

Abstract
Pneumocystis colonization has been associated with severity of chronic obstructive pulmonary disease (COPD). The relationship of Pneumocystis antibody status to COPD severity has not been investigated, but antibody levels might relate to both colonization susceptibility and COPD progression. We investigated anti-Pneumocystis antibody titers and airway obstruction in a cohort of patients with COPD. Undetectable anti-Pneumocystis antibody titer was an independent predictor of more-severe airway obstruction, although use of inhaled corticosteroids is a possible confounder of this effect.
Smoking is the primary risk factor for chronic obstructive pulmonary disease (COPD), but factors that determine which smokers will develop significant disease are largely unknown. Infectious agents might play a role in accelerating progression of airway obstruction or in perpetuating its progression after discontinuation of tobacco exposure. Pneumocystis jirovecii is a fungal pathogen that causes pneumonia in immunocompromised individuals. The presence of Pneumocystis in the lungs, even at low levels, produces inflammatory changes similar to those seen in COPD [1, 2]. Colonization is highly prevalent in patients with COPD and correlates with disease severity [3–5].
Host defense against Pneumocystis is complex and involves both the humoral and cellular immune responses [6]. CD4+ T cells have historically been implicated in susceptibility to colonization with Pneumocystis, but an antibody-mediated response is also likely to be important. Antibodies to the Pneumocystis endoprotease kexin (KEX1) may be particularly important, because immune responses to Pneumocystis kexin have been associated with control of Pneumocystis infection in animal models [7, 8].
The serum KEX1 antibody response in patients with COPD has not been investigated and might be important for further clarifying the role of Pneumocystis in COPD by indicating a mechanism by which patients with COPD become colonized and by serving as a noninvasive marker of susceptibility to Pneumocystis colonization. We performed a cross-sectional pilot study to determine the relationship of Pneumocystis KEX1 antibodies to severity of airway obstruction in a cohort of former and current smokers.
Patients, materials, and methods
Persons who were former or current smokers with a history of smoking at least 10 packs per year were randomly selected from individuals enrolled in the Emphysema/COPD Research Center at the University of Pittsburgh (Pittsburgh, PA). Participants were recruited for this registry from various areas of Pittsburgh and its suburbs. Exclusion criteria included current exacerbation, completely reversible airflow obstruction, a significant allergy history, or a history of clinical asthma. The University of Pittsburgh Institutional Review Board approved the study, and all participants provided informed consent.
Spirometry and measurement of single breath carbon monoxide diffusing capacity (DLCO) were performed at entry into the Emphysema/COPD Research Center, according to American Thoracic Society criteria [9]. The percentage of forced predicted expiratory volume in 1 s (FEV1), forced vital capacity (FVC), and DLCO were calculated with use of standard reference equations [10, 11]. Plasma samples were obtained from patients at enrollment in the Emphysema/COPD Research Center registry and were stored at −80°C.
A partial fragment of the macaque-derived Pneumocystis kexin gene in the pBAD expression vector (gift from C. G. Haidaris, University of Rochester) was used to produce recombinant KEX1. Escherichia coli Top10 (Invitrogen), containing the pBAD-KEX1 plasmid, was grown overnight at 37°C in Luria-Bertani broth, supplemented with 100 μg/mL of carbenicillin, diluted 1:20 in fresh Luria-Bertani broth with 100 μg/mL of carbenicillin, and grown at 37°C to log phase (optical density of liquid medium at 600 nm, 0.7–0.8). KEX1 expression was induced by the addition of L-arabinose (0.01% final concentration) and continued culture for 4.5 h at 37°C. Cells were centrifuged for 10 min at 4000 g, and cell pellets were frozen at −80°C until use. Cells were lysed by thawing in extraction buffer (6 mol/L guanidine-hydrogen chloride, 50 mmol/L disodium hydrogen orthophosphate, and 300 mmol/L sodium chloride; pH, 7.0) at room temperature for 20 min. After centrifugation (20 min at 7240 g), the supernatant fluid was applied to Talon metal affinity resin (Clontech Laboratories), and KEX1 was eluted with 150 mmol/L imidazole. Purified protein was subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to nitrocellulose, blocked in 5% nonfat milk with 1% bovine serum albumin and 0.05% Tween 20 in PBS, and incubated with a plasma sample obtained from a macaque with Pneumocystis pneumonia. Microtiter plates (Immunolon 4HBX; Thermo Fisher Scientific) were coated with 5 μg/mL of purified KEX1 in sodium bicarbonate (pH, 9.5). Heat-inactivated plasma was diluted 1:100 in blocking buffer (PBS with 5% nonfat milk). Fifty microliters of plasma were plated into KEX1-coated wells, and serial dilutions up to 1: 12,800 were made to determine end point titers. Goat antihuman immunoglobulin-conjugated horseradish peroxidase (1: 10,000 for IgG; Sigma-Aldrich) was used for detection, and plates were developed by standard methods. Normal human plasma samples (Pneumocystis negative by antibody titer assay) were used as negative controls. The reciprocal end point titer was calculated as the highest dilution at which the optical density was the same or less than that of the control.
To determine whether patients with low KEX1 levels had a generalized defect in humoral immunity, plasma samples were also tested for antibodies to influenza with use of the hemagglutinin inhibition assay, adapted from the Centers for Disease Control and Prevention laboratory-based influenza surveillance manual [12]. Antibody titers against A/Fujian/411/2002 (H3N2) and A/Wisconsin/65/2005 (H3N2) were determined.
Stata, version 8 (Stata), was used for analysis, and statistical significance was determined at P<05. Variables were analyzed with use of either Student’s t test and the Wilcoxon rank-sum test or the χ2 test and Fisher’s exact test. Demographic variables and antibody levels were determined for the entire cohort. Because we hypothesized that COPD severity would be associated with decreased antibody titers, we then examined the relationship of antibody levels as a continuous variable to pulmonary function parameters. Univariate analyses were also performed to determine clinical variables related to an undetectable KEX1 antibody titer (defined as a KEX1 titer <1:100). Multivariate linear regression was performed to determine independent predictors of FEV1%, FEV1-to-FVC ratio, and DLCO by including variables hypothesized to be causally related to the outcomes or that were statistically significant at P=.1 in univariate analyses. Models were run with log-transformed absolute anti-Pneumocystis reciprocal end point titers and with antibody status as a dichotomous variable (detectable vs. undetectable antibody titer). We explored interactions between model variables, and normality of model residuals was assessed. Similar models were performed for influenza antibody titers.
Results
One hundred fifty-three patients were included in the analysis (table 1). The median anti-KEX1 antibody titer was 375 (range, 1–12,800), and 96 patients (62.7%) had detectable titers. Patients with undetectable antibody titers were similar to those with detectable titers with regard to age, sex, and number of patients who were currently smoking (table 1). Patients with undetectable titers tended to have a lower pack-year smoking history (defined as the average number of packs smoked per day multiplied by the number of years that the person smoked), compared with patients with detectable titers (50.2 pack-years vs. 58.6 pack-years; P=.06). Despite a somewhat lower pack-year smoking history, the patients with undetectable titers had a significantly lower FEV1-to-FVC ratio (0.46 vs. 0.51; P=.04) and tended to have a lower FEV1% predicted, compared with patients with detectable titers (49.9% vs. 57.8%; P=.08). The DLCO% predicted did not differ between groups. Compared with patients with detectable antibody titers, patients with undetectable titers were more symptomatic, according to the modified Medical Research Council dyspnea index scale (2.0 vs. 1.5; P=.04), and were more likely to be using inhaled corticosteroids (52.6% vs. 32.3%; P=.01). Patients with undetectable titers also had a lower mean (±SD) body mass index (defined as weight in kilograms divided by the square of the height in meters) than did patients who had a detectable antibody response (25.9 ±4.2 vs. 28.4 ±5.8); however, most patients had a normal body mass index. Only 5 patients had an abnormally low body mass index (<19), and there was no relationship between a low body mass index and Pneumocystis antibody status. Data on injection or inhaled illicit drug use was not available; however, the population consisted of older, HIV-uninfected patients, and thus, the prevalence of drug use was likely to have been low. There were no differences in the percentages of patients who had been hospitalized or had experienced exacerbation in the previous year according to antibody status.
Table 1.
Characteristics of patients with chronic obstructive pulmonary disease (COPD), by anti-Pneumocystis antibody status.
| Patients
|
|||||
|---|---|---|---|---|---|
| Characteristic | All (n= 153) | With undetectable antibody (n = 57) | With detectable antibody (n = 96) | OR (95% CI) | P |
| Age, mean years ± SD | 63.3 ± 8.0 | 63.6 ± 8.4 | 63.1 ± 7.8 | … | |
| Sex | |||||
| Male | 86 (56.2) | 30 (52.3) | 56 (58.3) | … | |
| Female | 67 (43.8) | 27 (47.7) | 30 (41.7) | … | |
| Current smoker | 42 (27.5) | 16 (28.1) | 26 (27.1) | … | |
| Smoking history, median pack-years (range)a | 55.4 (26.8) | 50.2 (18.3) | 58.6 (30.5) | 1.01 (1.00–1.03) | .06 |
| Mean FEV1% predicted ±SD | 54.9 ± 26.4 | 49.9 ± 25.6 | 57.8 ± 26.6 | 3.20 (0.88–11.5) | .08 |
| Mean FEV1:FVC ± SD | 0.49 ± 0.17 | 0.46 ± 0.17 | 0.51 ± 0.17 | 8.23 (1.07–63.2) | .04 |
| Mean DLCO% predicted ± SD | 45.8 ± 19.0 | 45.2 ± 17.4 | 44.6 ± 20.2 | … | |
| Median MMRC (range) | 2 (0–4) | 2 (0–4) | 1.5 (0–4) | 0.72 (0.53–0.97) | .04 |
| Used inhaled corticosteroids | 61 (39.9) | 30 (52.6) | 31 (32.3) | 0.43 (0.22–0.84) | .01 |
| Used oral corticosteroids | 9 (5.9) | 4 (7.0) | 5 (5.2) | … | |
| Mean BMI ± SD | 27.5 ± 5.4 | 25.9 ± 4.2 | 28.4 ± 5.8 | … | .009 |
| Exacerbation within the previous year | 37 (24.2) | 13 (22.8) | 24 (25.) | … | |
| Hospitalization within the previous year | 29 (19.0) | 14 (24.6) | 15 (15.7) | … | |
NOTE. Data are no. (%) of patients, unless otherwise indicated. BMI, body mass index (calculated as weight in kilograms divided by the square of the height in meters); DLCO, diffusing capacity for carbon monoxide; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; GOLD, Global Initiative on Obstructive Lung Diseases; MMRC, modified Medical Research Council dyspnea index.
Pack-years is defined as the average number of packs smoked per day multiplied by the number of years that the person smoked.
Multivariate analyses revealed that a low antibody titer, when analyzed either as an undetectable level or as a continuous value, was an independent predictor of a low FEV1% predicted (adjusted for age, current smoking status, and pack-year smoking history; P=.03) (figure 1A). Similarly, both an undetectable antibody titer and a low antibody titer were independently associated with a low FEV1-to-FVC ratio (adjusted for age, sex, current smoking status, and pack-year smoking history; P=.01). There was no relationship between DLCO and antibody titers. Similar analyses for influenza antibody titers demonstrated no statistically significant relationship to pulmonary function outcomes, suggesting that a generalized defect in the humoral response did not explain the relationship of KEX1 to airway obstruction (figure 1B).
Figure 1.
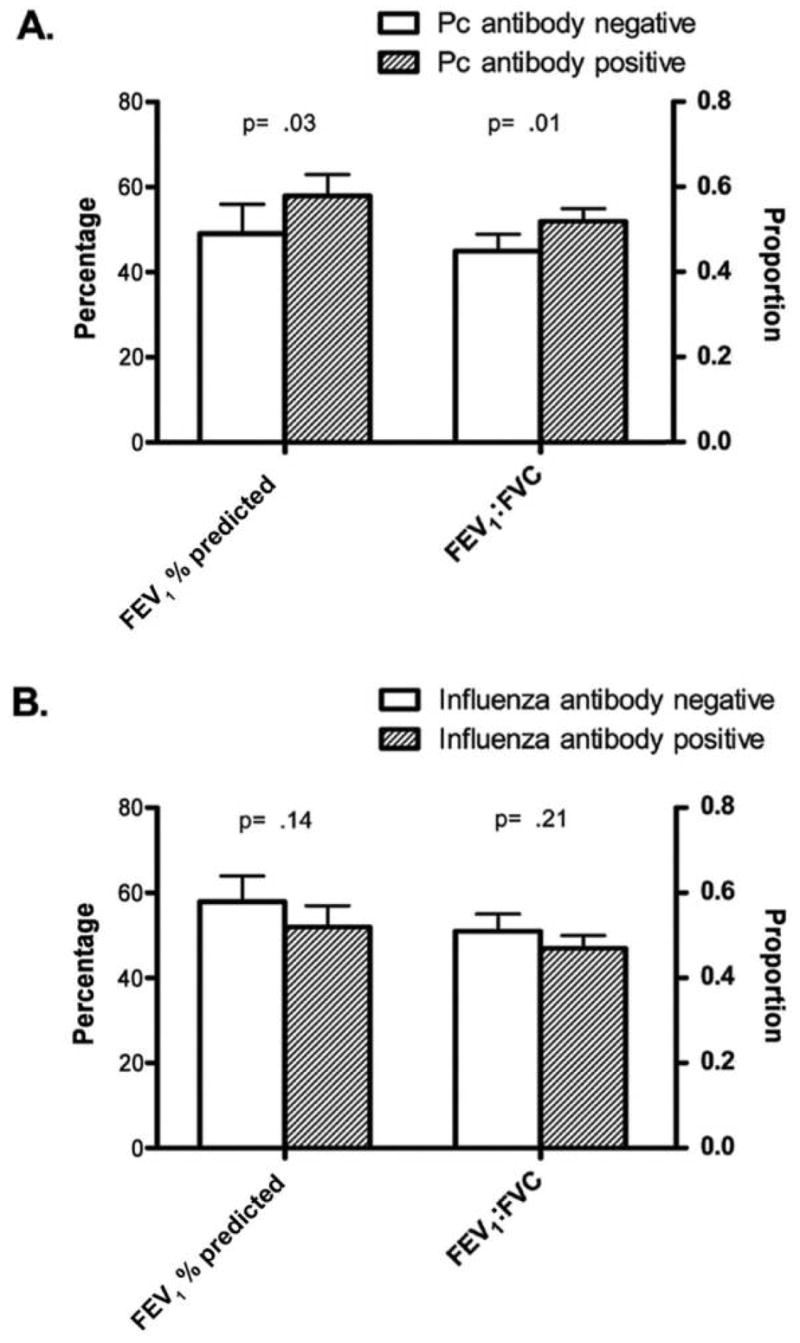
A, Adjusted mean spirometry values and 95% CIs for patients, by Pneumocystis (Pc) antibody status. B, Adjusted mean spirometry values and 95% CIs for patients, by anti-influenza antibody status. Data were adjusted for age, pack-year smoking history (defined as the average number of packs smoked per day multiplied by the number of years that the person smoked), current smoking status (forced expiratory volume in 1 s [FEV1] and FEV1-to–forced vital capacity [FVC] ratio), and sex (FEV1-to-FVC ratio only).
Discussion
To our knowledge, this study is the first to report the relationship between anti-Pneumocystis antibodies and the degree of COPD in smokers. We found that a low or undetectable anti-KEX1 antibody titer was an independent predictor of more-severe airway obstruction. We did not find a similar relationship with anti-influenza antibody titers, which suggests that the Pneumocystis antibody association is not merely a marker of a poor humoral immune response. This finding lends additional support to the hypothesis that Pneumocystis is involved in the pathogenesis or progression of COPD and suggests that the KEX1 antibody assay is a useful test in humans.
Previous studies demonstrated that an intact antibody response is important for protection from infection due to Pneumocystis. Studies have reported that HIV-infected patients have lower anti-Pneumocystis antibody levels than do HIV-uninfected blood donors [13, 14]. Patients who have repeated episodes of P. jirovecii pneumonia fail to mount an antibody response to the Pneumocystis major surface glycoprotein [15]. Although data regarding the anti-KEX1 antibody response in humans are limited, studies of rodent vaccine have indicated that immunization with KEX1 can result in a protective antibody response [7, 8]. The current study is, to our knowledge, the first to report that the majority of adults have a detectable anti-KEX1 titer and that this titer relates to COPD. Future studies will be necessary to determine whether a particular breakpoint can be used to distinguish patients colonized with Pneumocystis from those who are not colonized.
The association of low anti-Pneumocystis antibody titer with severity of airway obstruction is intriguing with regard to susceptibility to Pneumocystis colonization and progression of COPD. Data from nonhuman primates infected with chimeric simian immunodeficiency virus/HIV indicate that low or undetectable baseline anti-KEX1 titers predict subsequent susceptibility to colonization with Pneumocystis (H. M. Kling, T. W. Shipley, S. Patil, A. Morris, K. A. Norris, unpublished observation). We previously demonstrated that the prevalence of colonization with Pneumocystis is increased in patients with COPD, compared with the rate among those with other types of end-stage lung diseases, and that colonization with Pneumocystis is associated with COPD severity, independent of smoking history [5]. Although we did not have direct data on colonization with Pneumocystis in these patients, the current findings suggest that low or undetectable anti-Pneumocystis antibody titers might increase susceptibility to colonization with Pneumocystis, which in turn might stimulate pulmonary inflammation and worsen obstruction.
This study had several limitations. First, antibody levels were measured at a single time, and we did not have corresponding data on colonization with Pneumocystis. We also were unable to determine the cause and effect of Pneumocystis infection in patients with COPD in our study. The organism might worsen disease or might be an indicator of disease severity. Also, Pneumocystis is likely to be one of several pathogens involved in the pathogenesis and progression of COPD, and other organisms, such as Haemophilus influenzae and adenovirus, may be important [16, 17]. Despite these limitations, our study involved necessary preliminary work to demonstrate that KEX1 antibodies are detectable in humans and are related to COPD. Future studies examining the time course of antibody response in patients with COPD, linking antibody levels to detection of Pneumocystis colonization in respiratory specimens, and determining the role of Pneumocystis in disease progression will be informative.
The use of inhaled corticosteroids was a confounding factor in the analysis and interpretation of the Pneumocystis antibody response. Patients with a low antibody response were more likely than others to be using inhaled corticosteroids, although use of oral corticosteroids had no relationship to antibody levels. Because patients with more-severe COPD are more likely to receive prescriptions for inhaled corticosteroids, it is impossible to completely separate this effect. We do not think that inhaled corticosteroid use affected the ability to mount a systemic humoral immune response, because use of inhaled corticosteroids was not associated with influenza antibody response; however, we cannot rule out the possibility that these medications altered the immune environment of the lung, thereby decreasing the Pneumocystis antibody response. Nonetheless, a decrease in the ability to generate an antibody response that is the result of inhaled corticosteroid use might still increase the risk of colonization with Pneumocystis and potentially result in worsening of COPD.
In summary, we found that adult smokers have a detectable anti-KEX1 Pneumocystis titer and that lower antibody levels are independently associated with more-severe airway obstruction. These findings lend additional support to a potential role of Pneumocystis in the progression of COPD and suggest that decreased antibody response might be an important mechanism by which smokers become colonized. If future studies correlate antibody response with susceptibility to colonization with Pneumocystis, serum antibody titer could be used as a noninvasive marker of risk of Pneumocystis colonization to identify patients susceptible to such colonization.
Acknowledgments
We thank Donald Carter and Chad Karoleski for technical assistance.
Financial support. National Institutes of Health (HL07837 to A.M. and P50 HL084948 to F.S.).
Footnotes
Potential conflicts of interest. All authors: no conflicts.
References
- 1.Di Stefano A, Capelli A, Lusuardi M, et al. Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am J Respir Crit Care Med. 1998;158:1277–85. doi: 10.1164/ajrccm.158.4.9802078. [DOI] [PubMed] [Google Scholar]
- 2.Saetta M, Di Stefano A, Turato G, et al. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;157:822–6. doi: 10.1164/ajrccm.157.3.9709027. [DOI] [PubMed] [Google Scholar]
- 3.Calderon EJ, Regordan C, Medrano FJ, Ollero M, Varela JM. Pneumocystis carinii infection in patients with chronic bronchial disease. Lancet. 1996;347:977. doi: 10.1016/s0140-6736(96)91468-3. [DOI] [PubMed] [Google Scholar]
- 4.Probst M, Ries H, Schmidt-Wieland T, Serr A. Detection of Pneumocystis carinii DNA in patients with chronic lung diseases. Eur J Clin Microbiol Infect Dis. 2000;19:644–5. doi: 10.1007/s100960000329. [DOI] [PubMed] [Google Scholar]
- 5.Morris A, Sciurba FC, Lebedeva IP, et al. Association of chronic obstructive pulmonary disease severity and Pneumocystis colonization. Am J Respir Crit Care Med. 2004;170:408–13. doi: 10.1164/rccm.200401-094OC. [DOI] [PubMed] [Google Scholar]
- 6.Steele C, Shellito JE, Kolls JK. Immunity against the opportunistic fungal pathogen Pneumocystis. Med Mycol. 2005;43:1–19. doi: 10.1080/13693780400015360. [DOI] [PubMed] [Google Scholar]
- 7.Zheng M, Ramsay AJ, Robichaux MB, et al. CD4+ T cell–independent DNA vaccination against opportunistic infections. J Clin Invest. 2005;115:3536–44. doi: 10.1172/JCI26306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wells J, Haidaris CG, Wright TW, Gigliotti F. Active immunization against Pneumocystis carinii with a recombinant P. carinii antigen. Infect Immun. 2006;74:2446–8. doi: 10.1128/IAI.74.4.2446-2448.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller MR, Hankinson J, Brusasco V, et al. Standardisation of spirometry. Eur Respir J. 2005;26:319–38. doi: 10.1183/09031936.05.00034805. [DOI] [PubMed] [Google Scholar]
- 10.Crapo RO, Morris AH. Standardized single breath normal values for carbon monoxide diffusing capacity. Am Rev Respir Dis. 1981;123:185–9. doi: 10.1164/arrd.1981.123.2.185. [DOI] [PubMed] [Google Scholar]
- 11.Crapo RO, Morris AH, Gardner RM. Reference spirometric values using techniques and equipment that meet ATS recommendations. Am Rev Respir Dis. 1981;123:659–64. doi: 10.1164/arrd.1981.123.6.659. [DOI] [PubMed] [Google Scholar]
- 12.Bright RA, Carter DM, Crevar CJ, et al. Cross-clade protective immune responses to influenza viruses with H5N1 HA and NA elicited by an influenza virus–like particle. PLoS ONE. 2008;3:e1501. doi: 10.1371/journal.pone.0001501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daly KR, Fichtenbaum CJ, Tanaka R, et al. Serologic responses to epitopes of the major surface glycoprotein of Pneumocystis jiroveci differ in human immunodeficiency virus–infected and uninfected persons. J Infect Dis. 2002;186:644–51. doi: 10.1086/341565. [DOI] [PubMed] [Google Scholar]
- 14.Bishop LR, Kovacs JA. Quantitation of anti–Pneumocystis jiroveci antibodies in healthy persons and immunocompromised patients. J Infect Dis. 2003;187:1844–8. doi: 10.1086/375354. [DOI] [PubMed] [Google Scholar]
- 15.Daly KR, Huang L, Morris A, et al. Antibody response to Pneumocystis jirovecii major surface glycoprotein. Emerg Infect Dis. 2006;12:1231–7. doi: 10.3201/eid1208.060230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Retamales I, Elliott WM, Meshi B, et al. Amplification of inflammation in emphysema and its association with latent adenoviral infection. Am J Respir Crit Care Med. 2001;164:469–73. doi: 10.1164/ajrccm.164.3.2007149. [DOI] [PubMed] [Google Scholar]
- 17.Sethi S. Bacterial infection and the pathogenesis of COPD. Chest. 2000;117:286S–91S. doi: 10.1378/chest.117.5_suppl_1.286s. [DOI] [PubMed] [Google Scholar]