

Abstract
DNA sequence context has emerged as a critical determinant of the location and quantity of nucleobase damage caused by many oxidizing agents. However, the complexity of nucleobase and 2-deoxyribose damage caused by strong oxidants such as ionizing radiation and the Fenton chemistry of Fe2+-EDTA/H2O2 poses a challenge to defining the location of nucleobase damage and the effects of sequence context on damage chemistry in DNA. To address this problem, we developed a gel-based method that allows quantification of nucleobase damage in oxidized DNA by exploiting Escherichia coli exonuclease III to remove fragments containing direct strand breaks and abasic sites. The rigor of the method was verified in studies of guanine oxidation by photooxidized riboflavin and nitrosoperoxycarbonate, for which different effects of sequence context have been demonstrated by other approaches (Margolin, Y., Cloutier, J. F., Shafirovich, V., Geacintov, N. E., and Dedon, P. C. (2006) Nat. Chem. Biol. 2, 365–366). Using duplex oligodeoxynucleotides containing all possible three-nucleotide sequence contexts for guanine, the method was used to assess the role of DNA sequence context in hydroxyl radical-induced guanine oxidation associated with γ-radiation and Fe2+-EDTA/H2O2. The results revealed both differences and similarities for G oxidation by hydroxyl radicals and by one-electron oxidation by riboflavin-mediated photooxidation, which is consistent with the predominance of oxidation pathways for hydroxyl radicals other than one-electron oxidation to form guanine radical cations. Although the relative quantities of G oxidation produced by hydroxyl radicals were more weakly correlated with sequence-specific ionization potential than G oxidation produced by riboflavin, damage produced by both hydroxyl radical generators and riboflavin within two- and three-base runs of G showed biases in location that are consistent with a role for electron transfer in defining the location of the damage products. Furthermore, both γ-radiation and Fe2+-EDTA/H2O2 showed relatively modest effects of sequence context on the proportions of different damage products sensitive to E. coli formamidopyrimidine DNA glycosylase and hot piperidine, although GT-containing sequence contexts displayed subtle biases in damage chemistry (formamidopyrimidine DNA glycosylase/piperidine ratio). Overall, the results are consistent with the known chemistry of guanine oxidation by hydroxyl radical and demonstrate that charge migration plays a relatively minor role in determining the location and chemistry of hydroxyl radical-mediated oxidative damage to guanine in DNA.
With implications for mutagenesis and carcinogenesis, DNA damage caused by strong oxidizing agents such as ionizing radiation and Fenton chemistry (e.g. Fe2+-EDTA, Cu+/H2O2) affects both the base and sugar moieties (1, 2). This complexity poses a challenge to understanding the mechanisms of formation of the ultimate stable oxidation products and their relative amounts, especially in terms of defining the effects of DNA sequence context on the quantity and location of the damage. For example, ionizing radiation causes DNA damage by oxidation through both “direct” (non-scavengeable) and “indirect” (scavengeable) pathways, in roughly equal proportions that affect both the base and 2-deoxyribose components (2–4). The indirect pathway is mediated mainly by products of water radiolysis such as hydroxyl radicals that react with DNA by hydrogen atom abstraction from the 2-deoxyribose moiety to produce strand breaks and abasic sites, and by addition to nucleobases to form a variety of damage products (5–7). The direct effect of γ-radiation entails energy deposition that leads to ionization of the DNA nucleobases and the sugar-phosphate backbone, again leading to base damage and strand breaks (5).
This complicated mixture of sugar and base damage produced by strong
oxidants in DNA has hampered studies of the sequence selectivity of nucleobase
oxidation, which is critical for defining the contributions of charge transfer
to the spectrum of DNA lesions and for quantifying influences of local DNA
structure on the final spectrum of damage products. The role of charge
transfer in oxidatively damaged DNA has been well characterized in model
systems involving direct photooxidation reactions
(8) and those mediated by
agents such as pterins (9),
anthraquinones (10), rhodium
complexes (11), and riboflavin
(12). These agents oxidize
mainly DNA nucleobases by a common mechanism that is presumed to involve
one-electron oxidation of guanine (G) to form a guanine radical cation
(G ),3
because of the favorable redox potential for G relative to the other
nucleobases (1.29 V versus NHE for the guanine neutral radical,
G(-H)·; 1.58 V versus NHE for
G; see Ref.
13). The resulting electron
hole migrates through the π-stack of B-DNA in competition with trapping to
form stable products
(10–12),
with the common observation of damage “hot spots” at sites
containing multiple adjacent guanines (e.g. GG, GGG). Ab
initio calculations by Saito et al.
(12,
14) and Senthilkumar et
al. (15) revealed
sequence-specific variations in the ionization potentials (IP) of G, with the
lowest guanine IPs occurring in runs of G. Saito et al.
(12) further correlated
sequence-specific IP with the reactivity of G toward riboflavin-mediated
photooxidation and derived the expected inverse correlation between reactivity
and IP, an observation later verified directly by photoelectron spectroscopy
(8).
),3
because of the favorable redox potential for G relative to the other
nucleobases (1.29 V versus NHE for the guanine neutral radical,
G(-H)·; 1.58 V versus NHE for
G; see Ref.
13). The resulting electron
hole migrates through the π-stack of B-DNA in competition with trapping to
form stable products
(10–12),
with the common observation of damage “hot spots” at sites
containing multiple adjacent guanines (e.g. GG, GGG). Ab
initio calculations by Saito et al.
(12,
14) and Senthilkumar et
al. (15) revealed
sequence-specific variations in the ionization potentials (IP) of G, with the
lowest guanine IPs occurring in runs of G. Saito et al.
(12) further correlated
sequence-specific IP with the reactivity of G toward riboflavin-mediated
photooxidation and derived the expected inverse correlation between reactivity
and IP, an observation later verified directly by photoelectron spectroscopy
(8).
Similar arguments for charge migration have been made for the direct effect
of ionizing radiation. Although electron gain from the direct type effects
occurs mainly with the formation of cytosine and thymine anions
(16–19),
electron loss occurs most frequently at Gs, with
G being the predominant
species formed with γ-radiation
(13,
17,
20). However, direct type
effects also produce a significant amount of ionization of the sugar-phosphate
backbone (e.g. Refs.
21–24),
with the ensuing formation of strand breaks and abasic sites comprising
∼25% of the total damage
(23). This high background of
strand breaks complicates analysis of the sequence selectivity of base
oxidation and necessitates the use of indirect approaches in studies of charge
transfer with ionizing radiation, such as the use of easily ionized base
analogs. For example, Doddridge et al.
(26) demonstrated that
7,8-dihydro-8-oxoguanine (8-oxo-G), with its lower reduction potential than G
(0.74 V versus NHE; see Ref.
25), served as a damage hot
spot when placed in an oligodeoxynucleotide that was subjected to
γ-irradiation under dry film conditions that obviate the indirect
effects. The use of such base analogs in solution studies with ionizing
radiation, however, is complicated by artifacts such as the generation of
secondary radical-mediated oxidizing species from the quenching agents used to
obviate the indirect effects
(27).
DNA damage by strong oxidants yields a mixture of strand breaks and base
damage, thereby greatly complicating sequence-specific analysis of nucleobase
oxidation. To address this problem, we have developed a gel-based method that
allows quantification of nucleobase damage in oxidized DNA by exploiting
Escherichia coli exonuclease III (ExoIII) to remove fragments
containing direct strand breaks and abasic sites. The rigor of the method was
verified in studies of G oxidation by riboflavin-mediated photooxidation or
nitrosoperoxycarbonate (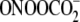 ), for
which dramatically different effects of sequence context have been
demonstrated by other approaches
(12,
28). Using γ-radiation
and Fe2+-EDTA, the method was applied to test the hypothesis that
hydroxyl radical-induced guanine oxidation would not be affected by sequence
context because of the predominance of oxidation pathways other than
one-electron oxidation to form
G radical cations.
), for
which dramatically different effects of sequence context have been
demonstrated by other approaches
(12,
28). Using γ-radiation
and Fe2+-EDTA, the method was applied to test the hypothesis that
hydroxyl radical-induced guanine oxidation would not be affected by sequence
context because of the predominance of oxidation pathways other than
one-electron oxidation to form
G radical cations.
EXPERIMENTAL PROCEDURES
Materials—Phosphorothioate-modified oligodeoxynucleotides were purchased from Integrated DNA Technologies (Coralville, IA) and gel-purified. All other reagents and chemicals were used without further purification. [γ-32P]ATP with activity of 6000 Ci/mmol was purchased from PerkinElmer Life Sciences. Piperidine, ferrous sulfate, riboflavin, and 30% (w/w) hydrogen peroxide solution were purchased from Sigma. All enzymes were purchased from New England Biolabs (Ipswich, MA). Chelex-100 was purchased from Bio-Rad. Potassium phosphate, sodium bicarbonate, and EDTA were purchased from VWR (West Chester, PA). Peroxynitrite solution was purchased from Cayman Chemical (Ann Arbor, MI). Distilled and deionized water was purified using a Milli-Q system from Millipore (Bedford, MA) and was used for all experiments.
Labeling and Annealing of Oligodeoxynucleotides—Purified single-stranded oligodeoxynucleotides were 5′-end-labeled with 32P by incubation at 37 °C for 1 h in a reaction that contained 0.2 nmol of 5′ ends, 0.1 mCi of [γ-32P]ATP, and 40 units of T4 PNK in 1× PNK buffer, in a total volume of 100 μl. Excess label was removed by gel filtration using Sephadex G-25 columns (Roche Diagnostics) that were washed four times with a volume of 300 μl of Chelex-treated 175 mm potassium phosphate buffer (pH 7.4). For annealing reaction, a total of 0.4 nmol of unlabeled complement was added to each purified labeled oligodeoxynucleotide; the mixture was heated at 95 °C for 5 min and was then allowed to slowly cool to room temperature over the course of 2 h.
Analysis of Damage in Double-stranded
Oligodeoxynucleotides—All damage analyses and controls were
performed in three separate experiments. Each Fenton reaction contained 2
mm hydrogen peroxide (concentration determined using an extinction
coefficient of 39.4 m-1 cm-1), 20 pmol of
labeled, double-stranded oligodeoxynucleotide, and 0.1 mm
FeSO4/EDTA (in the ratio of 1:1.1, freshly prepared before each
experiment), in 175 mm Chelex-treated potassium phosphate buffer
(pH 7.4) and a total volume of 50 μl. The DNA was always added last, and
the controls contained potassium phosphate buffer instead of
FeSO4/EDTA solution. The reactions were incubated at 37 °C for
2 h, after which Fenton reagent was removed by filtration through Sephadex
G-25 columns. For γ-irradiation experiments, the samples containing a
total of 20 pmol of labeled, double-stranded oligodeoxynucleotide in a total
of 50 μl of Chelex-treated 175 mm potassium phosphate buffer (pH
7.4) were irradiated in a 60Co source for a total dose of 50 gray.
Following treatment, the samples were incubated at ambient temperature for 20
min before purification by filtration using Sephadex G-25 columns. Studies of
oligodeoxynucleotide damage by 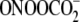 and riboflavin were performed according to the procedure described previously
(28).
and riboflavin were performed according to the procedure described previously
(28).
Damaged and purified oligodeoxynucleotides were treated with 5 units of ExoIII in 1× NE buffer 1 (New England Biolabs, Ipswich, MA) at 37 °C for 1 h in a total volume of 60 μl. These conditions were sufficient to remove all direct strand breaks generated during damage reactions, as determined by control experiments (results not shown). For hot piperidine treatment, oligodeoxynucleotides were desalted by gel filtration, incubated with 1 m piperidine at 90 °C in a total volume of 120 μl, lyophilized, and dissolved in a total of 5 μl of formamide gel loading buffer (28). To remove ExoIII activity prior to Fpg reactions, oligodeoxynucleotides were incubated with 20 mm EDTA to chelate Mg2+ present in 1× NE buffer 1 and passed through protein-binding Micropure-EZ filters (Millipore, Bedford, MA). The oligodeoxynucleotides were subsequently treated with 8 units of Fpg at 37 °C for 1 h, precipitated with ethanol, and dissolved in a total of 5 μl of formamide gel loading buffer.
A total of 2 μl of each sample was resolved on a 20% polyacrylamide gel containing 8 m urea and was subjected to PhosphorImager analysis (ImageQuant, GE Healthcare). Relative reactivities of Gs in each oligodeoxynucleotide sequence were determined as described previously (28).
RESULTS
Development of an Exonuclease Digestion Method for Quantifying Nucleobase Damage in Oligodeoxynucleotides—One approach to quantifying the sequence specificity of nucleobase damage employs controlled reactions in 32P-labeled, double-stranded oligodeoxynucleotides, with sequencing gel analysis of strand breaks formed when nucleobase lesions are expressed as strand breaks by treatment with hot alkali (e.g. piperidine) or with DNA N-glycosylase repair enzymes that recognize damaged nucleobases (e.g. E. coli formamidopyrimidine DNA glycosylase, Fpg) (12, 28). The major limitation of this approach is interference from direct strand breaks and labile abasic sites arising from 2-deoxyribose oxidation with agents such as γ-radiation and Fenton chemistry that damage both the base and sugar moieties (29). To resolve this problem, we devised a method to remove interfering direct strand breaks and abasic sites and thus allow for the direct analysis of nucleobase oxidation, as illustrated in Fig. 1A. The method utilizes double-stranded oligodeoxynucleotides (Table 1) containing three consecutive phosphorothioate linkages at the 3′ end of each strand to protect the strands from degradation by the 3′-to-5′-exonuclease activity of ExoIII (30, 31). Upon treatment of damaged oligodeoxynucleotides with the combined 3′-to-5′-exonuclease and AP endonuclease activities of ExoIII (30, 31), the oligodeoxynucleotides that contain abasic sites and direct strand breaks with unprotected 3′ ends are degraded, and the resulting 32P-containing small fragments are either lost during processing of the oligodeoxynucleotides or during sequencing gel electrophoresis (see supplemental Fig. 1 for a typical gel). The remaining full-length, 5′-32P-labeled oligodeoxynucleotides are protected from ExoIII degradation by the phosphorothioates at the 3′ end and are either undamaged or contain only nucleobase damage. Following inactivation (chelation of Mg2+ cofactor by EDTA; see Ref. 32) and removal of the enzyme by passing the reaction mixture through protein-binding filters, the nucleobase damage is converted to strand breaks by treatment with hot piperidine or Fpg. Sequencing gel analysis now reveals the location and quantity of nucleobase damage caused by the oxidizing agent, without interference from direct strand breaks or abasic sites.
FIGURE 1.

ExoIII-mediated removal of DNA containing strand breaks for sequencing gel analysis of G oxidation. A, schematic presentation of the ExoIII approach to strand break removal. B, demonstration of strand break removal with ExoIII. A 32P-labeled double-stranded oligodeoxynucleotide (S1-S in Table 1) was treated with Fe2+-EDTA/H2O2 and subsequently with ExoIII. Base damage was converted to strand breaks using hot piperidine, and the resulting fragments were resolved by denaturing PAGE (20%) as described under “Experimental Procedures.”
TABLE 1.
Sequences of oligodeoxynucleotides used for damage analysis Target guanines are in boldface type, and trinucleotide motifs are underlined. The invariant TGG sequence used for normalizing the quantity of damage is italicized, and asterisks denote phosphorothioate linkages.
| Oligodeoxynucleotide | Sequence (5′ → 3′) |
|---|---|
| S1-S | CGTACTCTTTGGTTGATGGGTTCTTTC*T*A*T |
| S2-S | CGTACTCTTTGGTCGGTTGCTTCTTTC*T*A*T |
| S3-S | CGTACTCTTTGGTAGTTGGATTCTTTC*T*A*T |
| S4-S | CGTACTCTTTGGTAGGTTGTTTCTTTC*T*A*T |
| S5-S | CGTACTCTTTGGTCGCTCGATTCTTTC*T*A*T |
| S6-S | CGTACTCTTTGGTAGCTAGATTCTTTC*T*A*T |
| S8-S | CGTACTCTTTGGTGGCTCGTTTCTTTC*T*A*T |
| S12-S | CGTACTCTTTGGTAGCTGGTTTCTTTC*T*A*T |
Validation of the ExoIII Method—The method was validated in
two ways. The first set of studies was designed to assess the effects of
ExoIII digestion on the nucleobase damage produced by two oxidants,
riboflavin-mediated photooxidation and
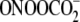 , that cause well defined G
oxidation in terms of quantity and location and that cause few strand breaks
or abasic sites (28). The
studies were conducted in a series of oligodeoxynucleotides (shown in
Table 1) with the following
general structure:
5′-CGTACTCTTTGGTX1G1Y1TX2G2Y2TTCTTTC*T*A*T-3′.
Each contains three consecutive phosphorothioate linkages (asterisks), two Gs
in variable three-nucleotide sequence contexts
(XiGiYi), and an
invariant TGG damage site for normalizing damage levels across all of the
oligodeoxynucleotides (12,
28). Following treatment of
duplex oligodeoxynucleotides with either 3 mm
, that cause well defined G
oxidation in terms of quantity and location and that cause few strand breaks
or abasic sites (28). The
studies were conducted in a series of oligodeoxynucleotides (shown in
Table 1) with the following
general structure:
5′-CGTACTCTTTGGTX1G1Y1TX2G2Y2TTCTTTC*T*A*T-3′.
Each contains three consecutive phosphorothioate linkages (asterisks), two Gs
in variable three-nucleotide sequence contexts
(XiGiYi), and an
invariant TGG damage site for normalizing damage levels across all of the
oligodeoxynucleotides (12,
28). Following treatment of
duplex oligodeoxynucleotides with either 3 mm
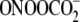 or 30 μm
riboflavin/366 nm irradiation
(28), samples were split, and
one-half was treated with ExoIII for 60 min. Following inactivation and
removal of ExoIII, the oligodeoxynucleotides were treated with hot piperidine,
and the resulting strand breaks were quantified by sequencing gel analysis, as
illustrated in Fig.
1B. The bar graphs shown in
Fig. 2 reveal that the quantity
of G oxidation detected by the sequencing gel method is not affected by ExoIII
treatment, which indicates that the ExoIII digestion step did not introduce
artifacts into the quantification of sequence-selective G oxidation in the
oligodeoxynucleotides. The results are also consistent with our previous
studies, in which the level of riboflavin-mediated oxidation of G varied
inversely with the sequence-specific IP of the central G in a three-nucleotide
context (relative G oxidation in decreasing order CGG > GGC > CGT >
TGC), whereas the opposite was true for
or 30 μm
riboflavin/366 nm irradiation
(28), samples were split, and
one-half was treated with ExoIII for 60 min. Following inactivation and
removal of ExoIII, the oligodeoxynucleotides were treated with hot piperidine,
and the resulting strand breaks were quantified by sequencing gel analysis, as
illustrated in Fig.
1B. The bar graphs shown in
Fig. 2 reveal that the quantity
of G oxidation detected by the sequencing gel method is not affected by ExoIII
treatment, which indicates that the ExoIII digestion step did not introduce
artifacts into the quantification of sequence-selective G oxidation in the
oligodeoxynucleotides. The results are also consistent with our previous
studies, in which the level of riboflavin-mediated oxidation of G varied
inversely with the sequence-specific IP of the central G in a three-nucleotide
context (relative G oxidation in decreasing order CGG > GGC > CGT >
TGC), whereas the opposite was true for
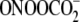 (TGC ∼ GGC > CGT >
CGG) (28).
(TGC ∼ GGC > CGT >
CGG) (28).
FIGURE 2.

Verification of the ExoIII digestion method with sequence-selective G
oxidation by 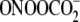 and
riboflavin. 32P-Labeled double-stranded oligodeoxynucleotides
containing four representative G sequence contexts
(Table 1) were treated with
and
riboflavin. 32P-Labeled double-stranded oligodeoxynucleotides
containing four representative G sequence contexts
(Table 1) were treated with
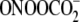 (A) or photo-activated
riboflavin (B), as described under “Experimental
Procedures.” The sample was subsequently split, with one-half treated
with ExoIII to remove direct strand breaks (black bars) and the other
half left untreated as a control (white bars). Base damage in both
samples was then expressed as strand breaks resulting from hot piperidine
treatment, and the resulting fragments were resolved on by denaturing PAGE
(20%), as described under “Experimental Procedures.”
(A) or photo-activated
riboflavin (B), as described under “Experimental
Procedures.” The sample was subsequently split, with one-half treated
with ExoIII to remove direct strand breaks (black bars) and the other
half left untreated as a control (white bars). Base damage in both
samples was then expressed as strand breaks resulting from hot piperidine
treatment, and the resulting fragments were resolved on by denaturing PAGE
(20%), as described under “Experimental Procedures.”
A second approach to validating the ExoIII digestion method entailed characterization of G oxidation in an oligodeoxynucleotide treated with Fe2+-EDTA/H2O2, an agent well known to oxidize 2-deoxyribose to produce strand breaks and abasic sites at all positions in DNA (29, 33) and to oxidize nucleobases (7, 34). As shown in Fig. 1B, Fe2+-EDTA/H2O2 produced a significant quantity of strand breaks at all positions in the oligodeoxynucleotide (Fig. 1B, lane 2), with the doublet band pattern in the gel image arising from differential migration of oligodeoxynucleotide fragments containing 3-phosphate and 3′-phosphoglycolate ends (35, 36). Incubation of the damaged oligodeoxynucleotide with ExoIII prior to hot piperidine treatment substantially diminished the background of direct strand breaks and revealed damage mainly at Gs (Fig. 1B, lane 4). The combined results of these validation studies indicate that ExoIII is effective at removing strand breaks and oxidized abasic sites without interfering with nucleobase damage.
Assessment of the Sequence Selectivity of G Oxidation Produced by γ-Radiation and Fe+2-EDTA/H2O2—The ExoIII digestion and sequencing gel method was employed to define the role of sequence context in hydroxyl radical-mediated G oxidation, with comparison to one-electron oxidation by riboflavin-mediated photooxidation. The studies were initiated by performing damage reactions with each of the 32P-labeled, double-stranded oligodeoxynucleotides shown in Table 1 in 175 mm metal-free phosphate buffer at pH 7.4. Optimal doses for γ-irradiation (60Co, 1.1 gray/min) and Fe2+-EDTA/H2O2 treatment were chosen to produce a range of quantifiable damage signals from the sequencing gels and to cause a maximum of one detectable base oxidation event per oligodeoxynucleotide molecule, which, according to a Poisson distribution, occurs when less than 30% of the parent duplex is consumed following removal of strand breaks by ExoIII treatment and base damage expression by hot piperidine or Fpg. In light of the uncertainty about the exact nature of iron-based Fenton chemistry (37), we chose reaction conditions that would maximize hydroxyl radical production in an air-saturated solution as follows: 0.1 mm Fe2+-EDTA, 2 mm H2O2 in 175 mm Chelex-treated potassium phosphate buffer (pH 7.4) for 2 h at 37 °C (37).
As shown in Fig. 3 for
piperidine-labile G damage products, the reactivity at each G was determined
relative to the central G of the TGG normalization sequence contained in each
oligodeoxynucleotide (Table 1).
The ln(relative reactivity) data were plotted as a function of the
sequence-specific IP of the G (open circles in
Fig. 3; see supplemental Fig. 1
for a typical gel used for quantification). In the case of XGG,
GGX, and GGG sequences, the relative damage level was determined for
the central G to be consistent with our previous studies of G oxidation by
photooxidized riboflavin and
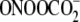 (28). For both
γ-radiation and Fe2+-EDTA/H2O2, the
ln(relative activity) of G varied only modestly with sequence context, within
a range of 0.60–1.0 for γ-radiation and 0.71–0.96 for
Fe2+-EDTA/H2O2. Similar trends were observed
with Fpg-labile G oxidation for both agents (closed circles in
Fig. 3; see supplemental Fig.
1), although relatively lower reactivity was observed for
5′-CGT-3′, 5′-AGT-3′, 5′-TGT-3′,
5′-GGT-3′, and 5′-GGC-3′ (0.4–0.6 for
γ-radiation and 0.5–0.6 for Fe2+-EDTA).
(28). For both
γ-radiation and Fe2+-EDTA/H2O2, the
ln(relative activity) of G varied only modestly with sequence context, within
a range of 0.60–1.0 for γ-radiation and 0.71–0.96 for
Fe2+-EDTA/H2O2. Similar trends were observed
with Fpg-labile G oxidation for both agents (closed circles in
Fig. 3; see supplemental Fig.
1), although relatively lower reactivity was observed for
5′-CGT-3′, 5′-AGT-3′, 5′-TGT-3′,
5′-GGT-3′, and 5′-GGC-3′ (0.4–0.6 for
γ-radiation and 0.5–0.6 for Fe2+-EDTA).
FIGURE 3.

Relative reactivities of Gs in different sequence contexts as a function of their sequence-specific ionization potentials. Labeled oligodeoxynucleotides with sequences shown in Table 1 were subjected to oxidation by 50 gray of γ-irradiation (A), 0.1 mm Fe2+-EDTA/2 mm H2O2 (B), and riboflavin-mediated photooxidation (C). Subsequent to treatment with ExoIII to remove the background of 2-deoxyribose oxidation, the oligodeoxynucleotides were treated with hot piperidine (○) or Fpg (•) to convert oxidized bases to strand breaks and subjected to sequencing gel electrophoresis. For all three oxidants, the relative reactivity of each G was calculated as the percentage of total radioactivity in the corresponding band, relative to the damage induced within the central G of the invariant TGG sequence, quantified after autoradiography as described under “Experimental Procedures.” Data for riboflavin-mediated photooxidation were taken from Margolin et al. (28). Data in all three panels were plotted on the same y axis scale for purposes of comparison and represent the mean ± S.D. for three experiments. Dashed lines represent linear regression fits of combined piperidine and Fpg data for γ-irradiation (y =-0.469x + 2.88; r2 = 0.22) and Fe-2+EDTA/2 mm H2O2 (y =-0.376x + 2.28; r2 = 0.16) and of riboflavin-mediated photooxidation data from Margolin et al. (28) (y =-2.30x + 14.5; r2 = 0.61); all lines have statistically significant negative slopes, as noted in the text.
Assessment of the Sequence Selectivity of G Oxidation Produced by
γ-Radiation and
Fe+2-EDTA/H2O2 within
Runs of G—In addition to the correlation between IP and sequence
context-dependent G reactivity as evidence of charge transfer, G oxidation by
classical one-electron oxidants such as photoactivated riboflavin has also
been observed to localize at the 5′-most G in GG motifs, and variably at
the 5′- or middle G of GGG
(38), as a result of charge
transfer. Although the sequence context studies revealed no significant
preference for damage in runs of G by γ-radiation or
Fe2+-EDTA/H2O2 compared with other sequence
contexts (Fig. 3), we sought to
determine whether the hydroxyl radical was site-selective within GG and GGG
motifs. This was accomplished by quantifying the relative amount of damage
sustained by G in four representative
5′-TXGXT-3′ sequence contexts as a result of
exposure to γ-radiation and
Fe2+-EDTA/H2O2, with the relative reactivity
expressed as the fraction of total G oxidation in a pair or trio of Gs. As
shown in Fig. 4, the quantity
of damage produced by photooxidized riboflavin exhibits an inverse correlation
with the calculated G IP values, which is consistent with the previous studies
of Saito and co-workers (12,
14), most notably for the
TGGGT sequence context (38).
The results with 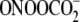 reveal a
weaker dependence on the IP of G, as shown in
Fig. 4 and in our previously
published studies (28). In the
case of the TG1G2CT sequence,
reveal a
weaker dependence on the IP of G, as shown in
Fig. 4 and in our previously
published studies (28). In the
case of the TG1G2CT sequence,
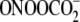 was highly selective for the G2
position adjacent to C, which is consistent with the direct relationship
between G IP and reactivity observed for
was highly selective for the G2
position adjacent to C, which is consistent with the direct relationship
between G IP and reactivity observed for
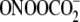 (28). The results with
γ-radiation and Fe2+-EDTA/H2O2 indicate
that oxidation is slightly favored at the 5′-G in GG sequence contexts
except in TG1G2G3T, where there G2
and G1 sustain similar levels of damage. These results are similar
to the riboflavin data, with less pronounced selectivity.
(28). The results with
γ-radiation and Fe2+-EDTA/H2O2 indicate
that oxidation is slightly favored at the 5′-G in GG sequence contexts
except in TG1G2G3T, where there G2
and G1 sustain similar levels of damage. These results are similar
to the riboflavin data, with less pronounced selectivity.
FIGURE 4.

Relative oxidative damage sustained by individual Gs within four representative G runs for four oxidizing agents. Numbers on the columns for riboflavin-mediated photooxidation correspond to the sequence-specific calculated ionization potential (eV) for each G identified in the sequence above each graph and along the z axis (12). The y axis corresponds to the fraction of the total γ-radiation-induced oxidative damage sustained by all Gs in the run of Gs. The data represent the mean for three experiments; the standard deviation for all of the data is <10%.
Estimation of the Sequence Dependence of G Oxidation Chemistry Caused
by γ-Radiation and
Fe+2-EDTA/H2O2—In
addition to the effects on the quantity of G oxidation, sequence context may
also affect the chemistry of the damage. One approach to assessing the
sequence dependence of G oxidation chemistry entails quantification of the
differential sensitivity of the lesions to hot piperidine and Fpg treatments.
It is well established that the reactivity of different G oxidation products
with piperidine and Fpg is variable and depends upon the structures of the
lesions (39) with 8-oxo-dG and
the 2,6-diamino-5-formamido-4-hydroxypyrimidine form of dG (Fapy-dG) sensitive
to Fpg and resistant to piperidine treatment
(39–42).
The opposite is true for the G oxidation product
2,2-diamino-4-[(2-deoxy-β-d-erythro-pentofuranosyl)-amino]-5(2H)-oxazolone
(oxazolone; Ox), the stable degradation product of the 2-aminoimidazolone
lesion (39,
43,
44). The diastereomeric
spiroiminodihydantoin lesions, a major product of G oxidation by carbonate
radical anion (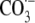 ) in DNA
(45) and a minor product of
8-oxo-dG oxidation in riboflavin-mediated photooxidation
(46), are Fpg-sensitive but
only partially alkali-labile
(47). Because the major G
oxidation products arising from γ-irradiation of DNA include, among a
minority of other products, the Fpg-sensitive Fapy-dG and 8-oxo-dG and the
piperidine-sensitive Ox (48,
49), we set out to estimate
the sequence specificity of G oxidation chemistry using the approach of
Spassky and Angelov (39) by
measuring differences in the proportions of Fpg- and piperidine-sensitive
lesions in the various sequence contexts.
) in DNA
(45) and a minor product of
8-oxo-dG oxidation in riboflavin-mediated photooxidation
(46), are Fpg-sensitive but
only partially alkali-labile
(47). Because the major G
oxidation products arising from γ-irradiation of DNA include, among a
minority of other products, the Fpg-sensitive Fapy-dG and 8-oxo-dG and the
piperidine-sensitive Ox (48,
49), we set out to estimate
the sequence specificity of G oxidation chemistry using the approach of
Spassky and Angelov (39) by
measuring differences in the proportions of Fpg- and piperidine-sensitive
lesions in the various sequence contexts.
As shown in Fig. 5, the
ratios of Fpg- to piperidine-sensitive lesions produced by γ-radiation
and Fe2+-EDTA/H2O2 in all sequence contexts
vary over an ∼2-fold range. This range of ratios is similar to that
observed with riboflavin-mediated photooxidation
(Fig. 5), but it stands in
contrast to the 5.5-fold variation in the ratios of Fpg- to
piperidine-sensitive lesions produced by
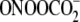 (28). As noted earlier, Gs
within 5′-GT-3′ motifs (5′-CGT-3′,
5′-AGT-3′, 5′-GGT-3′, and 5′-TGT-3′) are
characterized by Fpg/piperidine ratios that are significantly lower than the
other ratios (p < 0.02) and range from 0.5 to 0.7 for
γ-radiation and from 0.6 to 0.8 for
Fe2+-EDTA/H2O2. In addition, the
5′-GGC-3′ sequence context is also characterized by a relatively
low Fpg/piperidine ratio of 0.8 in the case of damage produced by
Fe2+-EDTA/H2O2.
(28). As noted earlier, Gs
within 5′-GT-3′ motifs (5′-CGT-3′,
5′-AGT-3′, 5′-GGT-3′, and 5′-TGT-3′) are
characterized by Fpg/piperidine ratios that are significantly lower than the
other ratios (p < 0.02) and range from 0.5 to 0.7 for
γ-radiation and from 0.6 to 0.8 for
Fe2+-EDTA/H2O2. In addition, the
5′-GGC-3′ sequence context is also characterized by a relatively
low Fpg/piperidine ratio of 0.8 in the case of damage produced by
Fe2+-EDTA/H2O2.
FIGURE 5.

Ratios of relative amounts of Fpg- to piperidine-sensitive oxidized G
lesions produced by Fe2+-EDTA/H2O2 (white
bars) and γ-radiation (black bars) (A) and
riboflavin-mediated photooxidation (white bars) and
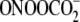 (black bars; data from
Ref. 28) within
different sequence contexts (B). Values represent mean ±
S.D. for three experiments. The sequence contexts are organized from
left to right as a function of increasing IP.
(black bars; data from
Ref. 28) within
different sequence contexts (B). Values represent mean ±
S.D. for three experiments. The sequence contexts are organized from
left to right as a function of increasing IP.
DISCUSSION
The complexity of nucleobase and sugar damage caused by strong oxidizing
agents such as ionizing radiation and the Fenton chemistry typified by
Fe2+-EDTA/H2O2 poses a challenge to defining
the location of nucleobase damage and the effects of sequence context on the
damage. To address this problem, we developed a gel-based method that allows
quantification of nucleobase damage in oxidized DNA by exploiting ExoIII to
remove fragments containing direct strand breaks and abasic sites. The
feasibility of this approach was proven in two sets of studies. The first
entailed corroboration of the sequence selectivity of G oxidation by
riboflavin-mediated photooxidation and by
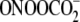 (Fig. 2). These agents were
previously observed to cause G oxidation with widely differing sequence
selectivity; the level of riboflavin-mediated oxidation of G decreases as a
function of the sequence-specific IP of the central G in a three-nucleotide
context (relative G oxidation in decreasing order CGG > GGC > CGT >
TGC), whereas the opposite is true for
(Fig. 2). These agents were
previously observed to cause G oxidation with widely differing sequence
selectivity; the level of riboflavin-mediated oxidation of G decreases as a
function of the sequence-specific IP of the central G in a three-nucleotide
context (relative G oxidation in decreasing order CGG > GGC > CGT >
TGC), whereas the opposite is true for
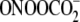 (TGC ∼ GGC > CGT >
CGG) (28). This behavior is
identical to that observed in studies employing ExoIII
(Fig. 2). Furthermore, ExoIII
digestion of oligodeoxynucleotides treated with
Fe2+-EDTA/H2O2 caused a significant reduction
in the direct strand breaks and abasic sites produced by this agent, with the
remaining damage confined to G bases (Fig.
1B), as expected from previous studies
(7,
34).
(TGC ∼ GGC > CGT >
CGG) (28). This behavior is
identical to that observed in studies employing ExoIII
(Fig. 2). Furthermore, ExoIII
digestion of oligodeoxynucleotides treated with
Fe2+-EDTA/H2O2 caused a significant reduction
in the direct strand breaks and abasic sites produced by this agent, with the
remaining damage confined to G bases (Fig.
1B), as expected from previous studies
(7,
34).
The ExoIII approach to removing strand breaks and abasic sites can be used to probe sequence selectivity of nucleobase oxidation by other oxidants that generate substantial amounts of 2-deoxyribose oxidation, such as ionizing radiation, peroxynitrite (ONOO-) (44), and peroxyl radicals (50). Previous approaches to studying nucleobase oxidation in Fenton reactions induced by Cu+ and Fe2+ in the presence of H2O2 and ascorbate addressed the strand break problem by adding sucrose to the damage reactions to suppress 2-deoxyribose oxidation (51). However, this approach generates sucrose-derived radical species that could lead to alterations of G oxidation chemistry (52).
The method was applied to define the role of sequence context in G
oxidation by hydroxyl radicals, with γ-radiation and
Fe2+-EDTA/H2O2 as model hydroxyl
radical-generating systems. Both of these oxidizing agents produce a
substantial quantity of 2-deoxyribose oxidation in DNA
(2,
33), which would otherwise
confound a gel-based analysis of G oxidation by adding 2-deoxyribose-derived
strand breaks to those derived from nucleobase damage. Although nucleobase
oxidation mediated by the direct effect of γ-radiation is associated
with charge transfer as a result of the formation of
G radicals
(13,
17,
20), the relatively dilute DNA
concentration employed in the present studies leads to a predominance of
hydroxyl radicals (i.e. the indirect effect) as the main DNA-damaging
species generated during γ-irradiation. In conducting these experiments,
we were mindful of the proposed formation of oxidizing species other than the
hydroxyl radical by the Fe2+-EDTA complex
(53–55),
specifically oxidizing iron-oxo complexes such as the perferryl
([Fe2+-O2 ↔
Fe3+-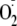 ])
and ferryl ([Fe4+ = O]) species
(37). Although these species
are strong oxidants, they are less reactive than hydroxyl radical with
molecules such as benzoate, tert-butyl alcohol, acetate ion,
arginine, and serine (53,
54), presumably because of
their lower reduction potentials relative to that of hydroxyl radical
(56). It has been observed
that the relative yields of hydroxyl radical and the iron-oxo complexes are
strongly dependent on factors such as pH, solvent polarity, buffer components,
and the metal chelator (55,
57,
58). The concentration of
H2O2 in the reaction mixture was shown to have a
significant effect on the efficiency of ·OH generation, with
low concentrations (≤ μm) favoring iron-oxo species and high
concentrations (mm) favoring hydroxyl radical
(37,
57,
58). Taking into account the
complicated chemistry of Fe2+-EDTA, we conducted
Fe2+-EDTA-mediated DNA oxidation reactions in the presence of 2
mm H2O2, which leads to predominant formation
of hydroxyl radical (37).
])
and ferryl ([Fe4+ = O]) species
(37). Although these species
are strong oxidants, they are less reactive than hydroxyl radical with
molecules such as benzoate, tert-butyl alcohol, acetate ion,
arginine, and serine (53,
54), presumably because of
their lower reduction potentials relative to that of hydroxyl radical
(56). It has been observed
that the relative yields of hydroxyl radical and the iron-oxo complexes are
strongly dependent on factors such as pH, solvent polarity, buffer components,
and the metal chelator (55,
57,
58). The concentration of
H2O2 in the reaction mixture was shown to have a
significant effect on the efficiency of ·OH generation, with
low concentrations (≤ μm) favoring iron-oxo species and high
concentrations (mm) favoring hydroxyl radical
(37,
57,
58). Taking into account the
complicated chemistry of Fe2+-EDTA, we conducted
Fe2+-EDTA-mediated DNA oxidation reactions in the presence of 2
mm H2O2, which leads to predominant formation
of hydroxyl radical (37).
As shown in Figs. 3,
4,
5, the G oxidation patterns
observed for γ-radiation and
Fe2+-EDTA/H2O2 showed strong similarities for
the two agents, as expected for the predominant role of hydroxyl radicals in
their mechanisms of DNA oxidation
(2,
33,
37). However, there were
differences from the one-electron oxidation mediated by photoactivated
riboflavin, which is presumed to be dominated by
G radicals
(13,
17,
20). Linear regression
analysis revealed a much weaker correlation (r2 = 0.2)
between the level of G oxidation and the sequence-specific GIPfor
γ-radiation and Fe2+-EDTA/H2O2
(Fig. 3, A and
B) compared with the stronger correlation observed for
riboflavin here (Fig.
3C, r2 = 0.6) and in previous studies
(12,
28). However, the analysis
confirmed statistically significant negative slopes for the plots shown in
Fig. 3 for both hydroxyl
radical generating agents and riboflavin (p < 0.02 and <0.0004,
respectively), which is consistent with a weak negative correlation between IP
and G reactivity.
A stronger similarity of the behavior of hydroxyl radicals and riboflavin
in terms of sequence-selective G oxidation was apparent in the observed biases
in damage location within GG and GGG motifs. In general, the relationship
between the level of G oxidation produced by hydroxyl radicals and the
calculated IP of G in the four GG- and GGG-containing sequences was less
pronounced than that associated with riboflavin, but an inverse correlation
between reactivity and IP is still apparent
(Fig. 4). This behavior stands
in contrast to the varying pattern of G oxidation in GG and GGG sequences by
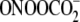 (Fig. 4).
(Fig. 4).
These observed effects of sequence context on hydroxyl radical-induced G
oxidation raise questions about the basis for both the similarities and the
differences with respect to one-electron oxidants such as riboflavin. The most
obvious difference involves the initial step in G oxidation, with addition of
the hydroxyl radical to C4 or C8 of G to produce 8-hydroxy- and 4-hydroxy-G
radical species (8-OH-G· and 4-OH-G·;
Scheme 1)
(59) as opposed to
one-electron removal from G to produce
G with riboflavin-mediated
photooxidation. The additional complication of 2-deoxyribose oxidation by
hydroxyl radicals will be discussed shortly. The reactions subsequent to the
initial electron removal by photoactivated riboflavin fit a widely accepted
general model of charge transfer mediated by the
G radical, with migration of
the hole to sequence contexts that confer the lowest IP to G
(Fig. 3). Furthermore, the
reactions of riboflavin-mediated
G within runs of G
(e.g. GG and GGG contexts) have been explained by a more complicated
model to account for differences observed with longer range sequence contexts,
such as TGGG versus CGGG
(38).
SCHEME 1.

The mechanistic basis for the observed sequence context effects of hydroxyl
radical-induced G oxidation is not as well defined as in the case of
riboflavin-mediated photooxidation. This can be accounted for by the following
considerations. As opposed to the predominance of
G as the initial product of
one-electron oxidation of G, hydroxyl radical reacts to form
8-OH-G· and 4-OH-G· radicals. The
subsequent transformations of 8-OH-G· yield the stable
products 8-oxo-G and Fapy-G
(60). The loss of a water
molecule from 4-OH-G· at physiological pH results in
G(-H)· (59)
that appears to undergo conversion mainly to the stable damage product Ox
(61)
(Scheme 1). The complicating
factor here is the balance between the fates of 8-OH-G·,
4-OH-G·, and G(-H)·. A bias toward
8-OH-G· would lead to a predominance of stable damage
products (i.e. 8-oxo-G and Fapy-G) formed at the initial site of G
oxidation, whereas higher proportions of 4-OH-G· and its
dehydration product G(-H)· could lead to migration of the
radical to neighboring G bases, as discussed shortly. This may explain the
diminished sequence context effects on hydroxyl radical-mediated G oxidation
relative to that of riboflavin.
This weak sequence dependence may arise from contributions of the
G(-H)· radical. It is well established that this species can
arise from both dehydration of 4-OH-G· and from
deprotonation of G, so it
represents a common intermediate for G oxidation caused by riboflavin and
hydroxyl radicals (62). This
does not explain, however, the role of G(-H)· in determining
the location of the final damage products in runs of two or three Gs. At one
extreme, G(-H)· could simply represent an intermediate on
the path to final damage products, with the sequence selectivity for damage
products determined by the stability of the G(-H)· radical
(38). In this case,
G(-H)· does not participate in electron transfer reactions
with neighboring bases and is consumed by reactions such as recombination with
superoxide and the subsequent formation of species such as Ox
(Scheme 1)
(61) at the site of initial G
oxidation. According to this model, the predominance of
8-OH-G·, 4-OH-G·, and
G(-H)· in the case of γ-radiation and
Fe2+-EDTA/H2O2 treatment would explain the
less pronounced sequence specificity of G oxidation product formation
(Fig. 3, A and
B) if hydroxyl radicals are relatively unselective in
their initial oxidation of Gs at different sites in DNA. Tullius and
co-workers (36) have
demonstrated that the reactivities of hydroxyl radicals at different positions
in 2-deoxyribose are governed in part by the solvent exposure of the hydrogen
atoms at these positions, which would suggest some degree of
“selectivity” of hydroxyl radical reactivities for solvent-exposed
Gs as well. This may explain the weak sequence dependence of hydroxyl
radical-induced G oxidation (Fig. 3,
A and B) that is significantly less pronounced
than the strong charge transfer-mediated sequence dependence for the
distribution of final damage products with one-electron oxidants such as
riboflavin (Fig. 3C)
(12,
28).
The G(-H)· may also participate in electron transfer in
DNA. The deprotonation of the
G cation in DNA in solution
occurs with a rate constant of ≥3 × 106 s-1
(63), suggesting that
intra-strand proton-coupled hole transfer can occur between
G(-H)· and a neighboring G in DNA on slower time scales
(64). G(-H)·
is a relatively strong oxidant, only 0.29 V weaker than
G (1.29 and 1.58 V
versus NHE, respectively; see Ref.
13), so it is reasonable to
assume that it can remove an electron from a neighboring G, albeit with a
slower rate than G because of
this energy difference. This would explain the similarities between the
hydroxyl radical generators and riboflavin-mediated photooxidation in terms of
relative reactivity between different G sequence contexts
(Fig. 3) and within runs of G
(Fig. 4). Just as with
G, the most probable position
of the G(-H)· (i.e. the most stable position)
determines the eventual final location of damage products in a run of Gs. This
is consistent with the studies of Saito and co-workers
(38), who correlated ab
initio molecular orbital calculations with damage product analysis and
concluded that the predominance of damage at the G2 of
TG1G2G3 correlates well with the greater
stability of the G(-H)· at G2.
Although there are similarities between the hydroxyl radical generators and the one-electron oxidant, there are clear differences. One factor possibly contributing to these differences is the oxidation of the 2-deoxyribose moiety by hydroxyl radical but not by one-electron oxidants such as riboflavin. It is well known that hydroxyl radicals cause hydrogen atom abstraction from the 2-deoxyribose in DNA, with the resulting carbon-centered radicals capable of reacting with the C8 of purines (65, 66) and with molecular oxygen to form peroxyl radicals and a host of degradation products (29, 67). Although the ExoIII digestion removes the final strand break and abasic site products of the sugar radical species, the sugar radicals could confound the mechanism of base oxidation by reacting with neighboring nucleobases to generate base radical species. Precedent for this activity comes from the observations of 5′,8-cyclopurine nucleotides arising from addition of a 5′-radical at the C8 position of purines (65, 66).
In addition to the weak sequence dependence of hydroxyl radical-mediated G
oxidation, we also observed limited sequence effects on the chemistry of the G
damage (Fig. 5). The ratio of
the quantity of Fpg-sensitive to hot piperidine-sensitive lesions has been
shown to be an accurate estimate of site-specific guanine oxidation chemistry
(28,
39). Indeed, the higher
Fpg-to-piperidine ratios that were observed with riboflavin
(Fig. 5) are consistent with
the higher proportion of Fpg-sensitive lesions associated with riboflavin
compared with γ-radiation
(68). The major lesions
observed to date with riboflavin consist of Fpg-sensitive 8-oxo-G and
piperidine-sensitive Ox (39,
40,
68–70),
along with smaller amounts of spiroiminodihydantoin, which is sensitive to Fpg
(46,
71) and partially labile in
piperidine (47). The major
stable G oxidation products in hydroxyl radical-induced DNA damage
(7,
72,
73) include 8-oxo-G and Ox,
along with Fapy-G, which is sensitive to both Fpg and piperidine
(40). The ∼2-fold
sequence-dependent variation in the proportions of Fpg- and
piperidine-sensitive lesions produced by the hydroxyl radical generators and
riboflavin is smaller than the ∼5-fold range for
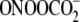 -induced G oxidation
(28) and points to the modest
role of sequence context in determining the spectrum of G damage products. The
stronger sequence dependence of G oxidation chemistry by
-induced G oxidation
(28) and points to the modest
role of sequence context in determining the spectrum of G damage products. The
stronger sequence dependence of G oxidation chemistry by
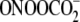 may reflect the more
complicated set of reactive intermediates arising from this agent as follows:
may reflect the more
complicated set of reactive intermediates arising from this agent as follows:
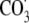 and nitrogen dioxide radical
(
and nitrogen dioxide radical
(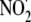 )
(74). These studies with
sequence-dependent variation in Fpg- and piperidine-sensitive G oxidation
products provide testable models for analysis of damaged spectra by chemically
specific methods such as mass spectrometry.
)
(74). These studies with
sequence-dependent variation in Fpg- and piperidine-sensitive G oxidation
products provide testable models for analysis of damaged spectra by chemically
specific methods such as mass spectrometry.
As noted earlier, the results with the 5′-GT-3′ contexts
present a special case of context-dependent G oxidation chemistry. The
XGT sequence motifs are consistently characterized by the lowest
Fpg-to-piperidine ratios for γ-radiation and
Fe2+-EDTA/H2O2, as well as riboflavin and
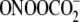 (Fig. 5). This phenomenon is
not general to Gs positioned next to pyrimidines since XGC motifs
show Fpg-to-piperidine ratios similar to other sequence contexts
(Fig. 5). There are many
possible explanations for the unusual behavior of the XGT sequences.
One involves the propensity for hydroxyl radical-mediated formation of tandem
lesions at GT sequences involving both 8-oxo-G and a thymine-derived formyl
residue (49,
75,
76). The tandem lesions appear
to arise by attack of a thymine peroxyl radical at the C8 of G
(49). However, any model must
also account for the fact that the unusual Fpg-to-piperidine ratios are also
observed with
(Fig. 5). This phenomenon is
not general to Gs positioned next to pyrimidines since XGC motifs
show Fpg-to-piperidine ratios similar to other sequence contexts
(Fig. 5). There are many
possible explanations for the unusual behavior of the XGT sequences.
One involves the propensity for hydroxyl radical-mediated formation of tandem
lesions at GT sequences involving both 8-oxo-G and a thymine-derived formyl
residue (49,
75,
76). The tandem lesions appear
to arise by attack of a thymine peroxyl radical at the C8 of G
(49). However, any model must
also account for the fact that the unusual Fpg-to-piperidine ratios are also
observed with 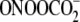 and
riboflavin-mediated photooxidation. Based on the ratio of Fpg and piperidine
sensitivities, the XGT context effects suggest the existence of a
local structure that is unfavorable to formation of 8-oxo-G (piperidine
resistance) or favorable to formation of Ox (Fpg-resistant; Fapy-G is
sensitive to both Fpg and piperidine). The established intrinsic flexibility
of DNA sequences containing alternating purine and pyrimidine bases
(77) may favor a backbone
conformation that promotes addition of the hydroxyl radical to the C4 position
of G and produces a greater number of Ox lesions at the expense of 8-oxo-G.
This shift to a greater proportion of piperidine-sensitive lesions is also
consistent with a model of local helical instability proposed by Spassky and
Angelov (39) to explain
differential Fpg and piperidine sensitivities of G oxidation products. The
results with the Fpg and piperidine sensitivity warrant further study to
identify the spectrum of G oxidation products and the role of sequence context
in defining the damage spectrum for both hydroxyl radicals and one-electron
oxidants.
and
riboflavin-mediated photooxidation. Based on the ratio of Fpg and piperidine
sensitivities, the XGT context effects suggest the existence of a
local structure that is unfavorable to formation of 8-oxo-G (piperidine
resistance) or favorable to formation of Ox (Fpg-resistant; Fapy-G is
sensitive to both Fpg and piperidine). The established intrinsic flexibility
of DNA sequences containing alternating purine and pyrimidine bases
(77) may favor a backbone
conformation that promotes addition of the hydroxyl radical to the C4 position
of G and produces a greater number of Ox lesions at the expense of 8-oxo-G.
This shift to a greater proportion of piperidine-sensitive lesions is also
consistent with a model of local helical instability proposed by Spassky and
Angelov (39) to explain
differential Fpg and piperidine sensitivities of G oxidation products. The
results with the Fpg and piperidine sensitivity warrant further study to
identify the spectrum of G oxidation products and the role of sequence context
in defining the damage spectrum for both hydroxyl radicals and one-electron
oxidants.
The observations made with ionizing radiation and Fe2+-EDTA/H2O2 have implications for the biological impact of G oxidation and DNA damage in general. For example, each of the various G oxidation products has a different mutagenic potential in terms of both the type of mutation and the frequency (78). This is further complicated by the recognized role of sequence context as a determinant of polymerase fidelity not only in copying normal DNA but also in translesion synthesis (79). Sequence-dependent variation in the spectrum of DNA oxidation products may thus influence the spectrum of mutations arising in oxidatively stressed cells.
In conclusion, we have developed an approach to quantifying
sequence-selective nucleobase oxidation in DNA in the presence of strand
breaks and have used it to demonstrate a more limited sequence selectivity for
both the quantity and chemistry of G oxidation produced by the hydroxyl
radical generators γ-radiation and
Fe2+-EDTA/H2O2 compared with the one-electron
oxidants riboflavin-mediated photooxidation and
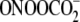 . The results are consistent
with the formation of neutral G radical species (8-HO·-G,
4-HO·-G, G(-H)·) by hydroxyl radicals as
compared with the formation of
G by one-electron oxidants,
but also point to the potential for intramolecular electron transfer with
G(-H)· as the means to account for the correlation between G
reactivity and IP in GG and GGG sequences.
. The results are consistent
with the formation of neutral G radical species (8-HO·-G,
4-HO·-G, G(-H)·) by hydroxyl radicals as
compared with the formation of
G by one-electron oxidants,
but also point to the potential for intramolecular electron transfer with
G(-H)· as the means to account for the correlation between G
reactivity and IP in GG and GGG sequences.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants CA110261 (NCI) and ES002109 (NIEHS Center grant). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1.
Footnotes
The abbreviations used are:
G, guanine radical cation;
NHE, normal hydrogen electrode; Ox, oxazolone,
2,2-diamino-4-[(2-deoxy-β-d-erythro-pentofuranosyl)-amino]-5(2H)-oxazolone;
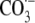 , carbonate radical anion;
8-oxo-G, 7,8-dihydro-8-oxoguanine; Fpg, E. coli formamidopyrimidine
DNA glycosylase; ExoIII, E. coli exonuclease III; Fapy-G,
2,6-diamino-5-formamido-4-hydroxypyrimidine; G, guanine;
G(-H)·, guanine neutral radical; 4-OH-G·,
4-hydroxyguanine neutral radical; 8-OH-G·, 8-hydroxyguanine
neutral radical; IP, ionization potential;
, carbonate radical anion;
8-oxo-G, 7,8-dihydro-8-oxoguanine; Fpg, E. coli formamidopyrimidine
DNA glycosylase; ExoIII, E. coli exonuclease III; Fapy-G,
2,6-diamino-5-formamido-4-hydroxypyrimidine; G, guanine;
G(-H)·, guanine neutral radical; 4-OH-G·,
4-hydroxyguanine neutral radical; 8-OH-G·, 8-hydroxyguanine
neutral radical; IP, ionization potential;
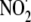 , nitrogen dioxide radical;
, nitrogen dioxide radical;
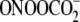 , nitrosoperoxycarbonate;
ONOO-, peroxynitrite.
, nitrosoperoxycarbonate;
ONOO-, peroxynitrite.
References
- 1.Breen, A. P., and Murphy, J. A. (1995) Free Radic. Biol. Med. 18 1033-1077 [DOI] [PubMed] [Google Scholar]
- 2.von Sonntag, C. (1987) The Chemical Basis of Radiation Biology, Taylor & Francis, New York
- 3.Roots, R., and Okada, S. (1975) Radiat. Res. 64 306-320 [PubMed] [Google Scholar]
- 4.Jones, G. D., Boswell, T. V., Lee, J., Milligan, J. R., Ward, J. F., and Weinfeld, M. (1994) Int. J. Radiat. Biol. 66 441-445 [DOI] [PubMed] [Google Scholar]
- 5.Becker, D., and Sevilla, M. D. (1993) Adv. Radiat. Biol. 17 121-180 [Google Scholar]
- 6.Cadet, J., Delatour, T., Douki, T., Gasparutto, D., Pouget, J. P., Ravanat, J. L., and Sauvaigo, S. (1999) Mutat. Res. 424 9-21 [DOI] [PubMed] [Google Scholar]
- 7.Frelon, S., Douki, T., Favier, A., and Cadet, J. (2002) J. Chem. Soc. Perkin Trans. I 2866-2870 [Google Scholar]
- 8.Yang, X., Wang, X. B., Vorpagel, E. R., and Wang, L. S. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 17588-17592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ito, K., and Kawanishi, S. (1997) Biochemistry 36 1774-1781 [DOI] [PubMed] [Google Scholar]
- 10.Henderson, P. T., Jones, D., Hampikian, G., Kan, Y., and Schuster, G. B. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 8353-8358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hall, D. B., Holmlin, R. E., and Barton, J. K. (1996) Nature 382 731-735 [DOI] [PubMed] [Google Scholar]
- 12.Saito, I., Nakamura, T., Nakatani, K., Yoshioka, Y., Yamaguchi, K., and Sugiyama, H. (1998) J. Am. Chem. Soc. 120 12686-12687 [Google Scholar]
- 13.Steenken, S., and Jovanovic, S. V. (1997) J. Am. Chem. Soc. 119 617-618 [Google Scholar]
- 14.Sugiyama, H., and Saito, I. (1996) J. Am. Chem. Soc. 118 7063-7068 [Google Scholar]
- 15.Senthilkumar, K., Grozema, F. C., Guerra, C. F., Bickelhaupt, F. M., and Siebbeles, L. D. A. (2003) J. Am. Chem. Soc. 125 13658-13659 [DOI] [PubMed] [Google Scholar]
- 16.Cullis, P. M., Davis, A. S., Malone, M. E., Podmore, I. D., and Symons, M. C. R. (1992) J. Chem. Soc. Perkins Trans. II 1409-1412 [Google Scholar]
- 17.Sevilla, M. D., Becker, D., Yan, M., and Summerfield, S. R. (1991) J. Phys. Chem. 95 3409-3415 [Google Scholar]
- 18.Bernhard, W. A. (1989) J. Phys. Chem. 93 2187-2189 [Google Scholar]
- 19.Cullis, P. M., Evans, P., and Malone, M. E. (1996) Chem. Commun. 985-986
- 20.Gregoli, S., Olast, M., and Bertinchamps, A. (1982) Radiat. Res. 89 238-254 [PubMed] [Google Scholar]
- 21.Milligan, J. R., Aguilera, J. A., and Ward, J. F. (1993) Radiat. Res. 133 151-157 [PubMed] [Google Scholar]
- 22.Ito, T., Baker, S. C., Stickley, C. D., Peak, J. G., and Peak, M. J. (1993) Int. J. Radiat. Biol. 63 289-296 [DOI] [PubMed] [Google Scholar]
- 23.Purkayastha, S., Milligan, J. R., and Bernhard, W. A. (2007) Radiat. Res. 168 357-366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Purkayastha, S., and Bernhard, W. A. (2004) J. Phys. Chem. B 108 18377-18382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steenken, S., Jovanovic, S. V., Bietti, M., and Bernhard, K. (2000) J. Am. Chem. Soc. 122 2373-2374 [Google Scholar]
- 26.Doddridge, Z. A., Cullis, P. M., Jones, G. D. D., and Malone, M. E. (1998) J. Am. Chem. Soc. 120 10998-10999 [Google Scholar]
- 27.Ding H., and Greenberg, M. M. (2007) J. Am. Chem. Soc. 129 772-773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Margolin, Y., Cloutier, J. F., Shafirovich, V., Geacintov, N. E., and Dedon, P. C. (2006) Nat. Chem. Biol. 2 365-366 [DOI] [PubMed] [Google Scholar]
- 29.Dedon, P. C. (2008) Chem. Res. Toxicol. 21 206-219 [DOI] [PubMed] [Google Scholar]
- 30.Putney, S. D., Benkovic, S. J., and Schimmel, P. R. (1981) Proc. Natl. Acad. Sci. U. S. A. 78 7350-7354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greenberg, M. M., Weledji, Y. N., Kim, J., and Bales, B. C. (2004) Biochemistry 43 8178-8183 [DOI] [PubMed] [Google Scholar]
- 32.Kow, Y. W. (1989) Biochemistry 28 3280-3287 [DOI] [PubMed] [Google Scholar]
- 33.Price, M. A., and Tullius, T. D. (1992) Methods Enzymol. 212 194-219 [DOI] [PubMed] [Google Scholar]
- 34.Murata-Kamiya, N., Kamiya, H., Muraoka, M., Kaji, H., and Kasai, H. (1997) J. Radiat. Res. 38 121-131 [DOI] [PubMed] [Google Scholar]
- 35.Dedon, P. C., and Goldberg, I. H. (1992) Chem. Res. Toxicol. 5 311-332 [DOI] [PubMed] [Google Scholar]
- 36.Balasubramanian, B., Pogozelski, W. K., and Tullius, T. D. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 9738-9743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qian, S. Y., and Buettner, G. R. (1999) Free Radic. Biol. Med. 26 1447-1456 [DOI] [PubMed] [Google Scholar]
- 38.Yoshioka, Y., Kitagawa, Y., Takano, Y., Yamaguchi, K., Nakamura, T., and Saito, I. (1999) J. Am. Chem. Soc. 121 8712-8719 [Google Scholar]
- 39.Spassky, A., and Angelov, D. (1997) Biochemistry 36 6571-6576 [DOI] [PubMed] [Google Scholar]
- 40.Haraguchi, K., Delaney, M. O., Wiederholt, C. J., Sambandam, A., Hantosi, Z., and Greenberg, M. M. (2002) J. Am. Chem. Soc. 124 3263-3269 [DOI] [PubMed] [Google Scholar]
- 41.Gasparutto, D., Ravanat, J.-L., Gerot, O., and Cadet, J. (1998) J. Am. Chem. Soc. 120 10283-10286 [Google Scholar]
- 42.Tchou, J., Kasai, H., Shibutani, S., Chung, M. H., Laval, J., Grollman, A. P., and Nishimura, S. (1991) Proc. Natl. Acad. Sci. U. S. A. 88 4690-4694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tretyakova, N. Y., Wishnok, J. S., and Tannenbaum, S. R. (2000) Chem. Res. Toxicol. 13 658-664 [DOI] [PubMed] [Google Scholar]
- 44.Tretyakova, N. Y., Burney, S., Pamir, B., Wishnok, J. S., Dedon, P. C., Wogan, G. N., and Tannenbaum, S. R. (2000) Mutat. Res. 447 287-303 [DOI] [PubMed] [Google Scholar]
- 45.Joffe, A., Geacintov, N. E., and Shafirovich, V. (2003) Chem. Res. Toxicol. 16 1528-1538 [DOI] [PubMed] [Google Scholar]
- 46.Ravanat, J. L., Saint-Pierre, C., and Cadet, J. (2003) J. Am. Chem. Soc. 125 2030-2031 [DOI] [PubMed] [Google Scholar]
- 47.Crean, C., Lee, Y. A., Yun, B. H., Geacintov, N. E., and Shafirovich, V. (2008) ChemBioChem 9 1985-1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pouget, J. P., Frelon, S., Ravanat, J. L., Testard, I., Odin, F., and Cadet, J. (2002) Radiat. Res. 157 589-595 [DOI] [PubMed] [Google Scholar]
- 49.Douki, T., Riviere, J., and Cadet, J. (2002) Chem. Res. Toxicol. 15 445-454 [DOI] [PubMed] [Google Scholar]
- 50.Paul, T., Young, M. J., Hill, I. E., and Ingold, K. U. (2000) Biochemistry 39 4129-4135 [DOI] [PubMed] [Google Scholar]
- 51.Rodriguez, H., Holmquist, G. P., D'Agostino, R., Jr., Keller, J., and Akman, S. A. (1997) Cancer Res. 57 2394-2403 [PubMed] [Google Scholar]
- 52.Luo, G. M., Qi, D. H., Zheng, Y. G., Mu, Y., Yan, G. L., Yang, T. S., and Shen, J. C. (2001) FEBS Lett. 492 29-32 [DOI] [PubMed] [Google Scholar]
- 53.Rush, J. D., and Koppenol, W. H. (1986) J. Biol. Chem. 261 6730-6733 [PubMed] [Google Scholar]
- 54.Wink, D. A., Nims, R. W., Saavedra, J. E., Utermahlen, W. E., Jr., and Ford, P. C. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 6604-6608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sawyer, D. T., Kang, C., Llobet, A., and Redman, C. (1993) J. Am. Chem. Soc. 115 5817-5818 [Google Scholar]
- 56.Koppenol, W. H., and Liebman, J. F. (1984) J. Phys. Chem. 88 99-101 [Google Scholar]
- 57.Reinke, L. A., Rau, J. M., and McCay, P. B. (1994) Free Radic. Biol. Med. 16 485-492 [DOI] [PubMed] [Google Scholar]
- 58.Welch, K. D., Davis, T. Z., and Aust, S. D. (2002) Arch. Biochem. Biophys. 397 360-369 [DOI] [PubMed] [Google Scholar]
- 59.Candeias, L. P., and Steenken, S. (2000) Chemistry 6 475-484 [DOI] [PubMed] [Google Scholar]
- 60.Cadet, J., Douki, T., and Ravanat, J. L. (2008) Acc. Chem. Res. 41 1075-1083 [DOI] [PubMed] [Google Scholar]
- 61.Misiaszek, R., Crean, C., Joffe, A., Geacintov, N. E., and Shafirovich, V. (2004) J. Biol. Chem. 279 32106-32115 [DOI] [PubMed] [Google Scholar]
- 62.Giese, B., Amaudrut, J., Kohler, A.-K., Spormann, M., and Wessely, S. (2001) Nature 412 318-320 [DOI] [PubMed] [Google Scholar]
- 63.Kobayashi, K., and Tagawa, S. (2003) J. Am. Chem. Soc. 125 10213-10218 [DOI] [PubMed] [Google Scholar]
- 64.Shafirovich, V., and Geacintov, N. E. (2004) in Long-Range Charge Transfer in DNA II (Schuster, G. B., and Beratan, D., eds) pp. 129-157, Springer-Verlag, Berlin
- 65.Cadet, J., and Berger, M. (1985) Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 47 127-143 [DOI] [PubMed] [Google Scholar]
- 66.Dizdaroglu, M., Dirksen, M. L., Simic, M. G., and Robbins, J. H. (1986) Fed. Proc. 45 1626 [Google Scholar]
- 67.von Sonntag, C. (2006) Free Radical-induced DNA Damage and Its Repair: A Chemical Perspective, Springer-Verlag, Berlin
- 68.Douki, T., and Cadet, J. (1999) Int. J. Rad. Biol. 75 571-581 [DOI] [PubMed] [Google Scholar]
- 69.Kino, K., and Sugiyama, H. (2001) Chem. Biol. 8 369-378 [DOI] [PubMed] [Google Scholar]
- 70.Matter, B., Malejka-Giganti, D., Csallany, A. S., and Tretyakova, N. (2006) Nucleic Acids Res. 34 5449-5460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Leipold, M. D., Muller, J. G., Burrows, C. J., and David, S. S. (2000) Biochemistry 39 14984-14992 [DOI] [PubMed] [Google Scholar]
- 72.Cadet, J., Douki, T., Gasparutto, D., and Ravanat, J. L. (2003) Mutat. Res. 531 5-23 [DOI] [PubMed] [Google Scholar]
- 73.Cadet, J., Berger, M., Buchko, G. W., Joshi, P. C., Raoul, S., and Ravanat, J.-L. (1994) J. Am. Chem. Soc. 116 7403-7404 [Google Scholar]
- 74.Dedon, P. C., and Tannenbaum, S. R. (2004) Arch. Biochem. Biophys. 423 12-22 [DOI] [PubMed] [Google Scholar]
- 75.Box, H. C. (2001) Radiat. Res. 155 634-636 [DOI] [PubMed] [Google Scholar]
- 76.Box, H. C., Dawidzik, J. B., Wallace, J. C., Hauer, C. R., Stack, R. F., Rajecki, M. J., and Budzinski, E. E. (1999) Radiat. Res. 152 575-582 [PubMed] [Google Scholar]
- 77.Packer, M. J., Dauncey, M. P., and Hunter, C. A. (2000) J. Mol. Biol. 295 85-103 [DOI] [PubMed] [Google Scholar]
- 78.Delaney, J. C., and Essigmann, J. M. (2008) Chem. Res. Toxicol. 21 232-252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McCulloch, S. D., and Kunkel, T. A. (2008) Cell Res. 18 148-161 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.