used. Dioxane was used as an internal concentration standard to calculate the nanomoles of 13C incorporated per milligram protein.14 Chemical shifts were reported relative to the dioxane peak at 67.4 ppm, and peak assignments were made by comparison with literature values14 and with pure standards run under the same conditions. The amount of 13C in each peak was corrected for natural abundance (1.1% of all carbon atoms).
Calculation of Percent Enrichment
The total concentration of lactate or specific amino acids (determined by HPLC) was used to calculate percent enrichment for each isotope isomer (isotopomer) of lactate, glutamate, or glutamine15 according to the following equation:
Statistical Analysis
One-way ANOVA followed by Tukey's post hoc analysis was used for statistical analysis of 13C labeling of brain samples and blood glucose levels in the 3 experimental groups. A value of P<0.05 was considered significant. Values are expressed as mean±SEM for n=7 nonischemic controls, 7 hyperoxic animals, and 6 normoxic animals.
Results
Canine Cardiac Arrest and Resuscitation
There were no significant differences in baseline (preischemic) values for pO2, temperature, systolic arterial pressure, or blood glucose levels between animal groups. However, the pO2 at 30 minutes and at 1 hour reperfusion for hyperoxic animals was significantly greater (P<0.001) than the pO2 of normoxic resuscitated animals (Table 1). In addition, the blood glucose at 30 and 60 minutes of reperfusion in animals subjected to cardiac arrest and resuscitation under either hyperoxic or normoxic conditions was significantly higher (P<0.001) than in nonischemic animals (Table 1), as an expected outcome of the cardiac arrest and the administration of epinephrine during resuscitation. Because both ischemic animal groups were hyperglycemic by 30 minutes of reperfusion (before the [1-13C]glucose infusion) and nonischemic animals infused with the same amount of [1-13C]glucose did not become hyperglycemic, it follows that the postischemic hyperglycemia was attributable to cardiac arrest and resuscitation rather than glucose infusion. No differences among hyperoxic and normoxic groups were found in any other physiologic parameters.
TABLE 1. Physiologic Variables Before and After Cardiac Arrest and Resuscitation.
| Nonischemic | Hyperoxic | Normoxic | |
|---|---|---|---|
| pO2, mm Hg | |||
| Baseline | 106.3±5.2 | 109.8±4.9 | 100.3±1.7 |
| 30 minutes | 103.0±6.6 | 506.3±63.6*† | 96.5±23.7 |
| 60 minutes | 95.9±4.7 | 487.0±35.7*† | 88.7±4.0 |
| Glucose, mg/dL | |||
| Baseline | 80.6±3.6 | 79.4±3.2 | 79.8±3.5 |
| 30 minutes | 82.0±4.4 | 220.7±26.0* | 229.7±24.0* |
| 60 minutes | 97.1±2.1 | 326.4±12.8* | 304.2±28.0* |
| SAP, mm Hg | |||
| Baseline | 148.2±2.5 | 166.6±8.5 | 143.8±8.5 |
| 30 minutes | 145.4±15.0 | 127.0±18.3 | 140.5±20.1 |
| 60 minutes | 145.0±6.1 | 124.6±4.2 | 131.3±8.8 |
| Temperature, °C | |||
| Baseline | 37.7±0.1 | 37.7±0.1 | 37.7±0.1 |
| 30 minutes | 37.6±0.4 | 37.8±0.4 | 37.7±0.2 |
| 60 minutes | 37.7±0.1 | 37.9±0.2 | 37.5±0.1 |
SAP (systolic arterial pressure), arterial blood glucose, pO2, and rectal temperature were measured immediately before and at 30 and 60 minutes after cardiac arrest and resuscitation under either the hyperoxic or normoxic protocol (Methods). Values are mean±SE for n=7 nonischemic, 7 hyperoxic, and 6 normoxic animals.
Significantly different from nonischemic control animals (P<0.001).
Significantly different from normoxic resuscitated animals (P<0.001).
[1-13C]Glucose Metabolism
The amount of unmetabolized [1-13C]glucose was significantly increased (P<0.05) in both the hippocampus and cortex of animals resuscitated under hyperoxic conditions, compared with nonischemic animals (Figure 2A). There was no significant difference in the amount of unmetabolized glucose in normoxic brain compared with controls. A strong trend toward increased incorporation of 13C into [3-13C]lactate in the hippocampus (P=0.06) of animals reperfused under hyperoxic conditions (Figure 2B) suggested a shift from oxidative energy metabolism toward increased anaerobic metabolism. There was a significant increase in the percent enrichment in the C3 position of lactate in the hyperoxic group (4.6±0.6%, P<0.05) compared with nonischemic controls (2.6±0.6%), indicating that oxidative cerebral energy was affected in this model of global ischemia. The enrichment of lactate in the normoxic group (3.8±0.4%) was not different from that of hyperoxic or control animals. There were no differences in either the percent enrichment or incorporation of 13C into lactate in the cortex, suggesting that energy metabolism in the cortex was less affected than in the hippocampus.
Figure 2.

Hyperoxic reperfusion leads to increased unmetabolized glucose and increased [3-13C]lactate in the hippocampus and cortex. A, The amount of unmetabolized 13C-glucose was significantly higher in the hippocampus and cortex of animals resuscitated under hyperoxic conditions compared with nonischemic controls. The trend toward increased unmetabolized [1-13C]glucose in animals resuscitated under normoxic conditions was not significant (P=0.1). B, Although not significant (P=0.06), there was a trend toward elevated 13C labeling in the C3 position of lactate in the hippocampus of animals reperfused under hyperoxic, but not normoxic, conditions compared with nonischemic controls. There was no difference in 13C incorporation into lactate in the cortex. Values are mean±SE for n=7 nonischemic controls, 7 hyperoxic animals, and 6 normoxic animals. *Significantly different from nonischemic controls; 1-way ANOVA with Tukey post hoc analysis, P<0.05.
Incorporation of 13C Into Glutamate and Glutamine in the Hippocampus
The labeling pattern of brain metabolites after infusion of [1-13C]glucose is well documented.15 [1-13C]glucose enters glycolysis forming [3-13C]pyruvate, which can be converted to lactate or metabolized via the TCA cycle thereby giving rise in the first turn to [4-13C]α-ketoglutarate which is converted to [4-13C]glutamate and [4-13C]glutamine, and to [3-13C]glutamate and glutamine during the second turn of the cycle. Incorporation of 13C into brain metabolites was found in all animals infused with [1-13C]glucose, whereas in animals that were not infused with labeled glucose, few peaks larger than background noise were observed, indicating the low natural abundance of 13C in the canine brain (not shown). To the best of our knowledge, this is the first ex vivo study using [1-13C]glucose to evaluate canine cerebral energy metabolism. However, the amount of 13C glucose incorporated into glutamate and glutamine isotopomers in the hippocampus of the anesthetized canine (Figure 3A) is within the same order of magnitude as that of postanesthetized rat brain.15
Figure 3.

Hyperoxic reperfusion decreases hippocampal but not cortical metabolism of [1-13C]glucose to 13C-glutamate. A, Metabolism of [1-13C]glucose to glutamate but not glutamine is decreased in the hippocampus of animals resuscitated under hyperoxic conditions after a 10-minute cardiac arrest and 2-hour reperfusion paradigm. Decreased incorporation of 13C into glutamate C4 indicates decreased oxidative metabolism in neurons, possibly due to decreased PDHC activity and TCA cycle metabolism. Decreased labeling in the C2 of glutamate indicates impaired metabolism via the pyruvate carboxylase pathway in astrocytes. B, In the cortex, there was no difference in metabolism of 13C-glucose to either glutamate or glutamine after ischemia/reperfusion. Values are mean±SE for n=7 nonischemic controls, 7 hyperoxic animals, and 6 normoxic animals. *Significantly different from nonischemic controls; ANOVA with Tukey test, P<0.05.
Comparisons of the incorporation of 13C from the metabolism of glucose into glutamate and glutamine in nonischemic controls and animals after ischemia and 2-hour reperfusion are shown in Figure 3. Compared with nonischemic animals, incorporation of 13C into the C4 and C3 positions of glutamate in the hippocampus of canines resuscitated under hyperoxic conditions was decreased by 51% and 53.1%, respectively (Figure 3A). Reduced labeling in these isotopomers is indicative of reduced aerobic glucose metabolism through the pyruvate dehydrogenase pathway and TCA cycle. This difference was also evidenced in subsequent turns of the TCA cycle, because incorporation of 13C into the C1 position was also decreased by 79.7% in hyperoxic animals (Figure 3A). In addition, hyperoxic reperfusion decreased the incorporation of label into [2-13C]glutamate by 81.7%, indicating that metabolism via the pyruvate carboxylase pathway in astrocytes was also inhibited. There were no significant differences in incorporation of label in the normoxic reperfusion group compared with nonischemic controls. Although there was a consistent trend toward higher labeling in the normoxic (suggesting improved metabolism) compared with the hyperoxic samples, there were no significant differences in incorporation of label from the metabolism of [1-13C]glucose into glutamate in hippocampus of these groups. In contrast to the effects of hyperoxic reperfusion on incorporation of 13C label into glutamate, no significant difference in 13C labeling of glutamine was observed between the nonischemic, hyperoxic, and normoxic groups.
Incorporation of 13C Into Glutamate and Glutamine in the Cortex
Although the labeling pattern was similar to that observed in the hippocampus, there was considerably higher incorporation of label from the metabolism of [1-13C]glucose into glutamate and glutamine in the cortex of all groups of animals. However, in contrast to the results with the hippocampus, there were no differences in incorporation of 13C into glutamate or glutamine in the cortex of animals resuscitated under either hyperoxic or normoxic conditions when compared with nonischemic controls (Figure 3B).
Analysis of Amino Acids by HPLC in the Hippocampus
Analysis of amino acids by HPLC (Table 2) revealed an increased concentration of alanine in hyperoxic brain compared with nonischemic controls and animals resuscitated under normoxic conditions. No significant differences were found in the concentrations of aspartate, glutamate, glutamine, or γ-aminobutyric acid (GABA) after ischemia/reperfusion under different oxygen tensions. There were significant reductions in the percent enrichment of the C4, C3, C2, and C1 positions of glutamate in the hyperoxic group compared with nonischemic control animals (Figure 4), indicating that incorporation of 13C from glucose into glutamate was affected more than overall glutamate utilization. No change in 13C enrichment of glutamine was found (not shown).
TABLE 2. Hippocampal Amino Acid Concentrations (nmol/mg protein) After Cardiac Arrest and 2-Hour Reperfusion.
| Nonischemic | Hyperoxic | Normoxic | |
|---|---|---|---|
| Aspartate | 111.9±18.3 | 101.1±13.2 | 96.2±24.0 |
| Glutamate | 670.9±90.3 | 785.2±86.4 | 683.3±73.6 |
| Glutamine | 171.8±70.7 | 334.2±73.9 | 235.7±67.5 |
| GABA | 75.2±13.0 | 127.9±21.3 | 117.1±10.9 |
| Alanine | 38.5±6.7 | 84.4±16.9* | 77.1±7.5 |
Amino acids were determined by HPLC on extracts of frozen hippocampi from sham-operated, nonischemic animals and from animals after 10 minutes of cardiac arrest and 2-hour reperfusion using hyperoxic or normoxic resuscitation (Methods). Values are mean±SE for n=7 nonischemic, 7 hyperoxic, and 6 normoxic animals.
Significantly different from nonischemic controls (P<0.05).
Figure 4.

Hyperoxic reperfusion decreases 13C percent enrichment of glutamate in the hippocampus. Percent enrichment was calculated as described in Methods. Hyperoxic reperfusion significantly decreased percent enrichment in all isotopomers of glutamate. Values are mean±SE for n=7 nonischemic controls, 7 hyperoxic animals, and 6 normoxic animals. *Significantly different from nonischemic controls; ANOVA with Tukey test, P<0.05.
Discussion
This study is the first to report direct evidence that hyperoxic resuscitation and reperfusion after cardiac arrest impairs cerebral aerobic energy metabolism. Decreased incorporation of [1-13C]glucose into [4-13C]glutamate (and subsequently into [3-13C] and [1-13C] glutamate) in the hippocampus indicates altered metabolism via a pathway that uses the PDHC for entry into the TCA cycle. Because incorporation of label from infused glucose first appears in brain glutamate and later in glutamine16 and because acetyl CoA derived from glucose predominantly enters the neuronal TCA cycle, the decreased incorporation of 13C into [4-13C]glutamate primarily reflects an impairment in neuronal, rather than astrocytic, metabolism. Moreover, hippocampal 13C enrichment of all isotopomers of glutamate (Figure 4), but not glutamine, was reduced after hyperoxic reperfusion, providing further evidence that neuronal metabolism is impaired. In addition, because the total amounts of glutamate and glutamine analyzed by HPLC were unchanged in the different experimental groups, the reduced 13C glutamate enrichment can be attributed to decreased energy production and not other metabolic processes, such as changes in glutamate utilization. For several glutamate isotopomers, there was also a trend toward lower incorporation in the normoxic animals compared with nonischemics and a trend toward higher incorporation after normoxic compared with hyperoxic resuscitation. The variability in these results is not surprising, considering the large number of enzymatic and transport activities required for conversion of systemically administered glucose into glutamate and other metabolites within the brain. Future studies using this approach will therefore require a larger number of animals per group to reach a power sufficient to detect significant differences.
Postischemic impairment of PDHC enzyme activity and reduction in subunit immunoreactivity have been documented.2,3,6,17–20 In particular, previous results from our laboratory showed a 33% decrease in total hippocampal PDHC activity after hyperoxic reperfusion compared with nonischemic control animals.3 Cardell et al19 reported a 50% reduction in PDHC activity within 15 minutes of recirculation in a rat model of cerebral ischemia, attributable most likely to increased protein phosphorylation rather than a loss of total activity. The decreased incorporation of 13C into the C4 position of glutamate in the present study is consistent with the reduction in PDHC activity, but it could be caused by impairment of other metabolic enzymes.
Hyperoxic reperfusion after cardiac arrest exacerbates oxidative stress leading to increased lipid oxidation1 and elevated hippocampal nitrotyrosine immunoreactivity.6 Prooxidant reactive species, eg, the hydroxyl radical and peroxynitrite that cause these molecular alterations, can also inactivate important enzymes in energy metabolism, like PDHC3 or components of the electron transport chain. The findings that the brain NAD(P)H redox state is hyperoxidized during the first hour after ischemia and that this hyperoxidized state is exacerbated by hyperoxia suggest that reactions prior to the electron transport chain limit postischemic aerobic energy metabolism.21 The increased percent enrichment of [3-13C]lactate after hyperoxic reperfusion in the present study shows that glycolysis is not impaired.
In the present study, hyperoxic reperfusion decreased the incorporation of label from the metabolism of [1-13C]glucose into [4-13C]glutamate to 51% of that observed in nonischemic controls (Figure 3A). Studies of focal cerebral ischemia in rat models have also shown decreased incorporation of [1-13C]glucose into [4-13C]glutamate, indicating alterations in neuronal TCA cycle metabolism.15,22 Oxidation of glutamate via a partial TCA cycle is known to occur in neurons under various pathologic conditions13 in response to impaired aerobic glucose metabolism.23 Pascual et al23 showed increased glutamate oxidation after focal cerebral ischemia in the rat, consistent with intact metabolism via α-ketoglutarate dehydrogenase and use of glutamate as an alternative energy substrate in brain.
Although multiple turns of the TCA cycle lead to incorporation of label from [1-13C]glucose into [2-13C]glutamate, this isotopomer is primarily labeled by metabolism via the pyruvate carboxylase pathway in astrocytes.24 This pathway, which adds net carbon to the astrocytic TCA cycle, leads to de novo synthesis of glutamine in astrocytes and has an important role in providing glutamine to neurons to replenish the glutamate released during neurotransmission.15,25 Therefore, the decreased [2-13C]glutamate observed in the present study after hyperoxic reperfusion reflects both decreased metabolism via pyruvate carboxylase (astrocytic) and decreased labeling from subsequent turns of the TCA cycle after initial metabolism via PDHC (predominantly neuronal), because both processes lead to labeling in the C2 position.
To discern the relative astrocytic and neuronal contributions to the alterations in metabolism after hyperoxic reperfusion, a study using both 13C-glucose and 13C-acetate is warranted. Labeled acetate is taken up and metabolized exclusively by astrocytes, allowing for a more direct comparison of changes in glial metabolism.4 Because reperfusion improves astrocytic metabolism after focal cerebral ischemia,22 monitoring astrocytic metabolism at multiple time points may be useful in determining the precise role of astrocytes in reperfusion-mediated changes in metabolism after global ischemia.
Although significant differences in incorporation of 13C label into glutamate in the hippocampus were found, no difference in incorporation of [1-13C]glucose into isotopomers of glutamine occurred during either hyperoxic or normoxic reperfusion compared with nonischemic controls. Whereas Haberg et al22 reported changes in 13C incorporation into glutamine after focal cerebral ischemia, these differences were detected in the rat brain after 120 minutes of ischemia. In addition, alterations in glutamine are generally attenuated compared with the striking decrease in 13C incorporation into glutamate.22 Because considerably less label from glucose is incorporated into glutamine than glutamate, it is more difficult to determine alterations in labeling of 13C-glutamine in anesthetized brain after a short period of ischemia.
A significant increase in percent enrichment of lactate and a trend toward elevated incorporation of [1-13C]glucose into [3-13C]lactate (P=0.06) were observed in the hippocampus of animals resuscitated under hyperoxic but not normoxic conditions compared with nonischemic controls. Similarly, in a rat focal cerebral ischemia model, Haberg et al22 reported decreased 13C-glucose metabolism in the penumbra but did not observe increased 13C incorporation into lactate.22 In the current study, there was no change in enrichment or 13C-lactate labeling in the cortex, providing further evidence that hippocampal metabolism is particularly vulnerable in this clinically relevant model of global ischemia/reperfusion. The recent report by Richards et al3 that there was no change in PDHC activity in the cortex, regardless of the reperfusion paradigm, is consistent with this concept.
Significantly elevated amounts of unmetabolized 13C-glucose were observed in both the hippocampus and cortex of animals resuscitated under hyperoxic conditions after ischemia/reperfusion, with a trend toward greater unmetabolized glucose in animals resuscitated under normoxic conditions. These findings are consistent with impaired postischemic metabolism of [1-13C]glucose in rat models of focal cerebral ischemia.15,22,23 In the present study impaired metabolism of [1-13C]glucose to 13C-glutamate was only observed in the hippocampus after hyperoxic resuscitation. This finding indicates that the canine hippocampus is selectively vulnerable to the high oxygen tension used during hyperoxic reperfusion, which is consistent with earlier findings in our model3,6 and in rodent models of cerebral ischemia.10,26
Summary
This study represents the first experiments using [1-13C]glucose in conjunction with ex vivo NMR spectroscopy to evaluate metabolic alterations after global cerebral ischemia in the clinically relevant canine model. The significant changes in 13C NMR spectra observed after 2 hours of reperfusion after 10 minutes of cardiac arrest indicate the power of 13C NMR to detect early alterations in metabolism that may contribute to postischemic delayed neuronal cell death and neurologic impairment. Our results also provide the first direct evidence that hyperoxic resuscitation and reperfusion after cardiac arrest exacerbate impairment of hippocampal oxidative energy metabolism. Current standards of care after cardiac arrest encourage the use of 100% O2 during resuscitation and for an undefined period thereafter. However, the results of this study, taken together with those demonstrating improved neurologic outcome and reduced hippocampal neuronal death in normoxic compared with hyperoxic reperfusion, further question the indiscriminate use of 100% O2 immediately after cardiac resuscitation.
Acknowledgments
The authors would like to thank Peter Baab, Kyni Jones, and Dr Cynthia Cotta-Cumba for expert technical assistance.
Sources of Funding: This study was supported by NIH NS055450, NS34152, NS049425, HD16596, and US Army DAMD 17-99-1-9483.
Footnotes
Disclosures: None.
References
- 1.Liu Y, Rosenthal RE, Haywood Y, Miljkovic-Lolic M, Vanderhoek JY, Fiskum G. Normoxic ventilation after cardiac arrest reduces oxidation of brain lipids and improves neurological outcome. Stroke. 1998;29:1679–1686. doi: 10.1161/01.str.29.8.1679. [DOI] [PubMed] [Google Scholar]
- 2.Bogaert YE, Rosenthal RE, Fiskum G. Postischemic inhibition of cerebral cortex pyruvate dehydrogenase. Free Radic Biol Med. 1994;16:811–820. doi: 10.1016/0891-5849(94)90197-x. [DOI] [PubMed] [Google Scholar]
- 3.Richards EM, Rosenthal RE, Kristian T, Fiskum G. Postischemic hyperoxia reduces hippocampal pyruvate dehydrogenase activity. Free Radic Biol Med. 2006;40:1960–1970. doi: 10.1016/j.freeradbiomed.2006.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sonnewald U, Kondziella D. Neuronal glial interaction in different neurological diseases studied by ex vivo 13C NMR spectroscopy. NMR Biomed. 2003;16:424–429. doi: 10.1002/nbm.837. [DOI] [PubMed] [Google Scholar]
- 5.Rosenthal RE, Williams R, Bogaert YE, Getson PR, Fiskum G. Prevention of postischemic canine neurological injury through potentiation of brain energy metabolism by acetyl-l-carnitine. Stroke. 1992;23:1312–1317. doi: 10.1161/01.str.23.9.1312. discussion 1317–1318. [DOI] [PubMed] [Google Scholar]
- 6.Vereczki V, Martin E, Rosenthal RE, Hof PR, Hoffman GE, Fiskum G. Normoxic resuscitation after cardiac arrest protects against hippocampal oxidative stress, metabolic dysfunction, and neuronal death. J Cereb Blood Flow Metab. 2006;26:821–835. doi: 10.1038/sj.jcbfm.9600234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenthal RE, Silbergleit R, Hof PR, Haywood Y, Fiskum G. Hyperbaric oxygen reduces neuronal death and improves neurological outcome after canine cardiac arrest. Stroke. 2003;34:1311–1316. doi: 10.1161/01.STR.0000066868.95807.91. [DOI] [PubMed] [Google Scholar]
- 8.Park LC, Zhang H, Sheu KF, Calingasan NY, Kristal BS, Lindsay JG, Gibson GE. Metabolic impairment induces oxidative stress, compromises inflammatory responses, and inactivates a key mitochondrial enzyme in microglia. J Neurochem. 1999;72:1948–1958. doi: 10.1046/j.1471-4159.1999.0721948.x. [DOI] [PubMed] [Google Scholar]
- 9.Petito CK, Feldmann E, Pulsinelli WA, Plum F. Delayed hippocampal damage in humans following cardiorespiratory arrest. Neurology. 1987;37:1281–1286. doi: 10.1212/wnl.37.8.1281. [DOI] [PubMed] [Google Scholar]
- 10.Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- 11.Zwemer CF, Whitesall SE, D'Alecy LG. Cardiopulmonary-cerebral resuscitation with 100% oxygen exacerbates neurological dysfunction following nine minutes of normothermic cardiac arrest in dogs. Resuscitation. 1994;27:159–170. doi: 10.1016/0300-9572(94)90009-4. [DOI] [PubMed] [Google Scholar]
- 12.Fiskum G, Liu Y, Bogaert YE, Rosenthal RE. Acetyl-l-Carnitine stimulates cerebral oxidative metabolism and inhibits protein oxidation following cardiac arrest in dogs. In: Krieglstein J, Oberpichler-Schwenk H, editors. Pharmacology of Cerebral Ischemia. Stuttgart: Wissenschaftliche Verlagsgesellschaft mbH; 1992. pp. 487–491. [Google Scholar]
- 13.Sonnewald U, McKenna M. Metabolic compartmentation in cortical synaptosomes: influence of glucose and preferential incorporation of endogenous glutamate into GABA. Neurochem Res. 2002;27:43–50. doi: 10.1023/a:1014846404492. [DOI] [PubMed] [Google Scholar]
- 14.McKenna MC, Sonnewald U. GABA alters the metabolic fate of [U-13C]glutamate in cultured cortical astrocytes. J Neurosci Res. 2005;79:81–87. doi: 10.1002/jnr.20309. [DOI] [PubMed] [Google Scholar]
- 15.Haberg A, Qu H, Saether O, Unsgard G, Haraldseth O, Sonnewald U. Differences in neurotransmitter synthesis and intermediary metabolism between glutamatergic and GABAergic neurons during 4 hours of middle cerebral artery occlusion in the rat: the role of astrocytes in neuronal survival. J Cereb Blood Flow Metab. 2001;21:1451–1463. doi: 10.1097/00004647-200112000-00010. [DOI] [PubMed] [Google Scholar]
- 16.Fitzpatrick SM, Hetherington HP, Behar KL, Shulman RG. The flux from glucose to glutamate in the rat brain in vivo as determined by 1H-observed, 13C-edited NMR spectroscopy. J Cereb Blood Flow Metab. 1990;10:170–179. doi: 10.1038/jcbfm.1990.32. [DOI] [PubMed] [Google Scholar]
- 17.Zaidan E, Sheu KF, Sims NR. The pyruvate dehydrogenase complex is partially inactivated during early recirculation following short-term forebrain ischemia in rats. J Neurochem. 1998;70:233–241. doi: 10.1046/j.1471-4159.1998.70010233.x. [DOI] [PubMed] [Google Scholar]
- 18.Bogaert YE, Sheu KF, Hof PR, Brown AM, Blass JP, Rosenthal RE, Fiskum G. Neuronal subclass-selective loss of pyruvate dehydrogenase immunoreactivity following canine cardiac arrest and resuscitation. Exp Neurol. 2000;161:115–126. doi: 10.1006/exnr.1999.7250. [DOI] [PubMed] [Google Scholar]
- 19.Cardell M, Koide T, Wieloch T. Pyruvate dehydrogenase activity in the rat cerebral cortex following cerebral ischemia. J Cereb Blood Flow Metab. 1989;9:350–357. doi: 10.1038/jcbfm.1989.53. [DOI] [PubMed] [Google Scholar]
- 20.Fukuchi T, Katayama Y, Kamiya T, McKee A, Kashiwagi F, Terashi A. The effect of duration of cerebral ischemia on brain pyruvate dehydrogenase activity, energy metabolites, and blood flow during reperfusion in gerbil brain. Brain Res. 1998;792:59–65. doi: 10.1016/s0006-8993(98)00121-8. [DOI] [PubMed] [Google Scholar]
- 21.Feng ZC, Sick TJ, Rosenthal M. Oxygen sensitivity of mitochondrial redox status and evoked potential recovery early during reperfusion in post-ischemic rat brain. Resuscitation. 1998;37:33–41. doi: 10.1016/s0300-9572(98)00031-8. [DOI] [PubMed] [Google Scholar]
- 22.Haberg A, Qu H, Sonnewald U. Glutamate and GABA metabolism in transient and permanent middle cerebral artery occlusion in rat: importance of astrocytes for neuronal survival. Neurochem Int. 2006;48:531–540. doi: 10.1016/j.neuint.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 23.Pascual JM, Carceller F, Roda JM, Cerdan S. Glutamate, glutamine, and GABA as substrates for the neuronal and glial compartments after focal cerebral ischemia in rats. Stroke. 1998;29:1048–1056. doi: 10.1161/01.str.29.5.1048. discussion 1056–1057. [DOI] [PubMed] [Google Scholar]
- 24.Shank RP, Bennett GS, Freytag SO, Campbell GL. Pyruvate carboxylase: an astrocyte-specific enzyme implicated in the replenishment of amino acid neurotransmitter pools. Brain Res. 1985;329:364–367. doi: 10.1016/0006-8993(85)90552-9. [DOI] [PubMed] [Google Scholar]
- 25.Sibson NR, Mason GF, Shen J, Cline GW, Herskovits AZ, Wall JE, Behar KL, Rothman DL, Shulman RG. In vivo 13C NMR measurement of neurotransmitter glutamate cycling, anaplerosis and TCA cycle flux in rat brain during [2-13C]glucose infusion. J Neurochem. 2001;76:975–989. doi: 10.1046/j.1471-4159.2001.00074.x. [DOI] [PubMed] [Google Scholar]
- 26.Konaka K, Ueda H, Li JY, Matsumoto M, Sakoda S, Yanagihara T. N-acetylaspartate to total creatine ratio in the hippocampal CA1 sector after transient cerebral ischemia in gerbils: influence of neuronal elements, reactive gliosis, and tissue atrophy. J Cereb Blood Flow Metab. 2003;23:700–708. doi: 10.1097/01.WCB.0000071888.63724.56. [DOI] [PubMed] [Google Scholar]