


Abstract
We have adapted an electrophoretic mobility shift assay (EMSA) to isolate genomic DNA fragments that bind the archaeal transcription initiation factors TATA-binding protein (TBP) and transcription factor B (TFB) to perform a genome-wide search for promoters. Mobility-shifted fragments were cloned, tested for their ability to compete with known promoter-containing fragments for a limited concentration of transcription factors, and sequenced. We applied the method to search for promoters in the genome of Methanocaldococcus jannaschii. Selection was most efficient for promoters of tRNA genes and genes for several presumed small non-coding RNAs (ncRNA). Protein-coding gene promoters were dramatically underrepresented relative to their frequency in the genome. The repeated isolation of these genomic regions was partially rectified by including a hybridization-based screening. Sequence alignment of the affinity-selected promoters revealed previously identified TATA box, BRE, and the putative initiator element. In addition, the conserved bases immediately upstream and downstream of the BRE and TATA box suggest that the composition and structure of archaeal natural promoters are more complicated.
INTRODUCTION
Relative to the explosive growth in DNA sequence data, our understanding of genomes, their dynamics and the organisms that are hosts to them seems to grow slowly. Although several sophisticated algorithms exist to predict protein-coding regions (e.g. 1–4), the identification of DNA elements that play crucial roles in their expression lags far behind. This is especially notable in the case of transcriptional promoters. The identification of promoters has been limited, by and large, to studies of individual genes. Traditional genetic, biochemical and structural studies of promoters, with their emphasis on identifying and characterizing individual promoters, cannot meet the challenge of the genomic era. These approaches—be they based on RNA analysis [e.g. nuclease protection (5) and reverse transcriptase run-off (6)] or based on DNA analysis [e.g. transcription factor or polymerase footprinting (7) and analysis of in vitro transcript run-offs (8)]—are not readily scalable to probe whole genomes.
There are few genomes with systematically annotated promoter features. Computational approaches to promoter identification are unreliable when the analysis is carried out a genome at a time (9). Comparative analysis of appropriately related genomes can identify important DNA sequences (10,11), but these methods are not specific to promoters. Even in organisms where promoters have been extensively studied, the available tools for in silico identification remain wanting. The situation is even more critical in the case of the Archaea, where there is little experimental data to provide the bases for the calibration of computational tools. Other genome-wide probing methods, such as RNA analyses with oligonucleotide arrays, can identify transcribed regions (under specific growth conditions), but do not locate the start site of the transcripts, nor do they provide information on cis-acting regulatory regions.
Archaeal transcription shares similarity with the eukaryal pol II transcription system, in terms of the enzyme and factors involved, as well as promoter architecture (12–15). Core components include the general transcription factors—TATA-binding protein (TBP) and transcription factor B (TFB), which are homologs of eukaryal TBP and TFIIB—and one multi-subunit RNA polymerase, similar to RNA pol II in overall architecture and subunit composition (16,17). Transcription initiation involves the recognition of promoter sequences by TBP, the rate-limiting step. A correctly oriented TBP–promoter complex is stabilized by binding of TFB, which is also responsible for recruiting RNA polymerase (14,15,18).
Mirroring the similarities in the transcription machineries, the available information supports the notion that core elements of archaeal promoters are similar to those of pol II promoters. A TATA box and an upstream purine-rich element (BRE) are key players in the assembly of the pre-initiation complex. The TATA box provides the initial recognition element for transcription factor binding, with the BRE stabilizing appropriate binding of TBP and being instrumental in defining transcription orientation. The formation of the tripartite archaeal pre-initiation complex has been confirmed experimentally using footprinting assays, electrophoretic mobility shift assays (EMSA) and base-specific cross-linking (19–23). Crystallography of the ternary complex has revealed base-specific contacts between promoter elements and transcription factors TBP and TFB (24,25). A third element, the initiator (Inr), located at the transcription start site, is similar to its eukaryal counterpart (26,27). In Archaea, it appears to be less important, since in vitro transcription initiates readily from new sites in artificial promoters recovered by in vitro evolution (20), or in promoters with insertions/deletions between the TATA box and Inr (28). Moreover, while this element is conserved in Methanococcales and Sulfolobales, it is not evident in Haloarchaea (29).
Of the elements comprising the pre-initiation complex in Archaea (and in Eucarya), the promoter is the least understood. To facilitate systematic analyses, we set out to identify and characterize archaeal promoters on a genome-wide basis. We devised an efficient method for isolating and identifying genomic sequences that specifically bind to the general transcription factors TBP and TFB. We chose to carry out the analysis in the archaeon Methanocaldococcus jannaschii, the first member of the Archaea to have a fully sequenced genome (30). Since binding of transcription initiation factors to promoter sequences can be readily assayed by EMSA (31,32), we postulated that this could be modified into a method to select protein binding sequences from a pool of random genomic DNA fragments. To identify the desired fragments, we use the polymerase chain reaction (PCR) to amplify DNA sequences with increased apparent molecular weight, followed by cloning and sequencing of random and/or screened clones. The procedure is very similar in spirit to in vitro evolution and ‘systematic evolution of ligands by exponential enrichment’ (SELEX) (33,34) or, more closely, to genomic SELEX (35–40) since we did not wish to alter the sequences or binding ability of the natural DNAs.
MATERIALS AND METHODS
Unless stated otherwise, all kits and enzymes were used as recommended by their manufacturers.
Genomic DNA library
Methanocaldococcus jannaschii cells were grown in a reactor vessel as previously described (41). Late exponential cells from 1 l of culture were lyzed by three cycles of freezing and thawing in the presence of 0.5% SDS and their genomic DNA was purified (42). One hundred micrograms of DNA, re-suspended in 1 ml 25% glycerol/1 M NaOAc (pH 5.5), was exhaustively fragmented using a Branson Sonifier Cell Disruptor by repeated 15 s pulses to yield fragments of 400–500-bp average size. Following ethanol precipitation, 45 µg of fragmented genomic DNA was treated with 1 U BAL31 nuclease (New England Biolabs, NEB) for 7 min at 30°C in a 100 µl volume. After extraction with phenol/chloroform and ethanol precipitation, the fragments were resolved in a 1.5% SeaPlaque (FMC Corp.) agarose gel. The region of the gel containing 200–300-bp fragments was excised and the DNA extracted (Qiagen Gel Extraction kit). DNA fragments measuring 320 ng were treated with 1 U T4 DNA polymerase (Invitrogen) for 30 min at 37°C and the DNA was ligated overnight at 16°C to 600 ng of SmaI-cleaved and dephosphorylated pUC18 vector (Invitrogen). The ligation product was treated with 1 U DNA polymerase I (NEB) for 1 h at 16°C to move (by nick-translation) the nick between the insert and the (initially dephosphorylated) plasmid, thereby expanding the region of intact duplex DNA. Based on the frequency of colonies recovered following electrotransformation into Escherichia coli strain XL1Blue-MRF', we estimate that this primary library (20 µl total volume) contains 4 × 106 recombinant molecules/µl. This is likely an underestimate of the effective complexity in the PCR-based procedures described subsequently.
DNA fragments for the selection
Aliquots of the primary library were used as template for generating a collection of random DNA fragments by PCR using M13 universal primers. Each 100 µl reaction contained 20 mM Tris–HCl (pH 8.4), 50 mM KCl, 2 mM MgCl2, 100 pmol of each primer, 40 nmol dNTPs, 1 µl library DNA and 3 U Taq DNA polymerase (Invitrogen). Following an initial denaturation at 94°C for 90 s, the library DNA was amplified for 25 cycles with denaturation at 95°C for 30 s, annealing at 55°C for 30 s and extension at 72°C for 30 s. The final elongation was 72°C for 10 min. The products of the amplification include 134 bp of plasmid DNA flanking the inserts. The amplified DNA was resolved in low-melting temperature agarose and the 300–400-bp DNA fragments were recovered as above.
For EMSA, DNA was end-labeled using [γ-32P]ATP and T4 polynucleotide kinase (Invitrogen). Labeled fragments were purified using a QIAquick Nucleotide Removal Kit (Qiagen).
Expression and purification of transcription factors
TBP from M. jannaschii was cloned as an N-terminal His6-tagged recombinant protein in the expression vector pQE31 (Qiagen). TFB, without intein, was cloned as a C-terminal His6-tagged recombinant protein in the vector pET29b (Novagen). The recombinant proteins were expressed in E. coli BL2-CodonPlus®(DE3)-RIPL. His6-tagged proteins were affinity-purified with QIAexpressionist™ (Qiagen). Cells were grown to OD600 of 0.5. Following a 1.5-h induction with 1 mM isopropyl-β-d-thiogalactopyronoside (IPTG), cells were collected by centrifugation and re-suspended in lysis buffer [50 mM Na·PO4 (pH 8.0), 300 mM NaCl and 10 mM imidazole], followed by two passages through a French pressure cell. The lysate was heat-treated at 70°C for 8 min, and spun at 15 000 g for 15 min to remove denatured host proteins. The cleared cell lysate was mixed with 1/6th volume of Ni2+-NTA agarose equilibrated with lysis buffer, loaded onto the column and eluted with an increasing concentration of imidazole. Fractions containing the target protein were pooled and dialyzed against 20 mM Tris–HCl (pH 7.8), 500 mM NaCl and 40% glycerol. Purified proteins were concentrated by ultrafiltration using an Amicon Centriprep YM10.
To increase the stability of TFB, a C-terminal, DNA-binding fragment (TFBc) was created by removing the first 137 amino acids. The resulting sequence was cloned into pET29b expression vector and expressed as a C-terminal His6-tagged recombinant protein. The purification procedure was slightly modified. Briefly, host cells were induced with 0.4 mM IPTG when OD600 reached 0.5, and harvested after 2 h. The cell pellet was re-suspended in lysis buffer [20 mM Tris–HCl (pH 8.3), 50 mM NaOAc, 180 mM KCl, 10 mM β-mercaptoethanol, 20 mM imidazole and 10% glycerol], and lyzed by sonication using a Branson sonifier fitted with a microtip (10 times for 30 s, with 30 s breaks). The dialysis step was omitted. The pooled fractions were concentrated in an Amicon-stirred ultrafiltration cell, and then passed through a Pharmacia PD10 column to remove imidazole.
Electrophoretic Mobility Shift Assays (EMSA)
For assaying the formation of the DNA/TFB/TBP complex, 20 µl reactions containing binding buffer [60 mM KCl, 20 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 9 mM (NH4)2SO4, 0.05 mM EDTA, 0.5 mM DTT, 0.05 mM phenylmethylsulfonylfluoride (PMSF), 5% glycerol, 2.5% PEG8000], 1 µg poly(dI-dC), 200 ng TBP, 400 ng TFB and 1 ng labeled DNA were incubated at 65°C for 20 min. Binding assays for DNA/TFBc/TBP contained TFBc binding buffer [150 mM KCl, 20 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 0.05 mM EDTA, 0.5 mM DTT, 0.1 mM PMSF, 5% glycerol], 20 ng TFBc, 50 ng TBP, 1 µg poly(dI-dC) and 1 ng labeled DNA; incubation was at 75°C for 30 min. After incubation, reactions were loaded on a 15 cm long 5% polyacrylamide gel containing 0.5 × TBE (45 mM Tris–borate, 1 mM EDTA, pH 8.3), 1 mM MgCl2 and 1% glycerol. Gels were run in 0.5 × TBE at 200 V for 2–3 h, exposed to X-ray film, or visualized by phosphorimaging, and quantitated with Image Gauge software.
Regions of the gel above the free probe containing the fraction of bound DNA (as inferred from their migration relative to that of tRNAVal promoter and TBP/TFB) were excised and the DNA extracted. Gel slices were soaked overnight at 37°C in buffer [0.5 M NH4OAc, 10 mM Mg(OAc)2, 1 mM EDTA (pH 8.0) and 0.1% SDS], followed by phenol and chloroform extractions and recovered by ethanol precipitation in the presence of carrier yeast tRNA. This material was amplified using M13 universal primers. Fifty microlitre PCR reactions were assembled containing 1/10th of gel-recovered DNA, 20 mM Tris–HCl (pH 8.4), 50 mM KCl, 2 mM MgCl2, 50 pmol primers, 20 nmol dNTPs and 3 U Taq DNA polymerase. The program used was denaturation at 94°C for 2.5 min, 26 cycles (95°C for 30 s, 55°C for 30 s, 72°C for 30 s) and a final extension at 72°C for 10 min. The resulting products were digested with KpnI and SalI, and cloned into correspondingly cut pUC18 plasmid.
Competition assay of selected DNAs versus a known promoter
The ability of the recovered fragments to compete effectively with the labeled tRNAVal reference promoter was tested in binding reactions as previously described, except for the addition of 200-ng unlabeled recombinant plasmid DNA. When binding reactions included TFBc (not TFB), the amount of unlabeled recombinant plasmid was raised to 400 ng. Gels were run as described and visualized by phosphorimaging. Labeled bound and free DNA were measured using Image Gauge, and the ratio of fb/f,i = DNAbound,i/DNAfree,i was calculated. The fb/f was first normalized using the ratio from a reference binding reaction without competitor DNA 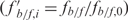 , and then referenced to the average
, and then referenced to the average 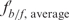 for the gel
for the gel 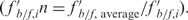 . The resulting
. The resulting 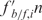 was used as a measure of the relative affinity to the transcription factors. The cut-off normalized score for promoters with ‘high affinity’ was set at 0.9.
was used as a measure of the relative affinity to the transcription factors. The cut-off normalized score for promoters with ‘high affinity’ was set at 0.9.
Hybridization screening of selected clones
For dot hybridizations, 30 ng plasmid DNA were denatured in 0.2 M NaOH at room temperature for 15 min, and transferred to Hybond-N+ nylon membranes. The membrane was rinsed briefly in 2× SSC (0.3 M NaCl and 30 mM sodium citrate), and the DNA fixed by vacuum drying at 80°C for 2 h. Pre-hybridization was at 68°C for 30 min in 10 ml buffer containing 0.25 M Na·PO4 (pH 7.2), 1.0 mM EDTA, 7% SDS and 1% BSA. Hybridizations were to a mixed probe-containing promoter DNA from 15 tRNA genes and one non-coding RNA gene that had been repeatedly selected in the initial screening. Promoter-containing DNA fragments were generated by PCR and cut with KpnI and BamHI; 50 ng of each were pooled. The fragment mix was labeled with [α-32P] dCTP in a fill-in reaction using Klenow fragment of E. coli DNA polymerase (Invitrogen), and purified with QIAquick Nucleotide removal kit (Qiagen). The probe was denatured at 95°C for 4 min before use. After overnight hybridization at 68°C in the same buffer, the membrane was washed with 2× SSC/0.1% SDS for 20 min, then 0.2× SSC/0.1% SDS for 20 min, followed by a final wash with 0.1× SSC/0.5%SDS for 20 min. Hybridization signal was measured by phosphorimaging, and a normalized signal intensity for each plasmid was calculated (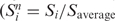 , where Saverage is the average signal strength from plasmids from the same preparation and the same membrane). The cut-off normalized score for ‘strong hybridization’ was set at 0.9.
, where Saverage is the average signal strength from plasmids from the same preparation and the same membrane). The cut-off normalized score for ‘strong hybridization’ was set at 0.9.
Alignment of promoter sequence and Logos
Collected sequences were inspected for the presence of conserved elements either manually or using MEME (43). Sequences were aligned by centering the conserved elements. Matrices of the base composition were derived at every position, and information content was calculated as:
where, I is information content of the position in bits, pi is random frequency of residue type i, ni is the number of instances of residue type i at the position and N is total number of sequences analyzed. This formula includes a pseudocount of pi for residue type i at each position, as small count correction. Sequence logos (44) were generated based on the local alignment. At each position, the height of the character for residue type i was determined by hi = I ni/N.
RESULTS
EMSA of a known promoter
It has been previously demonstrated that the binding of transcription initiation factors TBP and TFB to a DNA fragment containing an archaeal promoter can substantially reduce the mobility of the DNA on a gel (31,32). Typically, the mobility shift of a labeled DNA fragment is used as an assay for transcription factor binding. We reasoned that we could select promoter-containing DNA regions in the M. jannaschii genome by using EMSA to separate fragments of genomic DNA that bound transcription initiation factors TBP and TFB (and thus presumably contain a promoter) away from those that do not.
We first optimized conditions for transcription factor binding and gel shift assays using a 254-bp radiolabeled DNA fragment containing the tRNAVal gene promoter (PtRNAVal) from Methanococcus vannielii (28), a sequence that we had previously shown to be efficiently bound by the M. jannaschii transcription factors (32). Recombinant M. jannaschii TBP and TFB genes were expressed in E. coli, and the His6-tagged proteins purified. Efficient formation of the TBP/TFB/DNA complex was limited to the temperature range 55–80°C (data not shown). Only trace amounts of shifted DNA were detected following incubation at lower temperature, and no shifted band was observed following incubation at higher temperature. The low temperature limit agrees with the fact that M. jannaschii is a thermophile. The reason for the high temperature limit was not pursued (possibilities include protein instability, complex instability or denaturation of the high-A + T DNA fragment). Incubation pH has little effect on formation of the ternary complex in the range from 6.8 to 8.3 (data not shown). The condition most critical for efficient complex formation was the monovalent cation concentration; the optimal binding was found at 60–90 mM K+ (data not shown). Binding was specific, as demonstrated by the fact that it could be competed by addition of unlabeled cognate fragment, but not by addition of a DNA fragment of similar size containing the plasmid pUC18 polylinker region.
In the process of optimizing binding conditions, we noticed that upon incubation, purified TFB lost its binding ability precipitously. When the protein was pre-incubated in binding reaction conditions for 12 min at 60°C, only half of its binding ability was retained. When pre-incubation was carried out at 65°C for 15 min, the loss of binding activity was almost 80%. This loss of activity was observed over a wide range of pH values and ionic strengths. Instability of the M. jannaschii pre-initiation complex (consisting of TBP, TFB and RNA polymerase assembled on a suitable DNA substrate) has been reported (45), and our results suggest that it may be due to instability of TFB.
Archaeal TFB, like its eukaryal homologs, contains a C-terminal core domain responsible for interactions with TBP and DNA binding, and an N-terminal domain responsible for recruiting RNA polymerase. We evaluated the stability and binding ability of a truncated version of the protein lacking the N-terminal domain (referred to as TFBc). To construct TFBc, we aligned M. jannaschii TFB with homologs for which a crystal structure was available. The alignment revealed a conserved C-terminal domain of about 200 amino acids, corresponding to the core domain from Pyrococcus woesei (P. furiosus subsp. woesei) TFB used in crystallographic analyses (25). Based on these data, the first 137 amino acids of the protein were removed, and the C-terminal domain was expressed as a His6-tagged recombinant protein. TFBc formed the ternary DNA/TBP/TFBc complex with 5- to 10-fold greater efficiency than the full-length protein. Although a modest amount of non-specific binding was observed when reactions were carried out at 65°C, this was eliminated by raising the incubation temperature to 75°C and increasing the concentration of K+ to 150 mM. At least two factors are likely to contribute to the greater activity: TFBc is much more stable upon incubation than the full-length protein (only marginal loss was observed upon pre-incubation at 75°C for 30 min); and, it is possible that the N-terminal domain of the protein hinders the formation of the ternary complex, as has been observed in eukaryotes (46). Therefore, except for the first round of selection, all experiments below were conducted with TFBc, instead of the full-length protein. In contrast, TBP was stable after purification and did not lose activity measurably upon incubation in binding reaction conditions.
Selecting promoter-containing genomic DNAs by transcription initiation factor binding
We constructed a library of M. jannaschii DNA by ligating ∼250-bp random fragments into the SmaI restriction site of the plasmid pUC18. Vector sequences flanking the cloning site provided anchors for 25 cycles of PCR amplification of the DNA inserts to yield a population of ∼385-bp linear DNA molecules for the selection for TBP/TFB-binding sequences. To ensure a thorough sampling of genomic sequences in the selection, an aliquot of the primary library representing ∼30 genome equivalents was used as substrate for amplification. Because the inserts had been size-selected, they migrated as a well-defined band on an agarose gel (Figure 1A). For promoter selection, 12 ng (0.05 pmol) of labeled DNA fragments were incubated with 28 pmol each of TBP and TFB in a 60 µl reaction volume, and then resolved on a polyacrylamide gel (Figure 1B). No shifted band was directly observed, presumably because the amount of promoter-containing DNA constitutes only a small part of the entire population of fragments. Gel regions above the free DNA fragments were excised, the DNA extracted from them, and the resulting material subjected to 26 cycles of PCR amplification. Although the procedure was expected to recover the desired DNAs (those shifted to a lower mobility by the binding of transcription factors), it was also possible that the amplification products were due to a small amount of larger DNA contaminating the original library. Electrophoretic analysis of the amplified recovered DNAs verified that they were 350–400 bp, consistent with the original library (Figure 1C).
Figure 1.

Isolation of M. jannaschii genomic fragments with affinity to the transcription factors TBP and TFB. (A) PCR amplification of a small insert (200–300 bp) M. jannaschii genomic library. Left lane, 1-kb DNA ladder (Invitrogen); right lane, PCR product. (B) EMSA of TBP/TFB and labeled DNA. Fractions 1–4 contain transcription factor-bound DNA. (C) Agarose gel analysis of the re-amplification of transcription factor-bound DNA. Left lane, 1-kb DNA ladder; rightmost 4 lanes, product amplified from recovered DNA fractions 1 to 4 (Figure 1B). (D) Competition assay of recovered DNA fragments with the reference PtRNAVal promoter. Lane SP, specific competitor (plasmid containing the reference PtRNAVal promoter); lane NSP, non-specific promoter (empty vector); lanes C1 to C19, various recombinant plasmids harboring recovered genomic fragments.
Having demonstrated that the recovered DNA was of the desired size, we needed to verify the ability of individual DNA fragments to bind transcription factors. The PCR products were cut with the restriction enzymes KpnI and SalI and ligated into similarly cleaved pUC18 plasmid. Following transformation and growth, recombinant plasmids were tested for their ability to compete with a labeled fragment containing the tRNAVal promoter (PtRNAVal) for a limited supply of transcription factors (Figure 1D). Not only did this approach avoid the need to label the fragment from each clone, but it also provided a measure of each fragment's affinity for TBP and TFB relative to that of PtRNAVal.
Of 215 clone inserts tested in this way, 121 were judged to compete significantly with the PtRNAVal (Figure 2A). Of those judged to compete, 70 were sequenced and mapped onto the annotated M. jannaschii genome (30). Of these, 55 (almost 80%) included potential promoters upstream of tRNA-encoding genes (Figure 2A). These encompass 15 of the 23 tRNA genes that we predict to have their own promoter (E.L., unpublished data). Individual promoter regions were isolated 1–8 times (Tables S1 and S2). All sequenced fragments were different from one another, so multiple isolates of a given genomic region are independent events, not artifacts of duplicate clones. Five (7%) of the 70 sequences included potential promoters adjacent to G + C-rich islands with no known function or identified homologs in other organisms. We presume that these G + C-rich regions are small non-coding RNAs (ncRNAs), as other workers have suggested for similar regions (47,48). Three distinct putative ncRNAs are represented among the five clones. The presence of appropriately oriented transcription factor-binding sequences adjacent to these islands lends credence to their identification as transcribed genes (Table S1). Among the 70 clones, we uncovered only 3 (4%) putative promoters for protein-coding genes. The final eight fragments (11% of the 70) mapped to intragenic regions in the chromosome. Although it is tempting to regard these as false positives, this would be a potentially misleading designation in that they compete effectively with a tRNA gene promoter for transcription factors.
Figure 2.

Flow chart of screening strategies to characterize putative promoters isolated as transcription factor-bound genomic DNA fragments. (A) Initial selection without hybridization-based screening. (B) Selection with hybridization-based screening against most commonly isolated genome regions.
It appears that the competition screening introduced a heavy bias towards the isolation of strong promoters (and specifically tRNA promoters). For comparison, we performed additional competition assays against a protein-coding gene promoter. We used the promoter region of MJ1335, a gene annotated as encoding phosphoheptoisomerase. We had isolated this promoter region in the original selection and qualitatively characterized it as competing, but only weakly, with PtRNAVal. We tested 47 of the DNA fragments that did not compete effectively with PtRNAVal. Of these, 21 successfully competed with PMJ1335 and 18 of these were subsequently sequenced. Of these, five (28%) contained tRNA promoters, five (28%) contained presumptive ncRNA promoters and two (11%) contained a putative protein promoter. Finally, the number of fragments mapping to intragenic regions rose to 39%.
Our double competition criterion (competes with a protein gene promoter, but not a tRNA gene promoter) decreased substantially the number of tRNA promoters (from 80% to 28%). Unexpectedly, the recovery of promoters for putative ncRNAs increased dramatically.
To assess the bias introduced by our competition-based screens, we sequenced 24 random recovered DNA fragments that did not compete efficiently with PtRNAVal. Of these, 21% contained putative promoters (three for tRNA genes and two for protein-coding genes). One of the tRNA gene promoters found was not among those found previously, bringing the total to 16 of the expected 23 promoter regions. The remaining 79% mapped to intragenic regions. Because we have no confirmation of their binding of transcription initiation factors, some of them might be bona fide false positives from the selection.
Screening clones by hybridization
The recurring isolation of tRNA gene promoters led us to explore the use of a hybridization-based screening step against the tRNA promoter regions that we had already isolated. A preliminary screen using colony hybridization (49) was judged to be insufficiently reproducible for reliable results (data not shown). Clearer results were obtained when we used dot-blot hybridization to survey clones isolated by EMSA (Figure 2B). We analyzed 996 of these to determine how well they hybridized to a pool of already recovered and labeled tRNA promoters. For each clone, we also assessed the relative affinity for the transcription factors, as inferred from its ability to exclude the reference labeled tRNAVal promoter from the ternary complex in a competition assay (TFBc was used in these assays since it is more stable in solution than the full-length protein and yielded more reproducible data).
Therefore, each clone is identified by two coordinates—strength of hybridization to the pool of tRNA promoters recovered in the initial study, and relative affinity to the transcription factors (see Materials and Methods section for details). A double logarithmic scatter plot 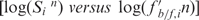 was generated (Figure 3). Clones were divided into four categories. Those that hybridized the mixed promoter probe and competed with PtRNAVal (34% of the total) were expected to be repeat isolations of tRNA promoters, and sequenced instances indicated this fraction was enriched with tRNA gene promoters (68% of total). Those that did not hybridize the mixed promoter probe and did not compete with PtRNAVal (25% of the total) were candidates for false positives (even though they were isolated by the gel shift). Eleven out of 12 sampled instances were found to be intergenic regions or be downstream of genes. The 297 clones that did not hybridize the mixed promoter, but did compete with PtRNAVal (30% of the total) were considered the best candidates for potentially unidentified promoters, and they were extensively sampled and sequenced. The composition of 167 sequenced clones is summarized in Figure 2B. Of particular note is the increase in the proportion of protein-coding gene promoters, showing that they are being gel shifted by the transcription factors, but much less efficiently than the tRNA and ncRNA promoters. Additional evidence that the screening helped to survey new promoters is that four additional tRNA genes were represented among the upstream regions of 37 tRNA genes, bringing up the total of recovered tRNA promoters to 20, out of 23 predicted.
was generated (Figure 3). Clones were divided into four categories. Those that hybridized the mixed promoter probe and competed with PtRNAVal (34% of the total) were expected to be repeat isolations of tRNA promoters, and sequenced instances indicated this fraction was enriched with tRNA gene promoters (68% of total). Those that did not hybridize the mixed promoter probe and did not compete with PtRNAVal (25% of the total) were candidates for false positives (even though they were isolated by the gel shift). Eleven out of 12 sampled instances were found to be intergenic regions or be downstream of genes. The 297 clones that did not hybridize the mixed promoter, but did compete with PtRNAVal (30% of the total) were considered the best candidates for potentially unidentified promoters, and they were extensively sampled and sequenced. The composition of 167 sequenced clones is summarized in Figure 2B. Of particular note is the increase in the proportion of protein-coding gene promoters, showing that they are being gel shifted by the transcription factors, but much less efficiently than the tRNA and ncRNA promoters. Additional evidence that the screening helped to survey new promoters is that four additional tRNA genes were represented among the upstream regions of 37 tRNA genes, bringing up the total of recovered tRNA promoters to 20, out of 23 predicted.
Figure 3.

Scatter plot of recombinant plasmids harboring transcription factor-binding genomic fragments. The axes represent relative affinity and hybridization strength. (I) High affinity and high hybridization; (II) Low affinity and high hybridization; (III) Low affinity and low hybridization; (IV) High affinity and low hybridization.
Although the introduction of screening schemes (both double competition assays, and prescreening of clones by hybridization) improved the recovery of novel putative promoter sequences, it is clear that the DNAs isolated in our selection are a non-random sample of the genome, being highly enriched in potential promoters for tRNAs and (probably) also for ncRNAs. These results suggest that—not surprisingly—promoters in M. jannaschii have different levels of affinity to the transcription factors TBP and TFB, and that the combination of the experimental conditions employed in the selection and subsequent screening are sensitive to these differences.
Core promoter elements in M. jannaschii
The sampling of promoters found is far from random, so it is premature to attempt an unbiased analysis of the M. jannaschii promoter sequences. However, analyses of the promoters from the 70 clones sequenced in the first round of selection and screening [representing 20 distinct intergenic regions (Table S1)] were interesting. BRE and TATA box sequences that could be recognized by eye or by computer-aided search with MEME (43) were aligned (Table S2). A sequence Logo (44) was generated in each of 2 ways. First, the individual sequences were weighted by the number of times they were isolated, giving emphasis to the strongest binding sequences (Figure 4B). Second, the consensus was calculated with each sequence counted equally (Figure 4A). The combined height of all letters stacked at a position is the information content at the position in bits (see Materials and Methods section). The relative heights of the letters stacked at a position reflect the relative frequency of the corresponding nucleotides. v
Figure 4.

Sequence Logo generated from aligned promoter sequences. (A) Logo of promoter sequences from the initial selection. (B) Same as A, but weighted for the frequency at which each sequence was selected. (C) Logo based on all alignable promoter sequences in this study.
The BRE and TATA box are obvious. The observed TATA box consensus of TWTATATA (W = A or T) differs little from the previously proposed TTTATATA consensus for methanogens (29). However, the Logos show conservation amongst these promoters that continues at least four, and perhaps seven or nine, residues beyond the 3' end of the canonical TATA box. This is most obvious in Figure 4B, but the heavy emphasis on the 15 tRNA promoter sequences makes interpretation beyond four positions questionable.
These promoters also display an unusually long BRE consensus, spanning 9–10 nucleotides (MRCCGAAAAG, where M = A or C, and R = A or G), rather than the more usual 6–7 (18,20, but compare 26). Because of the A + T-richness of the M. jannaschii genome, the random chance of a C or a G is lower than that of an A or a T, so a conserved C or G can be more informative. In Figure 4, this is reflected in the greater heights of some conserved C's and G's than of conserved A's and T's.
Another feature of some promoters in Archaea and Eucarya is a short initiator element (Inr), which includes the start site of transcription (26,27). Previous in vitro studies of the PtRNAVal showed that changes in this element, especially the start site residue, impaired promoter function (19). When aligned on the Inr (Table S3), our clones display a TGC consensus, with greatest conservation of the G, which is the start of the transcript (Figure S1). There are 19–24 bp of A + T-rich sequence between the canonical TATA box and the Inr (right side of Figure 4 and left side of Figure S1).
DISCUSSION
Genome-wide selection of archaeal promoters
We developed a genome-wide method to experimentally identify promoter sequences in M. jannaschii, based on the fact that archaeal promoter DNA is readily complexed with general transcription factors in vitro to form stable complexes that can be separated from unbound DNA by gel electrophoresis. The procedure was inspired by in vitro selection and evolution protocols (33–40), and it provided a simple and quick approach to isolate protein-bound DNA since no cross-linking or protein-specific antibody was required.
As in previous genomic SELEX work (35–40), we used a small-insert (200–300 bp) genomic DNA library; therefore, our pool of potential transcription factor-binding sequences was derived from an organism. This allowed the selection of only natural sequences, as opposed to ‘optimally interacting sequences’. The pitfalls inherent in searching for optimal binding sequences are apparent in previous studies. For example, ‘best’ –10 and –35 motifs for E. coli sigma factor σS were derived by SELEX using randomized DNA oligonucleotides; however, transcription from an artificial promoter containing them was weaker than from a promoter with the –35 consensus only (50). Similarly, in studies using SELEX to identify Lrp protein binding sites in M. jannaschii, the derived ‘optimal’ binding sequences performed poorly in identifying the natural binding sites for the proteins in the genome (51). Clearly, natural binding sites are not selected for maximal binding.
Some important modifications were adopted in our version of the affinity-based selection to improve efficiency. We used a simplified protocol to construct the genomic library (40). Genomic DNA fragments were size-fractioned and blunt-end ligated into the vector. The quality of the library was demonstrated by the independent isolation of multiple genomic DNA fragments covering the same region (Table S1). In addition, we performed only one round of binding, separation and recovery. Typically, in SELEX and genomic SELEX, multiple rounds are performed, resulting in the selection of sequences with progressively higher affinity, while lower-binding affinity sequences are effectively eliminated. In contrast, we reasoned that omitting repeated rounds of selection would allow us to recover a broader spectrum of promoter sequences, with varying affinities to the transcription factors; one potential problem associated with this strategy is that the background of (presumptive) false positives is increased. Subsequent screening was performed to isolate fragments with high affinity to the transcription factors, i.e. potential promoters from this archaeon. Criteria for the identification of putative promoters were based on genome context, as well as the frequency of recovered genomic DNA fragments (Figure S2).
Conserved motifs from the natural promoters were readily identified when recovered DNA sequences were aligned (Figure 4). In M. jannaschii, the TATA box (the binding site for TBP) is highly conserved, and nearly symmetrical as well, in agreement with the fact that the DNA-binding domain of archaeal TBP contains more nearly perfect repeats than does eukaryal TBP (29,52). This symmetry renders the TATA box alone incapable of determining the orientation of the transcription complex. Essential information regarding orientation is provided by the BRE (25), which also shows an extended region of conservation vis-a-vis its eukaryal element. The extended BRE indicates that the core promoter is more complex than expected. Base-specific cross-linking in the pre-initiation complex revealed close contact between this region and the C-terminal region of TFB (23). Cytosine at –7 (relative to TATA box), the most conserved position in the BRE extension, was identified as a preferred base in the selection of randomized DNA oligonucleotides using archaeal TBP and TFB (20). Interestingly, similar results (though with a conserved G instead of C at –7) were obtained when human TBP and TFIIB were used, indicating that this base may contribute to the interaction with TFIIB in eukaryal promoters as well (53). Moreover, a 3D structure of the human ternary TBP/TFB/DNA complex confirmed that G at position –7 makes water-mediated contacts with TFIIB (54). The conservation of position –7 across the archaeal–eukaryal lineage strongly suggests the relevance of this position, likely in mediating contact with TFB/TFIIB in both domains. Although we cannot assign a functional role to the other three bases in the extended BRE, their conservation suggests the extended BRE is an integral part of archaeal promoters. Interestingly, the extended BRE is most conserved in promoters with particularly high affinity for transcription factors; this may be relevant in recognizing functional distinctions among promoters. The extended consensus does not necessarily mean an extended binding site for these two factors; other molecules could bind the region.
Given the compact nature of the M. jannaschii genome it was not surprising that, in most cases, promoters were found close to the start site of a downstream ORF or RNA gene. We used this feature to define putative small ncRNAs. If the gap between a promoter and the downstream gene was large, and the G + C content in the region was higher than the average for the genome, these promoters were tentatively assigned to previously unidentified ncRNA genes. In thermophiles, high G + C content is a significant feature of structured RNAs, such as tRNAs and rRNAs. In silico studies have utilized the presence of high G + C islands to identify ncRNA genes in organisms with overall low genomic G + C content (47,48). We identified 10 presumptive ncRNA genes and their associated promoters from 27 gel shifted genomic DNA fragments. Four of these RNAs had previously been identified experimentally (47). Our results suggest that a systematic search for promoters is an effective strategy to identify novel transcripts. Further characterization of these ncRNA genes and their promoters is in progress.
Experimental concerns of in vitro selection of natural promoters
The dramatic difference between the isolation frequency of tRNA gene promoters and protein gene promoters makes it important to consider the sources of bias in the procedures employed, and how they might be circumvented. Foremost among our concerns is the effective concentration of the transcription initiation factors. The DNA-binding reactions prior to electrophoretic separation were ∼0.5 µM in each of the proteins. With the relative instability of TFB, we estimate that its effective concentration by the end of the incubation is <0.1 µM, about 20-fold lower than estimates of its in vivo concentration (Ying Jiang, C.I.R. and G.J.O., unpublished data). Given the demonstration that different promoters have different intrinsic affinities for the transcription factors (based on the competition assays), we presume that we are far below saturation binding of transcription factors to DNA. Although other factors could influence the distribution of promoters recovered, this effect is consistent with all of our observations. The most obvious solutions are to increase the stability of TFB by use of the TFBc fragment (as was done in later selections, although this could potentially introduce other biases), and to use higher concentrations of TFB (increasing solubility and other logistical problems).
A related issue is the possibility that transcription factor–DNA complexes formed, but that they dissociated during the electrophoretic separation. In particular, this could be aggravated by the use of a gel with (nominally) equal concentrations of Mg2+ and EDTA, in contrast to the binding reaction conditions in which Mg2+ is in excess. Although we have not observed decreases in shifted DNA with changes in gel conditions, nearly all of these observations are based upon the shifting of the strongest-binding promoters. Thus, it is possible that some classes of promoters (presumably those with lower affinity of the transcription factors) dissociate in the gel, and hence are not shifted. We have no direct measure of this, but we do observe a strong correlation between the efficiency with which weakly binding DNA fragments are mobility-shifted (as a function of transcription factor concentration) and the efficiency of the same fragment in competing for transcription factors in the binding reaction (measured by the gel shifting of a strongly-binding promoter).
Several of the isolated DNA regions were intragenic. We have already cautioned against classifying them as false positives. In organisms with compact genomes it is not unusual to find promoters inside genes. A comprehensive survey of 791 E. coli mRNA promoters indicated that 18% of them were positioned inside the preceding genes (55). Protein ORFs can also host, or overlap with, independently transcribed genes (particularly ncRNAs) as well. A variety of small non-messenger RNAs have been identified in a specialized cDNA library of the archaeon Archaeoglobus fulgidus (56); some of these RNAs are complementary to an ORF, some overlap an ORF and some are even internal to an ORF. In vitro, most of our ‘false positives’ were demonstrated to compete with bona fide promoters for transcription factors. While suggestive of in vivo binding and potential promoter activity, other explanations are possible, and our data are unable to distinguish among them.
Our protocol involves two PCR amplification steps, one before the promoter selection step and one after. At the end of the procedure, the combined errors of the two amplifications and the single-strand sequencing of recovered clones was ∼2.5 errors per 1000 bp DNA relative to the published genome sequence. Of these steps, only PCR errors introduced before the gel selection (at most 1.2 per 1000 bp, or about 1 per 4 genomic DNA fragments) could bias the selection. Only three selected clones had a sequence error in the presumed TATA or BRE, and these were excluded from the analyses reported above. Overall, there is no evidence that the PCR errors were sufficiently numerous to corrupt authentic promoters (false negatives) or create spurious promoter-like sequences (false positives).
A more problematic artifact was the creation of chimeric sequences. Overall, 12 (3.7%) of the sequenced clones included DNA from non-adjacent portions of the genome (again, they were excluded from the results reported). This artifact can be introduced either during the ligation step of library construction, or during the PCR amplification steps. Kanagawa (57) proposed two modes of chimera formation during multi-template PCR, either by template switching or premature termination of extension. Consistent with premature termination of extension, examination revealed homologous overlapping sequences at the junction point. Although this artifact was annoying, we cannot see any mechanism by which it could have altered our major observations. This said, PCR sequence errors could be reduced by decreasing the PCR cycles and using a proofreading polymerase; chimeras could be reduced by decreasing the PCR cycles and optimizing efficiency to minimize premature termination.
SUMMARY AND IMPLICATIONS
In conclusion, we report an experimental genome-wide approach to identify promoters; the procedure is based on the fact that promoter sequences are substrates for transcription factor binding, and thus could be selected in a manner reminiscent of SELEX. Our efforts resulted in the selection of a subset of promoter regions in the genome, most of them for genes that are highly expressed. Information extracted from these sequences allowed us to refine our knowledge of the composition and architecture of promoters in this organism, and to infer that strict adherence to canonical promoter elements (and their extensions) is a key feature of highly transcribed genes. We view this as a test case for the application of EMSA to a whole-genome screen of binding sites for a variety of proteins. The approach can be applied to any protein–DNA complex that is sufficiently stable to gel-shift the binding-site-containing DNA fragment.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank Ying Jiang for sharing her estimates of in vivo transcription factor concentrations. We thank Jian Zhang, Ying Jiang and other members of the laboratory for helpful discussions. We thank two anonymous referees for their insightful and helpful critiques. This work was supported by grants from the US Department of Energy (DE-FG02-01ER63201) and NASA (NAG 5-12334) to G.J.O. We thank Dr. Carl R. Woese for his help in supporting E.L. by sharing funds from the Stanley O. Ikenberry Chair, a position that he holds at the University of Illinois. Funding to pay the Open Access publication charges for this article has been waived by Oxford University Press.
Conflict of interest statement. None declared.
REFERENCES
- 1.Tiwari S, Ramachandran S, Bhattacharya A, Bhattacharya S, Ramaswamy R. Prediction of probable genes by Fourier analysis of genomic sequences. Comput. Appl. Biosci. 1997;13:263–270. doi: 10.1093/bioinformatics/13.3.263. [DOI] [PubMed] [Google Scholar]
- 2.Salzberg SL, Delcher AL, Kasif S, White O. Microbial gene identification using interpolated Markov models. Nucleic Acids Res. 1998;26:544–548. doi: 10.1093/nar/26.2.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Badger JH, Olsen GJ. CRITICA: coding region identification tool invoking comparative analysis. Mol. Biol. Evol. 1999;16:512–524. doi: 10.1093/oxfordjournals.molbev.a026133. [DOI] [PubMed] [Google Scholar]
- 4.Besemer J, Lomsadze A, Borodovsky M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001;29:2607–2618. doi: 10.1093/nar/29.12.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berk AJ, Sharp PA. Sizing and mapping of early adenovirus mRNAs by gel electrophoresis of S1 endonuclease-digested hybrids. Cell. 1977;12:721–732. doi: 10.1016/0092-8674(77)90272-0. [DOI] [PubMed] [Google Scholar]
- 6.Boorstein WR, Craig EA. Primer extension analysis of RNA. Methods Enzymol. 1989;180:347–369. doi: 10.1016/0076-6879(89)80111-9. [DOI] [PubMed] [Google Scholar]
- 7.Galas DJ, Schmitz A. DNase footprinting: a simple method for the detection of protein-DNA binding specificity. Nucleic Acids Res. 1978;5:3157–3170. doi: 10.1093/nar/5.9.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mackie GA, Parsons GD. Tandem promoters in the gene for ribosomal protein S20. J. Biol. Chem. 1983;258:7840–7846. [PubMed] [Google Scholar]
- 9.Qiu P. Recent advances in computational promoter analysis in understanding the transcriptional regulatory network. Biochem. Biophys. Res. Commun. 2003;309:495–501. doi: 10.1016/j.bbrc.2003.08.052. [DOI] [PubMed] [Google Scholar]
- 10.Gelfand MS, Koonin EV, Mironov AA. Prediction of transcription regulatory sites in Archaea by a comparative genomic approach. Nucleic Acids Res. 2000;28:695–705. doi: 10.1093/nar/28.3.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cliften P, Sudarsanam P, Desikan A, Fulton L, Fulton B, Majors J, Waterston R, Cohen BA, Johnston M. Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science. 2003;301:71–76. doi: 10.1126/science.1084337. [DOI] [PubMed] [Google Scholar]
- 12.Olsen GJ, Woese CR. Archaeal genomics: an overview. Cell. 1997;89:991–994. doi: 10.1016/s0092-8674(00)80284-6. [DOI] [PubMed] [Google Scholar]
- 13.Bell SD, Jackson SP. Transcription and translation in Archaea: a mosaic of eukaryal and bacterial features. Trends Microbiol. 1998;6:222–228. doi: 10.1016/s0966-842x(98)01281-5. [DOI] [PubMed] [Google Scholar]
- 14.Bell SD, Magill CP, Jackson SP. Basal and regulated transcription in Archaea. Biochem. Soc. Trans. 2001;29:392–395. doi: 10.1042/bst0290392. [DOI] [PubMed] [Google Scholar]
- 15.Ouhammouch M. Transcriptional regulation in Archaea. Curr. Opin. Genet. Dev. 2004;14:133–138. doi: 10.1016/j.gde.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 16.Langer D, Hain J, Thuriaux P, Zillig W. Transcription in Archaea: similarity to that in Eucarya. Proc. Natl Acad. Sci. USA. 1995;92:5768–5772. doi: 10.1073/pnas.92.13.5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Best AA, Olsen GJ. Evidence for similar subunit architectures between archaeal and eukaryal RNA polymerases. FEMS Microbiol. Lett. 2001;195:85–90. doi: 10.1111/j.1574-6968.2001.tb10502.x. [DOI] [PubMed] [Google Scholar]
- 18.Bell SD, Kosa PL, Sigler PB, Jackson SP. Orientation of the transcription preinitiation complex in Archaea. Proc. Natl Acad. Sci. USA. 1999;96:13662–13667. doi: 10.1073/pnas.96.24.13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hausner W, Wettach J, Hethke C, Thomm M. Two transcription factors related with the eukaryal transcription factors TATA-binding protein and transcription factor IIB direct promoter recognition by an archaeal RNA polymerase. J. Biol. Chem. 1996;271:30144–30148. doi: 10.1074/jbc.271.47.30144. [DOI] [PubMed] [Google Scholar]
- 20.Qureshi SA, Jackson SP. Sequence-specific DNA binding by the S. shibatae TFIIB homolog, TFB, and its effect on promoter strength. Mol. Cell. 1998;1:389–400. doi: 10.1016/s1097-2765(00)80039-8. [DOI] [PubMed] [Google Scholar]
- 21.Bell SD, Jackson SP. The role of transcription factor B in transcription initiation and promoter clearance in the archaeon Sulfolobus acidocaldarius. J. Biol. Chem. 2000;275:12934–12940. doi: 10.1074/jbc.275.17.12934. [DOI] [PubMed] [Google Scholar]
- 22.Bartlett MS, Thomm M, Geiduschek EP. Topography of the euryarchaeal transcription initiation complex. J. Biol. Chem. 2004;279:5894–5903. doi: 10.1074/jbc.M311429200. [DOI] [PubMed] [Google Scholar]
- 23.Renfrow MB, Naryshkin N, Lewis LM, Chen HT, Ebright RH, Scott RA. Transcription factor B contacts promoter DNA near the transcription start site of the archaeal transcription initiation complex. J. Biol. Chem. 2004;279:2825–2831. doi: 10.1074/jbc.M311433200. [DOI] [PubMed] [Google Scholar]
- 24.Kosa PF, Ghosh G, DeDecker BS, Sigler PB. The 2.1-Å crystal structure of an archaeal preinitiation complex: TATA-box-binding protein/transcription factor (II)B core/TATA-box. Proc. Natl Acad. Sci. USA. 1997;94:6042–6047. doi: 10.1073/pnas.94.12.6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Littlefield O, Korkhin Y, Sigler PB. The structural basis for the oriented assembly of a TBP/TFB/promoter complex. Proc. Natl Acad. Sci. USA. 1999;96:13668–13673. doi: 10.1073/pnas.96.24.13668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wich G, Hummel H, Jarsch M, Bar U, Böck A. Transcription signals for stable RNA genes in Methanococcus. Nucleic Acids Res. 1986;14:2459–2479. doi: 10.1093/nar/14.6.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomm M, Wich G. An archaebacterial promoter element for stable RNA genes with homology to the TATA box of higher eukaryotes. Nucleic Acids Res. 1988;16:151–163. doi: 10.1093/nar/16.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hausner W, Frey G, Thomm M. Control regions of an archaeal gene. A TATA box and an initiator element promote cell-free transcription of the tRNAVal gene of Methanococcus vannielii. J. Mol. Biol. 1991;222:495–508. doi: 10.1016/0022-2836(91)90492-o. [DOI] [PubMed] [Google Scholar]
- 29.Soppa J. Basal and regulated transcription in Archaea. Adv. Appl. Microbiol. 2001;50:171–217. doi: 10.1016/s0065-2164(01)50006-4. [DOI] [PubMed] [Google Scholar]
- 30.Bult CJ, White O, Olsen GJ, Zhou L, Fleischmann RD, Sutton GG, Blake JA, FitzGerald LM, Clayton RA, et al. Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii. Science. 1996;273:1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- 31.Qureshi SA, Khoo B, Baumann P, Jackson SP. Molecular cloning of the transcription factor TFIIB homolog from Sulfolobus shibatae. Proc. Natl Acad. Sci. USA. 1995;92:6077–6081. doi: 10.1073/pnas.92.13.6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Colón Gonzalez GM. Ph.D. Dissertation. University of Illinois; 2001. Archaeal basal transcription factor: DNA protein pnteractions. [Google Scholar]
- 33.Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 34.Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 35.Kinzler KW, Vogelstein B. Whole genome PCR: application to the identification of sequences bound by gene regulatory proteins. Nucleic Acids Res. 1989;17:3645–3653. doi: 10.1093/nar/17.10.3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singer BS, Shtatland T, Brown D, Gold L. Libraries for genomic SELEX. Nucleic Acids Res. 1997;25:781–786. doi: 10.1093/nar/25.4.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shtatland T, Gill SC, Javornik BE, Johansson HE, Singer BS, Uhlenbeck OC, Zichi DA, Gold L. Interactions of Escherichia coli RNA with bacteriophage MS2 coat protein: genomic SELEX. Nucleic Acids Res. 2000;28:E93. doi: 10.1093/nar/28.21.e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim S, Shi H, Lee DK, Lis JT. Specific SR protein-dependent splicing substrates identified through genomic SELEX. Nucleic Acids Res. 2003;31:1955–1961. doi: 10.1093/nar/gkg286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimada T, Fujita N, Maeda M, Ishihama A. Systematic search for the Cra-binding promoters using genomic SELEX system. Genes Cells. 2005;10:907–918. doi: 10.1111/j.1365-2443.2005.00888.x. [DOI] [PubMed] [Google Scholar]
- 40.Salehi-Ashtiani K, Luptak A, Litovchick A, Szostak JW. A genomewide search for ribozymes reveals an HDV-like sequence in the human CPEB3 gene. Science. 2006;313:1788–1792. doi: 10.1126/science.1129308. [DOI] [PubMed] [Google Scholar]
- 41.Giometti CS, Reich CI, Tollaksen S, Babnigg G, Lim H, Yates J.R., III, Olsen GJ. Structural modifications of Methanococcus jannaschii flagellin proteins revealed by proteome analysis. Eur. J. Mass Spectrom. 2001;7:195–205. doi: 10.1002/1615-9861(200108)1:8<1033::AID-PROT1033>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 42.Jarrell KF, Faguy D, Hebert AM, Kalmokoff ML. A general method of isolating high molecular weight DNA from methanogenic Archaea (archaebacteria) Can. J. Microbiol. 1992;38:65–68. doi: 10.1139/m92-010. [DOI] [PubMed] [Google Scholar]
- 43.Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- 44.Schneider TD, Stephens RM. Sequence Logos: A new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ouhammouch M, Werner F, Weinzierl RO, Geiduschek EP. A fully recombinant system for activator-dependent archaeal transcription. J. Biol. Chem. 2004;279:51719–51721. doi: 10.1074/jbc.C400446200. [DOI] [PubMed] [Google Scholar]
- 46.Roberts SG, Green MR. Activator-induced conformational change in general transcription factor TFIIB. Nature. 1994;371:717–720. doi: 10.1038/371717a0. [DOI] [PubMed] [Google Scholar]
- 47.Klein RJ, Misulovin Z, Eddy SR. Noncoding RNA genes identified in AT-rich hyperthermophiles. Proc. Natl Acad. Sci. USA. 2002;99:7542–7547. doi: 10.1073/pnas.112063799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schattner P. Searching for RNA genes using base-composition statistics. Nucleic Acids Res. 2002;30:2076–2082. doi: 10.1093/nar/30.9.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grunstein M, Hogness DS. Colony hybridization: a method for the isolation of cloned DNAs that contain a specific gene. Proc. Natl Acad. Sci. USA. 1975;72:3961–3965. doi: 10.1073/pnas.72.10.3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gaal T, Ross W, Estrem ST, Nguyen LH, Burgess RR, Gourse RL. Promoter recognition and discrimination by EσS RNA polymerase. Mol. Microbiol. 2001;42:939–954. doi: 10.1046/j.1365-2958.2001.02703.x. [DOI] [PubMed] [Google Scholar]
- 51.Ouhammouch M, Geiduschek EP. A thermostable platform for transcriptional regulation: the DNA-binding properties of two Lrp homologs from the hyperthermophilic archaeon Methanococcus jannaschii. EMBO J. 2001;20:146–156. doi: 10.1093/emboj/20.1.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marsh TL, Reich CI, Whitelock RB, Olsen GJ. Transcription factor IID in the Archaea: sequences in the Thermococcus celer genome would encode a product closely related to the TATA-binding protein of eukaryotes. Proc. Natl Acad. Sci. USA. 1994;81:4180–4184. doi: 10.1073/pnas.91.10.4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lagrange T, Kapanidis AN, Tang H, Reinberg D, Ebright RH. New core promoter element in RNA polymerase II-dependent transcription: sequence-specific DNA binding by transcription factor IIB. Genes Dev. 1998;12:34–44. doi: 10.1101/gad.12.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsai FT, Sigler PB. Structural basis of preinitiation complex assembly on human pol II promoters. EMBO J. 2000;19:25–36. doi: 10.1093/emboj/19.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huerta AM, Collado-Vides J. Sigma70 promoters in Escherichia coli: specific transcription in dense regions of overlapping promoter-like signals. J. Mol. Biol. 2003;333:261–278. doi: 10.1016/j.jmb.2003.07.017. [DOI] [PubMed] [Google Scholar]
- 56.Tang T-H, Bachellerie J-P, Rozhdestvensky T, Bortolin M.-L, Huber H, Drungowski M, Elge T, Brosius J, Hüttenhofer A. Identification of 86 candidates for small non-messenger RNAs from the archaeon Archaeoglobus fulgidus. Proc. Natl Acad. Sci. USA. 2002;99:7536–7541. doi: 10.1073/pnas.112047299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kanagawa T. Bias and artifacts in multitemplate polymerase chain reactions (PCR) J. Biosci. Bioeng. 2003;96:317–323. doi: 10.1016/S1389-1723(03)90130-7. [DOI] [PubMed] [Google Scholar]