

Abstract
UDP-N-acetyl-D-glucosamine (UDP-GlcNAc) is an essential precursor of peptidoglycan and the rhamnose-GlcNAc linker region of mycobacterial cell wall. In Mycobacterium tuberculosis H37Rv genome, Rv1018c shows strong homology to the GlmU protein involved in the formation of UDP-GlcNAc from other bacteria. GlmU is a bifunctional enzyme that catalyzes two sequential steps in UDP-GlcNAc biosynthesis. Glucosamine-1-phosphate acetyl transferase catalyzes the formation of N-acetylglucosamine-1-phosphate, and N-acetylglucosamine-1-phosphate uridylyltransferase catalyzes the formation of UDP-GlcNAc. Since inhibition of peptidoglycan synthesis often results in cell lysis, M. tuberculosis GlmU is a potential anti-tuberculosis drug target. In this study we cloned M. tuberculosis Rv1018c (glmU gene) and expressed soluble GlmU protein in E. coli BL21(DE3). Enzymatic assays showed that M. tuberculosis GlmU protein exhibits both glucosamine-1-phosphate acetyltransferase and N-acetylglucosamine-1-phosphate uridylyltransferase activities. We also investigated the effect on Mycobacterium smegmatis when the activity of GlmU is fully removed or reduced via a genetic approach. The results showed that activity of GlmU is required for growth of M. smegmatis as the bacteria did not grow in the absence of active GlmU enzyme. As the amount of functional GlmU enzyme was gradually reduced in a temperature shift experiment, the M. smegmatis cells became non-viable and their morphology changed from a normal rod shape to stubby-rounded morphology and in some cases they lysed. Finally a microtiter plate based assay for GlmU activity with an OD340 read out was developed. These studies therefore support the further development of M. tuberculosis GlmU enzyme as a target for new anti-tuberculosis drugs.
Keywords: Mycobacterium tuberculosis, Mycobacterium smegmatis, GlmU, glucosamine-1-phosphate acetyltransferase, N-acetylglucosamine-1-phosphate uridylyltransferase
1. Introduction
The emergence of drug resistant strains of bacteria has become a very serious problem in controlling infectious disease (Peterson, 2005). This is especially true in the case of tuberculosis. Tuberculosis (TB) remains a major threat to world health; the world-wide statistics of eight million people developing active tuberculosis and nearly one fourth of these dying from this disease is a stark reality (Dye et al.1999). (See also the Global Alliance for TB drug Development at www.tballiance.org.) TB, as a public health problem, has been complicated by the lack of a wide array of chemotherapeutics against its causative agent, Mycobacterium tuberculosis, and hence, the emergence of drug-resistant strains to the few front-line drugs has been a large problem in recent years. Although current first-line anti-TB drug regimens can achieve more than 99% efficacy, this is often reduced because of drug resistance (O'Brien and Nunn, 2001). Multiple drug resistant TB (MDR-TB), where the bacillus is resistant to several first line drugs used to be the most fearsome type of resistance; now, however, strains resistant to both first and second line antibiotics are appearing (Gandhi et al., 2006; Van Rie and Enarson, 2006); these have been dubbed extreme drug resistant TB (XDR-TB).
The mycobacterial cell wall core consists of two layers (McNeil and Brennan, 1991; Lee et al., 2006). The highly impermeable outer layer is composed of mycolic acids which are lipids containing 70 to 90 carbons. The inner layer consists of peptidoglycan. These two layers are covalently tethered via the connecting polysaccharide arabinogalactan (McNeil and Brennan, 1991; Lee et al., 2006), which is attached to the peptidoglycan through a disaccharide linker, α-L-rhamnosyl-(1,3)-α-D-GlcNAc-(phosphate). Thus, GlcNAc is an essential component of both peptidoglycan (as in most bacteria) and the rhamnose-GlcNAc linker of the cell wall in mycobacteria.
In E.coli (Mengin-Lecreulx and Vanheijenoort, 1994; Riley et al., 2006), three enzymes catalyze four sequential steps in the biosynthesis of UDP-GlcNAc, an activated nucleotide sugar of GlcNAc. The glutamine fructose-6-phosphate transferase, encoded by the glmS gene, catalyzes fructose-6-phosphate and glutamine to glucosamine-6-phosphate and glutamate, the phosphoglucosamine mutase, encoded by the glmM gene, converts glucosamine-6-phosphate (GlcNH2-6-P) to glucosamine-1-phosphate (GlcNH2-1-P). The GlmU encoded by glmU is a bifunctional enzyme. The first activity of GlmU, glucosamine-1-phosphate acetyltransferase, catalyzes the formation of N-acetylglucosamine-1-phosphate (GlcNAc-1-P) from glucosamine-1-phosphate using acetyl-CoA as the acetyl donor. The second activity of GlmU, N-acetylglucosamine-1-phosphate uridylyltransferase, catalyzes the formation of UDP-GlcNAc from GlcNAc-1-P and uridine triphosphate (UTP). The two active sites reside on the C and N terminal regions of the protein respectively (Olsen and Roderick, 2001). Analysis of the genome sequence of Mycobacterium tuberculosis H37Rv has shown that the protein encoded by M. tuberculosis Rv3436c, Rv3441c and Rv1018c are homologous to E. coli GlmS, GlmM, and GlmU, respectively. Therefore, we propose mycobacteria have the same biosynthetic pathway of UDP-GlcNAc with E. coli (Fig. 1). Antibiotics attacking peptidoglycan synthesis, such as β-lactams and glycopeptides often result in cell death typically via lysis (Holtje, 1995), and this is one reason why they are so attractive. GlmS and GlmM catalyze reactions identical or similar to those found in eukaryotes. However the first reaction catalyzed by GlmU is unique to prokaryotes and thus GlmU might be an important potential drug target.
Fig. 1.

(A) Biosynthetic pathway of UDP-GlcNAc formation in mycobacteria. GlmU (Rv1018c) is a bifunctional enzyme which has glucosamine-1-phosphate acetyl transferase and N-acetylglucosamine-1-phosphate uridylyl transferase activities. (B) The reactions of MurA and MurB which act on the UDP-GlcNAc product of GlmU and are used in the linked assay in which the loss of NADPH is monitored. In the bacterium, the product, UDP-MurNAc, is then converted to peptidoglycan.
In this study, we expressed M. tuberculosis GlmU protein in E. coli BL21(DE3) and identified GlmU to be a glucosamine-1-phosphate acetyltransferase and N-acetylglucosamine-1-phosphate uridylyltransferase. To determine the effect of active GlmU on mycobacterial growth, we prepared an M. smegmatis glmU knockout strain complemented with M. tuberculosis glmU on a rescue plasmid that was temperature sensitive for replication. Finally we developed a microtiter plate based assay for its activity.
2. Materials and methods
2.1 BACTERIAL STRAINS AND PLASMIDS
The characteristics of all bacterial strains and plasmids used in this study are described in Table 1. E. coli DH5α, NovaBlue and BL21(DE3) cells were grown in Luria-Bertani (LB) broth or LB agar at 37°C routinely. M. smegmatis mc2155 cells were grown in LB broth containing 0.05% Tween 80 or on LB agar at 37°C routinely. Sucrose was added to the LB agar at final concentration of 10% when selecting M. smegmatis glmU knockout strain ZW-2. Incubation of the M. smegmatis ZW-2 strain carrying a temperature sensitive rescue plasmid was at 30°C (permissive temperature) and 42°C (non-permissive temperature) respectively.
Table 1.
Bacterial strains and plasmids used in this study
| Strains/Plasmids | Description | Source |
|---|---|---|
| Strains | ||
| E. coli NovaBlue | Used for cloning and propagation of plasmids | Novagen |
| E. coli DH5α | Used for cloning and propagation of plasmids | Invitrogen |
| E. coli BL21(DE3) | Used for expressing M. tuberculosis GlmU protein | Invitrogen |
| E.coli ER2566 | Used for expressing E.coli MurA and MurB proteins | New England Biolabs’ |
| M. tuberculosis H37Rv | Pathogenic; used as a DNA template to amplify M. tuberculosis glmU gene | ATCC |
| M. smegmatis mc2155 | Wild-type; non-pathogenic; used as a DNA template to amplify M. smegmatis glmU and its upstream sequence | ATCC |
| M. smegmatis ZW-1 | M. smegmatis genome has the intact glmU gene and glum::kanR | This work |
| M. smegmatis ZW-2 (glmU knoukout strain) | M. smegmatis has only glmU::kanR in the presence of pZW-6 | This work |
| Plasmids | ||
| pBluescript II KS(−) | Carries ampR gene; used for DNA cloning | Strategene |
| pET16b | Carries ampR gene; used for expressing recombinant protein in E. coli | Novagen |
| pTYB4 | Carries ampR gene; used for expressing recombinant protein in E. coli | New England Biolabs’ |
| pBS-Mtb glmU | M. tuberculosis glmU gene was cloned to the SmaI site of pBluescript II KS(−) | This work |
| pET16b-Mtb glmU | M. tuberculosis glmU gene was cloned to the NdeI and XhoI sites of pET16b; the N-terminus of M. tuberculosis GlmU was fused with a histidine tag | This work |
| pTYB4-Ec murA | E.coli murA gene was cloned to the NcoI and SmaI sites of pTYB4 | This work |
| pTYB4-Ec murB | E.coli murB gene was cloned to the NcoI and SmaI sites of pTYB4 | This work |
| pMD18-T | Carries ampR gene; used for cloning PCR product with A’ at 3’ ends | Takara |
| pUC4K | Carries ampR gene and kanR cassette | GE Healthcare |
| pPR27-xylE | Carries genR, sacB and xylE genes; carries replication origin for E. coli and temperature-sensitive replication origin for mycobacteria | (Li et al., 2006) |
| pET23b-Phsp60 | Carries ampR gene; carries M. bovis BCG hsp60 promoter | (Li et al., 2006) |
| pCG76 | Carries strR gene; carries replication origin for E. coli and temperature-sensitive replication origin for mycobacteria. | (Guilhot et al.,1994) |
| pZW-1 | M. smegmatis glmU gene with its upstream sequence was cloned to the EcoRV site of pMD18-T | This work |
| pZW-2 | The kanR cassette was inserted to the ApaI site of M. smegmatis glmU in pZW-1 | This work |
| pZW-3 | sucide plasmid, glum::kanR was cloned to the NotI and SpeI sites of pPR27-xylE | This work |
| pZW-4 | M. tuberculosis glmU gene was cloned to the EcoRV site of pMD18-T | This work |
| pZW-5 | M. tuberculosis glmU gene was cloned to the NdeI and BamHI sites of pET23b-Phsp60 | This work |
| pZW-6 (pCG76-Mtb glmU) | rescue plasmid, Phsp60-Mtb glmU was cloned to the XbaI and BamHI sites of pCG76 | This work |
The final concentration of antibiotics used in this study are as follows: ampicillin (Amp), 100 µg ml−1; gentamicin (Gen), 5 µg ml−1 for E. coli and M. smegmatis; kanamycin (Kan), 50 µg ml−1 for E. coli and 25 µg ml−1 for M. smegmatis; and streptomycin (Str), 20 µg ml−1 for E. coli and 10 µg ml−1 for M. smegmatis.
2.2 CLONING AND GENETIC MANIPULATION
Cloning procedures were performed according to standard protocols (Sambrook and Russell, 2001). M. smegmatis mc2155 genomic DNA was prepared as previously described (Jackson et al., 2000). The Probe for Southern blotting was labeled with digoxygenin (DIG)-labeled dUTP using a DIG High Prime Labeling and Detection Starter Kit I (Roche). The SmaI-digested M. smegmatis genomic DNA fragments were separated by 0.8% agarose gel and transferred to Nytran membrane (Whatman Schleicher and Schuell) as described (Li et al., 2006). DNA hybridization and detection were performed according to Roche’s instructions.
2.3 CLONING, EXPRESSION AND PURIFICATION OF M. TUBERCULOSIS GLMU AND E. COLI MURA AND MURB
The M. tuberculosis glmU gene (Rv1018c) was amplified from M. tuberculosis H37Rv genomic DNA (supplied by Colorado State University through the NIH contract “Tuberculosis research materials and vaccine testing.”) by using Mtb glmU1 primer, 5’ CATATGACGTTTCCTGGTGACAC 3’ (underlined sequence is NdeI site) and Mtb glmU2 primer, 5’ CTCGAGTCACGGTGTCTGATCAGCGTC 3’ (underlined sequence is XhoI site). The amplified PCR product was ligated into pBluescript II KS(−) (Strategene) to generate a plasmid pBS-Mtb glmU (Table 1). Following sequence confirmation Mtb glmU gene was ligated into the NdeI and XhoI sites of pET-16b (Novagen), resulting in pET16b-Mtb glmU (Table 1). pET16b-Mtb glmU was transformed to E. coli BL21(DE3) for expression of GlmU protein.
E. coli BL21(DE3) containing pET16b-Mtb glmU was grown in 2 liters of LB broth containing Amp overnight at 37°C. The cells were transferred to a shaker at room temperature and induced overnight by the addition of isopropyl-D-thiogalactopyranoside (IPTG) to a final concentration of 0.1 mM. The cells were harvested and pellets resuspended in 20 ml lysis prep buffer (20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 20% glycerol) with 1 µM leupeptin, 1 µM pepstatin A, and 1 mM phenylmethylsulphonyl fluoride (PMSF). The cells were broken using the French Pressure method, followed by centrifugation at 20 000 x g for 40 minutes. The supernatant was applied to a 1.5 ml column volume HIS-Select HF affinity gel (Sigma). The column was washed with 40 ml wash buffer (lysis prep buffer with 20 mM imidazole, pH 8.0). The protein was eluted with 15 ml elute buffer (20 mM Tris-HCl, 500 mM NaCl, 20% glycerol, 200 mM imidazole, pH 7.0) with 1 µM leupeptin, 1 µM pepstatin A, and 1 mM PMSF, and the first 5 ml was collected for testing.
The purified protein was run on 12% SDS-PAGE and transferred to a nitrocellulose membrane (Whatman Schleicher and Schuell) in blotting buffer (20 mM Tris-base, 150 mM glycine and 20% methanol). The membrane was blocked with 2% BSA in TBST buffer (10 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.05% Tween 20) and incubated with (anti)-polyhistidine clone HIS-1 antibody (Sigma) at 1 to 6000 dilution followed by washing with TBST. The membrane was incubated with antimouse-IgG-AP conjugate (Sigma) and the protein band was visualized in BCIP/NBT solution.
The gene murA was amplified from the E. coli K12 genomic DNA with the following two primers incorporating NcoI and SmaI restriction enzyme sites (underlined): 5’ AACTCCATGGATAAATTTCGTGTTCAG 3’ and 5’ AGACCCGGGTTCGCCTTTCACACGC 3’. The PCR products were digested with NcoI and SmaI, ligated into the expression vector pTYB4, which had been previously digested with NcoI and SmaI. The E. coli murB gene was amplified from the E. coli genomic DNA with the following two primers incorporating NcoI and SmaI restriction enzyme sites (underlined) and cloned into pTYB4: GCAGCCATGGACCACTCCTTAAAACC and GTTATCCCGGGTGAAATTGTCTCCAC. pTYB4-Ec murA and pTYB4-Ec murB were transformed ER2566 respectively. The expressed E. coli MurA and E.coli MurB proteins were purified by IMPACT™ Protein Purification system according to New England Biolabs’ instructions.
2.4 DETERMINATION OF ACETYL TRANSFERASE AND URIDYLYLTRANSFERASE ACTIVITIES OF M. TUBERCULOSIS GLMU PROTEIN
The acetyl transferase and uridylyltransferase activities of M. tuberculosis GlmU protein were determined separately. For the acetyl transferase activity, the reaction mixture contained 100 µM each of acetyl CoA and glucosamine-1-phosphate in 10 mM Tris buffer (0.1 mM MgCl2 and 0.01% Triton X 100, pH 8). The transacetylation was carried out by the addition of 100 ng of GlmU (purified) or crude M. smegmatis extract and the reaction was incubated for 30 minutes at room temperature. A control experiment, without the addition of an enzyme source, and thus with only the substrates and buffer was also carried out. After the incubation, a part of the reaction mixture was dried in a cleaned glass tube and mild hydrolysis was carried out with 0.2 mM TFA (Trifluroacetic acid) at 80°C for 1 hour. The tubes were cooled to room temperature and dried. This drying was repeated twice in the presence of methanol. This was followed by acetylation with 1:1 of deutero acetic anhydride/pyridine, overnight. After drying the solvents, the acetylated products were extracted with D2O:CHCl3 (1:1) and the chloroform layer was subjected to GC/MS. The acetylated-derivatives were dissolved in chloroform prior to injection on a DB-5 column at an initial temperature of 50°C held for 1 min. The temperature was increased to 275°C at a rate of 5°C/min. The mass spectral analysis was carried in a positive CI mode with the collision gas being methane. The starting material showed an M+H-63 ion at 3 AMU higher than product due to the non-deuterated methyl group in the product.
To determine if the uridylyltransferase was active, the enzyme reaction was run in the reverse direction from PPi and UDP-GlcNAc back to GlcNAc-1-P and UTP (Mengin-Lecreulx and Vanheijenoort, 1994). The reaction was performed in 40 µl total volume containing 25 µM UDP-GlcNAc, 1 mM pyrophosphate (PPi) and 42 µg/ml GlmU protein in 50 mM Tris-HCl (pH 8.2) containing 1 mM MgCl2. After incubation at 37°C for 30 minutes, the reaction was then run on a Beckman HPLC with a 30 minute gradient starting with 100% 75 mM KH2PO4 and ending with 100% 500 mM KH2PO4.
For the microtiter plate assay, GlmU activity was linked with MurA and MurB from E. coli. The assay was run by combining 4 µg purified M. tuberculosis GlmU, 0.6 ìg purified E. coli MurA and 80 ng purified E. coli MurA in 80 µl 200 mM Tris containing 10 mM MgCl2, 200 µM NADPH, and 20 mM KCl in each well of a 96 well plate. The reaction was started by adding 20 µl 200 mM Tris, 10 mM MgCl2 containing 1 mM UTP, 1 mM acetyl CoA, 1 mM glucosamine-1-phosphate, and 1 mM phosphoenol pyruvate. The Absorbance at 340 nm was followed for 45 minutes at 30°C in a Benchmark Plus (Bio-Rad) plate reader and the data were analyzed using Microsoft Excel.
2.5 CONSTRUCTION OF SUICIDE PLASMID AND RESCUE PLASMID
The amino acid sequence of M. tuberculosis H37Rv GlmU protein (http://genolist.pasteur.fr/TubercuList/) was used as a query in BLASTP to identify the most homologous gene in M. smegmatis mc2155 genome. The M. smegmatis glmU gene (MSMEG_5426) was found to have 75% identical homology with Rv1018c. M. smegmatis glmU (1449 bp) with its upstream region (491 bp) was amplified from mc2155 genomic DNA by using LA Taq DNA polymerase (Takara), glmU1 primer (5’ACTAGTCTCGACACGGACCCGGGAGC 3’, underlined sequence is SpeI site) and glmU2 primer (5’TGCGGCCGCTCAGCTCTCGTCGCCCAACG3’, underlined sequence is NotI site). The PCR product of 1947 bp was purified and cloned into pMD18-T vector (Takara) to generate a plasmid a plasmid pZW-1 (Table 1). The glmU gene was disrupted by inserting the kanamycin resistance cassette (kanR) from pUC4K (GE Healthcare), yielding pZW-2 (Table 1). The pZW-2 was digested by NotI and SpeI and the resulting glmU::kanR fragment (3.1 kb) was ligated into the NotI and SpeI sites of pPR27-xylE (Li et al., 2006), resulting in a suicide plasmid pZW-3 (Table 1). The parent plasmid pPR27 (Pelicic et al., 1997) is a shuttle vector containing the replication origins for both E.coli and mycobacteria. Specifically, the replication origin for mycobacteria has mutations resulting in the plasmid replication being temperature sensitive in mycobacteria. Therefore, pZW-3 replicates at permissive temperature (30°C), but it can not replicate at non-permissive temperature (42°C), resulting in its integration to M. smegmatis genome where both intact glmU gene and glmU::kanR are present.
M. tuberculosis glmU gene was amplified from M. tuberculosis H37Rv genomic DNA by using Mtb glmU1 primer, 5’ CATATGACGTTTCCTGGTGACAC 3’ (underlined sequence is NdeI site)’ and Mtb glmU3 primer, 5’GGATCCTCACGGTGTCTGATCAGCGTC 3’ (underlined sequence is BamHI site). The PCR product of M. tuberculosis glmU was cloned into pMD18-T vector to generate a plasmid pZW-4 (Table 1). The M. tuberculosis glmU gene was ligated into the NdeI and BamHI sites of pET23b-Phsp60 (Li et al., 2006), yielding in pZW-5 (Table 1). The Phsp60-Mtb glmU was ligated to the XbaI and BamHI sites of pCG76 (Guilhot et al., 1994), resulting in rescue plasmid pZW-6 (Table 1). The M. tuberculosis glmU gene was transcribed from the promoter of heat shock protein 60 from M. bovis BCG and M. tuberculosis GlmU protein complemented glmU::kanR in M. smegmatis ZW-2. The parent plasmid pCG76 has the same temperature-sensitive mycobacterial replication origin as pPR27 and thus can replicate at 30°C, but is effectively lost at 42°C.
2.6 SELECTION OF M. SMEGMATIS GLMU KNOCKOUT STRAIN ZW-2
M. smegmatis electrocompetent cells were prepared as described (Pelicic et al., 1997) and suicide plasmid pZW-3 was electroporated to the competent cells with a Gene Pulser (Bio-Rad). Transformants were grown on LB agar plates containing Kan and Gen at 30°C. One colony was then propagated in LB broth containing 0.05% Tween 80, Kan and Gen at 30°C, and the cells were then plated onto an LB agar plate containing Kan and Gen and incubated at 42°C until colonies appeared. M. smegmatis ZW-1 (Table 1) with integrated pZW-3 at the glmU locus was selected using Southern blot.
The pZW-6 rescue plasmid was electroporated into ZW-1 cells. Transformants were grown on an LB agar plate containing Kan and Str, and incubated at 30°C. Transformants were then plated onto an LB agar plate containing Kan, Str and 10% sucrose. M. smegmatis ZW-2 (glmU gene knockout strain) (Table 1) was confirmed by using Southern blot.
2.7 MONITORING GROWTH OF M. SMEGMATIS ZW-2
The growth of M. smegmatis ZW-2 was followed by monitoring the optical density (OD) at 600 nm of cultures and measuring colony forming units (CFU). M. smegmatis ZW-2 was inoculated into 2 tubes containing 5 ml of LB broth with 0.05% Tween 80 and the appropriate antibiotics, and the cells were incubated at 30°C and 42°C respectively. M. smegmatis mc2155 and M. smegmatis mc2155 with pZW-6 were used as controls. The OD600 was detected at the interval of 24 hours and the growth curves at both 30°C and 42°C were obtained.
To measure CFU M. smegmatis ZW-2 cells were grown in LB broth containing 0.05% Tween 80 and Kan at 30°C until the OD600 reached 0.10, and the cells were transferred to a 42°C incubator. The OD600 of the cells grown at 42°C was determined at the interval of 24 hours. For CFU determinations (taken at the same time points), dilutions were spread on LB agar plates containing Str and Kan. The plates were incubated at 30°C before CFU were counted.
2.8 DETECTION OF M.TUBERCULOSIS GLMU PROTEIN IN M. SMEGMATIS ZW-2
M. smegmatis ZW-2 cells grown at 30°C (OD600 was 0.10) were switched to a 42°C incubator. The ZW-2 cells kept growing at 30°C were as controls. The cells were harvested at the interval of 24 hours and pellets resuspended in lysis prep buffer with 1 mM PMSF. The cells were broken by sonication followed by centrifugation at 20 000 x g for 30 minutes. The supernatants were run on 10% SDS-PAGE and Western blot was performed as described in 2.3. The membrane was incubated with the anti-M. smegmatis GlmU serum from mouse, which was prepared in our lab (unpublished data), then was incubated with antimouse-IgG-AP conjugate (Sigma). The protein band was visualized in BCIP/NBT solution.
2.9 MORPHOLOGY OF M. SMEGMATIS ZW-2 AT 42 °C
The effects of decreasing GlmU activity on the morphology of ZW-2 were examined by scanning electron microscopy (SEM) at the same time points that the CFUs were determined. The cells were washed three times in the 0.1 M Phosphate buffer (pH 7.4) and fixed by 2.5% glutaraldehyde and 1% OsO4 followed by dehydration through a graded series of ethanol (20, 40, 60, 70, 80, 90, 100%). The cell pellet was resuspended in 100% ethanol, applied to a silicon wafer slide, and gold coated to a thickness of 5 nm. The cells were examined with a JSM-6500F scanning electron microscope (at the Central Instrumentation Facility, Colorado State University) using an accelerating voltage of 15 kV.
3. Results
3.1. INITIAL IDENTIFICATION OF GLMU IN MYCOBACTERIA
The amino acid sequence encoded by M. tuberculosis H37Rv gene Rv1018c is expected to encoded GlmU due to is homology with other bacterial GlmU proteins. This sequence was used as a query in BLASTP to identify the most homologous gene in M. smegmatis mc2155 genome. The M. smegmatis glmU gene (MSMEG_5426) was found to have 75% identical homology with Rv1018c. The sequence alignment of the two mycobacterial GlmU proteins and the E. coli GlmU protein is shown in Fig. 2. To confirm the presence of GlmU in M. smegmatis, the conversion of GlcNH2-1-P to GlcNAc-1-P, the N-acetylase activity of GlmU was shown to occur in crude M. smegmatis extracts (Fig. 3c) using the GC/MS method shown in Fig. 3a.
Fig. 2.

Sequence alignment of glmU from E. coli, M. tuberculosis and M. smegmatis. The symbol “.” indicates where breaks have been inserted by the computer in order to obtain the best alignment. The sequence underlines indicate the uridylyl transferase active site (Kostrewa et al., 2001). The active site for the acetylation involves amino acids from two adjacent, interacting beta helices which is depicted as follows: bold letters are the Acetyl Coenzyme A binding amino acids, the bold italics letters are the amino acids which bind the glucosamine-1-phosphate (Olsen et al., 2007). The numbering at the end of each line for the three sequences refers to the amino acid number of each species’ sequence. Consensus 1 shows the homology when the M. tuberculosis and M. smegmatis sequences (second and third lines) are compared while Consensus 2 shows the homology when the sequence of all three organisms (first, second, and third lines) are compared. The symbols used in Consensus 1 and Consensus 2 are as follows: a “.” means no consensus was found; an upper case abbreviation of an amino acid means a perfect consensus is present; a lower case abbreviation of an amino acid means an imperfect consensus was found where this amino acid was present in all but one sequence. Other symbols indicate where closely related amino acids are found and thus: “!” is where I and V are found; “$” is where L or M are found; “%” is where F or Y are found; and “#” is where N, D, Q, or E are found.
Fig. 3.

Determination of GlmU acetyl transferase activity. (A) The preparation of tetra-O-perdeuterioacetyl-N-perdeuterioacetyl derivative from the starting GlcNH2-1-phosphate and tetra-O-perdeuterioacetyl-N-acetyl derivative from product GlcNAc-1-phosphate. D6Ac2O = hexadeuterioacetic anhydride. (B) The M+H-63 ion (63 is the weight of trideuterio acetic acid) detected by GC/MS (CI mode) analysis after derivatization of the reactants incubated in buffer with no enzyme (control reaction). (C) The M+H-63 ion detected by GC/MS (CI mode) analysis after derivatization of the reactants incubated in buffer with crude M. smegmatis enzymes. (D) The M+H-63 ion detected by GC/MS (CI mode) analysis after derivatization of the reactants incubated in buffer with M. tuberculosis GlmU expressed in E. coli. In both C and D the starting material was fully converted to product. The acetyl transferase activity incubations were in 200 µl of 10 mM Tris buffer (0.1 mM MgCl2 and 0.01% Triton X 100), pH 8 containing 100 µM each of acetyl CoA and glucosamine-1-phosphate. Either 100 ng of purified M. tuberculosis GlmU (D), or 0.5 µg of crude M. smegmatis extract (C), or no enzyme (B) was added and the reactions incubated for 30 minutes at room temperature before derivatization.
3.2 EXPRESSION AND PURIFICATION OF M. TUBERCULOSIS GLMU PROTEIN
The M. tuberculosis glmU gene was cloned into the NdeI and XhoI sites of pET16b yielding an N-terminal histidine tag on the GlmU fusion protein with an expected molecular weight of 54.10 kD. The M. tuberculosis GlmU was expressed as a soluble protein in E. coli BL21(DE3) after induction by 0.1 mM IPTG at room temperature and purified by Ni2+ affinity chromatography. Purified M. tuberculosis GlmU protein was determined to be the expected size by SDS-PAGE and Western blot analysis (Fig. 4).
Fig. 4.

Analysis of the purified M. tuberculosis GlmU protein by SDS-PAGE (A) and Western blot (B). Lane 1. PageRuler prestained protein ladder (Fermentas, from top to bottom: 170, 130, 100, 70, 55, 40, 35, 25 kD); Lane 2. purified M. tuberculosis GlmU protein (with histidine tag) (5 µg) with an expected molecular weight of 54.10 kD. The (anti)-polyhistidine clone HIS-1 antibody at 1 to 6000 dilution was used in Western blot analysis (B).
3.3 ACETYL TRANSFERASE AND URIDYLYLTRANSFERASE ACTIVITIES OF M. TUBERCULOSIS GLMU PROTEIN
The acetyl transferase of purified M. tuberculosis GlmU protein was demonstrated using the GC/MS assay as shown in Fig. 3d. The uridylyltransferase activity was shown in the reverse direction where UDP-GlcNAc is converted back to UTP and GlcNAc-1-P. Thus incubation of UDP-GlcNAc with PPi for 30 minutes lead to complete disappearance of UDP-GlcNAc (Fig. 5c) while a control reaction (Fig. 5b), in which PPi was not included in the reaction mixture, lead to no reaction.
Fig. 5.

The uridylyl transferase activity of GlmU enzyme as assayed by HPLC. (A) UDP-GlcNAc standard, (B) UDP-GlcNAc with GlmU and no PPi, and (C) UDP-GlcNAc with GlmU with PPi. The enzyme reactions were incubated at 37°C for 30 minutes in 40 µl in 50 mM Tris-HCl (pH 8.2) containing 1 mM MgCl2, 25 µM UDP-GlcNAc, 1 mM pyrophosphate (PPi) (in C only), and 42 µg/ml GlmU protein.
3.4 GENERATION OF M. SMEGMATIS GLMU GENE KNOCKOUT STRAIN ZW-2
M. smegmatis mc2155 transformants with pZW-3 were selected on LB agar plates containing Kan and Gen at 30°C and all colonies turned to yellow when catechol was sprayed on the plates. The yellow colonies were propagated in LB broth containing 0.05% Tween 80, Kan and Gen at 30°C, and then spread on LB agar plates containing Kan and Gen at 42°C. Since pZW-3 plasmid is unable to replicate at 42°C, the Kan resistant colonies on the plates have necessarily integrated glmU::kanR at the glmU locus in the M. smegmatis mc2155 genome. An M. smegmatis transformant that arose from a single homologous recombination event at the glmU locus (ZW-1) was confirmed by Southern blot.
After the rescue plasmid pZW-6 was electroporated to M. smegmatis ZW-1, the generation of M. smegmatis ZW-2 (glmU knockout strain) (can grow in LB agar containing Kan and 10% sucrose) was selected for. Six M. smegmatis ZW-2 strains were confirmed by Southern blot (Fig. 6). All six strains showed expected bands of 1.0 kb (faint), 1.59 kb and 1.79 kb. The band at 1.34 kb was missing, thus confirming the loss of intact glmU gene. The bands of 0.65 kb, 0.80 kb and 5.3 kb were from pZW-6 plasmid.
Fig. 6.

Southern blot analysis of M. smegmatis ZW-2 strains.
(A) The expected DNA fragments of M. smegmatis mc2155. (B) The expected DNA fragments of M. smegmatis ZW-2. (C) Confirmation of M. smegmatis ZW-2 strains by Southern blot. Lanes 1–6. M. smegmatis ZW-2 strains have the hybridized DNA bands of 1.0 kb, 1.59 kb, and 1.79 kb; Lane 7. pZW-6 has the hybridized DNA bands of 0.65 kb, 0.8 kb and 5.3 kb; Lane 8. M. smegmatis mc2155 wild type has the hybridized DNA bands of 1.34 kb and 1.79 kb. (D) The map of plasmid pCG76-Mtb glmU (pZW-6) rescue plasmid.
3.5 ESSENTIALITY OF GLMU GENE FOR MYCOBACTERIAL GROWTH
In order to determine whether GlmU is required for mycobacterial growth, growth curves of M. smegmatis ZW-2 at both 30°C and 42°C were obtained (Fig. 7A). The results clearly showed that M. smegmatis ZW-2 grew only at 30°C, and not at 42°C because there was no pZW-6 to replicate. In contrast, M. smegmatis mc2155 and M. smegmatis mc2155 containing pZW-6 grew at both 30°C and 42°C. Therefore, the results showed that M. tuberculosis glmU gene was essential for mycobacterial growth.
Fig. 7.
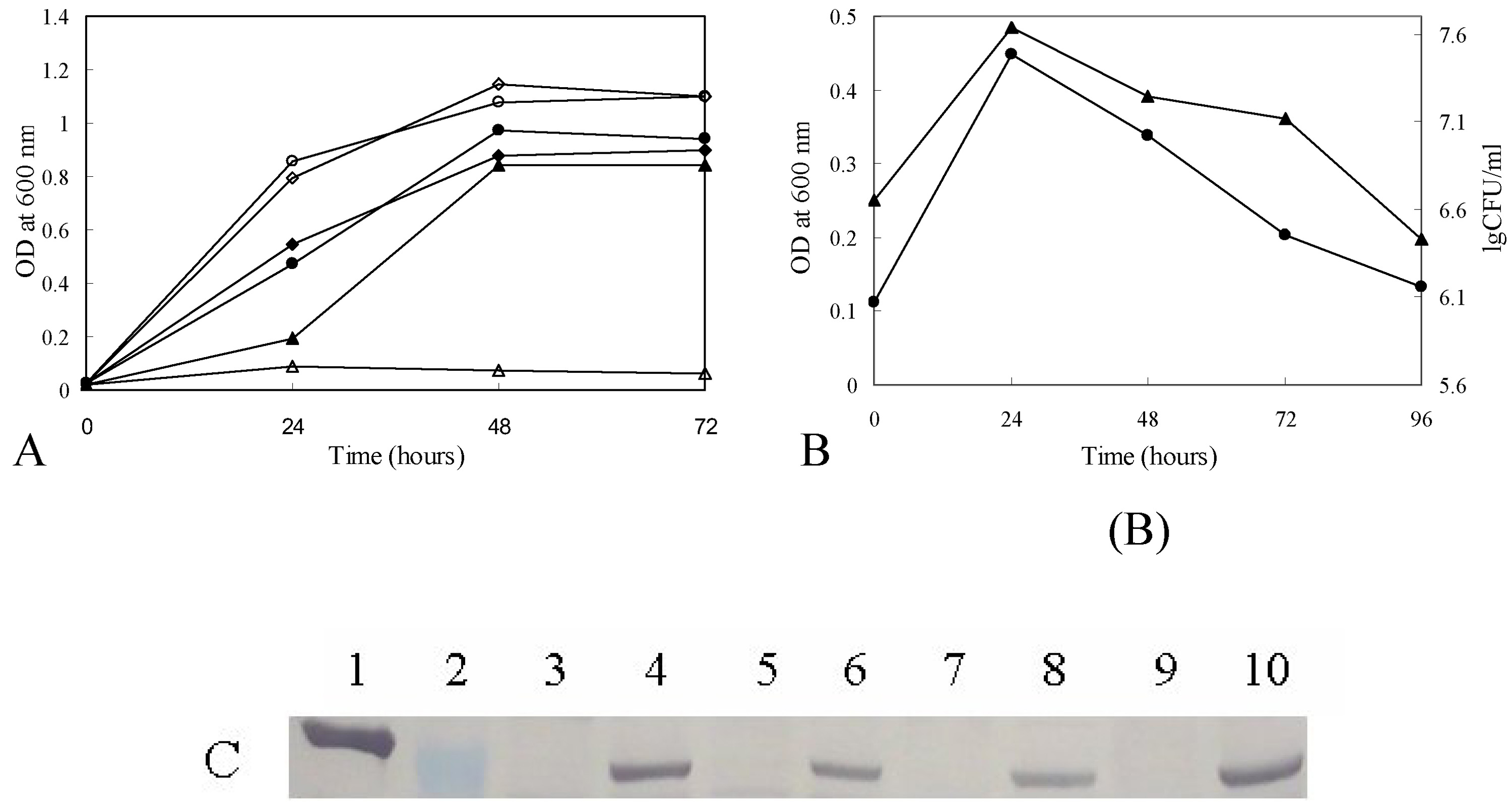
Growth curves of M. smegmatis ZW-2 strain.
(A) Growth curves of M. smegmatis ZW-2 strain at 30°C and 42°C. M. smegmatis ZW-2 at 30°C (▲); M. smegmatis ZW-2 at 42°C (□). M. smegmatis mc2155 at 30°C (●); M. smegmatis mc2155 at 42°C (○). M. smegmatis mc2155 carrying pZW-6 at 30°C (■); M. smegmatis mc2155 carrying pZW-6 at 42°C (□). (B) CFU and growth curve of ZW-2. The cells were grown at 30°C for 24 hours and then transferred to a 42°C incubator. CFU and OD600 of M. smegmatis ZW-2 grown at 42°C were determined at the interval of 24 hours. Log of the CFUs of M. smegmatis ZW-2 (▲); OD600 of M. smegmatis mc2155 ZW-2 at 42°C (●). (C) Western blot of GlmU showing its loss after the temperature shift. The antibody against M. smegmatis GlmU cross reacts with M. tuberculosis GlmU. Lanes are: 1. purified M. tuberculosis GlmU protein (with histidine tag) (54.10 kD); 2. PageRuler™ Prestained Protein Ladder (Fermentas), the MW of the protein visualized is 55 kD; 3, 5, 7, 9. M. smegmatis ZW-2 grown at 42°C for 1, 2, 3, 4 days after shifting the temperature from 30°C to 42°C; 4, 6, 8, 10. M. smegmatis ZW-2 grown at 30°C for 1, 2, 3 and 4 days. The molecular weight of the M. tuberculosis GlmU expressed from pCG76-Mtb glmU in M. smegmatis ZW-2 is 50.29 kD.
M. smegmatis ZW-2 was grown at 30°C to 0.10 OD600, and then the culture was switched to an incubator at 42°C. The CFU and OD600 over time were obtained as shown in Fig. 7B. Since functional GlmU protein was produced in M. smegmatis ZW-2 cells at 30°C, the cells multiplied for the first 24 hours after the temperature shift (Fig. 7B). Then dramatically, with increased incubation time at 42°C, the CFUs dropped as the amount of GlmU decreased. Western blot data (Fig. 7C) confirmed the expected loss of GlmU protein, indeed after 1 day insufficient GlmU for detection by western blot was seen (Fig 7C lane 3).
3.6 MORPHOLOGICAL CHANGES OF M. SMEGMATIS ZW-2 AT 42°C
Since UDP-GlcNAc is an important precursor for peptidoglycan of mycobacterial cell wall, we expected a change in morphology and possibly lysis of the bacteria as its biosynthesis decreased. Therefore, after the temperature was switched from 30°C to 42°C, the morphology of M. smegmatis ZW-2 grown at 42°C for 24, 48, 72, and 96 hours were observed by SEM. The morphology of M. smegmatis ZW-2 at 30°C just before switching to 42°C (Fig. 8A) is indistinguishable from M. smegmatis mc2155 wild type (Fig. 8G). With time, M. smegmatis ZW-2 cells at 42°C became dramatically shorter and rounder (Fig. 8B, C and D). Some of M. smegmatis ZW-2 cells at 42°C for 96 hours showed evidence of lysis (Fig. 8E and F). These SEM results suggest that lack of GlmU activity will result in profound negative effects on the bacterial cell morphology.
Fig. 8.

Scanning electron micrographs of M. smegmatis ZW-2 strain.
(A) M. smegmatis ZW-2 cells grown at 30°C for 24 hours just before transfer to 42°C (10 000X); (B–E) M. smegmatis ZW-2 cells at 42°C for 24; 48, 72 and 96 hours (10 000X); (F) M. smegmatis ZW-2 cells at 42°C for 96 hours (the same as in E) at higher magnification (30 000X); (G) Wild type M. smegmatis control. Arrows in (E) and (F) point to areas of possible lysis.
3.7 DEVELOPMENT OF A MICROTITER PLATE ASSAY FOR GLMU
Given the desirability of GlmU as a drug target we next designed a microtiter plate based assay to detect its activity. This assay (Fig. 1) relies on the enzymes MurA and MurB to convert UDP-GlcNAc into UDP-MurNAc with the concomitant reduction of NADPH. Thus E. coli MurA and MurB were cloned and expressed in E coli and the resulting proteins purified as described in material and methods. As is evident in Fig. 9, when all enzymes and substrates are present NADPH is oxidized with a concomitant decrease in OD340.
Fig. 9.

GlmU, MurA and MurB 96 well plate assay.
The enzymatic activity of M. tuberculosis GlmU was also determined in a 96 well plate format utilizing MurA and MurB, (both from E. coli) and NADPH. When all enzymes are present (◊) the A340 drops (average slope −0.0054, standard deviation 0.00013) due to the loss of NADPH. When substrates are omitted from the reaction (Δ-no acetyl Coenzyme-A (average slope −0.0007, standard deviation 0.00033), ○ - no glucosamine-1-phosphate (average slope −0.0005, standard deviation 0.00023), × - no phosphoenol pyruvate (average slope −0.0008, standard deviation 0.00018) and □ - no UTP (average slope −0.0006, standard deviation 0.00017)) the A340 does not decrease. Similar lack of activity was seen (data not presented in graph) when any of the three enzymes were left out (no GlmU (average slope −0.00094, standard deviation 0.000012), no MurA (average slope −0.00043, standard deviation 0.00012) and no MurB (average slope −0.0006, standard deviation 0.00019).
4. Discussion
UDP-GlcNAc is an essential precursor of peptidoglycan and rhamnose-GlcNAc linker region of the mycobacterial cell wall. The pathway for UDP-GlcNAc biosynthesis is significantly different in eukaryotes and prokaryotes. In eukaryotes, N-acetylation occurs on GlcNH2-6-P and not on GlcNH2-1-P. The second reaction, the formation of UDP-GlcNAc from UTP and GlcNAc-1-P, is the same in prokaryotes and eukaryotes. In eukaryotes, uridylyltransferase activity is carried by distinct monofunctional enzymes that show little sequence homology with GlmU. However, the tertiary structure of the active site of eukaryotic enzymes responsible for the uridylyltranserase activity is similar to the uridylyltransferase active site of the prokaryotic enzymes (Peneff et al., 2001a; Peneff et al., 2001b). Given this similarity, the acetylation reaction is considered the more attractive drug target for bacteria.
We have shown herein that M. tuberculosis GlmU protein has two distinct activities; glucosamine-1-phosphate acetyltransferase and N-acetylglucosamine-1-phosphate uridylyltransferase. For mycobacteria, the GlmU enzyme clearly is essential, because when it is knocked out M. smegmatis can only grow in the presence of a rescue plasmid containing the M. tuberculosis glmU gene, and it cannot grow when the rescue plasmid does not replicate. Insertional mutagenesis studies using transposon site hybridization (TraSH) methodology reported that M. tuberculosis also glmU was an essential gene (Sassetti et al.2003); this is now extended to determine the fate of the bacteria after the gene is turned off (Fig 7 & Fig 8). Our experimental approach is somewhat limited in that the GlmU enzyme cannot be abruptly turned off, but rather the replication of the rescue plasmid containing functional M. tuberculosis glmU gene is stopped. This results in a decrease in the amount of GlmU present in the cell over time. SEM data showed a dramatic change in morphology including what appeared to be lysis of cells in the latest time point. The cells are not cleanly broken in half as sometimes happens with β-lactams, but some cells appear to be ruptured (Fig. 8E and F). Therefore, both the essentiality of GlmU and the effect of the loss of its activity on mycobacterial cells strongly support its development as a target for new TB drugs. Potentially, such new drugs could have similar bactericidal properties as β-lactams. The assay developed herein for microtiter plates allows for inhibitors of both the acetyl transferase and the uridylyltransferase to be screened for.
Inhibitors of GlmU are also important in terms of inhibiting the linker region of the mycobacterium cell wall. We have previously demonstrated the effects of knocking out the rhamnosyl formation enzymes, RmlD (Ma et al., 2002) and Rml B and RmlC (Li et al., 2006). Assays for inhibitors RmlC and RmlD are proceeding at the NIH Roadmap screening center. This work now validates and develops techniques appropriate for similar screening of GlmU for inhibitors.
Acknowledgement
This work was supported by funds provided through National Basic Research Program of China (2006CB504400), National Natural Science Foundation of China (30670454) and National Institute of Health (USA) (AI 33706).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Dye C, Scheele S, Dolin P, Pathania V, Raviglione MC WHO Global Surveillance and Monitoring Project. Consensus statement. Global burden of tuberculosis: estimated incidence, prevalence, and mortality by country. The Journal of the American Medical Association. 1999;282:677–686. doi: 10.1001/jama.282.7.677. [DOI] [PubMed] [Google Scholar]
- Gandhi NR, Moll A, Sturm AW, Pawinski R, Govender T, Lalloo U, Zeller K, Andrews J, Friedland G. Extensively drug-resistant tuberculosis as a cause of death in patients co-infected with tuberculosis and HIV in a rural area of South Africa. Lancet. 2006;368:1575–1580. doi: 10.1016/S0140-6736(06)69573-1. [DOI] [PubMed] [Google Scholar]
- Guilhot C, Otal I, Van Rompaey I, Martin C, Gicquel B. Efficient transposition in mycobacteria: construction of Mycobacterium smegmatis insertional mutant libraries. Journal of Bacteriology. 1994;176:535–539. doi: 10.1128/jb.176.2.535-539.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtje JV. From growth to autolysis: the murein hydrolases in Escherichia coli. Archives of Microbiology. 1995;164:243–254. doi: 10.1007/BF02529958. [DOI] [PubMed] [Google Scholar]
- Jackson M, Crick DC, Brennan PJ. Phosphatidylinositol is an essential phospholipid of mycobacteria. Journal of Biological Chemistry. 2000;275:30092–30099. doi: 10.1074/jbc.M004658200. [DOI] [PubMed] [Google Scholar]
- Kostrewa D, D'Arcy A, Takacs B, Kamber M. Crystal structures of Streptococcus pneumoniae N-acetylglucosamine-1- phosphate uridyltransferase, GlmU, in apo form at 2.33 A resolution and in complex with UDP-N-acetylglucosamine and Mg(2+) at 1.96 A resolution. Journal Molecular Biology. 2001;305:279–289. doi: 10.1006/jmbi.2000.4296. [DOI] [PubMed] [Google Scholar]
- Lee A, Wu SW, Scherman MS, Torrelles JB, Chatterjee D, McNeil MR, Khoo KH. Sequencing of oligoarabinosyl units released from mycobacterial arabinogalactan by endogenous arabinanase: identification of distinctive and novel structural motifs. Biochemistry. 2006;45:15817–15828. doi: 10.1021/bi060688d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xin Y, McNeil MR, Ma Y. rmlB and rmlC genes are essential for growth of mycobacteria. Biochemical and Biophysical Research Communications. 2006;342:170–178. doi: 10.1016/j.bbrc.2006.01.130. [DOI] [PubMed] [Google Scholar]
- Ma Y, Pan F, McNeil MR. Determination that dTDP-rhamnose formation is essential for growth of mycobacteria. J.Bacteriol. 2002;184:3392–3395. doi: 10.1128/JB.184.12.3392-3395.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengin-Lecreulx D, Vanheijenoort J. Copurification of glucosamine-1-phosphate acetyltransferase and N-acetylglucosamine-1-phosphate uridylyltransferase activities of Escherichia coli: characterization of the glmU gene product as a bifunctional enzyme catalyzing two subsequent steps in the pathway for UDP-N-acetylglucosamine synthesis. Journal of Bacteriology. 1994;176:5788–5795. doi: 10.1128/jb.176.18.5788-5795.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil MR, Brennan PJ. Structure, function, and biogenesis of the cell envelope of mycobacteria in relation to bacterial physiology, pathogenesis, and drug resistance; some thoughts and possibilities arising from recent structural information. Research in Microbiology. 1991;142:451–463. doi: 10.1016/0923-2508(91)90120-y. [DOI] [PubMed] [Google Scholar]
- O'Brien RJ, Nunn PP. The need for new drugs against tuberculosis, Obstacles, opportunities, and next steps. American Journal of Respiratory and Critical Care Medicine. 2001;163:1055–1058. doi: 10.1164/ajrccm.163.5.2007122. [DOI] [PubMed] [Google Scholar]
- Olsen LR, Roderick SL. Structure of the Escherichia coli GlmU pyrophosphorylase and acetyltransferase active sites. Biochemistry. 2001;40:1913–1921. doi: 10.1021/bi002503n. [DOI] [PubMed] [Google Scholar]
- Olsen LR, Vetting MW, Roderick SL. Structure of the E. coli bifunctional GlmU acetyltransferase active site with substrates and products. Protein Science. 2007;16:1230–1235. doi: 10.1110/ps.072779707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelicic V, Jackson M, Reyrat JM, Jacobs WR, Jr, Gicquel B, Guilhot C. Efficient allelic exchange and transposon mutagenesis in Mycobacterium tuberculosis. Proceedings of the National Academy of Sciences. 1997;94:10955–10960. doi: 10.1073/pnas.94.20.10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peneff C, Mengin-Lecreulx D, Bourne Y. The crystal structures of Apo and complexed Saccharomyces cerevisiae GNA1 shed light on the catalytic mechanism of an amino-sugar N-acetyltransferase. Journal of Biological Chemistry. 2001;276:16328–16334. doi: 10.1074/jbc.M009988200. [DOI] [PubMed] [Google Scholar]
- Peneff C, Ferrari P, Charrier V, Taburet Y, Monnier C, Zamboni V, Winter J, Harnois M, Fassy F, Bourne Y. Crystal structures of two human pyrophosphorylase isoforms in complexes with UDPGlc(Gal)NAc: role of the alternatively spliced insert in the enzyme oligomeric assembly and active site architecture. The EMBO Journal. 2001;20:6191–6202. doi: 10.1093/emboj/20.22.6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson LR. Squeezing the antibiotic balloon: the impact of antimicrobial classes on emerging resistance. Clinical Microbiology and Infection. 2005;11:4–16. doi: 10.1111/j.1469-0691.2005.01238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley M, Abe T, Arnaud MB, Berlyn MK, Blattner FR, Chaudhuri RR, Glasner JD, Horiuchi T, Keseler IM, Kosuge T, Mori H, Perna NT, Plunkett G, III, Rudd KE, Serres MH, Thomas GH, Thomson NR, Wishart D, Wanner BL. Escherichia coli K-12: a cooperatively developed annotation snapshot-2005. Nucleic Acids Research. 2006;34:1–9. doi: 10.1093/nar/gkj405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell DG. Molecular Cloning: A Laboratory Manual. 3 rd ed. New York: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Molecular Microbiology. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- Van Rie A, Enarson D. XDR tuberculosis: an indicator of public-health negligence. Lancet. 2006;368:1554–1556. doi: 10.1016/S0140-6736(06)69575-5. [DOI] [PubMed] [Google Scholar]