


Abstract
DNA aptamers RT5, RT6 and RT47 form a group of related sequences that inhibit HIV-1 reverse transcriptase (RT). The essential inhibitory structure is identified here as bimodular, with a 5′ stem–loop module physically connected to a 3′-guanosine quadruplex module. The stem–loop tolerates considerable sequence plasticity. Connections between the guanosine triplets in the quadruplex could be simplified to a single nucleotide or a nonnucleic acid linker, such as hexaethylene glycol. All 12 quadruplex guanosines are required in an aptamer retaining most of the original loop sequence from RT6; only 11 are required for aptamer R1T (single T residue in intra-quadruplex loops). Circular dichroism (CD) spectroscopy gave ellipticity minima and maxima at 240 nm and 264 nm, indicating a parallel arrangement of the quadruplex strands. The simplified aptamers displayed increased overall stability. An aptamer carrying the original intra-quadruplex loops from RT6 inhibited RT in K+ buffers but not in Na+ buffers and displayed significant CD spectral broadening in Na+ buffers, while R1T inhibited RT in both buffers and displayed less broadening in Na+ buffers. The bimodular ssDNA aptamers inhibited RT from diverse primate lentiviruses with low nM IC50 values. These data provide insight into the requirements for broad-spectrum RT inhibition by nucleic acid aptamers.
INTRODUCTION
Antiviral chemotherapy has achieved spectacular results in prolonging the survival of patients infected with HIV-1. Morbidity and mortality related to HIV-1 have dramatically declined in developed countries, converting HIV infection into a treatable chronic disease. However, current antiviral drugs do not eradicate the virus, and prolonged treatment can have serious side effects and select drug-resistant viral strains (1). In addition, millions of new infections occur worldwide each year (2,3). Continued efforts toward the discovery of new antiviral strategies therefore remain imperative.
The reverse transcriptase (RT) of HIV-1 is a primary target for inhibition by current drugs, which include the nucleoside analog RT inhibitors (NRTIs, primarily chain terminators) and the nonnucleoside RT inhibitors (NNRTIs, noncompetitive allosteric inhibitors of polymerization by RT). Nucleic acid aptamers comprise a third class of RT inhibitors. Because many aptamers compete with the template/primer duplex for access to the enzyme (4–6), they have been referred to as TRTIs (template/primer analog RT inhibitors) (7). Aptamers are derived from the combinatorial method of in vitro selection, or SELEX (for Selective Evolution of Ligands by EXponential enrichment). Numerous aptamers have been identified that bind RT with high affinity and that inhibit its enzymatic activity in vitro (4,5,8–15) [reviewed in (16)]. Several of these aptamers have also been demonstrated to interfere with viral replication in cell culture (7,12,15,17,18). Clinical application of RNA aptamers may eventually take the form of gene therapy, wherein genes that direct the expression of the therapeutic aptamer are delivered to target cells (e.g. CD34+ stem cells) for intracellular expression. Direct clinical application of DNA aptamer inhibitors of RT will require further improvements in delivery to the appropriate target cells. However, both RNA and DNA aptamers are valuable research tools for dissecting the molecular mechanisms of viral replication and pathogenesis.
While both RNA and DNA aptamers to RT have been described, DNA aptamers offer several unique advantages and opportunities. (i) They can be synthesized at large scale cheaply and efficiently using technology that is available worldwide. (ii) DNA aptamers can be stored in desiccated form for years, then be refolded and fully activated upon rehydration, and their shelf-life can be further prolonged by storage in the presence of metal chelators, such as EDTA. (iii) Chemical derivatization can be readily accomplished by existing synthetic methods to adapt a given aptamer to a variety of delivery and diagnostic platforms. (iv) Nucleic acids are generally nonimmunogenic, so their repeated use is unlikely to induce an inflammatory immune response. (v) Several recent studies with RT mutants—including drug-resistant RT (19,20)—and with RT from phylogenetically diverse virus (14) suggest that the genetic threshold for the development of significant resistance to some ssDNA aptamers may be very high.
Five sets of ssDNA aptamers to HIV-1 RT have been described. The present study builds from the aptamer set selected by Schneider et al. (11), who identified at least seven sequence families from among 30 aptamer isolates. Several aptamers from this set strongly inhibit the RNA-dependent DNA polymerase activity of HIV-1 RT and bind RT with dissociation constant (Kd) values near 1 nM (11). We recently demonstrated that a 49-nt version of aptamer RT1, designated RT1t49, inhibits RT from diverse strains of HIV-1, HIV-2 and SIVcpz (a close relative of HIV-1 that infects chimpanzees) (14). Mutational and footprinting analysis, combined with molecular modeling, support a two-helix structure for RT1t49 in which one helix occupies the primer/template-binding cleft in the protein and the other helical structural unit is thought to interact with the back side of the fingers or thumb domain (5). Other work has evaluated the influence of specific RT point mutations on binding by RT1t49 and several other aptamers (19,20). Although some progress has been made in identifying how the RT1 family of ssDNA aptamers interacts with HIV-1 RT, relatively little is known about the other structural families from this set.
The second set of ssDNA aptamers, isolated by Andreola et al. (12), was targeted to interact with the RNase H domain of RT by utilizing alternate cycles of positive selection for binding to the p66/p51 heterodimer with cycles of negative selection to remove aptamers binding the p51/p51 homodimer. Two species from this selection, designated ODN93 and ODN112, inhibit both DNA polymerization and RNase H activities in vitro with half-maximal inhibitory values (IC50) of 500 nM. When added to cell culture simultaneously with virus, these same DNAs interfered with viral infectivity. Truncated version of these anti-RNase H aptamers, designated 93del and 112del, both form guanosine quadruplex structures. Both molecules also cross-react with HIV-1 integrase, blocking both ‘end-processing’ and strand-insertion activities by integrase, with IC50 values in these reactions as low as 10–100 nM (21).
The other three aptamer sets each have distinct features. DeStefano and Cristaforo (13) selected GC-rich duplexes with recessed 3′-ends that resemble the primer/template substrate utilized by the virus during (+)-strand genome synthesis. Somasunderam et al. (15) used combinatorial bead chemistry to identify sulfur-containing thioaptamers that bind RT with Kd values as low as 70 nM and that interfere with viral replication in cell culture. Several of these DNAs are G-rich and have the potential to form quadruplex structures (15). More recently, Bowser and colleagues (22) used capillary electrophoresis to effect the rapid selection of ssDNA aptamers with especially high affinity for HIV-1 RT (Kd = 0.18 nM).
The aptamers from the five selections above comprise a highly diverse collection of sequences and secondary structures, including several that form guanosine quadruplex structures. Quadruplexes are a highly stable, multi-layer fold in which four guanosines within a given layer hydrogen bond from the Watson–Crick face of each guanosine to the Hoogsteen face of another guanosine, and successive layers stack together [reviewed in (23)]. Sequences capable of forming quadruplex structures are found in human (24) and Oxytricha nova (25) telomeres, and in several promoter sequences (26–32). Recent advances in quadruplex aptamer identification, characterization and delivery have led to increased interest in developing them as tools for modulating biology. For example, ssDNA aptamers to the blood clotting factor thrombin (33) and to insulin (34) both form quadruplexes and have been studied extensively. AS1411 is a quadruplex-forming DNA aptamer that targets nucleolin and that is currently in clinical trials as a treatment for various cancers (35), and several DNA quadruplexes have shown potent inhibition of HIV-1 integrase (36,37).
In the present work, we reinvestigated several molecules from the Schneider set of ssDNA aptamers selected to bind HIV-1 RT (11). Three aptamers demonstrate bimodular structure comprising quadruplex and helical elements. Both elements are required for RT inhibition, and they must be physically connected. We find that there are relatively few sequence constraints within either element or in the connection between them, and the chemical nature of the linkages between the two major structural elements and within the quadruplex played no significant roles in determining inhibition of HIV-1 RT. Importantly, the bimodular aptamer exhibited potent inhibition of RT derived from phylogentically diverse HIV and SIV strains. Our findings aid in understanding the mechanism of HIV RT inhibition utilizing structured DNA inhibitors and reinforce the potential clinical and research value of quadruplex DNA aptamers.
MATERIALS AND METHODS
RT and DNA oligonucleotides
RT were expressed in Escherichia coli and purified as described in (4). Except where noted, all assays utilized RT from HIV-1 strain HXB2 (group M, subtype B). Aptamer RT6 was synthesized and purified by Operon Biotechnologies, Inc., (Huntsville, AL, USA; http://www.operon.com). All other aptamer oligonucleotides and primer/template substrates were synthesized and purified by Integrated DNA Technologies, Inc. (http://www.idtdna.com). The original full-length aptamers were 81 nt in length (11). The ‘full-length’ aptamers studied here were synthesized as 80-nt molecules by removing the 3′-terminal dG residue. This change was without functional consequence, as our IC50 values were similar to those measured previously (11). Computational predictions of DNA secondary structures with mfold 3.1 (http://www.rpi.edu/~zukerm/) (38,39) utilized only the nonquadruplex portions of each molecule. All oligonucleotides used in this study are given in Table 1.
Table 1.
Oligonucleotides used in this study
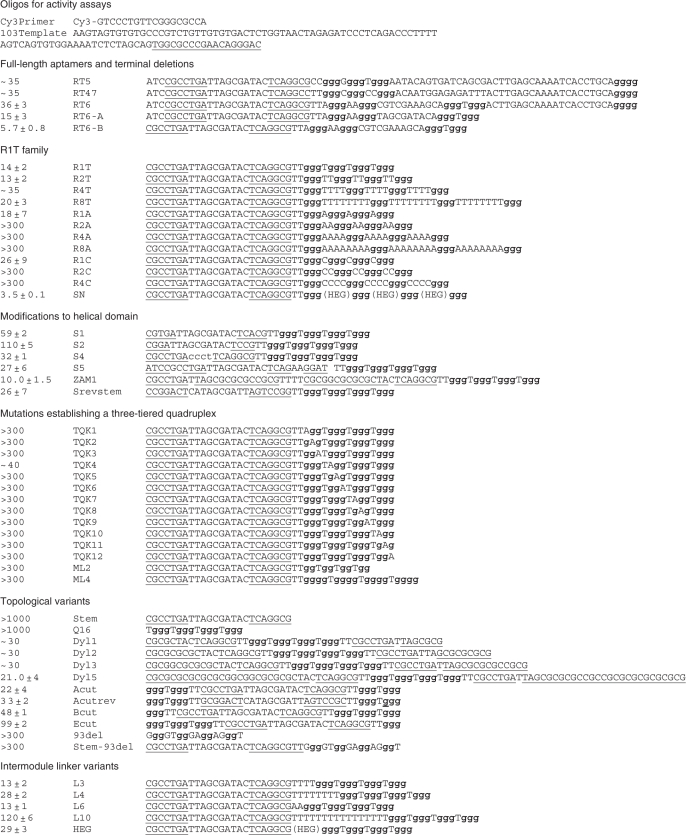 |
Underlined nucleotides form the base-paired helical element; bold nucleotides comprise the guanosine triplet repeats.
Inhibition of RT enzymatic activities
Aptamer DNA was denatured by heating to 95°C for 5 min, followed by slow cooling at a rate of 2° per minute to room temperature and stored in frozen condition. Subsequent dilutions were made from this refolded stock without additional refolding. Measurements of DNA-dependent DNA polymerization (DDDP) were carried out essentially as described (4). For DDDP assays, an 18-nt Cy3-labeled DNA primer was annealed to a 103-nt synthetic DNA template corresponding to the primer-binding sequence and U5 segments of HIV-1 strain HXB2. Reactions were assembled in reaction buffer (50 mM Tris–HCl pH 8.3, 75 mM KCl, 5 mM MgCl2, 10 mM DTT) with 30 nM primer, 45 nM template and 0.2 mM dNTPs. After annealing primer and template strands, DNA aptamers were added. Reactions were initiated by adding RT to a final concentration of 3 nM active site. For IC50 measurements, final aptamer concentrations were 0 nM, 0.3 nM, 1.0 nM, 3.0 nM, 10 nM, 30 nM, 100 nM and 300 nM. Additional reactions included 1 μM and 3 μM where indicated. After 10 min at 37°C, reactions were quenched by addition of 2 volumes (20 μl) of gel-loading buffer (95% formamide with 0.01% bromophenol blue). For reactions assessing monovalent ion dependence, 75 mM KCl was replaced with 75 mM NaCl where indicated.
Reaction products were separated using denaturing (8 M urea) 10% PAGE and scanned for fluorescence using a Fujifilm FLA5000 imaging system. RT activity data were collected using Fujifilm Multi Gauge V2.3 image analysis software. Competitive inhibition by aptamers is due to a decrease in the concentration of available enzyme, and is described by the two-state model for RT-aptamer binding [Equation (1)]:
| 1 |
where Y is the measured percent activity at a given inhibitor concentration. This equation can be rearranged to a convenient form [Equation (2)] for curve-fitting with GraphPad Prism software to obtain IC50 values for aptamer inhibition:
| 2 |
where X is the log of the inhibitor concentration. Error terms for reported IC50 values are the standard deviations among triplicate assays.
Circular dichroism spectroscopy
DNA aptamer samples (SN, RIT and RT6-B) were prepared essentially as for inhibition assays, by heating to 90°C and renaturing at 25°C for 20 min. The concentrations 4 μM or 2 μM were adjusted in the corresponding buffers (K-buffer = 50 mM Tris–HCl pH 8.3, 75 mM KCl, 5 mM MgCl2, 10 mM DTT; Na-buffer = 50 mM Tris–HCl pH 8.3, 75 mM NaCl, 5 mM MgCl2, 10 mM DTT). Near-UV circular dichroism (CD) spectra were acquired at 25°C using an Aviv 62DS spectrometer (Lakewood, NJ) at 1.0 nm intervals between 350 nm and 210 nm in a 1.0 mm quartz cuvette, averaging each data point for 10 s.
RESULTS
Identification of a novel inhibitory DNA structure within ssDNA aptamers RT6, RT5 and RT47
Aptamers RT5, RT6 and RT47 form a subgroup of closely related sequences within Family VI of the ssDNA aptamers identified by Schneider et al. (11). Consistent with previous results, we obtained strong inhibition of DNA polymerization by RT from a subtype B strain of HIV-1 by 80-nt versions of each of these three aptamers. The concentration required for half-maximal inhibition (IC50) by RT6 was 36 ± 3 nM and was similar for the other two (data not shown). All three aptamers share the same sequence in the first 5–6 nt of the initial 35 N random segment (Figure 1A), and this shared segment is complementary to nucleotides within the 5′-primer-binding constant region. The remainder of each molecule includes four or more stretches of guanosine tri-nucleotide repeats. Five such repeats are evident in RT6. Aptamer RT6-A was generated by removing the 25 nt on the 3′-side of the fourth repeat of RT6 and modifying 5 nt between the third and fourth repeat. Aptamer RT6-A was more than 2-fold more potent for RT inhibition relative to the original RT6 aptamer (IC50 = 16 ± 3 nM). Similar 3′ deletions from RT5 and RT47 inactivated these aptamers for RT inhibition unless three guanosines were appended onto the 3′-ends of the truncated species to restore their original termini (data not shown). Removing both the 3′- terminal 25 nt and the 5′-terminal three unpaired nucleotides (ACG) of RT6 generated a 53-nt species designated RT6-B that retained potent RT inhibition (IC50 = 5.7 ± 0.8 nM). Finally, the 41-nt aptamer variant R1T (IC50 = 14 ± 2 nM, Figure 1C) was generated in which the intra-quadruplex loop sequences connecting the guanosine triplets in RT6-B were replaced with single thymidine nucleotides. These data suggest a structural model in which each of these aptamers forms a 5′-helical domain connected to a 3′-guanosine quadruplex domain (Figure 1B).
Figure 1.

Quadruplex model for RT6 family of aptamers to HIV-1 RT. (A) Sequences of aptamers RT5, RT6 and RT47 from Schneider set (11). Primer-binding sites for library amplification are in lower case, 35N originally random nucleotides are in upper case, guanosine clusters are underlined. Note that all aptamers analyzed in this work are given in Table 1. (B) Generalized secondary structure of most aptamers studied in this work. Stem and connector sequences shown are those of R1T, SN and several other aptamers. Intra-quadruplex loops are shown as curved lines. (C) Inhibition of HIV-1 RT DDDP activity by aptamer R1T. Aptamer concentrations (nM) are indicated above the lanes. RT and primer/template concentrations used in these assays are 3 nM and 30 nM, respectively. Open and filled arrows on the left indicate positions of unextended primer and full-length product bands, respectively.
Mutational support for quadruplex formation
To establish the quadruplex positions at which guanosine is required, each of the nucleotides that are proposed to define the G-quadruplex structures within RT6-A was individually changed to adenosine. All 12 G-to-A substitutions abolished RT inhibition (data not shown). These data provide strong evidence that RT inhibition by RT6-A (and by the original RT6) requires a three-tiered quadruplex. When similar mutations were generated in R1T, 11 of the 12 G-to-A mutations were similarly disruptive. However, deleting the first guanosine of the second triplet or changing it to A, C or T only reduced RT inhibition by two- to threefold (TQK4, Figure 2A, and data not shown). These data suggest that only two tiers of the quadruplex may be absolutely required within R1T. However, inserting or removing one guanosine from each (TGGG) repeat in the original R1T quadruplex to yield (TGGGG)4 in ML4 and (TGG)4 in ML2 prevents aptamer-mediated RT inhibition (data not shown). The potential significance of these findings is discussed below.
Figure 2.

Sequence plasticity of stems and loops. Inhibition of RT DDDP activity by aptamer variants. Aptamer concentrations (nM) are indicated above the lanes. (A) Aptamer TKQ4 retains inhibition even though it carries a G-to-A mutation in the fourth of the 12 G's that comprise the quadruplex. All other G-to-A variants were inactive for inhibition (data not shown). (B) Full concentration range of aptamers from 0.3 nM to 300 nM was used in establishing IC50 values for R1T variants in which quadruplex loops are replaced with multiple thymidines or multiple adenosines. Similar titrations were carried out in triplicate for all aptamers for which IC50 values are reported. Aptamer R1T was included in subsequent panels to provide a consistent reference baseline of inhibition. (C) RT inhibition by aptamer SN, in which quadruplex loops are replaced with hexaethylene glycol. (D)and (E), RT inhibition by aptamers carrying mutations in helical stem domain, as detailed in text.
Sequence plasticity of quadruplex loops and stem
To define further the sequence requirements within intra-quadruplex loops, the single Ts in each loop of R1T were changed to other sizes and sequences. Replacement with single adenosines (R1A, IC50 = 18 ± 7 nM, Figure 2B) or cytidines (R1C, IC50 = 26 ± 9 nM) was well tolerated, while replacement with single guanosines was not (R1G, IC50≫250 nM). Expanding the loops with runs of two (R2T, IC50 = 13 ± 2 nM), four (R4T, IC50 ≈ 35 nM) or eight (R8T, IC50 = 20 ± 3 nM) thymidines was also well-tolerated. In contrast, expansion of the loops to two (R2A), four (R4A) or eight (R8A) adenosines or with two (R2C) or four (R4C) cytidines yielded aptamers with greatly reduced capacity for inhibiting RT (Figure 2B). To eliminate the possibility that RT-loop nucleotide contacts are essential for aptamer recognition, the intra-quadruplex loops were replaced with the relatively inert, nonnucleosidic hexaethylene glycol (HEG) to yield the SN aptamer. Interestingly, inhibition by the SN aptamer is more than 10 times more potent (IC50 = 3.5 ± 0.1 nM, Figure 2C) than the original RT6, and more than fourfold more potent than R1T. We conclude that specific loop nucleotides within the quadruplex region are not required for inhibition or for stability, and that the loops are only minimally constrained as to sequence.
Additional variants of R1T were generated to identify sequence constraints within the 7-bp stem comprising the 5′-structural domain. Deletion of 2 bp (S1, IC50 = 59 ± 2 nM) or 3 bp (S2, IC50 = 110 ± 5 nM) from the stem progressively compromised RT inhibition (data not shown), while lengthening the stem to 22 bp slightly improved inhibition (ZAM1, IC50 = 10.0 ± 1.5 nM). Reversing the sequence orientation within this domain (Srevstem, IC50 = 26 ± 7 nM) had minimal impact on IC50 (Figure 2D). Replacing the 10 nt at the end of the R1T stem with a CCCT tetraloop (S4, IC50 = 32 ± 1 nM, Figure 2E) had only a twofold effect on RT inhibition. These results establish that the 5′-domain requires a generic double-helical fragment stabilized by seven or more base pairs of essentially any sequence.
Loop sequences affect sensitivity to ionic environment
Potassium ions strongly favor quadruplex formation, while sodium ions are usually less stabilizing and lithium ions are destabilizing (40,41). We observed equivalent DNA polymerization activity by HIV-1 RT in buffers containing either K+ or Na+. RT inhibition by aptamers SN, R1T and RT6-B was therefore monitored in the original buffer containing K+ and also in a buffer in which K+ was replaced with Na+. The same RT was nearly inactive in buffers containing Li+, preventing analysis of inhibition in the presence of Li+. All three aptamers strongly inhibited RT in the presence of K+. Aptamers SN and R1T were also strongly inhibitory in the Na+-containing buffer, while aptamer RT6-B lost all inhibition under these conditions (Figure 3). Because these three aptamers differ only in the loop sequences connecting the individual guanosine triplets of the quadruplex, we conclude that the loop sequences in RT6-B render that aptamer sensitive to ionic destabilization, while the simplified structures of R1T and SN stabilize those aptamers to ionic substitution.
Figure 3.

Loop sequences modulate ionic sensitivity. RT inhibition by aptamers RT6-B, R1T and SN was carried out in normal buffer (potassium as the primary monovalent cation), or in buffers in which potassium was replaced with sodium. Note that in sodium, R1T effected the most potent inhibition.
Circular dichroism (CD) spectroscopy is a powerful analytical tool for identifying quadruplexes DNA structures. For quadruplexes in which all four strands are in the same orientation (parallel), CD minima and maxima are typically near 240 nm and 264 nm, respectively. For quadruplexes in which strands orientation alternates (antiparallel), the corresponding values are typically near 265 nm and 295 nm, respectively. The CD spectra for RT6-B and R1T both show maxima at 262 nm and minima at 242 nm at room temperature in the presence of K+ (Figure 4). In Na+ buffer the ellipticity is strongly flattened for RT6-B, especially at low DNA concentration, indicting destabilization of the quadruplex structure. Slight flattening is also evident in R1T, although to a lesser extent. Quadruplexes containing HEG loops, such as the one in aptamer SN, have previously been shown to be more stable than quadruplexes with single T loops in both K+ and Na+ buffers (42); thus, SN is also expected to retain its quadruplex in the Na+ buffer. These data provide independent confirmation of guanosine quadruplex formation within the RT6 family of aptamers, and they establish that the structural integrity of the simple quadruplex within R1T is less sensitive to ionic conditions, consistent with the inhibition data above.
Figure 4.

CD analysis of aptamer structure. Aptamers RT6-B and R1T were analyzed at either 2 μM or 4 μM, as indicated on the figure. Black traces indicate normal buffer containing potassium. Red traces indicate buffer in which potassium was replaced with sodium.
A generic, physical connection is required between the duplex and quadruplex modules
RT inhibition was measured for several variants of R1T to determine the importance of the physical connection between the two structural modules (Figure 5A). No inhibition was observed for a 28-nt oligo containing only the 5′ stem–loop motif (stem), nor for a 16-nt oligo containing the (TGGG)4 quadruplex motif (Q16). Similarly, no inhibition was observed when both oligos were added simultaneously at a ratio of 1:1, even when their individual concentrations were increased to 3000 nM (Figure 5B). A physical (covalent) connection between the DNA duplex and G-quadruplex is therefore required to effect inhibition.
Figure 5.

Topological requirements. (A) Schematic diagrams of topological variants described in the text, assuming three-layer, parallel quadruplex stacks. (B) RT inhibition assays utilizing separated subdomains of R1T (‘Stem’ and ‘Q16’). Inhibition is essentially eliminated when the two domains are not physically connected. ‘Mix’ refers to a 1:1 mixture of stem and Q16 oligos, each at the indicated concentrations. ‘Ctrl’ is an irrelevant control DNA: 5′ d(GCGGGACAATGGAGAGAGGG). (C) RT inhibition assays in which the connector domain is replaced with HEG. (D) RT inhibition assays in which inter-module connection is via two strands, with 5′ and 3′ termini in the stem module (Dyl5) or between the second and third guanosine triplets (Acut).
The stem–loop and G-quadruplex elements within aptamers RT6, RT5 and RT47 are joined by single-stranded segments with the sequences TTA, CTT and CC, respectively, while in R1T the connector is TT. To determine the effect of varying this connector sequence, RT inhibition was measured for variants of R1T in which the connector was changed to several other sequences. Changing those two thymidines to two adenosines (A2 connector in L6), or expanding it to four (T4 in L3) or eight (T8 in L4) thymidines had minimal effect on IC50 values (13 ± 1, 13 ± 2 and 28 ± 2 nM, respectively) (data not shown). To determine whether the negative charges or other nucleic acid-related features of these connector sequences are required for RT inhibition, the duplex and quadruplex modules were joined via HEG (Figure 5C). The IC50 value of the HEG-linked molecule (29 ± 3 nM, HEG) is similar to that of the T8-linked molecule. Thus, the primary role of the connector is to provide a physical connection between the two domains, rather than to provide a specific structural or functional role. However, there is an optimal range of lengths for the connector, as increasing it to T16 significantly weakened inhibition (L10, IC50 = 120 ± 6 nM).
Topological malleability of the connectivity between the structural elements
In all of the aptamer variants above, the two structural elements are connected via one flexible linker between the 3′-end of the helical element and the 5′-end of the quadruplex element. The 5′- and 3′-ends of the molecule are located, respectively, at the termini of the helical and quadruplex elements. Several variants of R1T were evaluated to determine whether alternative topologies are allowed. To accommodate relocating the 5′ and 3′ termini to other positions, the helical fragment was joined to the quadruplex by separate thymidine dinucleotides at each end of the quadruplex (Figure 5A). For variants Dyl1, Dyl2, Dyl3 and Dyl5, the 5′ and 3′ termini lie within the original loop at the distal end of the helical element. The helical portions of these molecules contain a total of 15 bp, 19 bp, 22 bp or 35 bp, including an internal mismatch derived from the original stem in RT6. All four variants strongly inhibit HIV-1 RT (e.g. IC50 = 21.0 ± 3.5 nM for Dyl5) (Figure 5D). The 5′ and 3′ termini were next relocated to internal positions within the quadruplex (Figure 5A). Placing the 3′-end after the second guanosine triplet (Acut, IC50 = 22 ± 4 nM, Figure 5D) yielded inhibition within a factor of two of the potency of R1T. Reversing the sequence within the central loop at the distal end of the helical element (Acutrev, IC50 = 33 ± 2 nM) had only a slight effect on IC50. Inhibitory potency was further reduced when the 3′-end was placed after the first guanosine triplet (Bcut, IC50 = 48 ± 1 nM) or after the third guanosine triplet (Ecut, IC50 = 99 ± 2 nM), likely reflecting a destabilization of the quadruplex module. Potent inhibitors based on R1T can therefore be generated by joining the helical fragment and quadruplex by either a single or a double linker between the helical and quadruplex elements.
Cross-clade inhibition of divergent primate lentiviral RT's
The ssDNA aptamers studied here were originally selected to bind RT from HIV-1 strain BH10 (group M, subtype B). The inhibition studies presented above all utilize RT from the closely related HIV-1 strain HXB2 (six amino acid differences in 560 positions, preserving 98.9% identity). To define the phylogenetic breadth of inhibition by these bimodular ssDNA aptamers, DNA-dependent DNA polymerization was monitored for a panel of five RT variants from diverse primate lentiviral strains from within HIV-1 group M (average 92% identity with RT from BH10) and three from outside this group (10). Aptamer SN was chosen for this analysis, as it was the most potent inhibitor among those studied here. All eight lentiviral RT's were inhibited. With the exception of the HIV-2 isolate, all IC50 values were (within error) within a factor of two of being identical to IC50 for inhibition of the subtype B enzyme (Table 2). The SN aptamer (and related forms) are therefore highly promising reagents for developing broad-spectrum anti-HIV agents.
Table 2.
Inhibition of phylogenetically diverse primate lentiviral reverse transcriptases
| Strain | Phylogenetic groupa | Percent identityb | IC50 (nM) |
|---|---|---|---|
| HXB2 | HIV-1 (M:B) | 98.9 | 3.5 ± 0.9 |
| 92UG021 | HIV-1 (M:A/D)c | 92.9 | 3.0 ± 0.3 |
| 98CN009 | HIV-1 (M:C) | 92.9 | 3.4 ± 1.1 |
| 94CY017 | HIV-1 (M:A) | 90.4 | 7.2 ± 0.7 |
| 93TH253 | HIV-1 (M:A/E)c | 90.5 | 6.1 ± 0.6 |
| MVP5180 | HIV-1 (O) | 78.2 | 4.9 ± 0.4 |
| TAN1 | SIVcpzP.t.s. | 72.7 | 2.1 ± 0.3 |
| EHO-287 | HIV-2 (B) | 60.5 | 12.0 ± 0.5 |
aHIV-1 phylogenetic groupings are designated by ‘(group:subtype)’.
bPercent amino acid identity in complete RT sequences in comparison with HIV-1 strain BH10.
cStrains 93TH243 and 92UG021 are intersubtype recombinants. RT from the former groups with subtype A; RT from the latter groups with subtype D (10).
DISCUSSION
‘Universalist’, bimodular, single-stranded DNA aptamers with stem-plus-quadruplex domains
This study demonstrates a novel structural paradigm for bimodular DNA inhibitors of HIV-1 RT, in which one module is a helical stem and the other is a guanosine quadruplex. We identified this architecture within aptamers RT5, RT6 and RT47—isolated previously (11)—and defined the sequence components required to achieve the functional structures by monitoring RT inhibition for a collection of 60 variants of RT6. Structural interpretations were further confirmed by CD spectroscopy. The most potent variant, designated SN, strongly inhibited not only RT from HIV-1 subtype B, which was the subtype originally used in generating this family of aptamers, but also several other HIV-1, SIVcpz and HIV-2 RT's ranging from approximately 60% to 92% amino acid identity with RT from subtype B. Inhibition of these RT and additional members of the RT diversity panel was previously monitored for four RNA pseudoknot aptamers (10) and for three ssDNA aptamers (14). ssDNA aptamer RT8 was a ‘specialist’, exhibiting strict specificity for RT from subtype B. In contrast, ssDNA aptamers RT1t49 and RT1t49(-5) were ‘universalists’ for primate lentiviral RTs, inhibiting every member of the panel. The four RNA aptamers displayed intermediate behavior, inhibiting two or more members of the panel while leaving more than half of the panel uninhibited. Thus, the ‘universalist’ inhibitory behavior of the bimodular aptamers studied here (especially SN) is similar to that of RT1t49 and RT1t49(-5).
Mutational analysis revealed a simple core structure in these bimodular aptamers. The helical module can be of any sequence. Internal bulges or mismatches are not required; converting them into fully base paired sites had no consequence on inhibition. Variants with seven or more base pairs in the stem gave similar RT inhibition, while variants with shorter stems were less effective. The distal end of this stem can either be a closed loop of any sequence, or it can contain the 5′ and 3′ termini. Mutations within the quadruplex module retained RT inhibition, so long as the altered sequence retained the capacity to form the quadruplex structure. Aptamer R1T (IC50 = 14 ± 2 nM), which may assume a two-layer quadruplex (see below), yielded slightly weaker inhibition than the three-layer quadruplex aptamers RT6-B (IC50 = 5.7 ± 0.8 nM) and SN (IC50 = 3.5 ± 0.1 nM). Replacing the T's with HEG (SN) gave the strongest inhibition among all variants tested here. We conclude that the duplex and quadruplex structural modules, linked by a short tether, are sufficient for potent inhibition of HIV-1 RT, and that sequence-specific recognition plays little or no role in specifying the protein–ssDNA interaction.
Interaction models for the helical element
RT contains a strongly electropositive trough running between the DNA polymerase and RNase H active sites (43–45), which binds helical nucleic acids in co-crystal structures and in modeled complexes (6,13,44,46–48). The binding interactions of a dual-stem aptamer, denoted RT1t49, have been modeled based on Fe(II)-mediated DNA cleavage mapping (5). Differences in the hydroxyl radical cleavage patterns in the presence of wild-type RT and active site mutants suggested that the 5′-end of RT1t49 is near the polymerase active site, and that the long stem of this aptamer extends towards the RNase H active site (5). This model places the second structural domain—a short duplex stem—on the back side of the fingers or thumb domain, perhaps close to the normal trajectory of the 5′ single-strand extension in the template strand (49). A similar mode of interaction by the helical domain of these bimodular aptamers would place the quadruplex module on the back side of the fingers or thumb domain of RT.
Structure of the quadruplex element
Our mutational and spectroscopic data support a quadruplex with parallel strands of repeated guanosines arranged into at least two stacked layers. All 12 guanosines were immutable within RT6-A, implying a three-layer stack in this context. By extension, three stacked layers are proposed for the original aptamers RT6-B, RT6, RT5 and RT47. Adding or removing a guanosine from each of the four repeats to shorten or extend the stack within R1T abolished inhibition, again implying a three-layer stack. In contrast, only 11 of the 12 guanosines were strictly required in R1T. RT inhibition was reduced but not abolished upon deletion or mutating the first guanosine of the second repeat within R1T to A, C or T, potentially indicating that the nucleotide in this position is translocated to the loop, and that only two stacked layers are absolutely required in this context. An alternative possibility is that the three remaining guanosines in this tier of the quadruplex may provide sufficient stabilization to allow the fourth nucleotide to stack onto the quadruplex so as to retain overall structure. Our results are also in agreement with other authors’ findings that quadruplex stability is affected by the sequence of the loops (42,50). In particular, the RT6-B aptamer inhibits RT only in buffer that contains K+ ions and shows considerable broadening of its CD spectrum in buffer that contains Na+, while the more compact R1T inhibits in both K+ and Na+, and shows less line broadening.
Some of the quadruplex modules included within these bimodular aptamers are analogous to quadruplexes that have been studied previously. A systematic survey of quadruplexes with differing numbers of repeated G's or with differing loop sequences identified (G3T)4 as being especially stable (51,52). The (G3T)4 quadruplex is very similar to the (TG3)4 quadruplex within R1T. CD spectroscopy of (G3T)4 indicated parallel strands within the stack (51), similar to the results obtained here for R1T and RT6-B. In analogy with aptamer SN, the sequence (G3-HEG)3G3 was previously shown to form a three-tiered quadruplex and displayed greater thermal stability than (G3T)4 (42). The (G3T)4 quadruplex—also referred to as T30923 and as T30695—has been studied for inhibition of HIV-1 integrase (53,54), in addition to structural studies (36,51–53). Importantly, an NMR structure of (G3T)4 shows only two layers of stacked guanosine tetrads with partially ordered ‘GT’ loops (36,53). These observations are consistent with the suggestion above that aptamer R1T may only require two layers of stacked quadruplex. However, the (TG3)4 quadruplex studied here (denoted Q16 when used in isolation) was unable to inhibit RT without a physical connection to the helical domain, even when used at 3000 nM concentration. The well-studied aptamer ‘TBA’ (GGTTGGTGTGGTTGG) forms a two-layer quadruplex that binds and inhibits thrombin in the low nanometer range (33). However, in preliminary assays, we observe no inhibition of thrombin-mediated cleavage of a model, chromogenic peptide even in the presence of 3000 nM aptamer SN (data not shown), suggesting that TBA may be a poor analog of SN. In sum, most of the quadruplex modules within these aptamers are best modeled as three-layered stacks of parallel strands, in some cases with single-nucleotide or nonnucleic acid loops, and we cannot rule out that two layers may be sufficient for R1T.
Interaction models for the quadruplex element
Aptamers with diverse quadruplex modules can accomplish RT inhibition, arguing for a promiscuous interaction with the quadruplex domain. In particular, the intra-quadruplex loops tolerate considerable plasticity. The loops in aptamers RT5, RT47 and RT6 are unrelated to each other. Inhibition was improved by simplifying the loops to single A (R1A), C (R1C) or T residues (R1T) and was further improved by replacing the loops with HEG (SN). For each of these four alterations, inhibition correlated with previously measured increases in the stabilities of isolated quadruplex DNAs (42,51,52). Loop substitutions with multi-homonucleotide sequences other than T (R2C, R2A, etc.) did not inhibit RT. Furthermore, aptamers with the phosphodiester chain termini located within the quadruplex retained inhibition. Thus, the loops are more likely to serve simply to stabilize the folded quadruplex rather than to interact directly with the protein.
The interaction model noted above for the helical domain based on RT1t49 (5) places the quadruplex module on the back side of the fingers or thumb domain of RT. An alternative interaction mode is suggested by studies of two quadruplexes, 93del and (G3T)4, which bind HIV-1 integrase. Integrase is in the same structural superfamily as RNase H, with the greatest similarity in the central catalytic core (IN residues 60–160) (55,56). In this alternative model, the quadruplex domains of the bimodular aptamers bind the RNase H domain of RT, effectively reversing the orientation of the helical domain relative to the model suggested above. Quadruplex 93del (GGGGTGGGAGGAGGGT) was originally selected as a longer ODN93 for its ability to bind mature heterodimer RT and not to bind a p51/p51 homodimer lacking the RNase H domain (21). An NMR structure of 93del shows an interlocking dimer of two three-layer quadruplexes, arranged in predominately parallel strands. Those authors modeled the 93del dimer as docking into the channel formed by HIV-1 IN, and they suggest potential interactions of the quadruplex loops with the protein (55). Quadruplex (G3T)4 also binds and inhibits HIV-1 IN (36,53,57,58). Shortening or extending the run of G's [(G2T)4 and (G4T)4, respectively] did not diminish IN inhibition, a result that was interpreted as indicating DNA–IN interaction through loops on the ends of the quadruplex, rather than a side-on interaction (53). Computational modeling of that interaction places the (G3T)4 quadruplex in the catalytic domain of HIV-1 IN. Interestingly, this NMR structure shows the guanosine triplets arranged into antiparallel strands, even though CD analysis shows an ellipticity maximum near 264 nm, indicative of a parallel strand arrangement. Computational docking models again suggest essential interactions between quadruplex loops and the integrase catalytic core, particularly Asp64, Asp116 and Glu152 (53).
The data and models from studies of these IN aptamers are at odds with our RT inhibition studies with the bimodular aptamers. Specifically, we observe similar degrees of RT inhibition for a multitude of loop analogs, and RT inhibition is disrupted by shortening [ML2, (TG2)4] or extending [ML4, (TG4)4] the run of G's, in contrast with the effects of similar alterations within IN-binding quadruplexes (55). Furthermore, a hybrid molecule carrying the stem and connector elements and 93del in place of the (TG3)4 quadruplex (stem-93del) did not inhibit RT in pilot reactions. Thus, R1T and the members of the RT6 family of ssDNA aptamers are structurally and functionally distinct from previously described quadruplex aptamers. While, it is possible that the quadruplex domains of the bimodular aptamers interact with the RNase H domain of RT, we conclude that the IN-binding quadruplex models are not appropriate models for defining this interaction. New experimental data are needed to address this question directly.
CONCLUSION
The bimodular RT-binding aptamers presented here represent a novel architecture for RT antagonists. We demonstrate that a helical fragment connected to diverse quadruplex domains is able to specify the correct localization and the strength of binding to RT protein, yielding competition with primer/template for access to the protein. The interactions appear distinct from those observed previously for quadruplex–integrase, quadruplex–thrombin and quadruplex–RNase H complexes. It is likely that the helical and quadruplex modules contact the protein directly, and that their physical connection can facilitate cooperativity of binding, with both modules interacting simultaneously. Further, it is possible that simultaneous binding at two sites, facilitated by a flexible linker, aids in the cross-clade recognition exhibited both by the stem-plus-quadruplex bimodular aptamers studied here and by the dual-stem aptamers studied previously (5,11,14). Current work seeks to determine whether the bimodular RT-binding aptamers described here confer antiviral bioactivity in cell culture and whether they interact with other cellular components. The results of those studies, and those presented here, will aid the design of RT antagonists for use in drug discovery and studies of HIV-1 replication.
FUNDING
National Institutes of Health (grant AI62513 to D.H.B.). Funding for open access charge: National Institutes of Health (grant AI074389 to D.H.B.).
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors wish to thank Drs Michael Henzl and Anmin Tan for their assistance with the CD analyses.
REFERENCES
- 1.Hariri S, McKenna MT. Epidemiology of human immunodeficiency virus in the United States. Clin. Microbiol. Rev. 2007;20:478–488. doi: 10.1128/CMR.00006-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.UNReport. HIV infection on the rise worldwide: U.N. report. Clin. Infect. Dis. 2007;44:3–4. doi: 10.1086/511966. [DOI] [PubMed] [Google Scholar]
- 3.Abdool Karim SS, Abdool Karim Q, Gouws E, Baxter C. Global epidemiology of HIV-AIDS. Infect. Dis. Clin. North Am. 2007;21:1–17. doi: 10.1016/j.idc.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 4.Held D, Kissel J, Saran D, Michalowski D, Burke D. Differential susceptibility of HIV-1 reverse transcriptase to inhibition by RNA aptamers in enzymatic reactions monitoring specific steps during genome replication. J. Biol. Chem. 2006;281:25712–25722. doi: 10.1074/jbc.M604460200. [DOI] [PubMed] [Google Scholar]
- 5.Kissel J, Held D, Hardy R, Burke D. Active site binding and sequence requirements for inhibition of HIV-1 reverse transcriptase by the RT1 family of single-stranded DNA aptamers. Nucleic Acids Res. 2007;35:5039–5050. doi: 10.1093/nar/gkm420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jaeger J, Restle T, Steitz TA. The structure of HIV-1 reverse transcriptase complexed with an RNA pseudoknot inhibitor. EMBO J. 1998;17:4535–4542. doi: 10.1093/emboj/17.15.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joshi P, Prasad VR. Potent inhibition of human immunodeficiency virus type 1 replication by template analog reverse transcriptase inhibitors derived by SELEX (systematic evolution of ligands by exponential enrichment) J. Virol. 2002;76:6545–6557. doi: 10.1128/JVI.76.13.6545-6557.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tuerk C, MacDougal S, Gold L. RNA pseudoknots that inhibit human immunodeficiency virus type 1 reverse transcriptase. Proc. Natl Acad. Sci. 1992;89:6988–6992. doi: 10.1073/pnas.89.15.6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burke DH, Scates LA, Andrews K, Gold L. Bent pseudoknots and novel RNA inhibitors of type 1 human immunodeficiency virus (HIV-1) reverse transcriptase. J. Mol. Biol. 1996;264:650–666. doi: 10.1006/jmbi.1996.0667. [DOI] [PubMed] [Google Scholar]
- 10.Held D, Kissel J, Thacker S, Michalowski D, Saran D, Ji J, Hardy R, Rossi J, Burke D. Cross-clade inhibition of recombinant HIV-1, HIV-2 and SIVcpz reverse transcriptases by RNA pseudoknot aptamers. J. Virol. 2007;81:5375–5384. doi: 10.1128/JVI.01923-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schneider DJ, Feigon J, Hostomsky Z, Gold L. High-affinity ssDNA inhibitors of the reverse transcriptase of type 1 human immunodeficiency virus. Biochem. 1995;34:9599–9610. doi: 10.1021/bi00029a037. [DOI] [PubMed] [Google Scholar]
- 12.Andreola M-L, Pileur F, Calmels C, Ventura M, Tarrago-Litvak L, Toulme J.-J, Litvak S. DNA aptamers selected against the HIV-1 RNase H display in vitro antiviral activity. Biochemistry. 2001;40:10087–10094. doi: 10.1021/bi0108599. [DOI] [PubMed] [Google Scholar]
- 13.DeStefano J, Cristofaro J. Selection of primer-template sequences that bind human immunodeficiency virus reverse transcriptase with high affinity. Nucleic Acids Res. 2006;34:130–139. doi: 10.1093/nar/gkj426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kissel J, Held D, Hardy R, Burke D. Single-stranded DNA aptamer RT1t49 inhibits RT polymerase and RNase H functions of HIV type 1, HIV type 2, and SIVCPZ RTs. AIDS Res. Hum. Retroviruses. 2007;23:699–708. doi: 10.1089/aid.2006.0262. [DOI] [PubMed] [Google Scholar]
- 15.Somasunderam A, Ferguson MR, Rojo DR, Thiviyanathan V, Li X, O'Brien WA, Gorenstein DG. Combinatorial selection, inhibition, and antiviral activity of DNA thioaptamers targeting the RNase H domain of HIV-1 reverse transcriptase. Biochemistry. 2005;44:10388–10395. doi: 10.1021/bi0507074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Held D, Kissel J, Patterson J, Nickens D, Burke D. HIV-1 inactivation by nucleic acid aptamers. Front Biosci. 2006;11:89–112. doi: 10.2741/1782. [DOI] [PubMed] [Google Scholar]
- 17.Chaloin L, Lehmann MJ, Sczakiel G, Restle T. Endogenous expression of a high-affinity pseudoknot RNA aptamer supresses replication of HIV-1. Nucleic Acids Res. 2002;30:4001–4008. doi: 10.1093/nar/gkf522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joshi PJ, North TW, Prasad VR. Aptamers directed to HIV-1 reverse transcriptase display greater efficacy over small hairpin RNAs targeted to viral RNA in blocking HIV-1 replication. Mol. Ther. 2005;11:677–686. doi: 10.1016/j.ymthe.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 19.Fisher TS, Joshi P, Prasad VR. Mutations that confer resistance to template-analog inhibitors of human immunodeficiency virus (HIV) type 1 reverse transcriptase lead to severe defects in HIV replication. J. Virol. 2002;76:4068–4072. doi: 10.1128/JVI.76.8.4068-4072.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fisher TS, Joshi P, Prasad VR. HIV-1 reverse transcriptase mutations that confer decreased in vitro susceptibility to anti-RT DNA aptamer RT1t49 confer cross resistance to other anti-RT aptamers but not to standard RT inhibitors. AIDS Res Ther. 2005;2:8. doi: 10.1186/1742-6405-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Soultrait V, Lozach P, Altmeyer R, Tarrago-Litvak L, Litvak S, Andreola M. DNA aptamers derived from HIV-1 RNase H inhibitors are strong anti-integrase agents. J. Mol. Biol. 2002;324:195–203. doi: 10.1016/s0022-2836(02)01064-1. [DOI] [PubMed] [Google Scholar]
- 22.Mosing R, Mendonsa S, Bowser M. Capillary electrophoresis-SELEX selection of aptamers with affinity for HIV-1 reverse transcriptase. Anal. Chem. 2005;77:6107–6112. doi: 10.1021/ac050836q. [DOI] [PubMed] [Google Scholar]
- 23.Burge S, Parkinson G, Hazel P, Todd A, Neidle S. Quadruplex DNA: sequence, topology and structure. Nucleic Acids Res. 2006;34:5402–5415. doi: 10.1093/nar/gkl655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dai J, Punchihewa C, Ambrus A, Chen D, Jones R, Yang D. Structure of the intramolecular human telomeric G-quadruplex in potassium solution: a novel adenine triple formation. Nucleic Acids Res. 2007;35:2440–2450. doi: 10.1093/nar/gkm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haider S, Parkinson G, Neidle S. Crystal structure of the potassium form of an Oxytricha nova G-quadruplex. J. Mol. Biol. 2002;320:189–200. doi: 10.1016/S0022-2836(02)00428-X. [DOI] [PubMed] [Google Scholar]
- 26.Halder K, Mathur V, Chugh D, Verma A, Chowdhury S. Quadruplex-duplex competition in the nuclease hypersensitive element of human c-myc promoter: C to T mutation in C-rich strand enhances duplex association. Biochem. Biophys. Res. Commun. 2005;327:49–56. doi: 10.1016/j.bbrc.2004.11.137. [DOI] [PubMed] [Google Scholar]
- 27.Ambrus A, Chen D, Dai J, Jones R, Yang D. Solution structure of the biologically relevant G-quadruplex element in the human c-MYC promoter. Implications for G-quadruplex stabilization. Biochemistry. 2005;44:2048–2058. doi: 10.1021/bi048242p. [DOI] [PubMed] [Google Scholar]
- 28.Etzioni S, Yafe A, Khateb S, Weisman-Shomer P, Bengal E, Fry M. Homodimeric MyoD preferentially binds tetraplex structures of regulatory sequences of muscle-specific genes. J. Biol. Chem. 2005;280:26805–26812. doi: 10.1074/jbc.M500820200. [DOI] [PubMed] [Google Scholar]
- 29.Cogoi S, Xodo L. G-quadruplex formation within the promoter of the KRAS proto-oncogene and its effect on transcription. Nucleic Acids Res. 2006;34:2536–2549. doi: 10.1093/nar/gkl286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo K, Pourpak A, Beetz-Rogers K, Gokhale V, Sun D, Hurley L. Formation of pseudosymmetrical G-quadruplex and i-motif structures in the proximal promoter region of the RET oncogene. J. Am. Chem. Soc. 2007;129:10220–10228. doi: 10.1021/ja072185g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qin Y, Rezler E, Gokhale V, Sun D, Hurley L. Characterization of the G-quadruplexes in the duplex nuclease hypersensitive element of the PDGF-A promoter and modulation of PDGF-A promoter activity by TMPyP4. Nucleic Acids Res. 2007;35:7698–7713. doi: 10.1093/nar/gkm538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hershman S, Chen Q, Lee J, Kozak M, Yue P, Wang L, Johnson F. Genomic distribution and functional analyses of potential G-quadruplex-forming sequences in Saccharomyces cerevisiae. Nucleic Acids Res. 2007;36:144–156. doi: 10.1093/nar/gkm986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Macaya RF, Schultze P, Smith FW, Roe JA, Feigon J. Thrombin-binding DNA aptamer forms a unimolecular quadruplex structure in solution. Proc. Natl Acad. Sci. 1993;90:3745–3749. doi: 10.1073/pnas.90.8.3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Christiansen J, Kofod M, Nielsen F. A guanosine quadruplex and two stable hairpins flank a major cleavage site in insulin-like growth factor II mRNA. Nucleic Acids Res. 1994;22:5709–5716. doi: 10.1093/nar/22.25.5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teng Y, Girvan A, Casson L, Pierce WJ, Qian M, Thomas S, Bates P. AS1411 alters the localization of a complex containing protein arginine methyltransferase 5 and nucleolin. Cancer Res. 2007;67:10491–10500. doi: 10.1158/0008-5472.CAN-06-4206. [DOI] [PubMed] [Google Scholar]
- 36.Jing N, Rando R, Pommier Y, Hogan M. Ion selective folding of loop domains in a potent anti-HIV oligonucleotide. Biochemistry. 1997;36:12498–12505. doi: 10.1021/bi962798y. [DOI] [PubMed] [Google Scholar]
- 37.Mazumder A, Neamati N, Ojwang J, Sunder S, Rando R, Pommier Y. Inhibition of the human immunodeficiency virus type 1 integrase by guanosine quartet structures. Biochemistry. 1996;35:13762–13771. doi: 10.1021/bi960541u. [DOI] [PubMed] [Google Scholar]
- 38.Mathews D, Sabina J, Zuker M, Turner D. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 39.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Töhl J, Eimer W. Interaction energies and dynamics of alkali and alkaline-earth cations in quadruplex-DNA-structures. J. Mol. Model. 1996;2:327–329. [Google Scholar]
- 41.Simonsson T. G-quadruplex DNA structures-variations on a theme. Biol. Chem. 2001;382:621–628. doi: 10.1515/BC.2001.073. [DOI] [PubMed] [Google Scholar]
- 42.Risitano A, Fox K. Influence of loop size on the stability of intramolecular DNA quadruplexes. Nucleic Acid Res. 2004;32:2598–2606. doi: 10.1093/nar/gkh598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arnold E, Jacobo-Molina A, Nanni R, Williams R, Lu X, Ding J, Clark A, Zhang A, Ferris AL, Clark P, et al. Structure of HIV-1 reverse transcriptase/DNA complex at 7 A resolution showing active site locations. Nature. 1992;357:85–87. doi: 10.1038/357085a0. [DOI] [PubMed] [Google Scholar]
- 44.Jacobo-Molina A, Ding J, Nanni RG, Clark ADJ, Lu X, Tantillo C, Williams RL, Kamer G, Ferris AL, Clark P, et al. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 Å resolution shows bent DNA. Proc. Natl Acad. Sci. 1993;90:6320–6324. doi: 10.1073/pnas.90.13.6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. Crystal structure at 3.5 Å resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science. 1992;256:1783–1790. doi: 10.1126/science.1377403. [DOI] [PubMed] [Google Scholar]
- 46.Huang H, Chopra R, Verdine G, Harrison S. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science. 1998;282:1669–1675. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- 47.Sarafianos SG, Das K, Tantillo T, Clark ADJ, Ding J, Whitcomb JM, Boyer PL, Hughes SH, Arnold ET. Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA:DNA. EMBO J. 2001;20:1449–1461. doi: 10.1093/emboj/20.6.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kvaratskhelia M, Miller JT, Budihas SR, Pannell LK, Le Grice SF. Identification of specific HIV-1 reverse transcriptase contacts to the viral RNA:tRNA complex by mass spectrometry and a primary amine selective reagent. Proc. Natl Acad. Sci. USA. 2002;99:15988–15993. doi: 10.1073/pnas.252550199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tuske S, Sarafianos S, Clark AJ, Ding J, Naeger L, White K, Miller M, Gibbs C, Boyer P, Clark P, et al. Structures of HIV-1 RT-DNA complexes before and after incorporation of the anti-AIDS drug tenofovir. Nat. Struct. Mol. Biol. 2004;11:469–474. doi: 10.1038/nsmb760. [DOI] [PubMed] [Google Scholar]
- 50.Risitano A, Fox K. Stability of intramolecular DNA quadruplex: comparison with DNA duplexes. Biochemistry. 2003;42:6507–6513. doi: 10.1021/bi026997v. [DOI] [PubMed] [Google Scholar]
- 51.Rachwal P, Findlow I, Werner J, Brown T, Fox K. Intramolecular DNA quadruplexes with different arrangements of short and long loops. Nucleic Acid Res. 2007;35:4214–4222. doi: 10.1093/nar/gkm316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rachwal P, Brown T, Fox K. Effect of G-tract length on the topology and stability of intramolecular DNA quadruplexes. Biochemistry. 2007;46:3036–3044. doi: 10.1021/bi062118j. [DOI] [PubMed] [Google Scholar]
- 53.Jing N, Marchand C, Liu J, Mitra R, Hogan M, Pommier Y. Mechanism of inhibition HIV-1 integrase by G-tetrad forming oligonucleotides in vitro. J. Biol. Chem. 2000;275:21460–21467. doi: 10.1074/jbc.M001436200. [DOI] [PubMed] [Google Scholar]
- 54.Jing N, De Clercq E, Rando R, Pallansch L, Lackman-Smith C, Lee S, Hogan M. Stability-activity relationships of a family of G-tetrad forming oligonucleotides as potent HIV inhibitors. J. Biol. Chem. 2000;275:3421–3430. doi: 10.1074/jbc.275.5.3421. [DOI] [PubMed] [Google Scholar]
- 55.Phan A, Kuryavyi V, Ma J.-B, Faure A, Andréola M-L, Patel D. An interlocked dimeric parallel-stranded DNA quadruplex: a potent inhibitor of HIV-1 integrase. Proc. Natl Acad. Sci., USA. 2005;102:634–639. doi: 10.1073/pnas.0406278102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jing N, Gao X, Rando R, Hogan M. Potassium-induced loop conformational transition of a potent anti-HIV oligonucleotide. J. Biomol. Struct. Dyn. 1997;15:573–585. doi: 10.1080/07391102.1997.10508967. [DOI] [PubMed] [Google Scholar]
- 57.Jing N, Hogan M. Structure-activity of tetrad forming oligonucleotides as a potential anti-HIV integrase therapeutic drug. J. Biol. Chem. 1998;273:34992–34999. doi: 10.1074/jbc.273.52.34992. [DOI] [PubMed] [Google Scholar]
- 58.Lyonnais S, Hounsou C, Teulade-Fichou M.-P, Jeusset J, Le Cam E, Mirambeau G. G-quartets assembly within a G-rich DNA flap. a possible event at the center of the HIV-1 genome. Nucleic Acid Res. 2002;30:5276–5283. doi: 10.1093/nar/gkf644. [DOI] [PMC free article] [PubMed] [Google Scholar]