

Abstract
Histone deacetylase (HDAC) inhibitors, including various benzamides and hydroxamates, are currently in clinical development for a broad range of human diseases, including cancer and neurodegenerative diseases. We recently reported the identification of a family of benzamide-type HDAC inhibitors that are relatively non-toxic compared with the hydroxamates. Members of this class of compounds have shown efficacy in cell-based and mouse models for the neurodegenerative diseases Friedreich ataxia and Huntington disease. Considerable differences in IC50 values for the various HDAC enzymes have been reported for many of the HDAC inhibitors, leading to confusion as to the HDAC isotype specificities of these compounds. Here we show that a benzamide HDAC inhibitor, a pimelic diphenylamide (106), is a class I HDAC inhibitor, demonstrating no activity against class II HDACs. 106 is a slow, tight-binding inhibitor of HDACs 1, 2, and 3, although inhibition for these enzymes occurs through different mechanisms. Inhibitor 106 also has preference toward HDAC3 with Ki of ∼14 nm, 15 times lower than the Ki for HDAC1. In comparison, the hydroxamate suberoylanilide hydroxamic acid does not discriminate between these enzymes and exhibits a fast-on/fast-off inhibitory mechanism. These observations may explain a paradox involving the relative activities of pimelic diphenylamides versus hydroxamates as gene activators.
The link between post-translational modifications by reversible histone acetylation and deacetylation and mRNA transcription has been shown to be one of the key mechanisms of epigenetic gene regulation (1). Acetylation of histone lysine residues, controlled by the histone acetyltransferases and histone deacetylases (HDACs),2 has been a subject of intense recent research (2–5). Generally, histone hypoacetylation causes transcriptional silencing, whereas histone hyperacetylation results in transcriptional activation of various genes (6–8). Eighteen HDACs have been identified in the human genome, including the zinc-dependent HDACs (class I, class II, and class IV), and the NAD+-dependent enzymes (class III or sirtuins) (9, 10). HDACs 1, 2, 3, and 8 belong to class I, showing homology to the yeast enzyme RPD3. Class II is further divided into class IIa (HDACs 4, 5, 7, and 9) and IIb (HDAC 6 and 10), according to their sequence homology and domain organization. HDAC11 is the lone member of class IV (9, 11). The sirtuins (class III) are related to the yeast Sir2 protein and are involved in regulation of metabolism and aging (10).
To date, a number of small molecule inhibitors of the zinc-dependent HDACs have been identified (12). These compounds can be broadly grouped in four chemical classes: the hydroxamates, the benzamides, butyrate analogs, and cyclic peptides, such as depsipeptide and related compounds (12, 13). Hydroxamate-based inhibitors, such as trichostatin A (TSA) and suberoylanilide hydroamic acid (SAHA; Fig. 1A) are believed to be pan-HDAC inhibitors (14–16); however, recent studies have shown that TSA and SAHA are actually class I-specific inhibitors, and previous results reporting inhibition of class II HDACs were due to class I HDAC contamination (17–19). Studies of benzamide-based HDAC inhibitors have shown that these compounds are class I-specific inhibitors, and have claimed distinct pharmacological properties of the benzamide HDAC inhibitors due to specific inhibition of class I HDACs (mainly HDAC1 and HDAC3) (20, 21). Some benzamides (BML-210 and related pimelic diphenylamides 4b and 106; Fig. 1A) have been shown to up-regulate specific genes, including the frataxin gene involved in the neurodegenerative disease Friedriech ataxia, at subtoxic inhibitor concentrations (22–24). In comparison, the hydroxamates SAHA and TSA were inactive as positive regulators of frataxin gene expression. Thus, the distinct pharmacological properties of the pimelic diphenylamides, as compared with the hydroxamates, cannot be explained by specificity for class I HDACs. Additionally, other benzamide HDAC inhibitors, such as MS-275, have reported IC50 values ranging from ∼740 nm to 8 μm for HDAC3, whereas the reported IC50 values for HDAC1 are more consistent, at around ∼200 nm (12, 25).
FIGURE 1.

Structures of HDAC inhibitors and time dependence on IC50 values. A, chemical structures of 106 (top) and SAHA (bottom). B, IC50 values for HDAC1 and HDAC3 with SAHA are independent of preincubation time (top). A preincubation time dependence of IC50 values for inhibitor 106 was observed for both HDAC1 and HDAC3. The inhibitors and enzymes were preincubated for the indicated times, followed by addition of the acetylated lysine substrate. Deacetylation reactions were run for 1 h, and 5 mg/ml (final concentration) of trypsin developer was added.
In this study, we investigated differences in the kinetic properties between the hydroxamate SAHA and the pimelic diphenylamide inhibitor 106 (Fig. 1A), with recombinant human HDACs. We discovered that the benzamide inhibitor 106, unlike the hydroxamate SAHA, is a slow, tight-binding inhibitor of HDACs 1, 2, and 3, with different inhibitory mechanisms and half-lives of the enzyme-inhibitor complexes. IC50 values for inhibitor 106 decreased significantly during preincubation with HDAC3, but this slow inhibition behavior is less pronounced for HDAC1 or -2. Hydroxamates, on the other hand, are fast-on/fast-off inhibitors of both HDAC1 and HDAC3. The kinetic parameters for these two classes of compounds were determined for HDACs 1 and 3, and compared with the cellular activities of 106 and SAHA.
EXPERIMENTAL PROCEDURES
Materials—Recombinant human HDAC1, HDAC2, HDAC3/N-CoR2, and HDAC8 expressed in baculovirus, were purchased from BPS Bioscience (San Diego, CA). Western blots were done to verify that no HDAC1 and HDAC3 cross-contamination was present in these enzyme preparations (supplemental Fig. S1). Acetylated-Lys(acetyl)-4-methylcoumarin-7-amide (acetyl-Lys(Ac)-AMC) was purchased from Biomol International (Plymouth Meeting, PA). Lys-C peptidase was purchased from EMD Chemicals (Gibbstown, NJ). For the class II HDACs, 4, 5, and 7, we obtained cDNAs for these human enzymes from Addgene in pcDNA3.1 with a C-terminal FLAG epitope. Protein overexpression was achieved by transfecting plasmids into HEK293t cells with 293-fectin reagent (Invitrogen). Cells were lysed at 72 h with whole cell lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.5% Triton X-100, 10% glycerol), and passed over FLAG-M2 affinity resin (Sigma) for purification. Following several washes, HDAC proteins were eluted with 5 bead volumes of 100 μg/ml FLAG peptide (Sigma), and assayed without further purification. HDAC inhibitor SAHA was purchased from Biomol International (Plymouth Meeting, PA) through a custom synthesis order, and 106 (N1-(2-aminophenyl)-N7-p-tolylheptanediamide) was synthesized as previous described (23) and provided by Repligen Corporation (Waltham, MA).
Assay of HDAC Activities and Inhibition Kinetics—The deacetylase activities of HDACs 1, 2, and 3 were measured by assaying enzyme activity using peptidase (Lys-C peptidase and trypsin) and the synthetic substrate acetyl-Lys(Ac)-AMC, as previously described (26, 27). Deacetylated lysine-AMC was released by the peptidase and free fluorogenic 4-methylcoumarin-7-amide (MCA) was generated. The fluorogenic MCA could then be read with an excitation wavelength of 370 nm and emission wavelength of 460 nm. Assays for class II HDACs were done using acetyl-Lys(trifluoroacetyl)-AMC under the same conditions (17) (synthesized at Scripps). All assays for HDAC activity on the acetylated lysine substrates were performed in 96-well, non-binding plates (Greiner Bio-one, NC) in 50 mm Tris-HCl buffer (pH 8.0), containing 137 mm NaCl, 1 mm MgCl2, 2.7 mm KCl, and 0.1 mg/ml bovine serum albumin (standard HDAC buffer) at ambient temperature. The final assay volume was 50 μl, except for the dilution experiment described below, which was at 100 μl. The amount of MCA generated was equal to deacetylated substrate and was normalized with a non-acetylated substrate standard (supplemental Fig. S2A).
Determination of the Inhibitor IC50 Values with Preincubation—Deacetylation assays were based on the homogenous fluorescence release assay, described above. Purified recombinant enzymes were incubated with serial-diluted inhibitors at the concentrations indicated in the figures, with preincubation times ranging from 0 to 3 h, in the standard HDAC buffer (as in Fig. 1B). Acetyl-Lys(Ac)-AMC substrate (at 10 μm, corresponding to the Km for both HDAC1 and HDAC3, supplemental Fig. S2B) was added after the preincubation period, and the reaction was allowed to run for 1 h. The trypsin peptidase developer, at a final concentration of 5 mg/ml, was added after 1 h, and the fluorescence emission was then measured using a Tecan M200 96-well plate reader (San Jose, CA). The IC50 was determined by fitting the data using the KaleidaGraph nonlinear regression program (Synergy Software, Reading, PA).
Slow, Tight-binding Kinetic Determination Using the Progression Method—Slow tight-binding kinetics of 106 with class I HDACs were evaluated by the progression curve approach described by Morrison and Walsh (28). In theory, there are three possible slow, tight-binding mechanisms, as previously described (29). The two most common ones are shown here as Mechanisms 1 and 2.
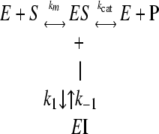 |
MECHANISM 1 |
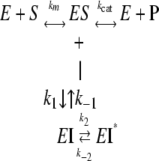 |
MECHANISM 2 |
To determine the mechanism and associated kinetic values, a series of inhibition progression curves for HDACs 1, 2, and 3, at different concentrations of inhibitor 106, were generated by adding 100 ng of each enzyme into separate reaction mixtures containing 50 μm acetyl-Lys(Ac)-AMC substrate (5 times the Km) and 2 milliunits of Lys-C peptidase developer. Lys-C was used in the progression curve method to prevent degradation of the HDAC enzymes during the assay, allowing a linear no-inhibitor control. The generation of the fluorogenic MCA, due to the deacetylation of the lysine substrate, was assessed continuously for up to 1 h at ambient temperature. Data from each progression curve, at different inhibitor concentrations, were fit using the nonlinear regression program KaleidaGraph to the integrated rate equation for slow-binding inhibitors:
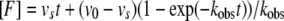 |
(Eq. 1) |
where [F] is the amount of MCA fluorophore generated, represented in arbitrary fluorescence units (r), which is proportion to the deacetylated substrate at time t. v0 and vs are the initial and the final steady-state velocities, respectively. kobs is the apparent first-order rate constant obtained by the best fit to the data. Because kobs is the only value that is not significantly altered by small systematic errors (29, 30), the kobs values were then plotted against the inhibitor concentrations for which each kobs value was obtained. For Mechanism 1, the relationship between kobs and the inhibitor concentration is linear,
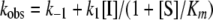 |
(Eq. 2) |
and
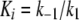 |
(Eq. 3) |
For Mechanism 2, the relationship between kobs and the inhibitor concentration is hyperbolic,
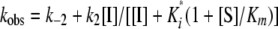 |
(Eq. 4) |
and
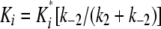 |
(Eq. 5) |
where Ki* is the stable complex forming constant and Ki is the overall final inhibitory constant for the entire process.
Determination of Ki for Fast On/Off Inhibitors—For a classical fast on/off competitive inhibitor, such as SAHA, the steady state HDAC enzyme velocities are achieved within seconds. The deacetylation rate in the presence (vi) and absence (v0) of the inhibitor are linear, with no time dependence. Therefore, the Ki of the inhibitor can easily be determined using the ratio of vi over v0 (single step fast on/off) according to the relationship (31, 32) in Equation 6.
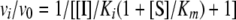 |
(Eq. 6) |
The velocity of each progression line was calculated, and the ratio of vi/v0 was plotted against the corresponding inhibitor concentration. The Ki was then determined by fitting the data to Equation 6, using the KaleidaGraph nonlinear regression program.
Comparative Disassociation through 100-fold Dilution—HDAC1 (10 μg) and HDAC3 (10 μg) were each incubated with 2 μm 106 or 100 nm SAHA, in standard deacetylase assay buffer, containing 0.1 mg/ml bovine serum albumin, for 1 h. After this preincubation time, 1 μl of each mixture was diluted into a final volume of 100 μl, containing 0.1 mg/ml bovine serum albumin, 50 μm acetyl-Lys(Ac)-AMC substrate, and 2 milliunits of Lys-C peptidase developer, without or with each inhibitor at the initial concentration (2 μm 106 or 100 nm SAHA). The amounts of HDAC1 and HDAC3 after dilution were 100 ng in the final assay solution. Progression curves were then measured for an additional hour.
Cell Culture, SDS-PAGE, and Western Blot Analysis—A lymphoblastoid cell line derived from a Friedreich ataxia patient (GM15850) was obtained from the NIGMS Genetic Repository (Coriell Medical Institute). Cells were cultured in RPMI 1640 media with 10% fetal bovine serum and 10 mm HEPES, at 37 °C in 5% CO2. After the 5th split, the cells were treated with inhibitor 106 (2 μm) or SAHA (2 μm) for 24 h, in culture medium. The treatment concentrations were determined based on growth inhibition through an MTS cell proliferation assay (Promega) (supplemental Fig. S3). These concentrations correspond approximately to the EC10 for 106, and 2 μm SAHA is near its EC50 for blocking cell proliferation. 24 h after treatment, the cells were washed twice with Hanks' balanced salts buffer (Invitrogen) to remove the inhibitor. A portion of the cell population was then harvested immediately after washing, and referred to as a time 0 point (24 h treatment point), and the rest of the cells were then re-cultured in cell culture media without added inhibitor. The re-cultured cells and controls were then harvested every hour for a total of 7 h. The cells were washed twice before lysis with a low salt lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 10% glycerol, 0.5% Triton X-100, and 1× protease inhibitors, Roche Diagnostics) for 30 min, and followed by a 15-s sonication pulse at 3 watts (Branson Sonifier 150, Branson, CT). The cell lysates were then denatured with LDS loading buffer (Invitrogen) and run on 4–12% SDS gradient polyacrylamide gels (Invitrogen). Total histone H3, acetylated histone H3 (K9+K14), and frataxin were visualized with primary histone H3 antibody from Abcam (Cambridge, MA), acetylated K9+K14 histone H3 antibody from Upstate (Temecula, CA), and anti-frataxin antibody from MitoSciences (Eugene, OR), respectively, followed by rabbit (for histone H3 and acetylated H3) or mouse (for frataxin) IgG-horseradish peroxidase conjugated secondary antibody (Cell Signaling, MA).
RESULTS
HDAC Inhibition Assays—We assayed each of the recombinant class I (HDACs 1, 2, 3, and 8) and representative class II (HDACs 4, 5, and 7) enzymes with the pimelic diphenylamide HDAC inhibitor 106 (Fig. 1A). For HDAC3, a recombinant fragment of the co-repressor protein N-CoR2 was co-expressed because the deacetylase activating domain of N-CoR is required for HDAC3 activity (reviewed in Ref. 33). Enzyme and inhibitor were preincubated for 1 to 3 h prior to addition of a fluorogenic substrate (see “Experimental Procedures”), and from compound titrations, IC50 values are calculated. Compound 106 exhibits good inhibitory activity against class I HDACs, with IC50 values of 150 nm for HDAC1 and 370 nm for HDAC3. 106 exhibits weaker inhibitory activities against HDAC2 and -8, 106 has an IC50 of 760 nm with HDAC2 and an IC50 of 5 μm after a 3-h preincubation with HDAC8 (supplemental Fig. S4). Because recent studies indicate that class II HDACs are not active on standard acetylated lysine peptide substrates (17), assays with recombinant class II HDACs 4, 5, and 7 used a trifluoracetylated lysine substrate (see Ref. 17), and we find that 106 has essentially no inhibitory activity against these enzymes (IC50 > 180 μm; supplemental Fig. S5). Previous studies have established that benzamide-type HDAC inhibitors are selective for class I HDAC enzymes and in particular MS-275 and other o-aminobenzamides show a ∼4 to 10-fold preference for HDAC1 over HDAC3 (12, 25, 34). Our results are thus consistent for 106 compared with other benzamides.
Effects of Preincubation Time on Observed IC50 Values—SAHA has been shown to be a competitive HDAC inhibitor (12). Dose-response curves of SAHA showed an average IC50 of 17.3 (± 2.1) nm for HDAC1 and 24.1 (± 3.7) nm for HDAC3, independent of preincubation time (Fig. 1B), showing that SAHA rapidly reaches equilibrium with these enzymes. 106 also inhibited the deacetylase activities of HDACs 1,2, 3, and 8; however, unlike SAHA, dose-response curves and calculated IC50 values for 106 vary with enzyme-inhibitor preincubation time (Fig. 1B for HDAC1 and HDAC3; supplemental Fig. S4 for HDAC2 and HDAC8). This was especially true for HDAC3. Even after 2 h of incubation, the IC50 for 106 with HDAC3 was still decreasing. Without preincubation, 106 exhibited an IC50 of 460 nm for HDAC1, and 5.8 μm for HDAC3. After a 15-min preincubation, the IC50 for HDAC1 came to equilibrium at 138 (± 38) nm. For HDAC3, however, the IC50 for 106 did not reach a steady-state value for several hours. After a 3-h preincubation, the IC50 decreased to 380 nm; a 15-fold decrease in the IC50 value compared with the IC50 measured without preincubation. Similar results were also observed for HDAC2 and HDAC8, with higher IC50 values (supplemental Fig. S4). These observations suggest that 106 is a slow, tight-binding inhibitor of class I HDACs. Also, there was a great difference in the on-rate (and presumably the off-rate) for 106 for HDAC1 and HDAC3, respectively. Thus, the IC50 of a particular enzyme/inhibitor pair is not a reliable measurement due to the variability with different procedures; i.e. preincubation periods. To better understand the mechanisms for inhibition of these enzymes and to determine the kinetic constants of slow, tight-binding inhibitors, progression curve experiments for class I HDACs were next performed.
HDAC Inhibitor 106 Is a Slow, Competitive Tight-binding Inhibitor of HDACs 1 and 2—HDAC1 deacetylation progression curves were measured in the presence of different concentrations of 106 (ranging from 0.5 nm to 20 μm), performed in triplicate (Fig. 2A). The data were fit with Equation 1, using the estimated v0 and vs values to determine the kobs for each run. The kobs was then plotted against the concentration of 106 used for each determination of kobs. A linear trend was observed, indicating a competitive tight-binding mechanism, as described by Mechanism 1 (Fig. 2A). The data were then fit with Equation 2, and Ki, k1, and k–1 were determined using Equations 2 and 3. For HDAC1 and inhibitor 106, k1 was 4.9 × 104 (± 1.2 × 104) m–1 min–1, and k–1 was 0.0072 (± 0.0017) min–1. Based on a presumed first-order decay, k–1 corresponds to a half-life of ∼1.5 h for the 106-HDAC1 complex. The Ki was then determined from triplicate experiments, using Equation 3. The Ki was found to be 148 (± 36) nm for HDAC1. The kinetics for HDAC2 were also investigated at various concentrations of 106, and found to be similar to HDAC1, suggesting that inhibition follows Mechanism 1, as described above (supplemental Fig. S4). From progression curves obtained at different concentrations of 106,a Ki of 102 nm was determined for HDAC2. Due to the high peptidase sensitivity of HDAC8 (data not shown), we were unable to perform a similar kinetic analysis.
FIGURE 2.

Time-dependent inhibition of HDAC1 and HDAC3 as a function of inhibitor 106 concentration. The reaction mixture contained 100 ng of enzyme in the standard HDAC Tris buffer (pH 8.0), 50 μm acetylated-lysine substrate, various concentrations of inhibitor 106 (indicated) and Lys-C peptidase (2 milliunits). A, the progress curves for HDAC1 in the presence of increasing concentrations of inhibitor 106 were measured for 1 h and fit with Equation 1 (left). The rate constant kobs as a function of inhibitor 106 concentration was plotted (right). B, the progress curves for HDAC3 in the presence of increasing concentrations of inhibitor 106 were measured for 1 h and fit with Equation 1 (left). The rate constant kobs as a function of inhibitor 106 concentration was plotted (right).
HDAC Inhibitor 106 Is a Slow, Competitive Tight-binding Inhibitor of HDAC3 with a Conformational Change in the Enzyme—HDAC3 deacetylation progression curves were measured at various concentrations of 106, as described above (Fig. 2B). Again, the data were fit with Equation 1, using the estimated v0 and vs values to determine kobs for each concentration of 106. The kobs values were then plotted against 106 concentration, and a hyperbolic relationship with an increasing inhibitor concentration was observed (Fig. 2B). This upward slope hyperbolic relationship was consistent with Mechanism 2, reflecting slow, tight-binding inhibition (having a slow step for forming a stable complex with a possible conformation change in the protein). The data were fit with Equations 4 and 5 to determine the kinetic parameters. The overall Ki value was determined to be 14 nm (± 3) nm, with Ki* of 224 nm (± 62 nm); k2 was 0.021 (± 0.002) min–1, and k–2 was 0.00143 (± 0.00197) min–1 for HDAC3. Based on a presumed first-order decay, this corresponds to a half-life of ∼8 h for the 106-HDAC3 complex.
Determination of Kinetic Constants and Ki for Inhibition of HDAC1 and HDAC3 with SAHA Using the Progression Method—For the fast-on/fast-off competitive inhibitors such as SAHA, Ki can also be determined using the progression method at a substrate concentration greater than the Km. For HDAC1 and HDAC3, deacetylation progression in the presence of various concentrations of SAHA is linear, confirming the fast-on/fast-off nature of the inhibitor. The ratio of v0/vi (where v0 is the deacetylase velocity in the absence of the inhibitor and vi is the deacetylase velocity in the presence of set concentrations of inhibitor) was plotted against inhibitor concentration (Fig. 3, A and B). The Ki values were then determined using Equation 6. The Ki values were 5.4 (± 0.1) and 7.8 (± 0.5) nm for HDAC1 and HDAC3, respectively.
FIGURE 3.

SAHA inhibition of HDAC1 and HDAC3. A, SAHA does not show a time-dependent inhibition of HDAC1 (left). The velocities of the reaction in the presence of different concentrations of SAHA (vi) over the velocity of the reaction in the absence of the inhibitor (v0) were plotted against inhibitor concentrations. The curves were then fit with Equation 6 to yield Ki values for HDAC1 (right). B, SAHA does not show a time-dependent inhibition of HDAC3 (left). The vi/v0 values were plotted and fit with Equation 6 to yield Ki values for HDAC3 (right). AFU, arbitrary fluorescence units.
Dilution Analysis of 106 and SAHA—If an inhibitor forms an exceptionally long-lived complex with its enzyme target, this complex may be stable to dilution. 10 μgof HDAC1 or HDAC3 were separately incubated with 2 μm 106, or with 100 nm SAHA for 1 h. After the preincubation period, the mixtures were diluted 100-fold in the presence of 50 μm substrate (5 times the Km) and 2 milliunits of Lys-C peptidase developer, with or without the inhibitors at their original concentrations. The results of enzyme progression curves for these reactions are shown in Fig. 4, A and B. In the case of HDAC1, 106 slowly loses its inhibitory activity over time (within the hour period of the enzyme reaction). SAHA lost the majority of its inhibitory activity immediately after dilution, at the beginning of the assay (Fig. 4A). For HDAC3, a 100-fold dilution of the 106/enzyme mixture did not significantly decrease the inhibitory activity of the compound over the 1-h reaction, whereas SAHA again lost most of its inhibitory activity at the beginning of the assay (Fig. 4B). These observations are consistent with the kinetic values determined with the progression method.
FIGURE 4.

Dilution experiment for determination of reversibility in vitro. A, the dilution protocol is described under “Experimental Procedures.” Upon 100-fold dilution, SAHA lost most of its activity against HDAC1. For inhibitor 106, the loss in inhibitory activity was significantly lower. B, upon 100-fold dilution, SAHA lost most of its activity against HDAC3. For inhibitor 106, essentially no loss in inhibitory activity was observed.
Prolonged Acetylation of Cellular Histone H3 with 106—We next monitored the effects of 106 and SAHA on acetylation of endogenous histone H3 in a lymphoblastoid cell line derived from a Friedreich ataxia patient (GM15850 cells). Cells were treated with 2 μm 106 or 2 μm SAHA for 24 h in separate cultures. The inhibitors were then removed through washing a fraction of the cells. An aliquot of these cells was collected for a zero time point, before subculturing the remaining cells in standard media without the inhibitors. Cells were then harvested at 0–7 h, and total cell lysates were prepared and subjected to SDS-PAGE. Total histone H3 and acetylated histone H3 (K9+K14) were then visualized through Western blotting with appropriate antibodies (Fig. 5). Hyperacetylation of histone H3 (at K9+K14) was clearly seen both in the zero time point and in the cell cultures where inhibitors were present (Fig. 5). Hyperacetylation of histone H3 due to the HDAC inhibitor 106 only decreased slightly after inhibitor removal, and did not fully return to basal levels until 6–7 h after the removal of the inhibitor. In contrast to the results for 106, histone H3 hyperacetylation due to SAHA was stronger in the presence of the inhibitor; however, after the removal of SAHA, histone H3 hyperacetylation disappeared after the first hour. In fact, a significant decrease in acetylation was even observed at the zero time point after washing the SAHA-treated cells, compared with the sample where SAHA was present.
FIGURE 5.

Prolonged histone H3 acetylation and increased frataxin protein by 106 in cells. GM15850 FRDA lymphoblast cells were either untreated (lane 1, marked 0 at top) or treated with either 106 (top panels) or SAHA (bottom panels) at 2 μm concentration for 24 h (lane 2, marked 2 μm at top), and then washed to remove the inhibitors. Cells were harvested at the indicated times (lanes 3–10, 0–7 h), protein extracts were prepared and subjected to Western blotting with antibody to unacetylated histone H3 as a loading control, antibody to acetylated histone H3 (K9 + K14), or frataxin antibody.
Up-regulation of Frataxin Protein Observed for Inhibitor 106-treated Cells but Not SAHA-treated Cells—Western blots were also performed to determine frataxin levels in cells after 24 h of treatment, followed by removal of the inhibitors 106 and SAHA, as described above. No increase in frataxin protein levels was observed with SAHA-treated cells (Fig. 5), consistent with previous results (22). Although an increase (∼2-fold) in frataxin protein was observed after 24 h of treatment with 106, a far more significant increase in frataxin levels was observed 1–2 h after removal of the inhibitor (>5-fold; Fig. 5). These results indicate that up-regulation of frataxin is more complex than simple inhibition of HDACs, and increases in histone acetylation.
DISCUSSION
HDAC inhibitor potency is commonly assessed with IC50 values. However, IC50 values can vary significantly with different experimental conditions (i.e. different concentrations of substrates, incubation and preincubation periods, inhibitory mechanism), as addressed in this study. Benzamide inhibitors were originally believed to be weak (high μm IC50 values) histone deacetylase inhibitors (35). In comparison to the highly active histone deacetylase inhibitors SAHA and TSA that exhibit nanomolar IC50 values, benzamides appear to be less effective. However, through phenotypic screening or targeted ex vivo cell culture screens, benzamide HDAC inhibitors have demonstrated their unique biological functions below their toxicity levels and effective deacetylase IC50 values (22–24).
In this study, we investigated the inhibitory properties of the pimelic diphenylamide 106, in comparison to the hydroxamate HDAC inhibitor SAHA. Our results show that unlike SAHA, the benzamide HDAC inhibitor 106 is a slow, tight-binding inhibitor of HDAC1, HDAC2, and HDAC3. 106 has a slow-on behavior for each of these HDACs, and IC50 values vary depending on the preincubation time period; hence it is extremely unlikely that IC50 values will yield consistent results unless care is taken to assure that the inhibitor and enzyme have reached equilibrium. This particular behavior likely contributes to the wide range of IC50 values reported for similar compounds, such as MS-275 (12, 25). It is noteworthy that MS-275 and TSA also behave similarly compared with 106 and SAHA, respectively, with HDAC1 and HDAC3, with properties attributable to their chemical class (i.e. benzamide versus hydroxamate, supplemental Fig. 6). In contrast to these findings, 106 is only a weak inhibitor of HDAC8 (IC50 = 5 μm).
Because Ki and the on- and off-rates are intrinsic properties of the particular enzyme-inhibitor pair, these values are constant, and should provide reliable estimates of inhibitory properties of the compounds for their target enzymes. The progression method was used to determine these kinetic values, and they are summarized in Table 1. Inhibitor 106 has a Ki value of 148 (±36) nm for HDAC1, ∼102 nm for HDAC2, and 14 (±3) nm for HDAC3. These data indicate that inhibitor 106 has a greater preference for HDAC3 (∼10-fold), a finding that the IC50 measurement failed to capture.
TABLE 1.
Kinetic values of inhibitor 106 and SAHA for HDAC1 and HDAC3
|
HDAC1
|
HDAC3
|
|||
|---|---|---|---|---|
| 106 | SAHA | 106 | SAHA | |
| Ki | 148 (± 36) nm | 5.4 (± 0.1) nm | 14 (± 3) nm | 7.8 (± 0.5) nm |
| Ki* | 224 (± 62) nm | |||
| k1 | 4.9 × 104 (± 1.2 × 104) m-1 min-1 | |||
| k-1 | 0.0072 (± 0.0017) min-1 | |||
| k2 | 0.021 (± 0.002) min-1 | |||
| k-2 | 0.00143 (± 0.00197) min-1 | |||
Although 106 is a slow, tight-binding inhibitor of each of the class I HDACs, kinetic measurements suggest that the inhibitory mechanisms are different for HDAC3 compared with HDACs 1 and 2. Inhibitor 106 behaves like a competitive tight-binding inhibitor for HDAC1 and HDAC2, whereas it appears to form a stable complex with HDAC3 through a slow step (Mechanisms 1 and 2, respectively). The results observed through the progression inhibition assays are also consistent with the results obtained from dilution experiments. Inhibitor 106 preferentially forms a stable complex with HDAC3 with k–2 of 0.00143 (±0.00197) min–1 (t½ ∼ 8 h). Upon 100-fold dilution of the enzyme/inhibitor mixture, the diluted mixture did not show any significant decrease in inhibitory capacity over 1 h. Unlike 106, SAHA and TSA inhibitory capacities are lost immediately (or within minutes) after dilution (Fig. 4, A and B, and supplemental Fig. S6), suggesting that these compounds rapidly dissociate from the HDAC enzymes. Studies aimed at providing a physical measurement of the half-life of the 106-HDAC3 complex are in progress.
The ability of 106 to prolong histone acetylation has also been observed in cell culture. Cells treated with 106 (at a sub-EC10 concentration as determined with an MTS cell proliferation assay) have hyperacetylated histone H3. These cells do not lose significant amounts of acetylated histone H3 even after removal of the inhibitor for hours (Fig. 5). In comparison, acetylation of histone H3 is lost in SAHA-treated cells within the first hour after removal of the drug (Fig. 5). The time frame for the loss of histone H3 hyperacetylation also correlates well with the in vitro kinetic data. This finding indicates that 106 likely behaves in a similar fashion in the context of the living cell. In addition, although sustained total histone H3 acetylation did not last over 7 h, it is likely that specific genes were up-regulated due to inhibition of HDAC3 and these genes could potentially still be active long after the elimination of inhibitor 106. It was surprising to find that full increases in frataxin protein were only observed after removal of compound 106 from the culture medium (Fig. 5). It is tempting to speculate that nonspecific inhibition of other HDACs, or unidentified enzymes, inhibits the full up-regulating potential of compound 106. After wash, this negative effect was removed and full up-regulation of frataxin protein was allowed. In this regard, a recent study has shown that inhibition of HDAC1 could potentially inhibit translation of some specific proteins (37).
The significant increase in the frataxin protein levels 1–2 h after the removal of inhibitor 106, and the inability of SAHA to up-regulate frataxin protein levels (Fig. 5), may explain a paradox raised by our study on the regulation of frataxin gene expression by HDAC inhibitors (22). In that study, we found that only pimelic diphenylamides increased expression of the frataxin gene in lymphocytes from Friedreich ataxia patients, and more recently in a mouse model for this disease (23), and the more active hydroxamate HDAC inhibitors SAHA and TSA were inactive (at their reported IC50 values). Our current findings showing that benzamide inhibitors that target HDAC3 form a stable complex with the enzyme and cause prolonged histone acetylation in cells might well resolve this paradox. Additionally, another related pimelic diphenylamide has shown efficacy in a mouse model for Huntington disease (36). Importantly, no apparent toxicity was noted in these studies, suggesting that the stability of the inhibitor-enzyme complex does not lead to animal toxicity.
From this study, it is clear that simply looking at relative inhibitory activities through IC50 determinations might not be a reliable way to screen for potentially useful and important HDAC inhibitors. Especially if the mechanism of inhibition is different from the expected fast-on/fast-off mechanism, IC50 values will not yield useful information, and such values might actually misguide a particular screening effort. Inhibitor 106 is not only a slow, tight-binding inhibitor of class I HDACs, it also appears to have different mechanisms of inhibition for different enzymes of this class, a finding that was completely unexpected. These results might also explain our observations for this class of HDAC inhibitors in both in vitro and in vivo studies (22, 23).
Supplementary Material
Acknowledgments
We thank Dr. James R. Rusche and Dr. Andrew Cooper for helpful discussions and materials. We also thank Dr. Ana Montero and Dr. M. Reza Ghadiri for the supply of acetyl-Lys(trifluoroacetyl)-AMC.
This work was supported, in whole or in part, by National Institutes of Health Grant R21-NS055781. This work was also supported by Repligen Corporation, Waltham, MA. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S6.
Footnotes
The abbreviations used are: HDAC, histone deacetylase; TSA, trichostatin A; SAHA, suberoylanilide hydroamic acid; MCA, 4-methylcoumarin-7-amide.
References
- 1.Jenuwein, T., and Allis, C. D. (2001) Science 293 1074–1080 [DOI] [PubMed] [Google Scholar]
- 2.Strahl, B. D., and Allis, C. D. (2000) Nature 403 41–45 [DOI] [PubMed] [Google Scholar]
- 3.Marmorstein, R. (2001) Nat. Rev. Mol. Cell Biol. 2 422–432 [DOI] [PubMed] [Google Scholar]
- 4.Wade, P. A. (2001) Hum. Mol. Genet. 10 693–698 [DOI] [PubMed] [Google Scholar]
- 5.Thiel, G., Lietz, M., and Hohl, M. (2004) Eur. J. Biochem. 271 2855–2862 [DOI] [PubMed] [Google Scholar]
- 6.Hassig, C. A., and Schreiber, S. L. (1997) Curr. Opin. Chem. Biol. 1 300–308 [DOI] [PubMed] [Google Scholar]
- 7.Kouzarides, T. (1999) Curr. Opin. Genet. Dev. 9 40–48 [DOI] [PubMed] [Google Scholar]
- 8.Grozinger, C. M., and Schreiber, S. L. (2002) Chem. Biol. 9 3–16 [DOI] [PubMed] [Google Scholar]
- 9.de Ruijter, A. J., van Gennip, A. H., Caron, H. N., Kemp, S., and van Kuilenburg, A. B. (2003) Biochem. J. 370 737–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blander, G., and Guarente, L. (2004) Annu. Rev. Biochem. 73 417–435 [DOI] [PubMed] [Google Scholar]
- 11.Gao, L., Cueto, M. A., Asselbergs, F., and Atadja, P. (2002) J. Biol. Chem. 277 25748–25755 [DOI] [PubMed] [Google Scholar]
- 12.Beckers, T., Burkhardt, C., Wieland, H., Gimmnich, P., Ciossek, T., Maier, T., and Sanders, K. (2007) Int. J. Cancer 121 1138–1148 [DOI] [PubMed] [Google Scholar]
- 13.Itoh, Y., Suzuki, T., and Miyata, N. (2008) Curr. Pharm. Des. 14 529–544 [DOI] [PubMed] [Google Scholar]
- 14.Butler, L. M., Agus, D. B., Scher, H. I., Higgins, B., Rose, A., Cordon-Cardo, C., Thaler, H. T., Rifkind, R. A., Marks, P. A., and Richon, V. M. (2000) Cancer Res. 60 5165–5170 [PubMed] [Google Scholar]
- 15.Richon, V. M., Emiliani, S., Verdin, E., Webb, Y., Breslow, R., Rifkind, R. A., and Marks, P. A. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 3003–3007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelly, W. K., O'Connor, O. A., Krug, L. M., Chiao, J. H., Heaney, M., Curley, T., MacGregore-Cortelli, B., Tong, W., Secrist, J. P., Schwartz, L., Richardson, S., Chu, E., Olgac, S., Marks, P. A., Scher, H., and Richon, V. M. (2005) J. Clin. Oncol. 23 3923–3931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lahm, A., Paolini, C., Pallaoro, M., Nardi, M. C., Jones, P., Neddermann, P., Sambucini, S., Bottomley, M. J., Lo Surdo, P., Carfi, A., Koch, U., De Francesco, R., Steinkuhler, C., and Gallinari, P. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 17335–17340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones, P., Bottomley, M. J., Carfi, A., Cecchetti, O., Ferrigno, F., Lo Surdo, P., Ontoria, J. M., Rowley, M., Scarpelli, R., Schultz-Fademrecht, C., and Steinkuhler, C. (2008) Bioorg. Med. Chem. Lett. 18 3456–3461 [DOI] [PubMed] [Google Scholar]
- 19.Jones, P., Altamura, S., De Francesco, R., Gallinari, P., Lahm, A., Neddermann, P., Rowley, M., Serafini, S., and Steinkuhler, C. (2008) Bioorg. Med. Chem. Lett. 18 1814–1819 [DOI] [PubMed] [Google Scholar]
- 20.Saito, A., Yamashita, T., Mariko, Y., Nosaka, Y., Tsuchiya, K., Ando, T., Suzuki, T., Tsuruo, T., and Nakanishi, O. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 4592–4597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bonfils, C., Kalita, A., Dubay, M., Siu, L. L., Carducci, M. A., Reid, G., Martell, R. E., Besterman, J. M., and Li, Z. (2008) Clin. Cancer Res. 14 3441–3449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herman, D., Jenssen, K., Burnett, R., Soragni, E., Perlman, S. L., and Gottesfeld, J. M. (2006) Nat. Chem. Biol. 2 551–558 [DOI] [PubMed] [Google Scholar]
- 23.Rai, M., Soragni, E., Jenssen, K., Burnett, R., Herman, D., Coppola, G., Geschwind, D. H., Gottesfeld, J. M., and Pandolfo, M. (2008) PLoS ONE 3 e1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gottesfeld, J. M. (2007) Pharmacol. Ther. 116 236–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu, E., Dul, E., Sung, C. M., Chen, Z., Kirkpatrick, R., Zhang, G. F., Johanson, K., Liu, R., Lago, A., Hofmann, G., Macarron, R., de los Frailes, M., Perez, P., Krawiec, J., Winkler, J., and Jaye, M. (2003) J. Pharmacol. Exp. Ther. 307 720–728 [DOI] [PubMed] [Google Scholar]
- 26.Wegener, D., Wirsching, F., Riester, D., and Schwienhorst, A. (2003) Chem. Biol. 10 61–68 [DOI] [PubMed] [Google Scholar]
- 27.Wegener, D., Hildmann, C., Riester, D., and Schwienhorst, A. (2003) Anal. Biochem. 321 202–208 [DOI] [PubMed] [Google Scholar]
- 28.Morrison, J. F., and Walsh, C. T. (1988) Adv. Enzymol. Relat. Areas Mol. Biol. 61 201–301 [DOI] [PubMed] [Google Scholar]
- 29.Duggleby, R. G., Attwood, P. V., Wallace, J. C., and Keech, D. B. (1982) Biochemistry 21 3364–3370 [DOI] [PubMed] [Google Scholar]
- 30.Cornish-Bowden, A. (1975) Biochem. J. 149 305–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tornheim, K. (1994) Anal. Biochem. 221 53–56 [DOI] [PubMed] [Google Scholar]
- 32.Koh, C. Y., Kazimirova, M., Trimnell, A., Takac, P., Labuda, M., Nuttall, P. A., and Kini, R. M. (2007) J. Biol. Chem. 282 29101–29113 [DOI] [PubMed] [Google Scholar]
- 33.Karagianni, P., and Wong, J. (2007) Oncogene 26 5439–5449 [DOI] [PubMed] [Google Scholar]
- 34.Siliphaivanh, P., Harrington, P., Witter, D. J., Otte, K., Tempest, P., Kattar, S., Kral, A. M., Fleming, J. C., Deshmukh, S. V., Harsch, A., Secrist, P. J., and Miller, T. A. (2007) Bioorg. Med. Chem. Lett. 17 4619–4624 [DOI] [PubMed] [Google Scholar]
- 35.Wong, J. C., Hong, R., and Schreiber, S. L. (2003) J. Am. Chem. Soc. 125 5586–5587 [DOI] [PubMed] [Google Scholar]
- 36.Thomas, E. A., Coppola, G., Desplats, P. A., Tang, B., Soragni, E., Burnett, R., Gao, F., Fitzgerald, K. M., Borok, J. F., Herman, D., Geschwind, D. H., and Gottesfeld, J. M. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 15564–15569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kawamata, N., Chen, J., and Koeffler, H. P. (2007) Blood 110 2667–2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.