

Abstract
The virally encoded integrase protein is an essential enzyme in the life cycle of the HIV-1 virus and represents an attractive and validated target in the development of therapeutics against HIV infection. Drugs that selectively inhibit this enzyme, when used in combination with inhibitors of reverse transcriptase and protease, are believed to be highly effective in suppressing the viral replication. Among the HIV-1 integrase inhibitors, the β-diketo acids (DKAs) represent a major lead for anti-HIV-1drug development. In this study, novel bifunctional quinolonyl diketo acid derivatives were designed, synthesized and tested for their inhibitory ability against HIV-1 integrase. The compounds are potent inhibitors of integrase activity. Particularly, derivative 8 is a potent IN inhibitor for both steps of the reaction (3′-processing and strand transfer) and exhibits both high antiviral activity against HIV-1 infected cells and low cytotoxicity. Molecular modeling studies provide a plausible mechanism of action, which is consistent with ligand SARs and enzyme photo-crosslinking experiments.
Introduction
Three different classes of chemotherapeutic agents are actually used to inhibit the replication of HIV-1, the etiological agent of acquired immunodeficiency syndrome (AIDS): the nucleoside (NRTI) and non-nucleoside (NNRTI) reverse transcriptase inhibitors, the protease inhibitors (PRI) and the inhibitors of the fusion of the virus with the host cell (for review see1). The highly active anti-retroviral therapy (HAART), which is based on the use of a combination of the cited drugs, effectively inhibits the replication cycle of HIV-1. The advent of HAART has made possible the suppression of the HIV-1 replication to such an extent that the virus becomes undetectable in the blood of infected persons. As a consequence, a decline of both mortality and morbidity due to the HIV-1 was reported in the recent years. However, HAART fails to eradicate the viral replication, which persists at a lower level in cellular reservoirs, despite the chemotherapy.2 The ability of HIV-1 to evolve drug resistance and the toxicity of HAART regimens makes the integrase (IN), which is the third virally encoded enzyme required for HIV-1 replication, a legitimate target for the development of new drugs. Moreover, IN has no cellular counterpart, and thus came into sight ten years ago as a new therapeutic opportunity.3–5 Hopefully, integrase inhibitors will become a potential additive to HAART or a salvage therapy for patients resistant to currently available anti-HIV drugs.
IN catalyzes the insertion of viral DNA into the host genome derived from reverse transcription of HIV RNA. Integration occurs via a multi-step sequence of reactions, including i) cleavage of a dinucleotide pair from the 3′-end of the viral DNA (termed “3′-processing”, 3′-P), ii) insertion of the resulting shortened strands into the host-cell chromosome (termed “strand transfer”, ST), iii) removal of the two unpaired nucleotides at the 5′-end of the viral DNA and gap-filling process (for review see 6).
Numerous compounds with different structural features have been reported as IN inhibitors6,7 of which the most important class is typified by an aryl β-diketo acid motif (DKAs)8 (compound 1,9 Figure 1). DKAs selectively inhibit the ST reaction of IN and exhibit antiviral activity against HIV-1-infected cells in a manner consistent with inhibition of integration.8,10 These compounds are also as useful tools to explore the molecular mechanism of IN.8,11,12
Figure 1.

Structures of selected HIV-1 IN inhibitors belonging to the mono and bifunctional DKAs.
More recently, some bifunctional DKAs (BDKAs) were reported, which are characterized by the presence of two diketo acid chains in the skeleton of the IN inhibitor (compounds 2–4, Figure 1).13–15 However, the BDKAs reported so far were less potent than mono functional counterparts and endowed with low antiviral activities. These properties could be ascribed to the lack of some important structural features in the reported BDKAs. In fact, structure-activity relationships on mono-functional aryl diketo acids led to conclude that the highest activity was obtained when the central aromatic ring is 1,3-disubstituted with the diketo acid chain and a benzyl moiety (see compound 1, Figure 1). In such a way, the angle between the two lines extended from the above groups is around 120°.9
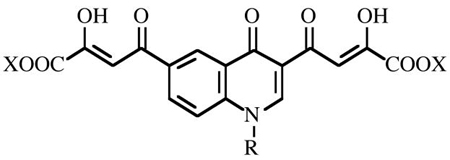
The aim of the present work was to obtain new BDKAs with improved activity against IN and HIV-1 infected cells. Thus, based on our prior experience16 we designed new inhibitors 5–8 based on the 4-(4(1H)-quinolinon-3-yl)-2,4-dioxobutanoic acid skeleton (Figure 2). This scaffold was chosen because i) it is easy to alkylate the 4(1H)-quinolinone at 1-position with a benzyl group to obtain a 1,3-disubstituted compound that well fits the geometric requirements for an optimal IN inhibitory activity; ii) a second diketo acid function can readily be introduced at position 6 of aromatic ring via acetyl intermediate. Moreover, with the aim to characterize the mode of binding of the novel compounds within the IN, docking studies were performed and a molecular basis of enzyme inhibition is proposed.
Figure 2.

Structural features of the newly designed bifunctional quinolonyl diketo acid derivatives.
Results and Discussion
Chemistry
Synthesis of derivatives 5–8 is outlined in Scheme 1. The key intermediate 3,6-diacetyl-4(1H)-quinolinone (9) was obtained by reaction of 4-acetylaniline with ethyl ethoxymethyleneacetoacetate, followed by thermic cyclization in diphenyl ether. 9 was then alkylated with 4-fluorobenzyl bromide in alkaline medium (K2CO3) to afford N-1 substituted quinolinone 10. Acetyl derivatives 9 and 10 underwent Claisen condensation with diethyl oxalate in presence of sodium ethoxide as a catalyst to afford the diketo esters 5 and 7, which were in turn hydrolyzed with NaOH 6N in the corresponding acid derivatives 6 and 8. Chemical and physical data of compounds 5–10 are reported in the experimental section; spectroscopic data of derivatives 5–8 are given in Supporting Information section.
Scheme 1a.

aReagents and conditions: (a) ethyl ethoxymethylene acetoacetate, neat, 120 °C 5 min, 73%; (b) Ph2O, reflux 50 min, 56%; (c) 4-flurobenzyl bromide, K2CO3, DMF, reflux 2h, 88%, (d) diethyl oxalate, C2H5ONa, THF C2H5OH, room temp 2 h, 5 100%, 45 min, 7 88%; (e) NaOH 1N, room temp 1.5 h, 6 52%, 8 77%.
Evaluation of biological activities
In vitro assays
Derivatives 5–8 were tested in vitro for ST inhibition in the presence of magnesium (Mg2+) using a novel plate-based electrochemoluminescent assay to generate IC50 values from triplicate experiments (Table 1). Compounds 5–8 were also tested for ST and 3′-P using gel-based assays in the presence of Mg2+ or Mn2+ (Figure 3). IC50 values were calculated using dose response curves (Figure 3B and 3C) and are summarized in Table 1. The newly synthesized BDKAs exhibit potent inhibitory activity against IN for both ST and 3′-P steps. The acid derivatives (6,8) are more potent than the corresponding esters (5,7), and the 1-p-F-benzyl substituted quinolinones (7,8) are more active than the unsubstituted counterparts (5,6). Derivative 8 is the most potent derivative with IC50 values for strand transfer around 15 nM (Figure 3 and Table 1). Compound 8 is selective for ST inhibiton with IC50 values approximately 20-fold lower for ST than for 3′-P (Figure 3 compare panels B and C and Table 1). Compound 8 also inhibits integrase with similar potency in the presence of Mg2+ or Mn2+ (Table 1). The activity in the presence of Mg2+ in addition to the selectivity for ST represents a trademark of the most potent DKAs.11 Compound 8 is to our knowledge, the most potent bifunctional diketo acid derivative reported to date.
Table 1.
Cytotoxicity, Antiviral and Anti-Integrase Activities of Derivatives 5–8
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Anti-IN Activity
|
Antiviral Activity
|
|||||||||
| Cpd | R | X | IC50a |
CC50d | EC50e | SI f | ||||
| ST
|
3′-P
|
|||||||||
| Mg2+b | Mg2+c | Mn2+c | Mg2+c | Mn2+c | ||||||
| 5 | H | C2H5 | 77 ± 13 | 39 | 80 | 28 | >333 | >200 | >50 | - |
| 6 | H | H | 9.1 ±1.2 | 2.9 | 0.43 | 22 | >4.1 | >200 | 36.3 | 5.5 |
| 7 | Bzg | C2H5 | 0.38 ± 0.01 | 0.29 | 0.25 | 21 | 40 | 191 | 20 | 9.6 |
| 8 | Bzg | H | 0.016 ± 0.004 | 0.017 | 0.012 | 0.44 | 0.20 | >200 | 4.29h | >47 |
| 2i | 1.83 | 7.8 | nrj | nr | - | |||||
| 3i | 6.5 | 82 | >25 | 61k | - | |||||
| 4i | 0.2 | 1.8 | 81 | 17 | 4.8 | |||||
Inhibitory concentration 50% (μM) determined from dose-response curves.
Experiments performed in triplicate using the BioVeris assay (ST assay in the presence of MgCl2).
Experiments performed on gels in the presence of MnCl2 or MgCl2 (see Figure 3).
Cytotoxic concentration 50% (μM).
Effective concentration 50% (μM).
Selectivity index = CC50/EC50.
Bz = CH2-4-F-Ph.
For this compound EC90 = 40 μM was determined.
nr: not reported.
Percentage of inhibition obtained at a concentration of 25 μM.
Figure 3.

Comparison of HIV-1 integrase inhibition by compound 7 and 8 in the presence of Mg2+. (A) Phosphorimager image showing a representatitve experiment. 21, 19 and STP correspond to the DNA substrate, the 3′-processing product and the strand transfer products, respectively. (B) HIV-1 integrase inhibition curve (derived from densitometric analysis of typical experiments) for 3′-processing. (C) HIV-1 integrase inhibition curve for strand transfer.
Cell-based assay
Antiviral activities are presented in Table 1. Compound 8 showed good antiviral efficacy in HIV-1 infected H9/HTLVIIIB cells (EC50 = 4.29 μM, EC90 = 40 μM) and low cytotoxicity (CC50 > 200 μM, SI > 46.6). 6 and 7 were 8 and 5 times, respectively, less potent than the parent compound 8, while 5 was inactive below 50 μM concentration. It is to note that the quinolonyl derivative 8 showed higher potency in inhibiting the replication cycle of HIV-1 in cell-based assays (EC50) and better SI than the reference derivatives 3 and 4.
These structure activity relationships led to emphasize the relevance of the free carboxylic acid group and the p-F-benzyl moiety for both tight binding with IN and high antiviral potency.
Docking studies
To elucidate the binding mode of the most active compound 8 within the IN catalytic core domain (CCD), docking calculations were carried out using the X-ray structure 1BIS (subunit B).17 A Mg2+ ion, was placed in the active site between the carboxylate oxygen atoms of residues D64 and D116, considering the geometry of the Mg2+ ion present in the PDB structure 1QS4 (subunit A).18
To take into account protein flexibility, 8 was docked to an ensemble of protein snapshots taken from a 1 ns molecular dynamics (MD) simulation, according to the Relaxed-Complex method.19 AutoDock 3.0.5 program was chosen because it utilizes a fully flexible ligand in its docking algorithm and because it has been shown to successfully reproduce many crystal structure complexes.20 Details of the docking setup and the MD simulation protocol are provided as Supporting Information. The protein turned out to be very stable throughout the MD simulation with the exception of the three loop regions 139–147, 165–172 and 185–196. The former, which we refer to “catalytic loop” is located close to the active site. Its importance for the catalytic activity together with the flexible nature are well-known.21 According to previous MD studies, the catalytic loop moves back and widens the active site region.22 The Mg2+ coordination by D64 and D116 residues was always conserved during the production run.
Docking of 8 to the MD snapshots, collected every 100 ps, revealed two predominant binding modes, which are shown in Figure 4. In the binding orientation showed in Figure 4a, the carboxylate group of one diketo acid chain of the ligand chelates the Mg2+ metal ion, whereas the other one inserts between residues K156 and K159, forming H-bonds with both side chains and with N155 CO backbone. The p-F-benzyl group points towards a hydrophobic pocket formed by the catalytic loop residues Y143, P142, I141, G140 and by residues I60, Q62, V77, V79, H114, G149, V150, I151, E152, S153, M154. Particularly, a favorable electrostatic interaction occurs between the fluorine atom on the benzyl ring and the amide group of Q62 residue. The quinoline ring of the molecule forms a stacked amide-aromatic interaction with the N155 side chain, while the quinoline carbonyl oxygen enables a hydrogen bond to the T66 side chain. In our model, T66, S153 and M154, whose mutations were found to confer resistance to DKAs, are all in close proximity of the ligand.8
Figure 4.

The two predominant binding modes of 8 (yellow) to MD snapshots of IN core domain. Residues lining the ligand position are highlighted. The metal ion is represented as magenta sphere.
A significant alternative to the described binding mode is given by a docking position in which the metal ion is chelated in a bidentate manner by the quinoline carbonyl oxygen and by one oxygen of the diketo acid function, while the hydroxyl and the carboxylate groups of the same branch contact the D116 and the N117 side chains, respectively (Figure 4b). On the opposite side of the molecule, the other diketo acid arm elongates towards the K156 and K159 residues contacting exclusively the latter amino acid. The p-F-benzyl group entirely inserts in the hydrophobic pocket where it is stabilized by hydrophobic interactions with I151 and P142 side chains. Although the two binding modes differ in some details, it is worth noting that in both docking solutions, the ligand interacts with the same enzyme attachment points: i) the metal ion, ii) the K156 and/or K159 residues, and iii) the hydrophobic pocket.
The reduced inhibitory potencies of diketo esters 5 and 7 compared to their respective acids 6 and 8 is due to the ability of both carboxylic functionalities to chelate the metal ion and to interact with the above mentioned lysine residues, whereas the diketo esters weakly bind to these portions of the enzyme. Thus the free carboxylic functions are necessary for a strong interaction with the IN enzyme, particularly the metal ion. On the other hand, the drop in activity of 6 compared to 8 in both ST and 3′-P (see Table 1) can be clearly attributed to the absence of the p-F-benzyl moiety, which in our model inserts into the hydrophobic pocket leading to a more efficient binding.
According to previous findings on BDKAs activity,13 8 is effective on both ST and 3′-P processes catalyzed by HIV-1 IN and in this feature differs from the reported monofunctional DKAs, which are generally found selective toward the ST. In this respect, it was hypothesized that BDKAs can bind both DNA acceptor and donor site of HIV-1 IN being inhibitors of both ST and 3′-P processes, whereas the monofunctional DKAs selectively bind the acceptor site being effective on ST.13 Here, in an effort to gain more insight into the mechanism of action of the reported ligands, a re-evaluation of the above described docking results was performed with the aid of the theoretical model of HIV-1 IN tetramer complexed with both donor and acceptor DNA developed by McCammon et al.23 A visual inspection of 8 docked into the CCD-DNA complex (Figure 5) led us to the following considerations: i) the diketo acid arm, which coordinates the Mg++ metal ion, is clearly important in anchoring the ligand to the catalytic site. It is worth noting that while this arm seems not to interfere with the donor DNA binding, it may physically hamper the acceptor DNA binding. In our model, either the acceptor DNA and the ligand contact the N117 residue (see Figure 4b). This amino acid plays a crucial role in the enzyme function as its mutation leads to an IN preferentially defective in the ST step of integration;24 ii) the other diketo acid function, inserting between K156 and K159 residues, would interfere with the donor DNA recognition and binding. This result is in accordance with photo-crosslinking data, which reveal that the above-mentioned lysines are critical for donor DNA binding.25 This could explain the ability of 8 to inhibit the IN catalyzed 3′-P reaction; iii) the p-F-benzyl group, which inserts in the hydrophobic pocket, would reduce the catalytic loop mobility and physically hamper the binding of the acceptor DNA. In the proposed model, both DNA molecules are in proximity of the loop; particularly, the acceptor DNA is in close contact with I151 and P142, which contribute to the ligand binding (Figure 4b). This finding also makes sense with respect to the experimental observation that the benzyl group in DKAs ligands is the primary determinant of the ST inhibition.13
Figure 5.

Model of IN CCD-DNA complex with 8 (rendered as CPK) bound to the active site. Donor DNA is in red and acceptor DNA is in green.
Conclusions
We have designed, synthesized and tested both in enzyme- or in cell-based assays novel BDKAs as anti-HIV-1 agents targeted to IN. These compounds are potent inhibitors of both the ST and 3′-P steps and are endowed with antiviral activity. Docking experiments have helped to provide a framework for interpreting the inhibitory activity of our BDKAs. As more information will be available to better characterize the details of DNA-IN interaction, this issue will be resolved more definitively. Moreover, studies on mono-functional derivatives related to 5–8 are in progress, with the aim to elucidate differences in the mechanisms of action.
Experimental section
Chemistry
General
Melting points were determined with a Buchi 530 capillary apparatus and are uncorrected. Infrared (IR) spectra were recorded on a Spectrum-one spectrophotometer. 1H NMR spectra were recorded on a Bruker AC 400 spectrometer. Merck silica gel 60 F254 plates were used for analytical TLC. Column chromatographies were performed on silica gel Merck 70–230 mesh. Concentration of solutions after reactions and extractions involved the use of a rotatory evaporator operating at a reduced pressure of approximately 20 Torr. Analytical results agreed to within ±0.40% of the theoretical values.
2-[[(4-Acetylphenyl)amino]methylene]-3-oxobutanoic acid ethyl ester
A mixture of 4-aminoacetophenone (2.0 g, 0.015 mol) and 2-(ethoxymethylene)-3-oxobutanoic acid ethyl ester26 (2.8 g, 0.015 mol) was heated at 120 °C for 5 min. After cooling, the solid that formed was washed with n-hexane and with a small amount of cold CHCl3 to obtain pure 2-[[(4-acetylphenyl)amino]methylene]-3-oxobutanoic acid ethyl ester (3.0 g, 73%), mp 156–158 °C (benzene), which was used in the next reaction without further purification.
3,6-Diacetyl-4(1H)-quinolinone (9)
2-[[(4-Acetylphenyl)amino]methylene]-3-oxobutanoic acid ethyl ester (5.0 g, 0.018 mol) was dissolved in boiling diphenyl ether (50 mL) and refluxed for 50 min. After cooling, the mixture was treated with n-hexane (100 mL) and the precipitate that formed was collected and filtered on a short silica gel column (ethyl acetate and then chloroform/methanol 10:1 as eluent) to afford 9 (2.3 g, 56%); mp > 300 °C (DMF/water).
3,6-Diacetyl-1-(4-fluorophenylmethyl)-4(1H)-quinolinone (10)
A solution of 9 (250 mg, 1.1 mmol) in dry DMF (10 mL) was treated with anhydrous K2CO3 (210 mg, 1.5 mmol) and 4-fluorophenylmethyl bromide (610 mg, 3.3 mmol) and the resulting suspension was stirred for 2.5 hours at 100°C. After cooling, cold water (0–4 °C) was added (40 mL) and the precipitate was filtered, washed with water, light petroleum ether in turn and then dried under IR lamp to provide 10 (330 mg, 89%); mp 213–214 °C (toluene).
General Procedure for the Synthesis of Diketo Esters 5 and 7
Sodium ethoxide (390 mg, 5.5 mmol) was added into a well stirred mixture of 9 or 10 (2.7 mmol) and diethyl oxalate (790 mg, 5.4 mmol) in anhydrous THF (2.7 mL) under nitrogen atmosphere. The mixture was stirred at room temperature (2 h for 5; 45 min for 7), then was poured into n-hexane (50 mL). The collected precipitate was vigorously stirred for 30 minutes in 1N HCl (50 mL). The yellow solid that formed was filtered, washed with water and dried under IR lamp. 5: 100% yield; mp > 300 °C (DMF/water). Anal. (C21H19NO9) C, H, N. 7: 88% yield; mp 189–190 °C (toluene). Anal. (C28H24FNO9) C, H, N, F.
General Procedure for the Synthesis of Diketo Acids 6 and 8
A mixture of 1N NaOH (6.5 mL) and compound 5 or 7 (1.3 mmol) in THF/methanol 1:1 (12 mL) was stirred at room temperature for 18 hours and then poured onto crushed ice. The aqueous layer was separated and treated with 1N HCl until reaching pH 3 and the yellow solid that formed was collected by filtration, then washed with water, hot dry ethanol, and light petroleum ether. 6: 52% yield; mp > 300 °C (washed with hot acetone). Anal. (C17H11NO9) C, H, N. 8: 77% yield; mp 178–180 °C (washed with hot dry ethanol). Anal. (C24H16FNO9) C, H, N, F.
Biological Assays
Integrase Assays
Compounds 5–8 were tested for their ability to inhibit HIV-1 integrase in vitro using a gel-based assay in addition to a plate-based electrochemoluminescent assay.
In the gel assay, A 5′-end-labeled 21-mer double-stranded DNA oligonucleotide corresponding to the last 21 bases of the U5 viral LTR, is used to follow both 3′-processing and strand transfer steps of the integration reaction (for review see6). Briefly, a DNA-enzyme complex is preformed by mixing 500 nM of recombinant HIV-1 integrase and 20 nM of 5′-labeled double-stranded DNA template in a buffer containing 50 mM MOPS, pH 7.2, 7.5 mM MnCl2 and 14.3 mM β-mercaptoethanol for 15 minutes on ice. The integration reaction is then initiated by addition of the drug and continued in a total volume of 10 μL for 60 minutes at 37°C. The reaction samples are stopped by adding the same volume of electrophoresis denaturing dye and loaded on 20% 19/1 acrylamide denaturing gel. Gels were exposed overnight and analyzed using a Molecular Dynamics Phosphorimager (Sunnyvale, CA).
The plate-based electrochemoluminescent assay was performed using a BioVeris M-SERIES® Analyzer (Gaithersburg, MD). DNA substrates were obtained from BioVeris and used according to the manufacturer’s recommendations. Briefly, donor DNA is incubated for 30 minutes at 37°C in the presence of 250 nM of recombinant HIV-1 integrase. After addition of the drug, the integration reaction is initiated by addition of target DNA. Reaction is carried out for 60 minutes at 37°C and then read on the BioVeris M-SERIES® Analyzer.
Anti-HIV Assays in Cultured Cell Lines
The anti-HIV drug testing was performed in 96-well plates with a defined, previously titred inoculum of a laboratory strain (HTLV-IIIB) to minimize the inoculum effect.
In brief, all compounds were dissolved in dimethyl sulfoxide and diluted in cell culture medium at concentrations ranging from 0.1 to 50 μM. Exponentially growing human T lymphocytes (H9 cell line) were added at 5000 cell/well. After the infectivity of a virus stock (HTLV-IIIB) is quantified, an aliquot containing 1×105 50% tissue culture infectious dose (TCID 50) per 5000 H9 cells per well is used as inoculum in each set of in vitro infections of H9 cells. Uninfected cells with the compound served as a toxicity control, and infected and uninfected cells without the compound served as basic controls. Culture were incubated at 37 °C in a 5% CO2 atmosphere for 4 days. Supernatant fluid of infected wells (in the absence of drug and at each of a number of drug concentrations) is harvested. HIV p24 antigen is quantified and the 50% inhibitory concentration (IC50) of drug is determined using the median effect equation.
Cytotoxicity Assays
Cytotoxicity of test compounds has been evaluated on human histiocytic lymphoma (U937) cell line obtained from American type culture collection (ATCC, Rockville, MD). Cells were plated in a 96 well-plate at a concentration of 5×103/mL in RPMI-1640 without phenol red, supplemented with 20% Fetal Calf Serum and antibiotics. Two hours after plating, compounds were added at different concentrations ranging from 1 μg/mL to 200 mg/mL and cells were incubated at 37 °C for 24 hrs. Then, cells were incubated at 37°C for 3 hrs with 1 mg/ml of MTT (Sigma-Aldrich, Milan, Italy). After incubation, the remaining water insoluble formazan was solubilized in absolute isopropanol containing 0.1 N HCl. Absorbance of converted dye was measured in an ELISA plate reader at the wavelength of 570 nm. The cytotoxicity of the compounds was calculated as the percentage reduction of the viable cells compared with the drug free control culture. The drug concentration required to reduce the cell viability by 50% has been called IC50.
Molecular Modeling
Molecular Dynamics (MD) Simulation
The catalytic core domain (CCD) structure of HIV-1 integrase (IN) was taken from PDB structure 1BIS.17 The chain B of the X-ray structure was chosen for our studies, and a Mg2+ ion was placed in the active site between carboxylate oxygen atoms of amino acid residues D64 and D116 considering the geometry of the Mg2+ ion that was present in the subunit A of the PDB structure1QS4.18 To sample the conformational space of IN enzyme, a 1 ns MD simulation was carried out in explicit solvent. All the water molecules present in the X-ray structure of the enzyme were removed and all hydrogen atoms were added to the residues, considering residues Arg, Lys, Glu, and Asp in their charged form, while all His residues were considered neutral by default. To make the system electroneutral, a total of 2 neutralizing counterions (Cl−) were added with the aid of VEGA (vs 1.5.0) program.27 The enzyme was then soaked with a 10 Å water layer. The calculations were carried out with the DISCOVER module of the INSIGHT II program using CVFF force field.28 A multiple-step procedure was used. The complex was energetically minimized with 5000 steps of a steepest descent minimization, followed by 3000 steps of conjugate gradient minimization to adjust the water molecules and the counterions locally and to eliminate any residual geometrical strain, keeping the heavy atoms of the enzyme fixed. The minimized solvated system was used as initial structure for an equilibration stage (100 ps), followed by a production run (1 ns). In the equilibration stage, energy minimization of the protein side chains were achieved employing 5000 steps of steepest descent and 3000 steps of conjugate gradient algorithm. The system was then heated gradually starting from 10 to 300 K in 5 ps steps and was equilibrated with temperature bath coupling (300 K). During the equilibration and the production run, the backbone of the enzyme secondary structures was kept fixed. A cutoff of 13 Å was used for nonbonded interactions. Coordinates and energies were saved every 10 ps yielding 100 structures. For the purpose of automatic docking snapshot each 100 ps were taken.
Docking Simulations
Docking of 8 to IN CCD was carried out using the AutoDock program package version 3.0.5.20 The LGA algorithm, as implemented in the AutoDock program, was used applying a protocol with a maximum number of 1.5 × 10−6 energy evaluations, a mutation rate of 0.01, a crossover rate of 0.80, and an elitism value of 1. For the local search, the pseudo-Solis and Wets algorithm was applied using a maximum of 300 interactions per local search. Fifty independent docking runs in different enzyme MD snapshot were carried out for 8. Results differing by less than 1.5 Å in positional root-mean-square deviation (rmsd) were clustered together and represented by the result with the most favorable free energy of binding. The obtained complexes were energetically minimized using 3000 steps of steepest descent algorithm, permitting only the ligand and the side chain atoms of the protein within a radius of 5 Å around the ligand to relax. The geometry optimization was carried out employing the DISCOVER program with the CVFF force field.
(1) Ligand Setup
Structure of 8 was generated from the standard fragment library of the SYBYL software version 7.0.29 Geometry optimizations were achieved with the SYBYL/MAXIMIN2 minimizer by applying the BFGS algorithm30 with a convergence criterion of 0.001 kcal/mol and employing the TRIPOS force field. Partial atomic charges were assigned using Gasteiger and Marsili formalism31 as implemented in the SYBYL package 8 was modelled in its keto-enolic form with the negatively charged carboxylate groups. Six flexible torsions were specified: two around the quinoline ring, two about the benzyl group, and two allowing the hydroxyl groups to rotate.
(2) Protein Setup
The protein structure was set up for docking as follows: the unpolar hydrogens were removed, and Kollman united-atom partial charges were assigned. Solvation parameters were added to the protein-DNA complex using the ADDSOL utility of the AutoDock program. The grid maps were calculated with AutoGrid. The grids were chosen to be large enough to include a significant part of the protein around the catalytic site. In all cases, we used grid maps with 61 × 61× 61 points with a grid-point spacing of 0.375 Å. The center of the grid was set to be coincident with the Mg2+ ion in the active site of catalytic domain.
Supplementary Material
Supporting Information Available: spectroscopic data of compounds 5-10, elemental analyses for derivatives 5-8. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
The authors thank Prof. J. Andrew McCammon for the donation of the IN-DNA complex coordinates. We also thank Dr. M. Zancato (University of Padova, Italy) for performing elemental analyses. This project was supported by Ministero della Sanità, Istituto Superiore di Sanità, “Programma Nazionale di ricerca sull’AIDS” (grant n° 30F.19), and Italian MIUR (PRIN 2004).
References
- 1.Hammer SM. Clinical practice. Management of newly diagnosed HIV infection. N Engl J Med. 2005;353:1702–1710. doi: 10.1056/NEJMcp051203. [DOI] [PubMed] [Google Scholar]
- 2.Cohen J. Therapies. Confronting the limits of success. Science. 2002;296:2320–2324. doi: 10.1126/science.296.5577.2320. [DOI] [PubMed] [Google Scholar]
- 3.Fesen MR, Kohn KW, Leteurtre F, Pommier Y. Inhibitors of human immunodeficiency virus integrase. Proc Natl Acad Sci U S A. 1993;90:2399–2403. doi: 10.1073/pnas.90.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cushman M, Sherman P. Inhibition of HIV-1 integration protein by aurintricarboxylic acid monomers, monomer analogs, and polymer fractions. Biochem Biophys Res Commun. 1992;185:85–90. doi: 10.1016/s0006-291x(05)80958-1. [DOI] [PubMed] [Google Scholar]
- 5.Carteau S, Mouscadet J-F, Goulaouic H, Subra F, Auclair C. Inhibitory effect of the polyanionic drug suramin on the in vitro HIV DNA integration reaction. Arch Biochem Biophys. 1993;305:606–610. doi: 10.1006/abbi.1993.1468. [DOI] [PubMed] [Google Scholar]
- 6.Pommier Y, Johnson AA, Marchand C. Integrase inhibitors to treat HIV/AIDS. Nat Rev Drug Discovery. 2005;4:236–248. doi: 10.1038/nrd1660. [DOI] [PubMed] [Google Scholar]
- 7.Johnson AA, Marchand C, Pommier Y. HIV-1 integrase inhibitors: a decade of research and two drugs in clinical trial. Curr Top Med Chem. 2004;4:1059–1077. doi: 10.2174/1568026043388394. [DOI] [PubMed] [Google Scholar]
- 8.Hazuda DJ, Felock P, Witmer M, Wolfe A, Stillmock K, Grobler JA, Espeseth A, Gabryelski L, Schleif W, Blau C, Miller MD. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science. 2000;287:646–650. doi: 10.1126/science.287.5453.646. [DOI] [PubMed] [Google Scholar]
- 9.Wai JS, Egbertson MS, Payne LS, Fisher TE, Embrey MW, Tran LO, Melamed JY, Langford HM, Guare JP, Jr, Zhuang L, Grey VE, Vacca JP, Holloway MK, Naylor-Olsen AM, Hazuda D, Felock PJ, Wolfe AL, Stillmock KA, Schleif WA, Gabryelski LJ, Young SD. 4-Aryl-2,4-dioxobutanoic acid inhibitors of HIV-1 integrase and viral replication in cells. J Med Chem. 2000;43:4923–4926. doi: 10.1021/jm000176b. [DOI] [PubMed] [Google Scholar]
- 10.Espeseth A, Felock P, Wolfe A, Witmer M, Grobler JA, Anthony N, Egbertson M, Melamed JY, Young S, Hamill T, Cole JL, Hazuda DJ. HIV-1 integrase inhibitors that compete with the target DNA substrate define a unique strand transfer conformation for integrase. Proc Natl Acad Sci U S A. 2000;97:11244–11249. doi: 10.1073/pnas.200139397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grobler JA, Stillmock K, Hu B, Witmer M, Felock P, Espeseth AS, Wolfe A, Egbertson M, Bourgeois M, Melamed J, Wai JS, Young S, Vacca J, Hazuda DJ. Diketo acid inhibitor mechanism and HIV-1 integrase: implications for metal binding in the active site of phosphotransferase enzymes. Proc Natl Acad Sci USA. 2002;99:6661–6666. doi: 10.1073/pnas.092056199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marchand C, Johnson AA, Karki RG, Pais GC, Zhang X, Cowansage K, Patel TA, Nicklaus MC, Burke TR, Jr, Pommier Y. Metal-dependent inhibition of HIV-1 integrase by beta-diketo acids and resistance of the soluble double-mutant (F185K/C280S) Mol Pharmacol. 2003;64:600–609. doi: 10.1124/mol.64.3.600. [DOI] [PubMed] [Google Scholar]
- 13.Marchand C, Zhang X, Pais GCG, Cowansage K, Neamati N, Burke TR, Jr, Pommier Y. Structural determinants for HIV-1 integrase inhibition by β-diketo acids. J Biol Chem. 2002;277:12596–12603. doi: 10.1074/jbc.M110758200. [DOI] [PubMed] [Google Scholar]
- 14.Pais GCG, Zhang X, Marchand C, Neamati N, Cowansage K, Svarovskaia ES, Pathak VK, Tang Y, Nicklaus M, Pommier Y, Burke TR., Jr Structure activity of 3-aryl-1,3-diketo-containing compounds as HIV-1 integrase inhibitors. J Med Chem. 2002;45:3184–3194. doi: 10.1021/jm020037p. [DOI] [PubMed] [Google Scholar]
- 15.Long Y-Q, Jiang X-H, Dayam R, Sanchez T, Shoemaker R, Sei S, Neamati N. Rational design and synthesis of novel dimeric diketoacid-containing inhibitors of HIV-1 integrase: implication for binding to two metal ions on the active site of integrase. J Med Chem. 2004;47:2561–2573. doi: 10.1021/jm030559k. [DOI] [PubMed] [Google Scholar]
- 16.(a) Artico M, Di Santo R, Costi R, Novellino E, Greco G, Massa S, Tramontano E, Marongiu ME, De Montis A, La Colla P. Geometrically and conformationally restrained cinnamoyl-compounds as inhibitors of HIV-1 integrase: synthesis, biological evaluation and molecular modeling. J Med Chem. 1998;41:3948–3960. doi: 10.1021/jm9707232. [DOI] [PubMed] [Google Scholar]; (b) Di Santo R, Costi R, Artico M, Tramontano E, La Colla P, Pani A. HIV-1 integrase inhibitors that block HIV-1 replication in infected cells. Planning synthetic derivatives from natural products. Pure Appl Chem. 2003;75:195–206. [Google Scholar]; (c) Costi R, Di Santo R, Artico M, Massa S, Ragno R, Loddo R, La Colla M, Tramontano E, La Colla P, Pani A. 2,6-Bis(3,4,5-trihydroxybenzylidene) derivatives of cyclohexanone: novel potent HIV-1 integrase inhibitors that prevent HIV-1 multiplication in cell-based assays. Bioorg Med Chem. 2004;12:199–215. doi: 10.1016/j.bmc.2003.10.005. [DOI] [PubMed] [Google Scholar]; (d) Costi R, Di Santo R, Artico M, Roux A, Ragno R, Massa S, Tramomtano E, La Colla M, Loddo R, Marongiu ME, Pani A, La Colla P. 6-Aryl-2,4-dioxo-5-hexenoic acids, novel integrase inhibitors active against HIV-1 multiplication in cell-based assays. Bioorg Med Chem Lett. 2004;14:1745–1749. doi: 10.1016/j.bmcl.2004.01.037. [DOI] [PubMed] [Google Scholar]; (e) Di Santo R, Costi R, Artico M, Ragno R, Greco G, Novellino E, Marchand C, Pommier Y. Design, synthesis and biological evaluation of heteroaryl diketohexenoic and diketobutanoic acids as HIV-1 integrase inhibitors endowed with antiretroviral activity. Il Farmaco. 2005;60:409–417. doi: 10.1016/j.farmac.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 17.Goldgur Y, Dyda F, Hickman AB, Jenkins TM, Cragie R, Davies DR. Three new structure of the core domain of HIV-1 integrase: an active site that binds magnesium. Proc Natl Acad Sci USA. 1998;95:9150–9154. doi: 10.1073/pnas.95.16.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldgur Y, Craigie R, Cohen GH, Fujiwara T, Yoshinaga T, Fujishita T, Sugimoto H, Endo T, Murai H, Davies DR. Structure of the HIV-1 integrase catalytic domain complexed with an inhibitor: A platform for antiviral drug design. Proc Natl Acad Sci USA. 1999;96:13040–13043. doi: 10.1073/pnas.96.23.13040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin J-H, Perryman AL, Schames JR, McCammon JA. Computational drug design accommodating receptor flexibility: the relaxed complex scheme. J Am Chem Soc. 2002;124:5632–5633. doi: 10.1021/ja0260162. [DOI] [PubMed] [Google Scholar]
- 20.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 21.Greenwald J, Le V, Butler SL, Bushman FD, Choe S. The mobility of an HIV-1 integrase active site loop is correlated with catalytic activity. Biochemistry. 1999;38:8892–8898. doi: 10.1021/bi9907173. [DOI] [PubMed] [Google Scholar]
- 22.Lins RD, Briggs JM, Straatsma TP, Carlson HA, Greenwald J, Choe S, McCammon JA. Molecular dynamics studies on the HIV-1 integrase catalytic domain. Biophysical J. 1999;76:2999–3011. doi: 10.1016/s0006-3495(99)77453-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Podtelezhnikov AA, Gao K, Bushman FD, McCammon JA. Modeling HIV-1 integrase complexes based on their hydrodynamic properties. Biopolymers. 2003;68:110–120. doi: 10.1002/bip.10217. [DOI] [PubMed] [Google Scholar]
- 24.van Gent DC, Groeneger AA, Plasterk RH. Mutational analysis of the integrase protein of human immunodeficiency virus type 2. Proc Natl Acad Sci USA. 1992;20:9598–9602. doi: 10.1073/pnas.89.20.9598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jenkins TM, Esposito D, Engelman A, Cragie R. Critical contacts between HIV-1 integrase and viral DNA identified by structure-based analysis and photo-crosslinking. EMBO J. 1997;16:6849–6859. doi: 10.1093/emboj/16.22.6849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coates RM, Hobbs SJ. α-Alkoxyallylation of activated carbonyl compounds. A novel variant of the Michael reaction. J Org Chem. 1984;49:140–152. [Google Scholar]
- 27.Pedretti A, Villa L, Vistoli G. VEGA: a versatile program to convert, handle, and visualize molecular structure on windowsbased PCs. J Mol Graph. 2002;21:47–49. doi: 10.1016/s1093-3263(02)00123-7. [DOI] [PubMed] [Google Scholar]
- 28.Accelrys. San Diego, CA: 2001. [Google Scholar]
- 29.SYBYL Molecular Modeling System. TRIPOS Assoc; St. Louis, MO: [Google Scholar]
- 30.Head J, Zerner MC. A Broyden-Fletcher-Goldfarb-Shanno optimization procedure for molecular geometries. Chem Phys Lett. 1985;122:264–274. [Google Scholar]
- 31.Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity - a rapid access to atomic charges. Tetrahedron. 1980;36:3219–3228. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: spectroscopic data of compounds 5-10, elemental analyses for derivatives 5-8. This material is available free of charge via the Internet at http://pubs.acs.org.