

Abstract
The COOH-terminal polybasic region (PBR) of Rac1, a Rho family GTPase member, is required for Rac1 self-association, membrane localization, nuclear translocation, and interaction with downstream effectors. We previously demonstrated that phosphatidylinositol-4-phosphate 5-kinase, one of the effectors that requires the polybasic region for interaction, is necessary for efficient invasin-promoted uptake of Yersinia pseudotuberculosis by nonphagocytic cells. Here we further examined the role of this region in invasin-promoted uptake. Using fluorescence resonance energy transfer experiments (FRET), we determined that engagement of integrin receptors by invasin caused elevated levels of Rac1 self-association at the site of bacterial adhesion in a PBR-dependent fashion. Self-association could be disrupted using several strategies: translocation of the Yersinia YopT prenylcysteine protease into host cells, inactivation of the Rac1 isoprenylation signal that is required for membrane localization, and elimination of the PBR. Disruption in each case impaired invasin-promoted uptake. To determine if there is a role for the PBR in Rac1 effector signaling that was independent of its role in membrane localization or multimerization, we examined the effect of the PBR in the context of a Rac1 derivative that was targeted to the membrane via an NH2-terminal lipid tail. The membrane-targeted Rac1 derivative restored significant invasin-promoted bacterial uptake in a PBR-dependent manner and yet displayed no detectable self-association. This study indicates that, in addition to its role in promoting membrane localization, the PBR exerts a positive effect on Rac1-controlled bacterial uptake that is independent of Rac1 self-association, most likely due to signaling to downstream effectors.
Uptake of pathogenic bacteria by normally nonphagocytic cells is uniformly regulated by members of the Rho GTPase family, including Cdc42, Rac1, and RhoA (1). In the case of the Gram-negative enteropathogenic bacterium Yersinia pseudotuberculosis, Rac1 is required for uptake, whereas Cdc42 and RhoA play either no role or a negative role, respectively (2-4). Rac1 can facilitate bacterial uptake by remodeling the actin cytoskeleton through one of three mechanisms: 1) inducing actin filament nucleation and branching by activating the Arp2/3 complex via WAVE family members (5); 2) increasing phosphoinositol 4,5-bisphosphate concentrations in the plasma membrane, resulting in uncapping of actin filaments (6, 7); or 3) inhibiting actin depolymerization by activating LIM kinases, which deactivate cofilin (8).
Activation of Rac1 requires GTP loading by guanine nucleotide exchange factors (RacGEFs), which show specificity for subclasses of Rho family members (9). Exchange takes place simultaneously with release of Rac1 from RhoGDI proteins, which maintain Rho family GTPases in an inactive state in the cell cytoplasm (10). Release allows insertion of Rac1 in a target membrane via a prenyl group linked to the carboxyl terminus of the protein (11). After exchange and insertion into the membrane, active Rac1 is able to bind downstream effectors, many of which modulate the actin dynamics associated with bacterial uptake (12). The activation observed is often a response to engagement of cell surface receptors, resulting in interaction with downstream effectors (13, 14). One example of a group of host cell surface molecules that activate Rac1 in response to substrate engagement is the β1 integrin receptor family, the members of which bind envelope proteins encoded by a wide range of pathogenic microorganisms (15, 16).
Y. pseudotuberculosis undergoes high efficiency bacterial uptake after engagement of β1 integrin receptors by the bacterial cell surface protein invasin (17). Invasin binds integrins with a much higher affinity than natural ligands, such as fibronectin and laminin (18). Invasin is also able to form multimers, which is predicted to allow receptor clustering, thought to be a prerequisite for triggering intracellular signaling processes required for bacterial uptake (19). The combined activities of high affinity binding and multimerization by invasin are critical for high efficiency invasin-mediated bacterial uptake that is regulated by activated Rac1 (19, 20).
Engagement of β1 integrins by Y. pseudotuberculosis triggers efficient recruitment of Rac1 to nascent phagosomal membranes, resulting in localized accumulation of the activated GTPase, as determined by FRET4 analysis (2). Although the most attractive model for Rac1 function at the phagocytic cup is that localized activation of Rac1 occurs at sites of receptor engagement, it is possible that active Rac1 is simply delivered to these sites by release of the GTP-loaded form from their soluble RhoGDI-bound complexes in the host cell cytoplasm. The latter possibility was suggested from a study in which fibronectin-coated beads were used to challenge cultured cells (14). This raises the possibility that Rac1-GTP can be sequestered by cytosolic RhoGDI and then directly delivered to the site of receptor clustering without a membrane-dependent activation step.
An additional mechanism for regulating the activity of Rac1 has been proposed. Gel filtration studies and co-immunoprecipitation experiments indicated that the polybasic region (PBR) at the COOH terminus of Rac1 mediates self-association of Rac1 (21). This self-association is independent of the nucleotide status of Rac1. It has been suggested that PBR-mediated self-association potentiates Rac1-GTP to activate effectors, based on the observation that Rac1 derivatives lacking the PBR are defective for activation of the serine/threonine kinase PAK1 (21). If local engagement of β1 integrin receptors indeed triggers a localized RhoGDI release from Rac1, there should also be an induction of Rac1 self-association at the sites of integrin engagement.
In this report, we investigate the role of the PBR in supporting invasin-mediated uptake of Y. pseudotuberculosis and identify sequence elements that are important for Rac1 self-association. Using derivatives that allow membrane localization of Rac1 without the presence of the PBR, we provide evidence that the role of this sequence in the uptake process appears to be independent of its role in self-association, presumably because the PBR is necessary to interact with downstream effectors or guanine nucleotide exchange factors.
MATERIALS AND METHODS
Cell Culture, Transfection, and Plasmid Constructs—Culture and transfection of COS1 cells were performed as previously described (2). Mammalian expression plasmids pmCFP-Rac1 and pmYFP-Rac1 encoding the NH2-terminal fusion of monomeric cyan fluorescence protein (CFP) or yellow fluorescence protein (YFP) to Rac1 as well as Rac1 derivatives having the G12V, R66A, C189S, or 6Q (183KKRKRK → 183QQQQQQ) mutations have been described (22, 23). The K186E mutation in Rac1 was generated using the Stratagene (La Jolla, CA) QuikChange site-directed mutagenesis kit. pLyn-mCFP consists of a 10-amino acid myristoylation/palmitoylation sequence of Lyn kinase fused to the 5′-end of the mCFP gene (6). pLyn-mYFP was generated by replacing mCFP with mYFP. Rac1(C189S) or Rac1(6Q/C189S) devoid of the geranylgeranylation signal were inserted in frame into the COOH terminus of pLyn-mCFP to generate pLyn-mCFP-Rac1(C189S) or pLyn-mCFP-Rac1(6Q/C189S). mCFP-GerGer and mYFP-GerGer containing fluorescence protein fused to the CAAX geranylgeranylation signals, without the upstream polybasic region, were kindly provided by Dr. R. Tsien (University of California, San Diego) (24).
Plasmids encoding HA-mYFP, Lyn-HA-mYFP, Myc-mCFP, and Lyn-Myc-mCFP fusions were constructed by replacing the enhanced green fluorescent protein gene in pEGFP-C1 (Clontech) with each tag-encoded gene indicated. Various Rac1 alleles (WT, R66A, 6Q, C189S, and 6Q/C189S) were then cloned into all four plasmids. All plasmids were verified by sequencing. Oligonucleotide sequences are available upon request. Rac1 derivatives used in this study and their properties are described in Table 1.
TABLE 1.
Rac1 derivatives used in this study
| Constructions | Properties | Predicted localization |
|---|---|---|
| mCFP-Rac1 (WT) | Wild type | Cytoplasm and membrane |
| mYFP-Rac1 (WT) | Wild type | Cytoplasm and membrane |
| mYFP-Rac1 (6Q) | Missing Rac1 PBR | Cytoplasm |
| mCFP-Rac1 (G12V) | GTP hydrolysis-defective | Cytoplasm and membrane |
| mCFP-Rac1 (R66A) | Defective for RhoGDI binding | Predominantly membrane |
| mCFP-Rac1 (C189S) | Missing prenylation site | Nucleus and cytoplasm |
| mYFP-Rac1 (K186E) | Defective for PIP5K binding | Membrane and cytoplasm |
| mCFP-CAAX | Prenylated mCFP | Membrane |
| mYFP-CAAX | Prenylated mYFP | Membrane |
| mCFP-Rac1 (G12V) (6Q) | GTP hydrolysis-defective; missing PBR | Cytoplasm |
| mCFP-Rac1 (G12V) (C189S) | GTP hydrolysis defective; missing prenylation site | Cytoplasm and nucleus |
| mCFP-Rac1 (G12V) (K186E) | GTP hydrolysis-defective; defective for PIP5K binding | Membrane and cytoplasm |
| Lyn-mCFP-Rac1 (6Q) (C189S) | Myristoylated; missing PBR and prenylation site | Membrane |
| Lyn-mCFP-Rac1 (C189S) | Myristoylated; missing prenylation site | Membrane |
| Lyn-mYFP-Rac1 (C189S) | Myristoylated; missing prenylation site | Membrane |
| Lyn-mCFP-Rac1 (G12V) (K186E) | Myristoylated; GTP hydrolysis-defective; defective for PIP5K binding | Membrane |
| Lyn-mYFP-Rac1 (G12V) (K186E) | Myristoylated; GTP hydrolysis-defective; defective for PIP5K binding | Membrane |
| Lyn-mYFP-Rac1 (G12V) (C189S) | Myristoylated; GTP hydrolysis-defective; missing prenylation site | Membrane |
| Lyn-mCFP-Rac1 (G12V) (C189S) | Myristoylated; GTP hydrolysis defective; missing prenylation site | Membrane |
| Lyn-mCFP-Rac1 (6Q) (C189S) | Myristoylated; missing PBR and prenylation site | Membrane |
| Lyn-mYFP-Rac1 (6Q) (C189S) | Myristoylated; missing PBR and prenylation site | Membrane |
| Lyn-mCFP-Rac1 (G12V) (6Q) (C189S) | Myristoylated; GTP hydrolysis-defective; missing PBR and prenylation site | Membrane |
| Lyn-mYFP-Rac1 (G12V) (6Q) (C189S) | Myristoylated; GTP hydrolysis-defective; missing PBR and prenylation site | Membrane |
| Myc-mCFP-Rac1 (WT) | Wild type | Cytoplasm and membrane |
| HA-mYFP-Rac1 (WT) | Wild type | Cytoplasm and membrane |
| Myc-mCFP-Rac1 (6Q) | Missing PBR | Cytoplasm |
| Myc-mCFP-Rac1 (C189S) | GTP hydrolysis-defective; missing prenylation site | Cytoplasm and nucleus |
| Myc-mCFP-Rac1 (R66A) | Defective for RhoGDI binding | Predominantly membrane |
| Lyn-Myc-mCFP-Rac1 (6Q) | Myristoylated; missing PBR | Membrane |
| Lyn-HA-mYFP-Rac1 (6Q) | Myristoylated; missing PBR | Membrane |
| Lyn-HA-mYFP-Rac1 (C189S) | Myristoylated; missing prenylation site | Membrane |
| Lyn-Myc-mCFP-Rac1 (C189S) | Myristoylated; missing prenylation site | Membrane |
| Lyn-HA-mYFP-Rac1 (6Q) (C189S) | Myristoylated; missing PBR; missing prenylation site | Membrane |
| Lyn-Myc-mCFP-Rac1 (6Q) (C189S) | Myristoylated; missing PBR and prenylation site | Membrane |
| Lyn-Myc-mCFP | Myristoylated mCFP | Membrane |
| Lyn-HA-mYFP | Myristoylated mYFP | Membrane |
Culture of Y. pseudotuberculosis Infection of Mammalian Cells and Immunofluorescence Protection Assay of Bacterial Uptake—Conditions for growth of virulence plasmid-cured Y. pseudotuberculosis YPIII(p-) with or without YopE or YopT and infection of COS1 have been described (22). The plasmid-cured Y. pseudotuberculosis strain lacks the virulence plasmid (pYV) that encodes YopE and YopT and is efficiently internalized into host cells. Strains that harbor the YopE Rho family GAP (22) or the YopT family CAAX protease (kind gift of Dr. James Bliska, SUNY Stony Brook) contain plasmids encoding these proteins as well as the plasmid pYV (yopT-deficient yopE::kan yopH::cam; referred to as strain YP17) to allow translocation of these proteins via the bacterial type III secretion system (22).
For bacteria lacking the virulence plasmid, the YPIII(p-) strain was grown logarithmically in Luria Bertani broth at 26 °C until an A600 of 0.7, prior to inoculation onto cultured mammalian cells (22). For strains harboring the pYV plasmid and Yop-encoding plasmids, bacteria were grown with aeration at 26 °C overnight in broth supplemented with 2.5 mm CaCl2 and 100 μg/ml ampicillin and then subcultured and grown at 26 °C until A600 of 0.2. At this point, the cultures were shifted to 37 °C and aerated for 1 h. A multiplicity of infection of 50:1 was used for YPIII(p-) incubations, and a multiplicity of infection of 25:1 was used for other derivatives. For the pYopE-expressing plasmid, 0.1 mm isopropyl-β-d-thiogalactopyranoside was supplemented during infection to induce YopE expression.
Bacterial uptake was assayed using immunofluorescence protection as described (6). Briefly, bacteria appropriately cultured were incubated with transfected cells for 30 min at a multiplicity of infection of 50 at 37 °C. After incubation, the adherent cells were analyzed for internalized or surface-bound bacteria as described previously (6). The monolayers were fixed in 3% paraformaldehyde and probed with primary antibody directed against Y. pseudotuberculosis, followed by a fluorescent secondary antibody (anti-IgG conjugated to either Alexa Fluor 594 or Cascade Blue) to detect extracellular bacteria. The cells were then permeabilized (2) and probed with antibodies directed against the bacteria to allow detection of both intracellular and extracellular bacteria. The coverslips were then probed with appropriate secondary antibodies to detect intracellular bacteria.
FRET Measurements—The basis of the assay is that association between Rac1 derivatives fused to either monomeric CFP or monomeric YFP should be detected as a FRET readout. COS1 cells were first transfected with a combination of various derivatives of mCFP-Rac1 and mYFP-Rac1 and then challenged with bacteria. Infections were stopped by fixation after 30 min. The cells were then imaged and analyzed for sensitized FRET from CFP to YFP essentially as described (22), using correction factors for CFP (0.32) and YFP (0.18) for bleed-through from CFP emission and cross-YFP excitation by the FRET filter set. To measure FRET, images from YFP, CFP, and FRET filter sets (22) were captured, choosing regions of interest about nascent phagosomes. Sensitized FRET was then calculated from these regions, by subtracting the CFP and YFP correction factors, using exactly the same procedure as described previously (22). FRET signals were normalized by combination of mCFP and mYFP emissions calculated from the CFP and YFP filter sets using the following formula as described (25).
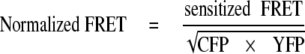 |
(Eq.1) |
Determination of mYFP-Rac1 Expression Levels Relative to Endogenous Rac1—COS1 cells were transfected with mYFP-Rac1 and cultured overnight. Transfected cells were lifted and subjected to flow cytometry using YFP fluorescence to sort cells into four fractions according to levels of fluorescence (YFP negative, low, medium, and high). A portion of the sorted cells were plated onto fibronectin-coated coverslips, allowed to adhere for ∼3 h, fixed in 4% paraformaldehyde, and imaged to quantify YFP fluorescence. YFP fluorescence quantification was performed as described above for FRET experiments. The remaining sorted cells were lysed in sample buffer; lysates were resolved by SDS-PAGE, blotted, and probed for Rac1 using a monoclonal anti-Rac1 antibody (clone 23A8; Sigma).
Immunoprecipitation of Myc/HA-tagged mCFP/mYFP Fusions—293T cells were transfected with 0.5 μg of each plasmid in 6-well dishes. Bait constructs were Myc-tagged, and prey constructs were HA-tagged. Cells were lysed 24 h post-transfection in 500 μl of lysis buffer (20 mm Tris, pH 7.5, 150 mm NaCl, 1% Nonidet P-40, and complete protease inhibitor mixture (Roche Applied Science)). Lysates were then incubated at 4 °C (with agitation) for 10 min and spun to remove debris. 50 μl of the cleared lysates was saved as the input fraction, and 400 μl was applied to washed anti-HA epitope affinity resin (monoclonal anti-HA; Sigma) and incubated at 4 °C (with agitation) for 1 h. The resin was washed three times in wash buffer (25 mm Tris, pH 7.5, 30 mm MgCl2, 40 mm NaCl, 0.1% Nonidet P-40), and bound proteins were eluted in 100 μl of sample buffer. 10 μl of eluted proteins (immunoprecipitate) as well as 10 μl of cleared lysates (input) were analyzed by SDS-PAGE and Western blotting. Input and immunoprecipitated proteins were detected using an anti-Myc epitope antibody (rabbit polyclonal; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) as well as an anti-HA epitope antibody (rabbit polyclonal; Santa Cruz Biotechnology).
RESULTS
Rac1 Self-associates in Cultured Cells—Y. pseudotuberculosis employs a variety of proteins to activate and misregulate Rac1 in host cells (22). In this report, we examined whether Y. pseudotuberculosis could also control self-association of Rac1 (21). Previously, self-association was demonstrated in vitro by gel filtration chromatography and immunoprecipitation of differentially tagged Rac1 derivatives in an event requiring the COOH-terminal PBR (Fig. 1A) (21). To determine if self-association occurs in cells and can be detected at spatially distinct sites within the cell, Rac1 self-association was analyzed by a FRET-based system, using monomeric CFP and YFP derivatives (mCFP-Rac1 and mYFP-Rac1) (Fig. 1C). When coexpressed in the same cells, mCFP-Rac1(WT) and mYFP-Rac1(WT) produced FRET throughout the cells in all regions except for the nucleus (Fig. 1, B and C). When normalized to the concentration of Rac1 at individual sites in the cell, FRET signals (normalized FRET) were notably stronger at membrane ruffles, indicating enhanced Rac1 self-association (Fig. 1C, arrows). These elevated FRET levels were specific to these sites, because at regions where no ruffles were evident, the lower levels of normalized FRET were independent of Rac1 concentrations.
FIGURE 1.

Rac1 self-association requires determinants important for plasma membrane localization. A, schematic of Rac1 derivatives used in this work. SW1 and SW2, Switch I and Switch II regions. Gly12 (G12), Arg66 (R66), and Lys186 (K186) are the sites of constitutively active, RhoGDI binding-defective, and PIP5Kα binding-defective mutations, respectively. KKRKRK, polybasic region (underlined). Cys189 (C189) is the site of Rac1 geranylgeranylation. B, reduced self-association in the absence of membrane localization or loss of the PBR. The mCFP- or mYFP-Rac1 derivatives noted in B were cotransfected into COS1 cells, and FRET measurements were determined (see “Materials and Methods”). Sensitized FRET from 20 regions of interest (see “Materials and Methods”) were plotted as a function of the intensity of the mCFP-Rac1 donor and mYFP-Rac1 acceptor. C-G, left, schematic diagrams for detecting Rac1-Rac1 self-association using FRET. Monomeric forms of CFP and YFP were fused to the NH2 terminus of Rac1, as displayed in schematic diagrams. Derivatives in which mYFP-Rac1 is defective for plasma membrane localization (e.g. Rac1 6Q and Rac1 C189S) are defective for interaction with wild-type Rac1 on the plasma membrane and thus failed to produce FRET. Right, COS1 cells were transfected with mCFP-Rac1 and mYFP-Rac1 having the indicated mutations. The cells were fixed and analyzed by FRET using fluorescence microscopy (see “Materials and Methods”). Sensitized FRET (sens. FRET) corresponds to the total amount of FRET, whereas normalized FRET (norm. FRET) corresponds to the amount of FRET per unit of fluorescent protein (see “Materials and Methods”). Both sensitized FRET and normalized FRET are displayed as color gradient look-up tables, with the appropriate scale bars below each panel. The arrows indicate membrane ruffles that were concentrated at sites of Rac1 recruitment. White scale bar in G, 10 μm (applies to all panels).
Consistent with biochemical data, self-association of Rac1, as detected by FRET, required the presence of the PBR on both partners. Co-transfection of mCFP-Rac1(WT) with mYFP-Rac1(6Q), which has each of the basic residues in the PBR replaced with Gln (26), produced a lower FRET signal (Fig. 1, B and D; 6Q), confirming the previous published findings that the PBR mediates Rac1 self-association in vitro (21).
Self-association Is Not Due to Overexpression of Rac1 Constructs—Since any self-association observed by FRET could result from aggregation of overexpressed protein, the concentration of Rac1 expressed from the transfected plasmids relative to endogenous levels of Rac1 was determined. COS1 cells were transfected with mYFP-Rac1 and sorted by flow cytometry to determine the concentration of Rac1 relative to the amount of YFP fluorescence observed by microscopy. The transfected cells showed a broad distribution of fluorescence (Fig. 2A), with a peak of untransfected cells and a shoulder of cells having increasing amounts of fluorescence. The total cell population was collected into four separate fractions consisting of cells with increasing amounts of YFP fluorescence (neg, lo, med, hi; Fig. 2A), and each population was analyzed to determine the amount of mYFP-Rac1 expression as well as the level of fluorescence using our standard microscopic techniques.
FIGURE 2.

FRET analysis of Rac1 self-association was performed at physiological Rac1 levels. A, mYFP-Rac1-transfected COS1 cells were sorted according to fluorescence (negative (neg), low fluorescence (lo), medium (med), and high (hi)). B, sorted cells were lysed and analyzed by Western blotting, probing with a monoclonal Rac1 antibody (top). Blots were scanned, and the extent of overexpression with respect to endogenous Rac1 levels was quantified by densitometry (bottom). C, sorted cells were plated onto fibronectin-coated coverslips and allowed to adhere. YFP fluorescence images were collected from low and medium fluorescence populations. Two representative images are displayed where the med image has been normalized to the fluorescence maximum of the lo image, as indicated by the look-up table. White scale bar, 10 μm. D, fluorescence intensity of the low and medium fluorescence populations was quantified exactly as the FRET analysis. Data are displayed as average YFP fluorescence ± S.E. on a per cell basis. Average YFP fluorescence from a typical FRET experiment is included in this graph for comparison purposes.
The expression of mYFP-Rac1 in the YFP-lo fraction was lower than that of endogenous Rac1, based on Western blotting with anti-Rac1 antibody. The levels of mYFP-Rac1 were about 50% of the levels of endogenous Rac1 in this population (Fig. 2B; YFP lo). In contrast, the other sorted fractions (Fig. 2B; YFP med and YFP hi) expressed much higher levels of YFP-Rac1 compared with endogenous Rac1. Transfectants plated onto coverslips from the YFP-lo population clearly had fluorescence levels similar to those being used for FRET analysis (Fig. 2, C and D; compare lo versus med). When the fluorescence was determined on individual cells from the mYFP-lo fraction, the average intensity of individual cells was ∼500 fluorescence units (Fig. 2D), which is about the mean intensity of all the cells used for FRET (Fig. 2D; mYFP-Rac1(FRET)). In fact, the cell populations that showed overexpression of YFP-Rac1 gave fluorescence intensities that were well beyond what was analyzed by FRET (Fig. 2D; med). Therefore, a large proportion of the cells analyzed by FRET expressed amounts of mYFP-Rac1 that are at or below endogenous levels of Rac1. We conclude that self-association is not due to overexpression of the fluorescence derivatives.
Plasma Membrane Localization of Rac1 Is Not Sufficient to Promote Self-association—The 6Q mutation that replaces the PBR has the secondary effect of interfering with plasma membrane localization of Rac1 (14) (Fig. 3, compare A with E). We therefore tested whether blocking plasma membrane localization would affect the ability of Rac1 to self-associate. The C189S mutation, immediately downstream of PBR, prevents geranylgeranylation of Rac1 at the carboxyl-terminal CAAX box and resulted in a large pool of the protein localizing in the nucleus, as previously described (27) (Fig. 3C). This lipid modification is essential for the Ras superfamily of small GTPases to anchor onto the plasma membrane (28). Based on the FRET assay, mCFP-Rac1(C189S) showed no ability to interact with mYFP-Rac1(WT) (Fig. 1, B and E; C189S), indicating the importance of plasma membrane localization for Rac1 self-association. The C189S mutation also blocks RhoGDI binding (29), raising the possibility that RhoGDI could play some role in Rac1 self-association. To rule out this possibility, the interaction of the Rac1(R66A) mutant was analyzed, which is unable to bind RhoGDI but maintains plasma membrane localization (22, 30). mCFP-Rac1(R66A) produced FRET with mYFP-Rac1(WT) as efficiently as was observed for the interaction between the fluorescent wild type derivatives of Rac (Fig. 1, B and F; R66A). Thus, plasma membrane localization, but not RhoGDI binding via the CAAX geranylgeranylation site, was necessary for Rac1 self-association.
FIGURE 3.

Rac1 plasma membrane localization requires prenylation and an intact PBR. mYFP-Rac1 fusions were expressed in COS1 cells and imaged microscopically for YFP fluorescence. A, mYFP-Rac1(WT); B, mYFP-Rac1(K186E), defective for effector (PIP5Kα) binding; C, mYFP-Rac1(C189S), lacking prenylation; D, Lyn-mYFP-Rac1(C189S), myristoylated derivative; E, mYFP-Rac1(6Q), defective PBR; F, Lyn-mYFP-Rac1(6Q)(C189S); G, mYFP; H, Lyn-mYFP, myristoylated mYFP. Note that Lyn myristoylation targets proteins localized nuclearly (C) or perinuclearly (E) to the plasma membrane (F and H, respectively). White scale bar in H, 20 μm (applies to all panels).
To determine whether plasma membrane localization was sufficient to cause intermolecular FRET in this particular assay, we targeted mCFP/mYFP onto the plasma membrane by either an NH2-terminal myristoylation signal from the Lyn protein (24) or the COOH-terminal geranylgeranylation site from Rac1 (see Fig. 3H for Lyn-mYFP localization). Neither of the two tags could enable the CFP/YFP pair to generate FRET (Fig. 4, compare A with B and C; see Fig. 5, A-C and I) even at high concentrations (Fig. 5, B and C). This lowered self-association was observed, in addition, with cells that were challenged with Y. pseudotuberculosis, since no FRET signal could be detected at regions of membrane aggregation resulting from invasin binding or at regions of membrane ruffling (see below; Fig. 7, B and C). We conclude that plasma membrane localization was not sufficient to cause protein self-association.
FIGURE 4.

The polybasic region is not sufficient to allow Rac1 self-association of membrane-localized protein. Schematic diagrams on the left represent the pairs of fluorescent proteins used for FRET analysis in COS1 cells. Derivatives used were mCFP-Rac1(WT) and mYFP-Rac1(WT) (A); the mCFP-GerGer and mYFP-GerGer pair having CAAX boxes at the carboxyl termini of the fluorescent proteins, lacking all other Rac1 sequences (24) (B); Lyn-mCFP and Lyn-mYFP, fluorescent proteins localized in the membrane via the NH2-terminal myristoylation site (C); Lyn-mCFP-Rac1-6Q(C189S) and Lyn-mYFP-Rac1-6Q(C189S), localizing hybrid proteins in the membrane via the NH2-terminal myristoylation site (D); and Lyn-mCFP-Rac1(C189S) and Lyn-mYFP-Rac1(C189S), in which CAAX box defective mutants with intact PBRs are forced into the plasma membrane by the Lyn myristoylation signal (E). Constructs encoding these proteins were transfected into COS1 cells, fixed the day after transfection, and subjected to FRET microscopy. Representative images of the CFP and YFP channels, along with the sensitized FRET (sens. FRET) are displayed as color gradient look-up tables, using the displayed scale. White scale bar in E, 10 μm (applies to all panels).
FIGURE 5.

Rac1 self-association of Rac1(WT) is independent of expression levels. A-H, COS1 cells were cotransfected with mCFP-Rac1 and mYFP-Rac1 constructs, and Rac1 self-association was determined by FRET in two regions of interest (ROI) within each cell. Each data point represents results from a single cell, and data are plotted for 20 cells per construct (see “Materials and Methods”). Sensitized FRET determinations were then normalized to the total concentration of both derivatives, calculated as (mCFP-Rac1 × mYFP-Rac1)1/2, and data were plotted as a function of the concentration of the derivatives in each of the regions of interest. Each panel shows data for the noted derivatives. I, summary of data from A-H. Data are plotted as the mean of the normalized FRET determinations ± S.E.; pooling data from all of the analyzed transfectants are displayed in A-H.
FIGURE 7.

Rac1 self-association is stimulated by Yersinia infection. COS1 cells were transfected with mCFP-Rac1 and mYFP-Rac1 constructs (A and D), mCFP-GerGer and mYFP-GerGer (B), or Lyn-mCFP-Rac1(C189S) and Lyn-mYFP-Rac1(C189S) (C). Transfected cells were challenged the next day with a 30-min incubation of YPIII(p-) (A-C) or YP17/pYopT (D), followed by immunostaining of extracellular bacteria (Ext. bacteria) bound onto host cells. Sensitized and normalized FRET readings (sens. FRET and norm. FRET, respectively) were determined as described (see “Materials and Methods”). The arrows denote nascent phagosomes. E, mean normalized FRET levels at nascent phagosomes were plotted comparing mCFP-Rac1/mYFP-Rac1 and mCFP-GerGer/mYFP-GerGer as described in the legend to Fig. 3I. White scale bar in D, 5 μm (applies to all panels).
Forcing plasma membrane localization of proteins that do not interact also was not sufficient to cause protein self-association. The addition of the Lyn myristoylation signal to either Rac1(C189S) (Fig. 3, compare C with D) or Rac1(6Q/C189S) (Fig. 3, compare E with F) resulted in plasma membrane localization of these derivatives. Neither Lyn-modified derivative generated a significant FRET signal for Rac1-Rac1 interaction (Fig. 4, D and E, and Fig. 5, D, E, and I). Therefore, the aberrant localization of these Rac1 derivatives to sites such as the nucleus and perinuclear locales (Fig. 3) does not explain the inability of these proteins to self-associate, since even when they are plasma membrane-localized, they could not self-associate. Additionally, GTPase activation does not cause self-association under these conditions, because constitutively active membrane-targeted Rac1 variants lacking the appropriate carboxyl-terminal sequences fail to generate a FRET signal (Fig. 5, F, G, and I).
Lowering Affinity for Effectors Reduces Rac1 Self-association—The PBR has been characterized to have a role in binding effectors, such as phosphatidylinositol-4-phosphate 5-kinase-α (PIP5Kα) and protein kinase C-related kinase 1 (7, 31). To determine whether disruption of effector recognition reduces Rac1 self-association, we introduced the K186E mutation into Rac1, which has been demonstrated to block the binding of PIP5Kα without affecting plasma membrane localization of Rac1 (7) (Fig. 3B). A lower level of mCFP-Rac1(K186E) self-association was observed relative to wild type (Figs. 1G and 5, H and I). Therefore, maximum Rac1 self-association depended on sequence determinants that are also important for effector binding (residue Lys186) as well as geranylgeranylation (residue Cys189).
Formation of a Stable Rac1-Rac1 Complex Requires the Presence of the PBR and Prenylation Signal—To support the data from the FRET assay, an independent test of Rac1 self-association was performed using a coimmunoprecipitation procedure (Fig. 6A). The power of the FRET readout is that it provides spatial information and may allow the detection of transient interactions. If the observed self-association is transient, then it may not survive other detection strategies, such as immunoaffinity techniques, that require slow dissociation rates. To determine if interaction could be observed using more restrictive conditions, several of the constructs used in the microscopic assay were tagged with either Myc or HA epitopes and expressed in mammalian cells (Fig. 6A). For each condition, two constructs were co-transfected into 293T cells, each having different tags to allow detection of self-association. Extracts from the transfectants were then subjected to immunoprecipitation with anti-HA, followed by immunoblotting with anti-Myc, to determine if a fraction of the Myc-tagged partner was found in the precipitate (see “Materials and Methods”). When HA-tagged CFP-Rac1(WT) was immunoprecipitated, it was able to pellet either Myc-tagged YFP-Rac1(WT) or Myc-tagged YFP-Rac1(R66A) based on immunoblotting, consistent with the FRET observations (Fig. 6, B-F). On the other hand, any disruption of the prenylation site (Rac1(C189S)) or the PBR (Rac1(6Q)) was sufficient to interfere with co-immunoprecipitation (Fig. 6, B-F). This loss of association was true even if only one of the partners was lacking the critical carboxyl-terminal sequences (Fig. 6, B and F; wt + 6Q and wt + C189S) or if membrane localization was forced by adding a Lyn myristoylation site (Fig. 6, B and F; Lyn constructs). Therefore, there is concordance between the two assays for detecting interaction, since Rac1 self-association is sufficiently long lived to survive detergent extraction and immunoprecipitation. Furthermore, these results provide direct evidence that Rac1 self-association requires the PBR and COOH-terminal prenylation signal.
FIGURE 6.

Stable self-association of Rac1 derivatives shows identical profile to FRET assay. A, HA (YFP derivatives) or Myc (CFP derivatives) epitope tags were introduced into each of the constructs used in the FRET assays and used in co-immunoprecipitation experiments to detect stable self-association. Displayed are maps of each of the constructs used. To measure self-association, the noted CFP and YFP derivatives were co-transfected into 293T cells, and lysates were subjected to immunoprecipitation (IP) with anti-HA IgG to precipitate YFP derivatives. Association with the noted CFP derivatives was detected after gel fractionation and Western blotting (IB) with anti-Myc IgG (B), and the total amount precipitated was determined by blotting with HA (D). As controls to determine relative efficiency of immunoprecipitation, the lysates were probed with anti-Myc (C) or anti-HA (E), loading 25% of the total fraction available for immunoprecipitation. IP, immunoprecipitate, noting antibody used. IB, immunoblot, noting antibody used. F, quantitation of the fraction of Rac1 that self-associates. Bands from B and C were subjected to densitometry scanning to determine the fraction of Myc-tagged protein that associated with HA-tagged Rac1.
Invasin Binding Results in Localized Rac1 Self-association—Engagement of β1 integrin receptors by the Y. pseudotuberculosis outer membrane protein invasin triggers the recruitment of Rac1 onto the phagosomal membrane (6). To examine the degree of self-association of Rac1 in response to invasin binding, we visualized Rac1 clustered about Y. pseudotuberculosis associated with COS1 after a 30-min incubation with bacteria. To analyze self-association, cells were challenged with a bacterial strain that expressed invasin as the only adhesin and lacks the ability to translocate Rac1-inactivating Yops (33). FRET analysis showed that the mCFP-Rac1 recruited around nascent phagosomes had elevated levels of self-association in comparison with the nearby Rac1 located at regions distal from the phagosomes (Fig. 7A). Rac1 on membrane ruffles displayed similar higher efficiency of self-association (data not shown; Fig. 1). This enhanced self-association was not due to simple recruitment of mCFP-Rac1 and mYFP-Rac1 to the active sites, because the FRET levels were still elevated after the FRET signals were normalized against the concentration of mCFP-Rac1, indicating that the fraction of Rac1 undergoing self-interaction is higher at the site of bacterial adhesion than at other sites (Fig. 7, compare A and B; see arrows). In addition, the increased FRET signal was dependent on the presence of Rac1 in the fusion constructs as well as the CAAX motif. When the simple mCFP-GerGer/mYFP-GerGer pair or the Lyn-mCFP-Rac1(C189S)/Lyn-mYFP-Rac1(C189S) pair was analyzed, neither was able to generate a FRET signal at any site in the host cells even on nascent phagosomes associated with sites of bacterial adhesion (Fig. 7, B and C; quantified in Fig. 7E). This is despite the fact that the CAAX-containing constructs showed evidence of concentration about the phagosomal cups (Fig. 7, A and B). Therefore, enhanced Rac1 self-association was not a result of increased membrane density resulting from invasin engagement of integrins but was a property of the presence of an intact Rac1 protein.
Removal of the CAAX Motif by Yersinia YopT Protease Destroys Rac1 Self-association—The above data argue that the CAAX prenylation motif and the PBR collaborate to promote Rac1 self-association. One possibility for how this occurs is that membrane insertion resulting from prenylation facilitates close contact between Rac1 monomers, stimulating PBR self-association. The other possibility is that insertion in the membrane is required to maintain self-association. To determine if membrane insertion was required for maintenance of the interaction, we analyzed the consequences of translocating the Y. pseudotuberculosis YopT protein into host cells on Rac1 self-association. The YopT protease activity targets membrane-localized Rac1 (13), cleaving just upstream of the CAAX box, releasing Rac1 from the membrane. If the role of the prenyl group is limited to initiating Rac1 association, then protein released from the membrane should continue to self-associate. To test this model, transfectants expressing the mCFP-Rac1/mYFP-Rac1 pair were challenged with a YopT-expressing Y. pseudotuberculosis strain, and the amount of FRET between the Rac1 pairs was determined (22). Within 1 h of incubation with Y. pseudotuberculosis, YopT had reduced Rac1 self-association, since no FRET signal could be detected between the Rac1 pairs (Fig. 7D). Therefore, because Rac1 rapidly lost self-association after release from the membrane, localization of the protein in the membrane via the prenyl groups was required to maintain this relationship. Furthermore, the loss of self-association after release from the membrane could not be due to competition for binding by RhoGDI, because removal of the prenyl group prevents recognition by RhoGDI (32).
Loss of Self-association Is Not the Cause of Defective Signaling That Results from Alterations in the Polybasic Region—To investigate the function of the Rac1 COOH terminus in invasin-promoted uptake, a strategy was pursued in which mutated plasmid-encoded Rac1 genes were introduced into cell lines that expressed endogenous Rac1. To this end, we took advantage of the Y. pseudotuberculosis YopE RhoGAP protein. YopE is translocated into mammalian cells and subsequently inactivates endogenous Rac1, thus inhibiting the phagocytosis of the bacterium. This uptake inhibition can be overcome by the exogenous expression of a GAP-insensitive (constitutively active) Rac1 allele. A number of Rac1 PBR mutants were introduced onto the constitutively active Rac1V12 allele and were transfected into COS1 cells. Cells were challenged with YopE-expressing Y. pseudotuberculosis (strain YP17/pYopE), and the extent of bacterial uptake was quantified.
As demonstrated previously, Rac1V12 restored significant bacterial uptake in the presence of YopE, allowing the effects of mutations to be compared (Fig. 8, mCFP-Rac1V12). Not surprisingly, the Rac1V12(C189S) derivative that lacks gerenylgerenylation, and therefore could not target to the membrane, was unable to bypass YopE activity (Fig. 8; mCFP-Rac1V12(C189S) versus Rac1V12; p ≤ 0.0004). Similarly defective was the RacV12(6Q) mutant, which also could not restore uptake in the presence of YopE (Fig. 8; mCFP-Rac1V12(6Q) versus mCFP-Rac1V12; p ≤ 0.004). Thus, invasin-mediated uptake required the PBR. To determine if the defect in uptake seen in these mutants was due to an absence of self-association or due to loss of effector interactions, the K186E effector binding mutation (34) that also lowers Rac1 self-association (Figs. 1 (B and G) and 5(H and I)) was analyzed. The effect of this mutation on bacterial uptake was similar to that seen with the 6Q mutation, since the two derivatives showed no statistical difference in being able to promote uptake (Fig. 8; mCFP-Rac1V12(K186E) versus Rac1V12(6Q); p = 0.47).
FIGURE 8.

Rac1 self-association is not required for promoting uptake. The indicated plasmids expressing various Rac1V12 mutants were used to transfect COS1 cells. All constructs except mCFP-Rac1(V12) were defective in self-association (Figs. 3 and 4), whereas only mCFP-Rac1(V12)(C189S) and mCFP-Rac1(V12)(6Q) were defective in localizing on the plasma membrane. 24 h after transfection, cells were challenged with YP17/pYopE for 30 min, fixed, and then immunostained for extracellular bacteria and internalized bacteria. Uptake efficiency of 50 CFP-positive cells from each of the three coverslips was determined and summarized as mean ± S.E. for each construct. Control, no plasmid included in transfection.
Since both the 6Q and C189S mutations reduce plasma membrane localization of Rac1, we could not determine if lowered uptake was a result of lowered self-association, effector interactions, or plasma membrane localization (14). To address this issue, uptake was analyzed under conditions in which plasma membrane localization of the mutant Rac1 proteins was forced, using the constructs that have a Lyn NH2-terminal myristoylation site (24). The addition of the Lyn myristoylation site significantly suppressed the defect observed in the C189S mutant (Fig. 8; mCFP-RacV12(C189S) versus Lyn-mCFP-RacV12(C189S); p < 0.004). Therefore, the loss of uptake that results from lack of prenylation appears to be almost totally due to defective membrane localization, because the Lyn construct lacking the prenylation site showed no evidence of self-association (Figs. 4E,5D, and 6) yet still could promote uptake (Fig. 8). On the other hand, if the 6Q mutation was introduced into the Lyn construct (Lyn-Rac1V12(6Q/C189S)), then uptake was as poor as observed in the construct lacking the Lyn site (Fig. 8; Rac1V12(C189S) versus Lyn-Rac1V12(6Q/C189S); p < 0.6). The defect in uptake resulting from the 6Q mutation was unlikely to be due to poor plasma membrane localization, because the myristoylation site was intact (Fig. 3F).
Since self-association of Rac1 does not appear to be essential for promoting invasin-mediated uptake in this system, and the 6Q mutation that replaces the PBR is defective even when plasma membrane localization is forced, the PBR must be required either for recognition by downstream effectors or RacGEF proteins. The importance of the latter could not be the cause of the defect in this assay, because Rac1V12 derivatives were used, which do not require guanine nucleotide exchange factors for activation. The importance of PBR recognition by effectors for invasin-dependent uptake is supported by results using the K186E mutant. Therefore, it appears that in this system, the PBR was required for downstream effector recognition.
DISCUSSION
In this report, we used FRET technology to demonstrate that Rac1 is able to self-associate in live cells and that the highest levels of this interaction occurred at sites documented to contain the highest concentrations of actin remodeling (14, 22), such as regions of cell surface ruffling and nascent phagosome formation during bacterial uptake. These are also the regions of the cell that have the highest levels of activated Rac1 (14, 22). This result supports the model that the Rac1 PBR is an important determinant of self-association (21). Our data indicate that the PBR is not sufficient for this interaction within host cells, however, since insertion of Rac1 into the membrane via its CAAX prenylation motif was required for detectable self-association. Using an assay for the ability of Rac1 to support uptake of Y. pseudotuberculosis, we demonstrated that Rac1 self-association was not required for driving actin polymerization events that lead to internalization of the microorganism. The PBR, however, was important for efficient uptake in this system under conditions in which uptake did not require self-association of Rac1, indicating that the PBR was probably required for interaction with downstream effectors. The nature of the assay used to analyze uptake involved using constitutive active derivatives of Rac1 and did not rule out the possibility that Rac1 self-association may be important to stimulate its activation.
Rac1 self-association was originally demonstrated using gel filtration experiments, which showed that the COOH-terminal PBR was required for the formation of high molecular weight species of Rac1 that had been purified as a nonprenylated protein (21). Our observations are consistent with the importance of this region in promoting self-association within mammalian cells (Fig. 1B), but unlike this biochemical assay, we could obtain no evidence for self-association in the absence of membrane localization. In particular, the orientation of the protein relative to the membrane was extremely important, since forcing membrane localization by an NH2-terminal myristoylation tag was not sufficient to generate a Rac1:Rac1 FRET signal (Figs. 4 and 5). It may be that an important nucleation event that initiates self-interaction requires a particular orientation of the protein in the membrane. Alternatively, prenylation could target Rac1 into microdomains on the plasma membrane that are required for close association of Rac1 monomers. The second possibility has support from a pair of observations. First, active Rac1 is preferentially localized on cholesterol-rich lipid raft domains, which could stimulate self-interaction (35). Second, Rac1 preferentially self-associated at the sites of actin polymerization observed in lamellipodia and nascent phagosomes. After recruitment to these sites, there could be back-signaling to membrane-localized Rac1 that stimulates self-association. In fact, the presence of a high concentration of active Rac1 at these sites (13, 14) could act as a further important stimulus of Rac1 self-interaction, perhaps by initiating cycles of signaling from membrane-localized Rac1 to downstream effectors and back to membrane-localized Rac1.
The self-association initiated at the host cell membrane is stable in the presence of gentle detergent, but it may have a limited lifetime within a host cell once Rac1 is liberated from the membrane. The FRET data obtained in this study could be reproduced using immunoprecipitation analysis, in which epitope-tagged Rac1 variants were used to identify self-interaction complexes (Fig. 6). Only Rac1 derivatives that showed self-association by FRET analysis were positive in the immunoprecipitation assay, so the FRET analysis was a good predictor of whether multimeric Rac1 complexes could survive the detergent extraction procedure. Release of Rac1 from the membrane by the Yersinia YopT protease, which removes the CAAX prenylation signal, disrupted self-interaction (Fig. 7). This could be due to binding of soluble proteins within the cell after release or the introduction of the protein into a microenvironment within the cell that stimulates dissociation. RhoGDI is the most obvious candidate for a protein that could stimulate dissociation of Rac1 monomers; however, the proteolytic product of YopT, nonprenenylated Rac1, is not a good substrate for RhoGDI (36). Release of Rac1 from the membrane by YopT results in PBR-dependent translocation of Rac1 into the nucleus, where at least a subpopulation of the protein is active (22). Although it is not clear what downstream effectors interact with the protein in this compartment, either the biochemical environment of the nucleus or its interacting partners could interfere with self-association.
The close correlation between results of the FRET assay and the co-immunoprecipitation of Rac1 derivatives indicated that the FRET analysis could be reproduced using traditional methods of detecting interaction. This connection was not predicted from previously published work using membrane-targeted CFP/YFP derivatives, which indicated that acylated proteins inserted into membranes associate with each other in the absence of any evidence of stable protein interactions (24). Using live cell imaging, Zacharias et al. (24) showed that the geranylgeranylated mYFP/mCFP pair generated a concentration-independent FRET signal. The FRET by proximity observed in that work, in the absence of any protein domains that promote interaction, is thought to be due to the formation of microdomains within membranes, resulting in close congregation of proteins (24). Although not investigated in that work, it is unlikely that such proximity interactions could have survived detergent extraction and allowed co-immunoprecipitation of protein pairs. Presumably, our fixation procedures prior to FRET (see “Materials and Methods”) prevented fluorophores that are located on closely oriented but noninteracting proteins from generating a signal, indicating that only strong protein-protein interactions can be detected. A recent study indicates that fixation can reduce the yield of FRET in selected circumstances, particularly when both fluorophores are fused on a single polypeptide. Mathematical modeling indicates that a likely explanation for this reduction is that with some FRET pairs, fixation can limit protein flexibility and lock the fluorophores in conformations that interfere with energy transfer (38). Although such interference appears irrelevant in the case of two physically unlinked proteins that have interacting regions (38), it is possible that FRET caused by proximally localized but noninteracting proteins requires a level of flexibility of the fluorophore that is blocked by fixation.
It has been suggested that PBR-mediated self-association of Rac1 might drive optimal effector signaling, based on the observation that a PBR-defective Rac1 derivative was highly defective in activating one of its downstream effectors, the serine/threonine kinase PAK1 (21). Arguing against this hypothesis were results from another study showing that NH2-terminal myristoylation could fully restore the ability of the PBR-defective Rac1 to activate the kinase activity of PAK1 (13). From our studies, we think it likely that the restoration of PAK1 signaling was due to the myristoyl group forcing membrane localization of the Rac1 mutant rather than due to self-association. Our results support the idea that the loss of membrane targeting that results from the disruption of the PBR is a more profound consequence than loss of self-association, because membrane targeting of Rac1 in the absence of self-association appeared sufficient to promote uptake of Y. pseudotuberculosis. A membrane-targeted myristoylated Rac1V12(C189S) did not self-associate, yet this activated Rac1 mutant was able to restore bacterial uptake under conditions in which the endogenous Rac1 was inactive, as long as the PBR was intact in such constructs.
In addition to promoting self-association and membrane targeting, the PBR is known to facilitate nuclear localization of Rac1 and binding to PIP5Kα as well as other downstream effectors of Rac1 (7, 31, 37, 39). Phosphatidylinositol 4,5-bisphosphate, the product of PIP5K, stimulates invasin-promoted uptake (6). Therefore, the importance of PBR could be explained by its ability to stimulate localized phosphoinositol 4,5-bisphosphate accumulation at the phagocytic cup. The fact that we could not bypass the requirement for the PBR in bacterial uptake by simply forcing localization of the protein into the membrane (Fig. 8) was very different from the result showing that PAK1 signaling was PBR-independent after localization of Rac1 in the plasma membrane (14). Therefore, stimulation of PAK1 is probably not sufficient to bypass the requirement of this sequence determinant for bacterial uptake. Consistent with this hypothesis was the finding that a less drastic change in the PBR that interferes with PIP5Kα binding (the Rac1(K186E) allele), was sufficient to interfere with the ability of a membrane-localized and constitutively active Rac1 variant to promote uptake.
Although the evidence presented here argue that loss of multimerization resulting from alterations in the PBR cannot explain the observed defects in invasin-mediated uptake, it should be pointed out that the Rac1 variants used to test uptake were activated forms of the protein (Fig. 8). It is possible that this approach bypasses a requirement for multimerization. At least for the integrin receptor, multimerization is important, although this does not mean that downstream signaling molecules require multimerization. Artificially dimerizing invasin greatly reduces the concentration of the protein required to promote uptake of particles (19). The evidence that downstream multimerization my play a role in regulating this event is that many GTPases are known to form oligomers, an event associated with self-stimulatory GAP activity (21). Although this latter observation argues that multimerization plays a negative role in signaling, self-inhibition of Rac1 may provide a mechanism to limit Rac1 activation that is important for successful completion of cytoskeletal activities. Such negative feedback may be critical at sites of local Rac1 activity, especially when GAP and RhoGDI activities are expected to be down-regulated. Since downstream signaling from Rac1 to other GTPases has complex consequences on invasin-mediated uptake (23), localized down-modulation of Rac1 signaling could play an important role in completing phagocytic events.
Future critical tests of the importance of multimerization in either down-modulation of Rac1 activity or stimulating downstream signaling will require the development of straightforward systems to analyze mutants at limiting and highly defined protein concentrations. Even so, this work demonstrates that multimerization and membrane localization promoted by the PBR do not encompass the entire spectrum of events that are modulated by this small region of the protein.
Acknowledgments
We thank Drs. Matt Heidtman, Matthias Machner, Molly Bergman, and Vicki Auerbuch for reviewing the manuscript and Drs. Jim Bliska, Ulla Knaus, and Kit Wong for supplying plasmids.
This work was supported, in whole or in part, by National Institutes of Health, NIAID, Grant R37AI23538 and NIDDK Program Project Award Grant P30DK34928. This work was also supported by the Howard Hughes Medical Institute. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Author's Choice—Final version full access.
Footnotes
The abbreviations used are: FRET, fluorescence resonance energy transfer; PBR, polybasic region; CFP, cyan fluorescent protein; mCFP, monomeric CFP; YFP, yellow fluorescent protein; mYFP, monomeric YFP; WT, wild type; GAP, GTPase-activating protein; HA, hemagglutinin; PIP5K, phosphatidylinositol-4-phosphate 5-kinase.
References
- 1.Gruenheid, S., and Finlay, B. B. (2003) Nature 422 775-781 [DOI] [PubMed] [Google Scholar]
- 2.Alrutz, M. A., Srivastava, A., Wong, K. W., D'Souza-Schorey, C., Tang, M., Ch'Ng, L. E., Snapper, S. B., and Isberg, R. R. (2001) Mol. Microbiol. 42 689-703 [DOI] [PubMed] [Google Scholar]
- 3.Black, D. S., and Bliska, J. B. (2000) Mol. Microbiol. 37 515-527 [DOI] [PubMed] [Google Scholar]
- 4.McGee, K., Zettl, M., Way, M., and Fallman, M. (2001) FEBS Lett. 509 59-65 [DOI] [PubMed] [Google Scholar]
- 5.Takenawa, T., and Miki, H. (2001) J. Cell Sci. 114 1801-1809 [DOI] [PubMed] [Google Scholar]
- 6.Wong, K. W., and Isberg, R. R. (2003) J. Exp. Med. 198 603-614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tolias, K. F., Hartwig, J. H., Ishihara, H., Shibasaki, Y., Cantley, L. C., and Carpenter, C. L. (2000) Curr. Biol. 10 153-156 [DOI] [PubMed] [Google Scholar]
- 8.Bierne, H., Gouin, E., Roux, P., Caroni, P., Yin, H. L., and Cossart, P. (2001) J. Cell Biol. 155 101-112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rossman, K. L., Der, C. J., and Sondek, J. (2005) Nat. Rev. Mol. Cell. Biol. 6 167-180 [DOI] [PubMed] [Google Scholar]
- 10.Dovas, A., and Couchman, J. R. (2005) Biochem. J. 390 1-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robbe, K., Otto-Bruc, A., Chardin, P., and Antonny, B. (2003) J. Biol. Chem. 278 4756-4762 [DOI] [PubMed] [Google Scholar]
- 12.Ridley, A. J. (2006) Trends Cell Biol. 16 522-529 [DOI] [PubMed] [Google Scholar]
- 13.del Pozo, M. A., Price, L. S., Alderson, N. B., Ren, X. D., and Schwartz, M. A. (2000) EMBO 19 2008-2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Del Pozo, M. A., Kiosses, W. B., Alderson, N. B., Meller, N., Hahn, K. M., and Schwartz, M. A. (2002) Nat. Cell Biol. 4 232-239 [DOI] [PubMed] [Google Scholar]
- 15.Rottner, K., Stradal, T. E., and Wehland, J. (2005) Dev. Cell. 9 3-17 [DOI] [PubMed] [Google Scholar]
- 16.Stewart, P. L., and Nemerow, G. R. (2007) Trends Microbiol. 15 500-507 [DOI] [PubMed] [Google Scholar]
- 17.Isberg, R. R., Voorhis, D. L., and Falkow, S. (1987) Cell 50 769-778 [DOI] [PubMed] [Google Scholar]
- 18.Van Nhieu, G. T., and Isberg, R. R. (1991) J. Biol. Chem. 266 24367-24375 [PubMed] [Google Scholar]
- 19.Dersch, P., and Isberg, R. R. (1999) EMBO J. 18 1199-1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leong, J. M., Morrissey, P. E., Marra, A., and Isberg, R. R. (1995) EMBO J. 14 422-431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang, B., Gao, Y., Moon, S. Y., Zhang, Y., and Zheng, Y. (2001) J. Biol. Chem. 276 8958-8967 [DOI] [PubMed] [Google Scholar]
- 22.Wong, K. W., and Isberg, R. R. (2005) PLoS Pathog. 1 125-136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong, K. W., Mohammadi, S., and Isberg, R. R. (2006) J. Biol. Chem. 281 40379-40388 [DOI] [PubMed] [Google Scholar]
- 24.Zacharias, D. A., Violin, J. D., Newton, A. C., and Tsien, R. Y. (2002) Science 296 913-916 [DOI] [PubMed] [Google Scholar]
- 25.Xia, Z., and Liu, Y. (2001) Biophys. J. 81 2395-2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Knaus, U. G., Wang, Y., Reilly, A. M., Warnock, D., and Jackson, J. H. (1998) J. Biol. Chem. 273 21512-21518 [DOI] [PubMed] [Google Scholar]
- 27.Kreck, M. L., Freeman, J. L., Abo, A., and Lambeth, J. D. (1996) Biochemistry 35 15683-15692 [DOI] [PubMed] [Google Scholar]
- 28.Hancock, J. F., Paterson, H., and Marshall, C. J. (1990) Cell 63 133-139 [DOI] [PubMed] [Google Scholar]
- 29.Michaelson, D., Silletti, J., Murphy, G., D'Eustachio, P., Rush, M., and Philips, M. R. (2001) J. Cell Biol. 152 111-126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gibson, R. M., and Wilson-Delfosse, A. L. (2001) Biochem. J. 359 285-294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Modha, R., Campbell, L. J., Nietlispach, D., Buhecha, H. R., Owen, D., and Mott, H. R. (2008) J. Biol. Chem. 283 1492-1500 [DOI] [PubMed] [Google Scholar]
- 32.Takai, Y., Kaibuchi, K., Kikuchi, A., and Sasaki, T. (1995) Methods Enzymol. 250 122-133 [DOI] [PubMed] [Google Scholar]
- 33.Logsdon, L. K., and Mecsas, J. (2006) Infect. Immun. 74 1516-1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tolias, K. F., Couvillon, A. D., Cantley, L. C., and Carpenter, C. L. (1998) Mol. Cell. Biol. 18 762-770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.del Pozo, M. A., Alderson, N. B., Kiosses, W. B., Chiang, H. H., Anderson, R. G., and Schwartz, M. A. (2004) Science 303 839-842 [DOI] [PubMed] [Google Scholar]
- 36.Shao, F., Vacratsis, P. O., Bao, Z., Bowers, K. E., Fierke, C. A., and Dixon, J. E. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 904-909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Williams, C. L. (2003) Cell. Signal. 15 1071-1080 [DOI] [PubMed] [Google Scholar]
- 38.Anikovsky, M. Dale, L., Ferguson, S., and Petersen, N. (2008) Biophys. J. 95 1349-1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lanning, C. C., Daddona, J. L., Ruiz-Velasco, R., Shafer, S. H., and Williams, C. L. (2004) J. Biol. Chem. 279 44197-44210 [DOI] [PubMed] [Google Scholar]