

Abstract
Human carbonyl reductase 1 (hCBR1) is an NADPH-dependent short chain dehydrogenase/reductase with broad substrate specificity and is thought to be responsible for the in vivo reduction of quinones, prostaglandins, and other carbonyl-containing compounds including xenobiotics. In addition, hCBR1 possesses a glutathione binding site that allows for increased affinity toward GSH-conjugated molecules. It has been suggested that the GSH-binding site is near the active site; however, no structures with GSH or GSH conjugates have been reported. We have solved the x-ray crystal structures of hCBR1 and a substrate mimic in complex with GSH and the catalytically inert GSH conjugate hydroxymethylglutathione (HMGSH). The structures reveal the GSH-binding site and provide insight into the affinity determinants for GSH-conjugated substrates. We further demonstrate that the structural isostere of HMGSH, S-nitrosoglutathione, is an ideal hCBR1 substrate (Km = 30 μm, kcat = 450 min-1) with kinetic constants comparable with the best known hCBR1 substrates. Furthermore, we demonstrate that hCBR1 dependent GSNO reduction occurs in A549 lung adenocarcinoma cell lysates and suggest that hCBR1 may be involved in regulation of tissue levels of GSNO.
Human carbonyl reductase 1 (hCBR1),3 an NADPH-dependent enzyme belonging to the short chain dehydrogenase/reductase family, has been shown to be involved in the metabolism of structurally diverse carbonyl-containing substances. This is in contrast to other members of the short chain dehydrogenase/reductase family, such as 11β-hydroxysteroid dehydrogenases 1 and 2 that interconvert cortisone and cortisol, and the 17β-hydroxysteroid dehydrogenases, which have well defined androgen and estrogen substrates. Previously reported substrates of hCBR1 include prostaglandins and xenobiotics such as the anti-cancer anthracyclin doxorubicin and the vitamin K2 precursor menadione (1). More recently, hCBR1 has been linked to the detoxification of reactive aldehydes such as 4-oxonon-2-enal and its GSH conjugate that are believed to play a central role in oxidative stress-related neurodegenerative disorders including Alzheimer and Parkinson diseases (2). Although extensive biological investigations of hCBR1, including RNA interference, pharmacology, and crystallography of human hCBR1 in complex with substrate mimics, have been carried out (3), the endogenous physiological substrate(s) of this enzyme remain to be defined. One well accepted aspect of hCBR1 substrate recognition is the presence of a GSH-binding pocket predicted to be in close proximity to the catalytic site. GSH conjugates of otherwise poorly recognized substrates such as prostaglandin A1 are reduced by hCBR1 (4), supporting this hypothesis. To identify additional physiological substrates of hCBR1, we initiated a structural biology effort to analyze the GSH-binding site of hCBR1, hypothesizing that cellular GSH adducts might serve as particularly good hCBR1 substrates.
We solved the x-ray co-crystal structure of hCBR1 in complex with GSH, demonstrating that the GSH-binding pocket lies within the catalytic pocket of the enzyme. To next assess the viability of other candidate substrates, we attempted to obtain crystals of hCBR1 with GSH adducts of reported hCBR1 substrates (e.g. prostaglandin A1-GSH and Menadione-GSH) as well as other cellular GSH adducts not known to be hCBR1 substrates like formaldehyde-GSH. We were able to obtain diffraction quality crystals and solve the structure of the GSH-formaldehyde conjugate, hydroxymethylglutathione (HMGSH), in complex with hCBR1, but we were unable to co-crystallize other reported GSH adduct substrates. Although HMGSH is a thio-hemiacetal that cannot be reduced by hCBR1 because of the low reduction potential of NADPH, we considered the possibility that other physiological isosteres of HMGSH might be efficient substrates. We found that the nitrogen-containing GSH adduct S-nitrosoglutathione (GSNO) is an efficient substrate of hCBR1, which implicates this enzyme in physiological GSNO catabolism.
EXPERIMENTAL PROCEDURES
Crystallization, Data Collection, and Refinement—Human carbonyl reductase 1 was overexpressed in Escherichia coli and purified as previously described (5). The crystals were obtained by the vapor diffusion method by growth in the presence of OH-PP (supplemental Fig. S1) as previously described (3). The crystals were soaked successively (three times) in precipitant solution containing 5 mm of either freshly prepared HMGSH4 or GSH. Crystals for hCBR1·NADP were grown from 20% polyethylene glycol 3350 and 0.2 m NaCl in space group P43212 with one molecule in the asymmetric unit. Single crystals were cryostabilized by rapid equilibration in precipitant solution containing 11.25% glycerol followed by flash freezing in a stream of nitrogen. The data set for hCBR1·NADP was measured in-house (Rigaku Raxis IV, UCSF). Data sets of hCBR1·NADP·OH-PP·GSH and hCBR1·NADP·OH-PP·HMGSH were measured at the 8.3.1 beamline of the Advance Light Source (Berkeley, CA). The data sets were integrated using DENZO and scaled with Scalepack (HKL2000 package (6)). The structures were solved by molecular replacement with CNS (7) or AMoRE (8). Starting coordinates were taken from hCBR1 in complex with OH-PP (Protein Data Bank code 1WMA) (3). Crystallographic refinement and electron density map calculations were carried out using REFMAC5 (9). The models of GSH and HMGSH were constructed and minimized using Moloc (10). Topology files were generated using the Dundee PRODRG2 server (11). Model building was accomplished using COOT (12). Detailed data and refinement statistics are given in Table 1. Atomic coordinates for hCBR1·NADP, hCBR1·NADP·OH-PP·GSH, and hCBR1·NAPD·OH-PP·HMGSH have been deposited to the Protein Data Bank (Protein Data Bank codes 3BHI, 3BHJ, and 3BHM). Refined structures were validated with PROCHECK (13). The figures were produced using PyMol 2002 (DeLano Scientific, San Carlos, CA).
TABLE 1.
Data collection and refinement statistics
| hCBR1·NADP | hCBR1·NADP·OH-PP·GSH | hCBR1·NADP·OH-PP·HMGSH | |
|---|---|---|---|
| Protein Data Bank code | 3BHI | 3BHJ | 3BHM |
| Data collection | |||
| Space group | P43 21 2 | P21 21 21 | P21 21 21 |
| Cell dimensions | |||
| a, b, c (Å) | 55.66, 55.66, 169.88 | 54.64, 55.47, 95.74 | 55.16, 55.94, 95.27 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 2.27-50.00 (2.27-2.35) | 1.77-27.00 (1.77-1.83) | 1.66-50.00 (1.66-1.72) |
| Rsym | 17.8 (40.2) | 2.9 (4.2) | 4.9 (32.1) |
| Completeness (%) | 99.7 (100.0) | 93.3 (91.7) | 97.1 (81.8) |
| Redundancy | 5.3 | 2.3 | 5.7 |
| I/σ | 9.29 (2.62) | 25.95 (16.13) | 27.19 (2.21) |
| Refinement | |||
| Resolution (Å) | 2.27-50.00 (2.27-2.33) | 1.77-27.00 (1.77-1.82) | 1.8-48.22 (1.80-1.85) |
| No. reflections | 13056/12416 | 27133/25749 | 27565/26182 |
| Rwork/Rfree | 20.2/28.2 | 17.3/22.3 | 19.7/26.9 |
| No. atoms | |||
| Protein | 2102 | 2103 | 2035 |
| Ligand | 48 | 142 | 112 |
| Water | 165 | 266 | 200 |
| Ion | 1 | 30 | 20 |
| B-factors | |||
| Protein | 17.8 | 13.8 | 16.8 |
| Ligand | 11.7 | 18.2 | 19.7 |
| Water | 21.1 | 27.1 | 28.9 |
| Ion | 21.5 | 35.8 | |
| Root mean square deviations | |||
| Bond lengths (Å) | 0.025 | 0.011 | 0.012 |
| Bond angles (°) | 2.076 | 1.334 | 1.346 |
| Ramachandran analysis | |||
| Residues in most favored regions (%) | 92.8 | 93.2 | 92.8 |
| Residues in additional allowed regions (%) | 6.8 | 6.4 | 7.2 |
| Residues in generously allowed regions (%) | 0.4 | 0.4 | 0 |
| Residues in disallowed regions (%) | 0 | 0 | 0 |
Km Determination for the Substrate GSNO—Human carbonyl reductase 1 activity was determined spectrophotometrically as previously described (3). The reactions contained 100 μm NADPH in 50 mm sodium phosphate (pH 6.8) and either 252 nm or 126 nm hCBR1. GSNO substrate was prepared by combining stoichiometric amounts (0.5 m each) of GSH, sodium nitrite, and HCl. Diethylenetriaminepentaacetic acid (1 mm) was added to prevent decomposition because of metal ion contamination. The yield of GSNO was verified spectrophotometrically by measuring the absorption of the created SNO group at 335 nm using an extinction coefficient of 0.92 mm-1 cm-1. The aqueous GSNO solution was added to samples to achieve concentrations of 151.4, 75.7, 37.85, 30.3, 22.7, and 15.1 μm. All of the reactions were performed in triplicate. The initial rates were calculated from the absorbance decrease (340 nm) using a combined extinction coefficient of 7.06 mm-1 cm-1 for GSNO and NADPH (14) and were applied to the Marquardt-Levenberg algorithm for Km determination. The kcat was determined at saturating substrate concentration (200 μm NADPH and 200 μm GSNO) using hCBR1 that had been purified by size exclusion chromatography (5) and never frozen.
Determination of NADPH and GSNO Stoichiometry—Reactions were prepared in the manner described for Km determination except substrate concentrations including 100 μm NADPH and 50 μm GSNO, 50 μm NADPH and 100 μm GSNO, or 50 μm NADPH and 50 μm GSNO. Stock solutions of GSNO contained diethylenetriaminepentaacetic acid (1 mm), and the concentration was verified spectrophotometrically (as above). The reactions were performed in triplicate, and the combined decrease in NAD(P)H and GSNO absorbance was determined at the end point for each reaction. Additionally, reaction stoichiometry was determined for human glutathione-dependent formaldehyde dehydrogenase (hFDH) (15) in an analogous manner using NADH.
Determination of Nitrite, Hydroxylamine, and Ammonia—The samples (1 ml) were prepared containing 250 mm sodium phosphate, 200 μm GSNO (prepared as above), and 200 μm of either NADH or NADPH. The reactions were initiated by the addition of FDH or hCBR1 respectively and allowed to continue for 1 h at 25 °C. Absorbance was monitored at 340 nm to verify that the reactions were complete. The samples were maintained at -80 °C until analyzed.
For all of the determinations, the standards were prepared in the assay buffer (250 mm sodium phosphate (pH 6.8). Nitrite concentration was determined by use of the modified Greiss reagent (Sigma) using the manufacturer's protocol. Hydroxylamine concentration was determined in a manner analogous to those previously described (14, 16). Briefly, 300 μl of 100 mm sodium phosphate (pH 4.4) was added to 100-μl sample. Then 200 μl of 1% (v/v) 8-hydroxyquinoline in 50% aqueous ethanol was added with mixing and followed by 200 μl of 1 m sodium carbonate. The samples were heated to 95 °C for 7 min and cooled on ice for 1 h, and the absorbance (750 nm) was determined. The hydroxylamine concentration was determined by comparing the sample absorbances to a standard curve prepared with samples containing hydroxylamine·HCl. Ammonia concentration was determined following instructions in the Sigma ammonia diagnostic kit. The standards were prepared using ammonium sulfate in assay buffer.
GSNO Reductase Activity from A549 Cell Lysates—A549 cells were cultured using F-12K medium containing 10% fetal bovine serum, penicillin, and streptomycin. Following removal of medium, the cells were washed twice with 1× phosphate-buffered saline and incubated with lysis buffer (150 mm NaCl, 1% Nonidet P-40, 50 mm Tris-Cl, 1 mm dithiothreitol, pH 8.0) at 4 °C for 20 min. The lysates were subsequently clarified by centrifugation (14,000 × g, 10 min), and the protein concentration was determined using the Bio-Rad DC protein assay. The activities were determined spectrophotometrically as above by subtracting the background rate of NADPH/GSNO reduction in the absence of lysate. The reactions were performed in triplicate using 1-cm-path length cuvettes containing 100 μm NADPH or NADH with and without the addition of 100 μm OH-PP-Me (supplemental Fig. S1) (3). Sodium phosphate (50 mm, pH 6.8) and GSNO (100 μm) were present in all reactions.
RESULTS AND DISCUSSION
Binding Mode of GSH and HMGSH to hCBR1—Human carbonyl reductase 1 is an enzyme with extremely broad substrate specificity capable of recognizing large anthracyclin anticancer agents such as daunorubicin as well as autocoids such as prostaglandins (1). The mechanism by which hCBR1 is thought to tolerate divergent carbonyl-containing substrates is through a common glutathione co-factor-binding site that enables recognition of GSH adduct substrates. We sought to first understand how GSH can deliver substrates to the catalytic machinery of hCBR1 using x-ray crystallography. We recently solved the crystal structure of hCBR1 in complex with a substrate-mimic inhibitor (OH-PP) of the enzyme (3). Although x-ray structures of both the porcine and hCBR1 enzymes have been reported, and both are known to contain functional GSH-binding sites, the location of the GSH-binding site has not been defined structurally.
Our attempts to co-crystallize hCBR1 with GSH did not yield protein crystals. Instead, we utilized soaking experiments to introduce GSH into hCBR1 crystals already containing NADP and the substrate mimic OH-PP (3). The crystals diffracted to 1.6 Å allowing high resolution analysis of hCBR1 with NADP, OH-PP, and GSH bound. The binding site for GSH is located in the cleft of the enzyme active site with the GSH-Cys oriented toward the nicotinamide moiety of the NADP co-factor and the OH-PP substrate mimic. Because GSH adducts are appended from the GSH-Cys, it appears that the orientation of GSH in these crystals is consistent with the putative mechanism of substrate recognition of GSH adducts (supplemental Fig. S2). Interactions between GSH and hCBR1 are mediated along the entire GSH tripeptide structure, predominantly by polar main chain interactions including the carboxylate of the γ-Glu to Ala192 NH and Tyr193 NH (2.9 Å and 2.8 Å), the cysteinyl NH to the Phe94 carbonyl (2.1 Å), and the glycine carboxylate to Val96 NH (2.9 Å) (Fig. 1, a and b). Interestingly, in addition to the Ala192 and Tyr193 main chain interactions, the α-carboxyl moiety of γ-Glu in GSH is positioned above the N-terminal end of a helix (Ser190–Arg209) and binds isostructurally to the sulfate ion found in the hCBR1·NADP·OH-PP structure (Protein Data Bank code 1WMA). The hydrogen bond between the α-amine of GSH and Q105 Oε1 (3.0 Å) is the only side chain interaction observed. Thus, the unique structural aspect of the iso-peptide bond between Glu and Cys in GSH is recognized through multiple protein ligand interactions in hCBR1. The structure of GSH bound to hCBR1 reveals the glutathione-binding pocket but does not further reveal how adducts of GSH can deliver structurally diverse carbonyl-containing compounds to the NADPH co-factor. To address this issue we made GSH adducts with menadione, prostaglandin A1, and formaldehyde, although the latter is not a competent substrate of the enzyme, and it does not contain a carbonyl moiety. The only complex that produced x-ray quality crystals was that of HMGSH, HO-PP, and NADP, which diffracted to 1.5 Å.
FIGURE 1.

The glutathione binding site of CBR1. a, electron density maps of hCBR1·OH-PP·NADP·GSH. b, binding mode of hCBR1·OH-PP·NADP·GSH. c, electron density maps of hCBR1·OH-PP·NADP·HMGSH. d, binding mode of hCBR1·OH-PP·NADP·HMGSH. For a and c, the 2Fo - Fc maps are contoured in brown at a level of 1σ. NADP, OH-PP, and either GSH or HMGSH (yellow, red, blue, and white) are shown occupying the CBR1 active site. For b and d, hydrogen bonding interactions and residues within a distance of 4.0 Å from the ligand are shown. Nonpolar residues (green), polar residues (purple), and charged residues (purple/blue) are indicated. The distances are given (Å).
The binding mode of HMGSH to hCBR1 was found to be isostructural to GSH (Fig. 1, c and d). Hydrogen bonding interactions are evident between Gln105 Oε1 and the α-amine of GSH (3.2 Å). The glutamyl carboxylate is anchored to Ala192 NH (2.9 Å) and Tyr193 NH (2.8 Å), the cysteinyl NH to Phe94 carbonyl (2.0 Å), and the glutamyl carboxylate to Val96 NH (2.8 Å) (Fig. 1, c and d). Interestingly, an intramolecular hydrogen bond between the hydroxyl of HMGSH and the glutamyl carbonyl (2.4 Å) is apparent.
HMGSH is not a carbonyl-containing adduct and is not a substrate for reduction by hCBR1. HMGSH has been shown to be degraded oxidatively to S-formylglutathione by the NADH-dependent enzyme FDH (17, 18). hCBR1 has not been shown to oxidize its substrates using NADP. Nonetheless, we asked whether hCBR1/NADP could oxidize HMGSH. We observed no catalysis of this reaction (data not shown).
The crystallographic effort to understand GSH substrate recognition unfortunately did not lead to a competent substrate-containing structure for hCBR1, leading us to focus on a crystal form of the enzyme that might be more catalytically relevant. In this regard, all of our crystals contained OH-PP, a carbonyl substrate mimic poised in contact with the catalytic triad of the active site (Ser139, Tyr193, and Lys197) and adjacent to the nicotinamide ring of NADP. Although the presence of this ligand greatly facilitated crystal formation, we sought a crystal without this ligand bound. Through extensive crystal screening, we were able to solve the structure of apo hCBR1 containing only the NADP co-factor. These crystals grew under different conditions in the space group P43212 and diffracted to 2.2 Å.
Comparison of the active site geometry of the OH-PP bound structures of hCBR1 with apo hCBR1 revealed no major loop movements or other major changes to the structure, with the exception of a significant side chain rotation of Cys226 (160°) lining the substrate-binding site. This cysteine movement is coincident with the movement of Phe267 adjacent to the Cys226. Interestingly, the former position of Cys226 Sγ is occupied by a chloride ion that is coordinated between the main chain amides of Ile140 (3.2 Å), Gly228 (3.3 Å), and Cys5 of the nicotinamide co-factor (3.5 Å) (Fig. 2).
FIGURE 2.

Comparison of hCBR1·NADP (a) and hCBR1·NADP·GSH (b) structures. Either a halogen (a) or the C226 Sγ thiol/thiolate (b) is stabilized by H-bonding interactions with main chain amines of Ile140 and Gly228. The anion-binding site is also at the N terminus of an α-helix.
The movement of Cys226 and occupancy of the Sγ position by a chloride ion explains a previously reported feature of an hCBR1 C226A mutant. The C226A mutant variant was reported to be catalytically inactive, but high concentrations of chloride (200 mm) rescued activity of the enzyme (4). Thus, apparently either a negatively charged halide (chloride) or cysteinyl thiolate in the vicinity of Ile140/Gly228 and the nicotinamide co-factor is necessary for catalytic activity. The anion-binding site is located at the N terminus of helix αF′-1. The dipole of this helix is likely to lower the pKa of the Cys226 side chain and to coordinate either the thiolate or chloride anion (see Fig. 3). By soaking experiments, the chloride could be replaced by bromide or iodide, which could be readily visualized crystallography (data not shown).
FIGURE 3.

Overlay of hCBR1·OH-PP·NADP (Protein Data Bank code 1WMA, pink), hCBR1·OH-PP·NADP·GSH (Protein Data Bank code 3BHJ, olive), hCBR1·OH-PP·NADP·HMGSH (Protein Data Bank code 3BHM, blue), and hCBR1·NADP (Protein Data Bank code 3BHI, yellow). NADP, HMGSH, and OH-PP are indicated. The flexible regions Lys95–Pro102 and Met234–Lys238 are shown.
The availability of four crystal structures of hCBR1 with different substrates and substrate mimetics allowed an assessment of relative flexibility of the binding pocket with respect to GSH binding. An overlay of four hCBR1 structures (Fig. 3) reveals flexibility in the loop region formed between Lys95 and Pro102 in four structures of hCBR1. This loop contains critical residues that form a tight complex upon GSH binding. For example the main chain movement of this loop displaces the Val96 NH by 1.0 Å upon introduction of GSH by soaking. The flexibility of the loop contacting GSH in multiple structures suggests there is plasticity in the ability to accommodate GSH substrate adducts of different sizes and is consistent with the broad substrate specificity of hCBR1. We cannot be certain, however, that binding orientation of GSH in the GSH structure with OH-PP represents the binding orientation of this GSH adducts when a substrate mimic inhibitor OH-PP is not present.
GSNO Reduction by hCBR1—Considering that the only adduct of GSH we were able to structurally visualize in the hCBR1 active site contains a hydroxymethylene substituent from formaldehyde and that this adduct (HMGSH) is not a substrate for the enzyme led us to consider isosteres of HMGSH as new substrates. A physiological GSH adduct, GSNO, involved in nitric oxide metabolism, is a very close structural analog of HMGSH. GSNO contains an N for CH2 substitution of HMGSH. Furthermore, the NADH-dependent enzyme FDH is able to reduce GSNO, suggesting that the NADPH-dependent hCBR1 would be capable of the same chemistry. GSNO reduction by hCBR1 was assessed spectrophotometrically by monitoring the decrease in GSNO and NADPH absorbance at 340 nm. GSNO was prepared from equimolar quantities of GSH, HCl, and NaNO2 and was efficiently reduced by hCBR1 in an NADPH-dependent manner (supplemental Fig. S3). hCBR1 appears specific for the GSNO S-nitrosothiol over similar S-nitrosothiols because reduction of S-nitrosocysteine is not catalyzed by hCBR1 (supplemental Fig. S4). The kinetic constants for GSNO reduction by hCBR1 (Table 2) are comparable with those of its best known substrate, menadione.
TABLE 2.
Comparison of kinetic constants for hCBR1- and hFDH-catalyzed GSNO reduction using NADPH and NADH, respectively
| Enzyme | Substrate | Km | kcat | kcat/Km |
|---|---|---|---|---|
| μm | min–1 | mm–1 min–1 | ||
| hCBR1 | GSNO | 30.1 ± 3.6 | 450 ± 85 | 14,950 |
| hCBR1 | Menadione (4)a | 22 | 402 | 18,272 |
| hFDH | GSNO (19) | 27 ± 8 | 2400 ± 400 | 90,000 |
The standard errors were reported to be <20% for menadione reduction by hCBR1 (19)
Comparison of hCBR1 and hFDH GSNO Reduction Products—hCBR1 has only been reported to reduce carbonyl groups to alcohols. Therefore, we investigated the products following reduction of the S-NO bond of GSNO by hCBR1 (Fig. 3). Multiple mechanisms of GSNO reduction by hFDH have been proposed (14, 19). In some cases the released GSH may participate in further reduction of intermediate products (Scheme 1).
SCHEME 1.

First, to distinguish stoichiometric reduction from other possibilities, we determined the reaction stoichiometry of hCBR1-catalyzed reduction of GSNO by NADPH. We found the reaction stoichiometry to be nearly 1:1 as determined by carrying out reactions with limiting amounts of either GSNO or NADPH; the same reaction stoichiometry was observed following NADH-dependent GSNO reduction by hFDH (supplemental Fig. S5). Given that the reaction stoichiometry is the same, we postulated that the products of both hFDH and hCBR1-catalyzed reactions might be similar.
Products formed from the NADH-dependent nitrosoglutathione reductase, hFDH, have been characterized (Scheme 1 and Ref. 14). With the hFDH enzyme, it is believed that hydride transfer occurs directly to the S-nitrosothiol nitrogen to form an “immediate” product that rearranges to form glutathione sulfinamide and can further be hydrolyzed to glutathione sulfinic acid and ammonia (14, 15). During these reactions, several measurable products, hydroxylamine, ammonia, and nitrite are produced.
We performed a side by side comparison of hFDH and hCBR1-catalyzed GSNO reduction with NADH and NADPH, respectively. Table 3 shows the concentrations of products of both enzyme-catalyzed reactions to be very similar. The most abundant product detected for both hFDH- and hCBR1-catalyzed reactions was hydroxylamine, whereas unaccounted product(s) comprise ∼84% of theoretical yield for both hFDH- and hCBR1-catalyzed reactions. More detailed observations on hFDH (14) suggest that this distribution of products reflects glutathione sulfinamide being the primary product. Given the similarity of product distributions and reaction stoichiometry of hCBR1- and hFDH-catalyzed GSNO reduction, it is likely that glutathione sulfinamide is also the primary product of hCBR1-catalyzed GSNO reduction.
TABLE 3.
Quantitation of hydroxylamine, ammonia, and nitrite for reactions prepared with 200 μm GSNO, 200 μm NADH (hFDH), or 200 μm NADPH (hCBR1) in 250 mm sodium phosphate pH 6.8
| NH2OH | NH3 | 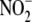 |
Unaccounted | |
|---|---|---|---|---|
| μm | μm | μm | ||
| hCBR1 | 30 ± 3 | 4.6 ± 0.4 | <15 | 165 |
| hFDH | 23 ± 5 | 5.7 ± 0.4 | <15 | 171 |
| No enzymea | 14 ± 10 | 1.0 ± 1.0 | 31 ± 1 | 154 |
The background measurements were made in the absence of diethylenetriaminepentaacetic acid
hCBR1-dependent GSNO Reductase Activity in Cell Lysates—To assess whether hCBR1 could contribute to cellular GSNO reduction, we assessed GSNO reduction in lung adenocarcinoma A549 cells, which have previously been used to investigate hCBR1 function (3). Because multiple enzymes could be responsible for observed GSNO reduction, we used the different co-factor (NADH versus NADPH) specificity of the reported GSNO reductase hFDH to differentiate between the two enzymes (hCBR1 utilizes NADPH exclusively, whereas hFDH exclusively uses NADH). Further, we needed to rule out nonenzyme-catalyzed reduction in the cell lysate, reduction caused by other enzymes like the thioredoxin system that may utilize NADPH for the homolytic cleavage of GSNO (20), and reduction by yet unidentified GSNO reductases. To distinguish between the hCBR1-dependent reductase activity in cell lysates from other NADPH-dependent GSNO reductases, we relied on an inhibitor of hCBR1, OH-PP-Me (supplemental Fig. S1) (3). Significant NADH-dependent GSNO reductase activity was measured in A549 cell lysates as measured by a decrease in absorbance at 340 nm (Fig. 4). This activity is likely due to hFDH activity. Consistent with this expectation, the hCBR1 inhibitor OH-PP-Me does not inhibit NADH-dependent GSNO reduction. We find significantly higher overall NADPH-dependent GSNO reductase activity in the A549 cell lysates. The addition of OH-PP-Me inhibits ∼30% of the NADPH-dependent activity, suggesting that this fraction of cellular GSNO reduction may be catalyzed by hCBR1. No decrease in NADH-dependent GSNO reduction was evident in the presence of OH-PP-Me (Fig. 4). Although nearly all NADH-dependent GSNO reductase activity in cells is attributable to hFDH (15), there is significant remaining NADPH-dependent activity when A549 cell lysates are treated with the hCBR1 inhibitor OH-PP-Me (Fig. 4). The source of this NADPH-dependent activity may be associated with homolytic cleavage by the thioredoxin system (20) or a yet undiscovered GSNO reductase.
FIGURE 4.

NADH- and NADPH-dependent GSNO reductase activity in A549 cell lysates with and without the presence of the selective hCBR1 inhibitor, OH-PP-Me (details under “Experimental Procedures”).
Relevance of the First Reported NADPH-dependent GSNO Reductase-hCBR1—Glutathione, which is present in cells at up to 10 mm concentration, has the ability to react with NO in cells to form GSNO, the concentration of which is tightly regulated and maintained in the micromolar range (21). The importance of regulating GSNO concentration is evident given its functions as a carrier of nitric oxide, which may include roles in microbial host defense, smooth muscle relaxation, bronchiolar dilation, and transcription factor regulation (22–24). Although several enzymes have been reported to degrade GSNO (25–27), only GSH-dependent FDH has been shown to regulate endogenous levels of GSNO (15) by use of a nicotinamide co-factor. This enzyme is unusual in that it utilizes NADH for reduction rather than the usual in vivo reductant, NADPH. Although the Km values of hCBR1 and hFDH for GSNO are similar, hFDH does exhibit a 5-fold greater kcat value. This increased catalytic efficiency of hFDH may allow the enzyme to function in the presence of low NADH levels.
The physiological relevance of these enzymes depends on the co-factor environment they operate in, and in that regard physiological levels of NADPH in cell have been found to be elevated with respect to NADP, whereas NAD+ levels are maintained higher than NADH. This implies NADPH is the universal reducing equivalent in vivo rather than NADH, and indeed, NAD+-dependent enzymes generally operate in the oxidizing direction (31). Although nicotinamide co-factor concentrations have not been determined in A549 cells, rat liver cytosolic analysis of these co-factors has indicated ratios of [NADH]/[NAD+] = 0.0015, and [NADPH]/[NADP] = 162.5 (28, 29). The total liver concentration of NAD+ and NADH has been reported to be 1.0 μmol/g, and the total NADP and NADPH concentration is ∼0.1 μmol/g (30). All of these data are consistent with a co-factor environment where NADPH is the general purpose reducing equivalent and is in sufficient concentration to support hCBR1 function. Unlike the thioredoxin system that consumes NADPH during homolytic cleavage of GSNO (releasing GSH and NO), hCBR1 represents the only known NADPH-dependent reductase that results in GSNO catabolism.
We have identified hCBR1 as an NADPH-dependent GSNO reducing enzyme (Km = 30 μm, kcat = 450 min-1). GSNO is reduced by hCBR1 with kinetic constants comparable with the best previously reported substrates of hCBR1, suggesting that GSNO may be as important an hCBR1 substrate as the many xenobiotic substrates of the enzyme. The fact that hCBR1 seems to be responsible for substantial fraction of the GSNO reductase activity in at least one cell type may suggest that this enzyme is at least partially responsible for regulation of NO/GSNO levels in tissues.
Supplementary Material
Acknowledgments
We thank Limin Liu for the generous contribution of recombinant human FDH enzyme.
The atomic coordinates and structure factors (codes 3BHI, 3BHJ, and 3BHM) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported, in whole or in part, by National Institutes of Health Grant RO1 AI44009 (to K. M. S.). This work was also supported by an award from the Sandler Program for Asthma Research (to K. M. S.) and by funds from the Deutsche Forschungsgemeinschaft (to D. R.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S6.
Footnotes
The abbreviations used are: hCBR1, human carbonyl reductase 1; GSNO, S-nitrosoglutathione; HMGSH, S-hydroxymethylglutathione; OH-PP, 3-(1-tert-butyl-4-amino-1H-indazol-3-yl)phenol; hFDH, human glutathione-dependent formaldehyde dehydrogenase.
HMGSH was produced in situ by incubating stoichiometric amounts of GSH and formaldehyde and yielded the desired linear conjugate rather than the previously found bicyclic adduct ((2S,7R)-7-(carboxymethylcarbamoyl)-5-oxo-9-thia-1,6-diaza-,bicyclo[4.4.1]undecane-2-carboxylic acid) (5).
References
- 1.Forrest, G. L., and Gonzalez, B. (2000) Chem. Biol. Interact 129 21-40 [DOI] [PubMed] [Google Scholar]
- 2.Doorn, J. A., Maser, E., Blum, A., Claffey, D. J., and Petersen, D. R. (2004) Biochemistry 43 13106-13114 [DOI] [PubMed] [Google Scholar]
- 3.Tanaka, M., Bateman, R., Rauh, D., Vaisberg, E., Ramachandani, S., Zhang, C., Hansen, K. C., Burlingame, A. L., Trautman, J. K., Shokat, K. M., and Adams, C. L. (2005) PLoS Biol. 3 764-776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tinguely, J. N., and Wermuth, B. (1999) Eur. J. Biochem. 260 9-14 [DOI] [PubMed] [Google Scholar]
- 5.Bateman, R., Rauh, D., and Shokat, K. M. (2007) Org. Biomol. Chem. 5 3363-3367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Otwinowski, Z., and Minor, W. (1997) Methods Enzymol. 276A 307-326 [DOI] [PubMed] [Google Scholar]
- 7.Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr. Sect. D Biol. Crystallogr. 54 905-921 [DOI] [PubMed] [Google Scholar]
- 8.Navaza, J. (2001) Acta Crystallogr. Sect. D Biol. Crystallogr. 57 1367-1372 [DOI] [PubMed] [Google Scholar]
- 9.Murshudov, G. N., Vagin, A. A., and Dodson, E. J. (1997) Acta Crystallogr. Sect. D Biol. Crystallogr. 53 240-255 [DOI] [PubMed] [Google Scholar]
- 10.Gerber, P. R., and Muller, K. (1995) J. Comput. Aided Mol. Des. 9 251-268 [DOI] [PubMed] [Google Scholar]
- 11.Schuttelkopf, A. W., and van Aalten, D. M. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 1355-1363 [DOI] [PubMed] [Google Scholar]
- 12.Emsley, P., and Cowtan, K. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 2126-2132 [DOI] [PubMed] [Google Scholar]
- 13.Laskowski, E. R. (1993) Mayo Clin. Proc. 68 1029-1030 [DOI] [PubMed] [Google Scholar]
- 14.Jensen, D. E., Belka, G. K., and Du Bois, G. C. (1998) Biochem. J. 331 659-668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu, L., Hausladen, A., Zeng, M., Que, L., Heitman, J., and Stamler, J. S. (2001) Nature 410 490-494 [DOI] [PubMed] [Google Scholar]
- 16.Arnelle, D. R., and Stamler, J. S. (1995) Arch. Biochem. Biophys. 318 279-285 [DOI] [PubMed] [Google Scholar]
- 17.Koivusalo, M., Lapatto, R., and Uotila, L. (1995) Adv. Exp. Med. Biol. 372 427-433 [DOI] [PubMed] [Google Scholar]
- 18.Uotila, L., and Koivusalo, M. (1997) Adv. Exp. Med. Biol. 414 365-371 [DOI] [PubMed] [Google Scholar]
- 19.Hedberg, J. J., Griffiths, W. J., Nilsson, S. J., and Hoog, J. O. (2003) Eur. J. Biochem. 270 1249-1256 [DOI] [PubMed] [Google Scholar]
- 20.Nikitovic, D., and Holmgren, A. (1996) J. Biol. Chem. 271 19180-19185 [DOI] [PubMed] [Google Scholar]
- 21.Gaston, B., Singel, D., Doctor, A., and Stamler, J. S. (2006) Am. J. Respir. Crit. Care. Med. 173 1186-1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fang, K., Johns, R., Macdonald, T., Kinter, M., and Gaston, B. (2000) Am. J. Physiol. 279 L716-L721 [DOI] [PubMed] [Google Scholar]
- 23.Que, L. G., Liu, L., Yan, Y., Whitehead, G. S., Gavett, S. H., Schwartz, D. A., and Stamler, J. S. (2005) Science 308 1618-1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hausladen, A., Privalle, C. T., Keng, T., DeAngelo, J., and Stamler, J. S. (1996) Cell 86 719-729 [DOI] [PubMed] [Google Scholar]
- 25.Hogg, N., Singh, R. J., Konorev, E., Joseph, J., and Kalyanaraman, B. (1997) Biochem. J. 323 477-481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hou, Y., Guo, Z., Li, J., and Wang, P. G. (1996) Biochem. Biophys. Res. Commun. 228 88-93 [DOI] [PubMed] [Google Scholar]
- 27.Trujillo, M., Alvarez, M. N., Peluffo, G., Freeman, B. A., and Radi, R. (1998) J. Biol. Chem. 273 7828-7834 [DOI] [PubMed] [Google Scholar]
- 28.Veech, R., Eggleston, L., and Krebs, H. (1969) Biochem. J. 115 609-619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williamson, D., Lund, P., and Krebs, H. (1967) Biochem. J. 103 514-527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reiss, P. D., Zuurendonk, P. F., and Veech, R. L. (1984) Anal. Biochem. 140 162-171 [DOI] [PubMed] [Google Scholar]
- 31.Ying, W. (2008) Antioxid Redox Signal 10 179-206 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.