

Abstract
Dopamine, serotonin and norepinephrine are essential for neurotransmission in the mammalian system. These three neurotransmitters have been the focus of considerable research since modulation of their production and their interaction at monoamine receptors has profound effects upon a multitude of pharmacological outcomes. Our interest has focused on neurotransmitter reuptake mechanisms in a search for medications for cocaine abuse. Herein we describe the synthesis and biological evaluation of an array of 2-aminopentanophenones. This array has yielded selective inhibitors of the dopamine and norepinephrine transporters with little effect upon serotonin trafficking. A subset of compounds had no significant affinity at 5HT1A, 5HT1B, 5HT1C, D1, D2, or D3 receptors. The lead compound, racemic 1-(4-methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one 4a, was resolved into its enantiomers and the S isomer was found to be the most biologically active enantiomer. Among the most potent of these DAT/NET selective compounds are the 1-(3,4-dichlorophenyl)- (4u) and the 1-naphthyl- (4t) 2-pyrrolidin-1-yl-pentan-1-one analogs.
Introduction
The endogenous monoamines, dopamine, serotonin and norepinephrine are essential for neurotransmission in the mammalian system. These three neurotransmitters, their biological receptors, and their reuptake mechanisms are the focus of considerable research since modulation of their production and their interaction at monoamine receptors has profound effects upon a multitude of pharmacological outcomes.1–8 Dopamine, serotonin and norepinephrine are released into the synapse where their concentrations are regulated, at least in part, by reuptake proteins located in the presynaptic membrane.9,10 These reuptake mechanisms have been termed the dopamine transporter (DAT), serotonin transporter (SERT), and the norepinephrine transporter (NET). The DAT is the target of numerous therapeutic agents such as Ritalin® (methylphenidate), Adderral® (amphetamine), Wellbutrin® or Zyban® (bupropion). Our interest has focused on the DAT in a search for medications for cocaine abuse2,11–14 since cocaine’s reinforcing and stimulant properties have long been associated with its propensity to bind to and inhibit monoamine transport systems, especially the DAT.15–24 Our work has concentrated on the design of compounds that inhibit all three monoamine uptake systems with different degrees of potency and selectivity. In the search for a new class of compounds that may provide a different access to agents that target the transport systems, our attention was drawn to bupropion (Figure 1), a compound marketed as an antidepressant (Wellbutrin®) as well as for smoking cessation (Zyban®). Bupropion is a 2-substituted aminopropiophenone,25,26 that has been explored extensively. Interestingly, and of relevance to the work which we describe later, the enantiomers of bupropion may not differ in their ability to inhibit biogenic amines.27 Bupropion is structurally closely related to a 2-substituted aminopentanophenone, pyrovalerone (Figure1).
Figure 1.

In 1992 Lancelot reported that pyrovalerone inhibits the DAT and the NET, and is a weak inhibitor of the SERT.28 Its synthesis was first reported by Heffe in 1964.29 Stille30 and Holliday31 confirmed its stimulant activity in animals and humans in 1963. In 1971 pyrovalerone was demonstrated to reduce symptoms of chronic fatigue in humans.32 Later studies in rat heart revealed that it inhibits NE uptake and effects the release of NE from storage or functional pools.25,33 In 1993 Vaugeois et al.34 reported that pyrovalerone stimulated locomotor activity in mice (2mg/Kg) for up to 1 hour and that this duration of action paralleled the time course of its DAT occupancy. Notwithstanding this early clinical interest, the literature reveals little SAR on pyrovalerone. Lancelot et al.28 reported the exchange of the phenyl ring for a thiophenyl ring. This exchange resulted in analogs of similar potency for both inhibition of DA and NE uptake. Further, an increase of size of the nitrogen containing ring from a 5-membered pyrrolidine to a 6-membered piperidine caused a substantial loss in binding potency at all uptake mechanisms. These researchers also reported that their analogs inhibited both DA and NE uptake but were less potent at inhibition at SERT, a finding very similar to that now reported for the analogs of the present study. Since then, one pharmacological study has appeared34 in which pyrovalerone was shown to occupy striatal sites labeled with GBR12783, and to manifest an increase in locomotor activity. However, there are no further reports concerning SAR or biological enantioselectivity of pyrovalerone or analogs. Consequently, there is little directly relevant SAR to guide the selection of pyrovalerone analogs for evaluation as potential cocaine medication.
Herein we describe the synthesis and biological evaluation of a family of analogs of 1-(4-methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one (pyrovalerone) 4a and show, in general, that these compounds are potent inhibitors of the dopamine transporter (DAT) and norepinephrine transporter (NET), but are relatively poor inhibitors of the serotonin transporter (SERT). In addition, certain compounds were evaluated for affinity at 5HT1A, 5HT1B, 5HT1C, D1, D2, and D3 receptors and were found to be inactive.
Chemistry
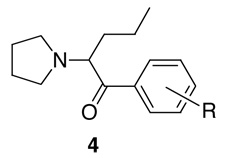
The general route of synthesis of pyrovalerone and close analogs (Scheme 1) is straightforward and was first published by Heffe in 1964.29 We have adopted this route wherever possible. The synthesis of target compounds 4 is presented in Scheme 1. Synthesis of 6, 7, 9f and 9g is shown in Scheme 2. Synthesis of compounds 9a–e is presented in Scheme 3. The ketones (Scheme 1) 2d–f are commercially available. Compound 2m was prepared from 2k. Ketones 2i–j and 2n were obtained from 2f according to a literature procedure.35 Other required ketones 2 were obtained either from aryl nitriles 1, or by Friedel-Crafts acylation of suitably substituted aryl precursors.
Scheme 1.

General Heffe synthesis of 1-(4-substitutedphenyl)-2-pyrrolidin-1-yl-pentan-1-ones, 4ab
aReagents and conditions: i) n-BuMgCl; ii) H2SO4; iii) AlCl3, Br2; iv) pyrrolidine
bThe Heffe synthesis was not followed for certain compounds. Synthetic details for those compounds are presented in the Experimental Section and are discussed in the text.
Scheme 2.

Synthesis of Analogs 6, 7, 9f and 9ga
aReagents and conditions: (i) AlCl3,Br2; (ii) Li2CO3, LiBr, DMF; (iii) pyrrolidine HCl, (HCHO)n; (iv) pyrrolidine; (v) n-BuNH2; (vi) piperidine
Scheme 3.

Synthesis of Compounds 9a–e
Thus, arylnitriles 1 were subjected to reaction with n-BuMgCl, followed by acidic hydrolysis to afford ketones 2h, 2p, 2r–u and 2w in excellent yields. Alternatively, ketones 2a, 2g and 2o were prepared by Friedel-Crafts acylation of toluene, iodobenzene and acetanilide respectively with valeroyl chloride. These ketones 2 were then brominated selectively with bromine in the presence of a catalytic amount of aluminum trichloride to provide the α-bromoketones 3 quantitatively. Ring bromination did not occur under these conditions. The α-bromoketones were then used without further purification in the subsequent reactions with pyrrolidine at room temperature to provide 4a, d–j, m–p, r–u and 4w. Compounds 4k and 4v were obtained by BBr3 demethylation of 4m and 4w respectively. Sonogashira coupling of 4g with propyne was used to prepare compound 4q, and Stille coupling with the respective stannylated heterocycles was employed to prepare compounds 4x–z from 4f. Nitro compound 4l was obtained by oxidation of compound 4o with H2O2/trifluoroacetic anhydride.
The resolution of racemic 1-(4-methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one 4a was accomplished by recrystallization from CH2Cl2/hexane of the diastereomeric salts obtained upon reaction with dibenzoyl-D-tartaric acid in refluxing ethanol (Scheme 4).
Scheme 4.

Resolution of 1-(4-methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one, 4aa
aReagents: (i) Dibenzoyl-D (or L)-tartaric acid, EtOH; (ii) Recrystallization (CH2Cl2/hexanes); (iii) Na2CO3. Et2O
This provided the (2R)-pyrovalerone dibenzoyl-D-tartrate salt. The purity was determined by 1H-NMR spectroscopy. The diastereomeric salt mixture showed two sets of triplets at δ = 0.73 and 0.69 ppm (CDCl3). These correspond to the ω-methyl protons of the pyrovalerone moieties of the (2S)-pyrovalerone dibenzoyl-D-tartrate and (2R)-pyrovalerone dibenzoyl-D-tartrate salts respectively. After four recrystallizations, the triplet at 0.73 ppm was no longer visible. The absence of the triplet attests to the diastereomeric purity of the compound, and this can be assumed to be >95% d.e. on the basis of the limits of sensitivity of the NMR experiment. It is noteworthy that the purified dibenzoyl-D-tartaric and L-tartaric acid diastereomeric salts of 4b and 4c are enantiomers and both resonate at δ 0.71 for the ω-methyl. The assignment of the absolute optical configuration of this diastereomer was confirmed by X-ray structural analysis as (2R) (optical rotation was [α]20D = +59.6° (c 1.06, EtOH)). Upon treatment with aqueous Na2CO3 and extraction into Et2O, then treatment with HCl, this diastereomeric salt gave (2R)-pyrovalerone 4c.
The (2S)-isomer 4b was then obtained from the enriched mother liquors by reaction with dibenzoyl-L-tartaric acid, recrystallization of the diastereomeric salts (optical rotation was [α]20D = −61.1° (c 1.07, EtOH)) and liberation of 4b upon treatment with aqueous sodium carbonate. The chiral center does not epimerize under these conditions. The enantiomeric purity of 4b and 4c can be anticipated to be >95% ee, that is the same as the diastereomeric purity of the precursor dibenzoyl tartrate salts. Enantiomeric purity was confirmed by HPLC chiral resolution using a Chiralpak AD column. Each isomer was thus confirmed to be >99% pure (ee > 98%).
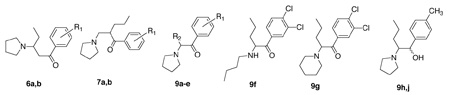
α,β-Unsaturated ketones 5a and 5 b were obtained (Scheme 2) by dehydrobromination of 3a and 3u with Li2CO3/LiBr in DMF. Reaction with pyrrolidine then gave 6a and 6b respectively. Compounds 7a and 7b were accessible via Mannich reaction of 3a and 3b with paraformaldehyde and pyrrolidine hydrochloride. Compound 3u was also used to provide 9f (reaction with butylamine) and 9g (reaction with piperidine). Compounds 9a and b were prepared (Scheme 3) by reaction of the appropriate α-bromoketones with pyrrolidine. Compounds 9c–e were prepared from the 2-pyrrolidinyl 8 29 by alkylation with propargyl bromide in the presence of sodium amide or by alkylation with allyl bromide followed by treatment with aqueous sodium hydroxide. Reduction of 4a with LiAlH4 gave 9h and 9j as a mixture of diastereomers, which were separated by flash column chromatography. All amines were converted to their HCl salts and recrystallized from EtOH/Et2O for biological assay with the exception of 4v which was isolated as its HBr salt.
Biology
The ligand affinities (Ki, nM) for inhibition of the dopamine, serotonin and norepinephrine transporters were determined in competition studies with [125I]RTI 55. Inhibition of monoamine uptake (IC50, nM) was evaluated in competition with [3H]dopamine, [3H]serotonin, and [3H]norepinephrine, and is presented in Table 1 and Table 2. In general, the analogs of 1-(4-methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one provide numerous examples of compounds that are potent inhibitors of the dopamine transporter and of dopamine reuptake. These compounds also inhibit NE reuptake with some potency, but are generally inactive at the SERT and for serotonin reuptake inhibition. One notable exception to this selectivity is the naphthyl analog 4t, which binds to all three transporters and inhibits reuptake at the nanomolar potency range. The lead compound, racemic pyrovalerone 4a, has been demonstrated here to be biologically enantioselective since the DAT inhibitory potency of the racemic mixture of 4a resides entirely with the 2S-enantiomer, 4b (DAT Ki = 18.1 nM; DA IC50 = 16.3 nM). Of these DAT/NET compounds, the most potent is the 3,4-dichlorophenyl substituted 4u, with DAT Ki= 11.5 nM and NET Ki = 37.8 nM. At this time it is unclear whether the inherent lipophilicity of both 4t and 4u is primarily responsible for their inhibitory potency. This question is currently being explored further.
Table 1.
The affinity of 4 ([125I]RTI 55) and its inhibition of uptake of [3H]dopamine, [3H]serotonin, and [3H]norepinephrine by HEK-hDAT, HEK-hSERT and HEK-hNET cellsa
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cpd | R | DAT | DA | DRb | SERT | SER | NET | NE | |
| 4 | Ki (nM) | Uptake (IC50) | Ki (nM) | Uptake (IC50) | Ki (nM) | Uptake (IC50) | |||
| Cocaine | 432 ± 29 | 461 ± 46 | 1.06 | 358 ± 24 | 494 ± 51 | 2150 ± 190 | 378 ± 48 | ||
| a | O-2371 | 4-CH3 (R/S) | 21.4 ± 4.6 | 52.0 ± 20 | 2.43 | 3770 ± 560 | 2780 ± 590 | 195 ± 26 | 28.3 ± 8.1 |
| b | O-2442 | 4-CH3 (S) | 18.1 ± 3.0 | 16.3 ± 2.3 | 0.91 | 2220 ± 550 | 1070 ± 230 | 109 ± 45 | 11.3 ± 2.4 |
| c | O-2440 | 4-CH3 (R) | 1330 ± 300 | 1790 ± 320 | 1.35 | >10µM | >10µM | ||
| d | O-2387 | H | 33.7 ± 5.4 | 52.3 ± 6.2 | 1.55 | >10µM | 199 ± 45 | 56.0 ± 13 | |
| e | O-2370 | 4-F | 82.0 ± 25 | 185 ± 62 | 2.26 | >10µM | 830±140 | 171 ± 35 | |
| f | O-2419 | 4-Br | 51.0 ± 6.7 | 39.5 ± 7.5 | 0.77 | 830 ± 190 | 1050 ± 90 | 386 ± 53 | 83.0 ± 30 |
| g | O-2493 | 4-I | 81.4 ± 9.2 | 32.0 ± 11 | 0.39 | 301 ± 26 | 197 ± 35 | 310 ± 34 | 46.5 ± 8.4 |
| h | O-2495 | 3-I | 109 ± 32 | 52.0 ± 16 | 0.48 | 1400 ± 120 | 1070 ± 170 | 670 ± 130 | 81.0 ± 20 |
| i | O-2575 | 4-CN | 5900 ± 1100 | 1000 ± 170 | 0.17 | >10µM | >10µM | ||
| j | O-2577 | 4-CH2OH | 48.7 ± 2.2 | 44.3 ± 8.4 | 0.91 | >10µM | 150 ± 23 | 12.4 ± 2.8 | |
| k | O-2418 | 4-OH | 125 ± 23 | 49.7 ± 3.4 | 0.40 | >10µM | 1290 ± 480 | 86.7 ± 7.5 | |
| l | O-2443 | 4-NO2 | 266 ± 32 | 1110 ± 340 | 4.17 | 2460 ± 290 | 1110 ± 450 | 2690 ± 530 | 531 ± 67 |
| m | O-2417 | 4-OCH3 | 329 ± 33 | 283 ± 66 | 0.86 | 4080 ± 410 | 2430 ± 720 | 2600 ± 1000 | 235 ± 8.7 |
| n | O-2558 | 4-CO2CH3 | 360 ± 140 | 154 ± 50 | 0.43 | 3950 ± 690 | 2350 ± 560 | 1140 ± 320 | 22.8 ± 3.3 |
| o | O-2439 | 4-NHCOCH3 | 30.2 ± 2.0 | 67.9 ± 8.4 | 2.25 | >10µM | 4000 ± 1100 | 317 ± 64 | |
| p | O-2481 | 4-CF3 | >10µM | 959 ± 92 | 1030 ± 340 | >10µM | |||
| q | O-2537 | 4-C≡CCH3 | 61.0 ± 16 | 11.8 ± 2.8 | 0.19 | 6700 ± 1100 | 3300 ± 1100 | 69.8 ± 5.4 | 19.3 ± 4.1 |
| r | O-2479 | 2-CH3 | 59.7 ± 9.0 | 63.0 ± 19 | 1.06 | 3720 ± 520 | 2020 ± 670 | 425 ± 63 | 19.7 ± 3.3 |
| s | O-2480 | 3-CH3 | 51.0 ± 14 | 62.9 ± 6.9 | 1.23 | 5900 ± 1600 | 4400 ± 1100 | 216 ± 38 | 9.4 ± 0.8 |
| t | O-2482 | Naphthyl | 20.1 ± 7.1 | 40.0 ± 13 | 1.99 | 33.1 ± 1.1 | 46.0 ± 5.5 | 136 ± 27 | 11.7 ± 0.9 |
| u | O-2390 | 3,4-Cl2 | 11.5 ± 1.4 | 43.0 ± 20 | 3.91 | 199 ± 50 | 600 ± 63 | 37.8 ± 3.2 | 21.0 ± 0.6 |
| v | O-2574 | 3,4-(OH) 2 | 84.0 ± 12 | 42.0 ± 11 | 0.50 | >10µM | 219 ± 71 | 7.6 ± 2.9 | |
| w | O-2512 | 3,4-(OCH3)2 | >10µM | 7460 ± 770 | 1540 ± 220 | >10µM | |||
| x | O-2441 | 4-Furan | 105 ± 17 | 122 ± 18 | 1.16 | 3330 ± 1200 | 2180 ± 440 | 95 ± 20 | 93 ± 38 |
| y | O-2438 | 4-Thiophene | 460 ± 120 | 539 ± 69 | 1.17 | 3320 ± 280 | 1960 ± 720 | 370 ± 160 | 263 ± 94 |
| z | O-2446 | 4-Mepyrrole | 3850 ± 330 | 5400 ± 1600 | 1.40 | >10µM | >10µM | ||
Numbers represent the means ± SEM from at least three independent experiments, each conducted with duplicate (for binding assays) or triplicate (for uptake assays) determinations. When the Ki or the IC50 for the test compound is greater than 10 µM, only two experiments were conducted and no standard error was reported. Data from Oregon Health & Science University and VA Medical Center, Portland, OR.
DR = Discrimination Ratio
Table 2.
The affinity of 6, 7 and 9 ([125I]RTI 55) and their inhibition of uptake of [3H]dopamine, [3H]serotonin, and [3H]norepinephrine by HEK-hDAT, HEK-hSERT and HEK-hNET cellsa
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cpd | R1 | R2 | DAT | DA | SERT | SER | NET | NE | |
| Ki(nM) | Uptake (IC50) | Ki(nM) | Uptake (IC50) | Ki(nM) | Uptake (IC50) | ||||
| 6a | O-2525 | 4-CH3 | >10µM | >10µM | >10µM | ||||
| 6a | O-2524 | 3,4-Cl2 | 8440 ± 310 | >10µM | 3900 ± 1000 | 1780 ± 220 | >10µM | ||
| 7a | O-2477 | 4-CH3 | >10µM | 4100 ± 1800 | 4800 ± 1200 | >10µM | |||
| 7a | O-2478 | 3,4-Cl2 | 1530 ± 520 | 2900 ± 1300 | 630 ± 110 | 710 ± 170 | >10µM | ||
| 9a | O-2384 | 3,4-Cl2 | CH2CH3 | 28.8 ± 2.1 | 55.0 ± 12 | 810 ± 150 | 441 ± 12 | 262 ± 36 | 18.5 ± 8.0 |
| 9b | O-2494 | 4-CH3 | CH2CH(CH3)2 | 13.7 ± 3.0 | 5.9 ± 2.3 | 2870 ± 10 | 2040 ± 150 | 259 ± 80 | 18.0 ± 5.0 |
| 9c | O-2556 | 4-CH3 | CH2CH=CH2 | 90.5 ± 3.1 | 55 ± 17 | >10µM | 1400 ± 370 | 88.0 ± 16 | |
| 9d | O-2557 | 3,4-Cl2 | CH2CH=CH2 | 39.9 ± 5.5 | 18.3 ± 3.7 | 1060 ± 170 | 440 ± 170 | 509 ± 100 | 24.9 ± 8.2 |
| 9e | O-2576 | 4-CH3 | CH2C≡CH | 2310 ± 110 | 231 ± 25 | >10µM | 4100 ± 1300 | 350 ± 120 | |
| 9f | O-2389 | 520 ± 110 | 1190 ± 58 | 5080 ± 60 | >10,000 | 4200 ± 1200 | 2520 ± 190 | ||
| 9g | O-2388 | 144 ± 48 | 666 ± 89 | 2460 ± 260 | >10,000 | 2350 ± 230 | 800 ± 200 | ||
| 9hb | O-2529-1 | >10µM | >10µM | >10µM | |||||
| 9jb | O-2529-2 | >10µM | >10µM | >10µM | |||||
Numbers represent the means ± SEM from at least three independent experiments, each conducted with duplicate (for binding assays) or triplicate (for uptake assays) determinations. When the Kior the IC50 for the test compound is greater than 10 µM, only two experiments were conducted and no standard error was reported. Data from Oregon Health & Science University and VA Medical Center, Portland, OR.
Compounds 9h and 9j are pure diastereomers
Discussion
The lead compound for these studies was racemic 1-(4-methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one (pyrovalerone) 4a (Table 1). In our assays this compound proved a potent inhibitor of both RTI 55 binding (Ki = 21.4 nM, about 20-fold more potent than cocaine as measured in the same assay) and of dopamine (DA) uptake (IC50 = 52 nM, about 9-fold more potent than cocaine). Its potency of RTI 55 inhibition of the NET (Ki = 195 nM) as well as of norepinephrine (NE) uptake (IC50 = 28.3 nM) was also marked. It was found to be more potent than cocaine in this assay by about 11-fold and 13-fold respectively. The discrepancy between the inhibition of RTI 55 binding at the NET compared with inhibition of NE uptake was seen throughout this series of compounds. This discrepancy was first reported by Eshleman et al. in 1999.36 They also noted that such differences were less evident in the case of the DAT and SERT. They suggested that this difference was likely a consequence of the ligand binding site on the NET being less closely linked to the sites of drug interactions with substrate and (NE) translocation than is the case for the DAT and the SERT.
Compound 4a was relatively inert at the SERT, with potency in the micromolar range. Therefore racemic 4a was potent at the DAT and NET, and selective against the SERT. Compound 4a exists as two enantiomers; only racemic 4a had been previously evaluated. The critical importance of absolute stereochemistry on biological function is well established. It is particularly relevant that both amphetamine (1-phenyl-2-aminopropane) and cathinone (1-phenyl-2-aminopropane-1-one) are biologically enantioselective with respect to their inhibition of DAT and NET.37,38 Indeed, the S-enantiomers are the eutomers in both cases. These two compounds bear strong structural similarities to the 1-aryl-2-pyrrolidin-1-yl-pentan-1-one analogs of this study, and therefore it was likely that their binding to, and thus inhibition of these transporters may likewise be similar. However, the structural similarity of the 1-aryl-2-pyrrolidin-1-yl-pentan-1-ones to the 2β-carbomethoxy-3α-aryl-8-azabicyclo[3.2.1]octane (tropane) class of DAT inhibitors is less clear. It is therefore interesting to note a comparison that utilized Dreiding models of WIN 35,428 (Figure 1) with enantiomers 2S-4b and 2R-4c (Scheme 4). The pyrrolidine nitrogens and the centroids of the aromatic rings were held coincident. In this rudimentary analysis the propyl side chain in the 2S-configuration overlapped with the C2-β-carbomethoxy of the tropane. However the 2R-configured compound had the propyl chain in a position similar to that of the 2α-carbomethoxy of the tropane. It has been well established that the 2α-carbomethoxy tropane analogs are less potent at the DAT than their 2β-carbomethoxy counterparts. On this basis we had postulated that 2S-4b might be the active enantiomer at the DAT. As shown in Table 1, enantiopure (2R-4c) is a poor inhibitor of RTI 55 binding at both DAT (Ki = 1,330 nM) and SERT (Ki >10 µM). In contrast, enantiopure (2S-4b) was quite potent at DAT (Ki = 18.1 nM) and selective (SERT: Ki > 2 µM). It was interesting that this relative potency of the 2S-4b enantiomer extended to the NET. Here the 2R-4c enantiomer was effectively inert at NET inhibition and NE uptake and the potency of racemic 4a resided exclusively in the 2S-4b enantiomer (NET: Ki = 109 nM; NE uptake: IC50 = 11.3 nM).
It is evident from the biological data (Table 1) that the inhibitory activities of these compounds cannot be easily correlated with varying electron density on the aromatic ring, nor with lipophilicity, or molecular refractivity. To this extent, this family of 1-aryl-2-pyrrolidin-1-yl-pentan-1-one analogs differs from other monoamine uptake inhibitors such as the 8-oxa-, 8-thia-, 8-aza- bicyclo[3.2.1]octanes,11,12,39,40 or methylphenidate analogs,41,42 where Structure Activity Relationships (SAR) were more easily discerned. Notwithstanding, certain relationships were evident among these analogs. Most clear was the fact that these 1-aryl-2-pyrrolidin-1-yl-pentan-1-one analogs were generally poor inhibitors of the SERT. Only two compounds (4t, 4u) manifested SERT Kis of <200 nM. The naphthyl analog 4t inhibited SERT with modest potency (Ki = 33.1 nM) and the high lipophilicity of this compound (cLogP = 4.77) may be partially responsible for this potency. However, the lipophilic dichlorophenyl analog 4u (cLog P = 5.01) manifested lesser SERT potency (Ki = 199 nM). Therefore lipophilicity was likely not the only factor that determined potency for 4t. Within the family of analogs evaluated, the 3,4-dichlorophenyl analog 4u was the most potent at DAT (Ki = 11.5 nM), followed by the 4-methyphenyl analog 4a. At NET, only 4q (Ki = 69.8 nM) and 4u (Ki = 37.8 nM) were potent inhibitors of RTI 55 binding, although many compounds manifested substantial inhibition of NE uptake (4a IC50 = 28.3; 4b IC50 = 11.3 nM; 4d IC50 = 56 nM; 4f IC50 = 83 nM; 4g IC50 = 46.5 nM; 4h IC50 = 81 nM; 4j IC50 = 12.4 nM; 4k IC50 = 86.7 nM; 4n IC50 = 22.8 nM; 4q IC50 = 19.3 nM; 4r IC50 = 19.7 nM; 4s IC50 = 9.4 nM; 4t IC50 = 11.7 nM; 4u IC50 = 21 nM; 4v IC50 = 7.6 nM; 4x IC50 = 93 nM; 9a IC50 = 18.5 nM; 9b IC50 = 18.0 nM; 9c IC50 = 88 nM; 9d IC50 = 24.9 nM).
It was particularly interesting that the catechol analog 4v was one of the most potent inhibitors of NE uptake (IC50 = 7.6 nM) of those evaluated. Protection as the dimethoxy compound 4w completely obliterated potency at all three monoamine transporters. The contrast between inhibition of the RTI 55 binding at the NET and inhibition of NE uptake is quite marked in the comparison of the disubstituted compounds 4u (3,4-dichloro substitution) and 4v (catechol moiety). In the former, the ratio of inhibition of NET binding to NE inhibition is about 2-fold, while in the latter this ratio is closer to 30-fold. The significance of this is unclear although this may again imply that the ligand binding site on the NET is only loosely associated with the site that effects NE translocation.36
The position of the methyl substituent on the aromatic ring influenced NE uptake potency in an opposite sense to its influence on DAT inhibition, although DA uptake inhibition was similar. Thus, while the 3-methyl analog 4s manifested an NE uptake IC50 = 9.4 nM, the 2-methyl 4r and 4-methyl 4a manifested IC50s = 19.7, 28.3 nM respectively. A comparison of 4-methyl 4a (DAT: Ki = 21.4 nM; NET: Ki = 195 nM), 2-methyl 4r (DAT: Ki = 59.7 nM; NET: Ki = 425 nM), and 3-methyl 4s (DAT: Ki = 51 nM; NET: Ki = 216 nM) 1-aryl-2-pyrrolidin-1-yl-pentan-1-ones showed that the 4-methyl 4a was at least twice as potent as the 2-methyl 4r and 3-methyl 4s at DAT. The 3-methyl 4s was about equipotent to the 4-methyl 4a at the NET, although the 2-methyl 4r remained about half as potent at the NET compared with 4a. The most DAT vs. NET selective compound in this series was the 4-acetamido derivative 4o with DAT Ki = 30.2 nM and NET Ki = 4 µM.
The search for medications for cocaine abuse has centered, primarily, about two approaches. The first is the design of compounds that can act as cocaine substitutes and that manifest, in contrast to cocaine, slow onset rates and long durations of action.11,43–45 The second approach has been to seek cocaine antagonists.13 These compounds would manifest high potency for inhibition of cocaine binding to the DAT and little or no effect on DA uptake (i.e. DA trafficking). This has been the focus of numerous studies, and Deutsch and Schweri46 have described the Discrimination Ratio (DR) as a guiding measure of potential cocaine antagonism. They defined the DR as the IC50 of DA uptake inhibition/Ki for inhibition of DA uptake by the test compound. They pointed out that a DR< 10 is of little significance owing to differences in conditions of each assay protocol. By this standard, none of the compounds here showed a DR > 5, and therefore none can be regarded as cocaine antagonists. Their use as potential medications for cocaine addiction may derive from onset and duration of action extensions, and these factors are currently under investigation.
Of note, the biaryl compounds 4x–z lacked impressive potency at all sites; this, again, is contrary to the effects of such substitution in the bicyclo[3.2.1]octane series in both the 8-aza-47 and 8-oxa series, as we shall report elsewhere.48
Table 2 presents an array of compounds that explored the displacement of the pyrrolidine ring along the butyl chain (6a, b), the introduction of different C2 side chains (9a,b) as well as introduction of side chain unsaturation (9c–e), the effects of opening the pyrrolidine ring (9f), as well as expanding it to the 6-membered piperidine (9g). Finally, reduction of the ketone to obtain both isomers (9h and 9j) is presented. The stereochemistry of these two diastereomers has not yet been determined. However, neither isomer shows any potency at DAT, SERT, and NET. A comparison of 6a with 4a, and 6b with 4u showed that essentially all inhibitory potency at all three transporters was lost when the pyrrolidine ring was moved one carbon along the chain. The nature of the pyrrolidine itself appears to be important since if it was opened (9f), or expanded (9g), the inhibitory potency was again much reduced compared with the parent compound 4u. Lancelot et al.28 had published a similar finding in their evaluation of 2-amino-1-(2-thienyl)-1-pentanones. Reduction of the ketone 4a to yield the diastereomeric alcohols 9h and 9j provided totally inactive compounds. Modification of the alkyl chain of 4a proved interesting. While a terminal acetylene (9e) resulted in a very substantial loss of potency at DAT, SERT, and NET, the allyl compounds 9c and 9d retained potency at DAT. The 3,4-dichloro compound 9d (DAT: Ki = 39.9 nM) was again the more potent of the two, although NET potency declined substantially (Ki = 509 nM) compared with the comparative compound 4u (Ki = 37.8 nM). Of these chain altered compounds, the isobutyl analog 9b proved most interesting with a DAT Ki = 13.7 nM, but DA uptake IC50 = 5.9 nM. This compares with data for 4a (DAT Ki = 21.4 and DA uptake IC50 = 52 nM). Thus the introduction of a branching methyl in the side chain has served to increase DA inhibition about 10-fold over the parent compound 4a. The possible significance of this is not clear at this time.
The biological selectivity within this class of compounds proved striking. Thirteen compounds (4b,f,k–m,o,p,r–t,y, 6a, and 6b) were evaluated for inhibition of 5HT1A, 5HT1B, 5HT1C, D1, D2, and D3 receptors. The compounds were essentially inactive (IC50 > 10 µM) in these assays. Two compounds (4o, which was a selective DAT inhibitor, and 4t, which had similar potency at DAT and SERT) were selected for evaluation of locomotor activity. Both manifested a time and dose dependent stimulation of locomotor activity (ED50 = 0.21 mg/Kg and 2.2 mg/Kg respectively) with a duration of action of > 8 hours.
Conclusion
A family of 38 analogs of a lead compound 1-(4-methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one (pyrovalerone) has been prepared. The biological activity at dopamine, serotonin and norepinephrine transporters has been determined. This family has yielded compounds that provide selective inhibitors of the dopamine and norepinephrine transporters with little effect upon serotonin trafficking. Furthermore, a subset of compounds selected for evaluation of their effect upon serotonin and dopamine receptors has shown them to be inactive at these sites. The lead compound 4a has been demonstrated to be biologically enantioselective and it remains to be determined whether this enantioselectivity extends to other members of this family of compounds. Two compounds 4o and 4t manifested a time and dose dependent stimulation of locomotor activity with a duration of action of > 8 hours.
The inhibitory potency, the neurotransmitter selectivity profile and the inactivity at selected receptor sites of 4 k and 4o have encouraged us to enter behavioral pharmacological evaluation in rat drug discrimination studies, and in vivo studies are currently ongoing.
Experimental Section
NMR spectra were recorded on a Jeol 300 NMR spectrometer (300.53 MHz for 1H and 75.58 MHz for 13C) with tetramethylsilane (TMS) as internal standard and DMSO-d6 as solvent, with the exception of compounds 2 and 3, which were measured in CDCl3. Optical rotations were measured on a Jasco P1010 polarimeter at room temperature. HPLC and MS data were obtained on an Agilent Series 1100 LC/MSD system. Melting points are uncorrected and were measured on a Mel-Temp melting point apparatus. Thin layer chromatography (TLC) was carried out on Baker Si 250F plates. Visualization was accomplished with iodine vapor, UV exposure or treatment with phosphomolybdic acid (PMA). Flash chromatography was carried out on Baker Silica Gel 40 µM (Silica gel). All reactions were conducted under an atmosphere of dry nitrogen. Elemental analyses were performed by Atlantic Microlab, Norcross, GA. Chemicals obtained from commercial sources were used as received. Room temperature is 22 °C +/− 2 °C. Yields have not been optimized.
General Procedure A. Preparation of intermediate ketones, 2
The ketones 2 were prepared (except where noted) by alkylation of the analogous commercially available nitrile compounds, followed by acidic hydrolysis. The nitrile (10 mmol) was added in several portions, over 0.5 h, to a solution of the n-BuMgCl (12 mmol) in toluene (20 mL). The reactions were monitored by TLC and heated where necessary. Generally, after 2 h stirring at room temperature, the reaction was complete. The reaction mixture was poured onto ice and concentrated H2SO4 (2 mL) was added. Hydrolysis of the intermediate imine usually occurred at room temperature after several minutes. However, for some substrates, heating was necessary to effect this transformation. The organics were extracted into Et2O, dried (MgSO4), filtered, and reduced in vacuo to an oil.
General Procedure B. Preparation of intermediate α-bromoketones, 3
Compounds 3 were prepared by α-bromination of ketones 2. The ketone (as a solution in Et2O, or CH2Cl2 (for less soluble substrates)) was cooled in an ice bath and anhydrous AlCl3 was added to the solution (1 – 5 mol%). Bromine (approximately 0.1 mol eq) was added to the solution all at once. Typically, after 10 min the solution changed from a light orange to colorless (if this change did not occur at 0 °C, then the mixture was warmed to room temperature). The remaining bromine (0.9 mol eq) was then added to the solution in a drop-wise manner over 5 min. The solution was neutralized (aqueous NaHCO3), separated, dried (MgSO4), filtered, and reduced to a lightly colored oil in vacuo. Yields were quantitative and the crude materials were sufficiently pure (1H NMR) for use in the subsequent step.
General Procedure C. 1-Aryl-2-pyrrolidin-1-yl-pentan-1-ones (4)
Compounds 4 were prepared employing General Procedure C except where noted. α-Bromoketone 3 (10 mmol) was dissolved in Et2O (10 mL) (EtOH is a suitable alternative solvent) and cooled on an ice bath. Pyrrolidine (22 mmol) was added all at once. The mixture became orange and an oil was observed to separate from the solution. After 1 – 24 h stirring at room temperature, the crude reaction mixture was partitioned between H2O (10 mL) and Et2O. The Et2O layer was separated and the aqueous layer was washed with Et2O (2 × 10 mL). The ether layer was extracted with 1 M aqueous HCl (2 × 10 mL), then backextracted into Et2O (3 × 10 mL) by basification to pH 8–9 with 20% aqueous Na2CO3 or 2 M aqueous NaOH. The Et2O extracts were dried (MgSO4) and filtered. The filtrate was treated with 2 M ethereal HCl (usually 5 – 10 mL) until precipitation of solid or oil had ceased. Solids (oils were triturated to give solids) were collected by filtration and recrystallized from EtOH/Et2O.
1-(4-Methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one hydrochloride (4a)
1-(4-Methylphenyl)pentan-1-one 2a, prepared by Friedel-Crafts acylation of toluene: 1H NMR δ 7.86 (dd, 2H), 7.25 (dd, 2H), 2.92 (m, 2H), 2.41 (s, 3H), 1.71 (m, 2H), 1.40 (m, 2H), 0.95 (t, 3H) was brominated (General Procedure B) to provide 2-bromo-1-(4-methylphenyl)-pentan-1-one 3a: 1H NMR δ 7.92 (d, 2H), 7.29 (d, 2H), 5.14 (dd, 1H), 2.43 (s, 3H), 2.25 - 2.05 (m, 2H), 1.65 - 1.35 (m, 2H), 0.98 (t, 3H). Compound 4a, obtained as a colorless solid, was prepared from 3a (General Procedure C). Yield 68%. Mp 180 °C (dec.); 1H NMR δ 10.8 - 10.65 (br, 1H), 8.01 (d, 2H), 7.44 (d, 2H), 5.56 (m, 1H), 3.7 - 3.55 (br, 1H), 3.55 - 3.4 (br, m, 1H), 3.35 - 3.2 (br, m, 1H), 3.15 - 3.0 (br, m, 1H), 2.42 (s, 3H), 2.15 - 1.85 (br, m, 6H), 1.4 - 1.2 (m, 1H), 1.15 - 0.95 (m, 1H), 0.78 (t, 3H); 13C NMR δ 196.1, 145.8, 132.1, 129.8, 129.0, 67.1, 53.5, 51.9, 31.8, 22.9, 21.3, 17.4, 13.7; APCI MS m/z 246 (M + 1); Anal. (C16H24ClNO.1/6H2O) C, H, N, Cl.
(1R)-1-(4-Methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one hydrochloride (4c) and (1S)-1-(4-Methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one hydrochloride (4b)
1-(4-Methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one hydrochloride, 4a, (10.0 g, 35.5 mmol) was extracted into Et2O from 20% aqueous Na2CO3 at pH 8–9. The ether was removed and the free base was dissolved in EtOH (50 mL) and heated to 70°C. Dibenzoyl-D-tartaric acid (12.7 g, 35.5 mmol) in hot ethanol (150 mL) was added all at once to the pale yellow solution of free base. The resulting colorless solution was refluxed for 1 min, cooled, and the solvent was removed in vacuo. The residue was dissolved in CH2Cl2 (530 mL) and hexane (700 mL) was added with swirling. After 3 d, the resulting crystalline solid (9.1 g) was collected by filtration. 1H NMR (CDCl3) showed a diastereomeric excess (d.e.) of 70 – 75%. Three consecutive recrystallizations from CH2Cl2/hexane (300 mL/400 mL) gave a single diastereoisomer (6.1 g, 61%). Mp 100 – 120 °C; 1H NMR δ 8.10 (d, 4H), 7.87 (d, 2H), 7.51 (t, 2H), 7.37 (t, 4H), 7.18 (d, 2H), 5.91 (s, 2H), 5.37 (t, 1H), 3.75 (br, m, 2H), 2.32 (s, 3H), 2.0 - 1.8 (br, m, 6H), 1.4 - 1.1 (br, m, 4H), 0.71 (t, 3H). X-ray structural analysis of this compound showed it to be the dibenzoyl-D-tartaric acid salt of (1R)-1-(4-methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one. [α]20D = +59.6° (c 1.06, EtOH).
The salt was dissolved in 20% aqueous Na2CO3 and extracted into Et2O. The Et2O layer was collected, dried and filtered. Ethereal 2M HCl was added to this solution to provide a white solid that was recrystallized from EtOH/Et2O to give pure (1R)-1-(4-methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one hydrochloride, 4c. The physical properties of this compound were identical with those of the racemic material 4a.
The residues from recrystallization of the dibenzoyl-D-tartaric acid (1R)-1-(4-methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one, were combined and the free base was liberated with 20% aqueous Na2CO3. The ethereal extracts were washed once with 20% aqueous Na2CO3, dried (MgSO4), filtered, and reduced in vacuo to an oil (5.2 g, 21 mmol). This oil was dissolved in hot EtOH (50 mL), and a solution of dibenzoyl-L-tartaric acid (7.5 g, 21 mmol) in hot EtOH (100 mL) was added with swirling. The mixture was refluxed for 1 min, cooled, and the solvent was removed in vacuo. Four recrystallizations, as described above, gave a single diastereoisomer (5.4 g, 50%). X-ray structural analysis confirmed the diastereomeric salt of dibenzoyl-L-tartaric acid (1S)-1-(4-methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one. [α]20D = −61.1 ° (c 1.07, EtOH).
The hydrochloride salt of (1S)-1-(4-methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one, 4b was then prepared as described above for (1R)-1-(4-methylphenyl)-2-pyrrolidin-1-ylpentan-1-one.
The enantiomeric purities of 4b and 4c were confirmed by chiral HPLC (Chiralpak AD, 0.46 × 25cm; Flow rate 1 mL/min; eluent 2–10% EtOH/hexanes + 0.1 % NEt3). 4b: tR = 6.77 min, purity 99.8%; 4c: tR = 5.85 min, purity 100%.
Single-crystal X-ray analysis of dibenzoyl-D-tartaric acid salt of (1R)-1-(4-methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one
Monoclinic crystals of the purified title compound were obtained from CH2Cl2/hexane. A representative crystal was selected and a 1.54178 Å data set was collected at 198 °K. Pertinent crystal, data collection and refinement parameters: crystal size, 0.32 × 0.12 × 0.03 mm; cell dimensions, a = 7.8458 (10) Å, b = 13.4366 (2) Å, c = 18.2054 (3) Å, α = 90°, β = 93.717 (10)°, γ = 90°; formula, C40H51NO9; formula weight = 689.82; volume = 1915.19 (5) Å3; calculated density = 1.196 g cm−3; space group = P21; number of reflections = 11525 of which 5630 were considered independent (R int = 0.0244). Refinement method was full-matrix least-squares on F2. The final R-indices were [I > 2σ (I)] R1 = 0.0520, wR2 = 0.1439.
Single-crystal X-ray analysis of dibenzoyl-L-tartaric acid (1S)-1-(4-methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one
Monoclinic crystals of the purified dibenzoyl-L-tartaric acid (1S)-1-(4-methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one were obtained from CH2Cl2/hexane. A representative crystal was selected and a 1.54178 Å data set was collected at 153 °K. Pertinent crystal, data collection and refinement parameters: crystal size, 0.58 × 0.16 × 0.05 mm; cell dimensions, a = 7.8456 (1) Å, b = 13.4605 (2) Å, c = 18.2956 (3) Å, α = 90°, β = 93.5910 (10)°, γ = 90°; formula, C40H51NO9; formula weight = 689.82; volume = 1930.88 (5) Å3; calculated density = 1.186 g cm−3; space group = P21; number of reflections = 9774 of which 5860 were considered independent (R int = 0.0317). Refinement method was full-matrix least-squares on F2. The final R-indices were [I > 2σ (I)] R1 = 0.0537, wR2 = 0.1410.
2-Pyrrolidin-1-yl-1-phenylpentan-1-one (4d)
Commercially available 2d was brominated (General Procedure B) to give 2-bromo-1-phenylpentan-1-one 3d: 1H NMR δ 8.02 (d, 2H), 7.62 (m, 1H), 7.49 (t, 2H), 5.15 (dd, 1H), 2.25 - 2.05 (m, 2H), 1.7 - 1.4 (m, 2H), 0.99 (t, 3H). Compound 4d, obtained as a colorless solid, was prepared from 3d (General Procedure C) (51% yield); Mp 173 °C; 1H NMR δ 10.9 - 10.6 (br, 1H), 8.11 (d, 2H), 7.78 (t, 1H), 7.64 (t, 2H), 5.62 (m, 1H), 3.64 (br, m, 1H), 3.49 (br, m, 1H), 3.26 (br, m, 1H), 3.10 (br, m, 1H), 2.15 - 1.85 (m, 6H), 1.4 - 1.2 (m, 1H), 1.2 - 0.95 (m, 1H), 0.78 (t, 3H); 13C NMR 196.7, 134.9, 134.5, 129.2, 128.8, 67.3, 53.6, 51.9, 31.7, 22.9, 17.4, 13.7; APCI MS m/z 232 (M + 1); Anal. (C15H22ClNO) C, H, N, Cl.
1-(4-Fluorophenyl)-2-pyrrolidin-1-yl-pentan-1-one hydrochloride (4e)
Commercially available 2e was brominated (General Procedure B) to give 2-bromo-1-(4-fluorophenyl)pentan-1-one 3e: 1H NMR δ 8.05 (dd, 2H), 7.16 (dd, 2H), 5.09 (dd, 1H), 2.25 - 2.05 (m, 2H), 1.7 - 1.35 (m, 2H), 0.99 (t, 3H). Compound 4e, obtained as a colorless solid, was prepared from 3e (General Procedure C) (84% yield). Mp 218 °C (dec.); 1H NMR δ 10.7 - 10.5 (br, 1H), 8.19 (m, 2H), 7.49 (t, 2H), 5.6 - 5.5 (br, m, 1H), 3.7 - 3.55 (br, 1H), 3.55 - 3.4 (br, 1H), 3.3 - 3.15 (br, m, 1H), 3.15 - 3.0 (br, 1H), 2.15 - 1.8 (br, m, 6H), 1.35 - 1.15 (m, 1H), 1.15 - 0.95 (m, 1H), 0.79 (t, 3H); 13C NMR δ 195.2, 132.2, 132.0, 131.3, 116.6, 116.3, 67.2, 53.5, 51.9, 31.7, 22.9, 17.4, 13.7; APCI MS m/z 250 (M + 1); Anal. (C15H21ClFNO) C, H, N, Cl.
1-(4-Bromophenyl)-2-pyrrolidin-1-yl-pentan-1-one hydrochloride (4f)
Commercially available 2f was brominated (General Procedure B) to give 2-bromo-1-(4-bromophenyl)pentan-1-one 3f: 1H NMR δ 7.88 (d, 2H), 7.63 (d, 2H), 5.06 (dd, 1H), 2.25 - 2.05 (m, 2H), 1.65 - 1.35 (m, 2H), 0.99 (t, 3H). Compound 4f, obtained as a colorless solid, was prepared from 3f (General Procedure C) (62% yield). Mp 200 °C (dec.); 1H NMR δ 10.7 - 10.5 (br, 1H), 8.03 (d, 2H), 7.87 (d, 2H), 5.56 (m, 1H), 3.7 - 3.55 (br, m, 1H), 3.55 - 3.4 (br, m, 1H), 3.35 - 3.1 (br, m, 1H), 3.1 - 3.0 (br, m, 1H), 2.1 - 1.8 (br, m, 6H), 1.4 - 1.2 (m, 1H), 1.15 - 0.95 (m, 1H), 0.78 (t, 3H); 13C NMR δ 196.0, 133.4, 132.4, 130.8, 129.4, 67.4, 53.7, 51.9, 31.6, 22.9, 17.3, 13.7; APCI MS m/z 312, 310 (M + 1); Anal. (C15H21BrClNO) C, H, N, Cl.
1-(4-Iodophenyl)-2-pyrrolidin-1-yl-pentan-1-one hydrochloride (4g)
1-(4-Iodophenyl)pentan-1-one 2g, prepared by Friedel-Crafts acylation of 4-iodobenzene and purified by distillation (Bp 112 °C, 0.1 mm Hg) and recrystallization from EtOH: (11% yield); 1H NMR δ 7.82 (d, 2H), 7.67 (d, 2H), 2.92 (t, 2H), 1.71 (m, 2H), 1.40 (m, 2H), 0.95 (t, 3H) was brominated (General Procedure B) to give 2-bromo-1-(4-iodophenyl)pentan-1-one 3g: 1H NMR δ 7.85 (d, 2H), 7.72 (d, 2H), 5.06 (dd, 1H), 2.25 - 2.05 (m, 2H), 1.65 - 1.35 (m, 2H), 0.98 (t, 3H). Compound 4g was prepared from 3g (General Procedure C) (37% yield); Mp 218 °C (dec.); 1H NMR δ 10.75 - 10.65 (br, 1H), 8.05 (d, 2H), 7.84 (d, 2H), 5.53 (m, 1H), 3.7 - 3.65 (br, 1H), 3.65 - 3.5 (br, m, 1H), 3.3 - 3.15 (br, m, 1H), 3.15 - 3.0 (br, m, 1H), 2.1 - 1.8 (br, m, 6H), 1.35 - 1.15 (m, 1H), 1.15 - 0.95 (m, 1H), 0.78 (t, 3H); 13C NMR δ 196.3, 138.2, 133.6, 130.3, 104.6, 67.3, 53.7, 51.9, 31.6, 22.9, 17.3, 13.7; APCI MS m/z 358 (M + 1); Anal. (C15H21ClINO) C, H, N, Cl.
1-(3-Iodophenyl)-2-pyrrolidin-1-yl-pentan-1-one hydrochloride (4h)
1-(3-Iodophenyl)pentan-1-one 2h, prepared in 29 % yield from 3-iodobenzonitrile (General Procedure A) and purified by column chromatography (3% EtOAc/hexane): Rf 0.25 (5% EtOAc/hexane); 1H NMR δ 8.28 (t, 1H), 7.90 (m, 2H), 7.21 (t, 1H), 2.93 (t, 2H), 1.71 (m, 2H), 1.40 (m, 2H), 0.96 (t, 3H); 13C NMR δ 199.1, 141.6, 138.8, 137.0, 130.3, 127.1, 94.4, 38.3, 26.2, 22.4, 13.9, was brominated (General Procedure B) to provide 2-bromo-1-(3-iodophenyl)pentan-1-one 3h: 1H NMR δ 8.33 (dd. 1H), 7.96 (ddd, 1H), 7.93 (ddd, 1H), 7.22 (d, 1H), 5.05 (dd, 1H), 2.25 - 2.05 (m, 2H), 1.7 - 1.35 (m, 2H), 0.98 (t, 3H). Compound 4h, obtained as a colorless solid, was prepared from 3h (General Procedure C) (20% yield); Mp 203 °C (dec.); 1H NMR δ 10.6 - 10.4 (br, 1H), 8.39 (s, 1H), 8.14 (d, 1H), 8.07 (d, 1H), 7.44 (t, 1H), 5.51 (m, 1H), 3.7 - 3.55 (br, m, 1H), 3.55 - 3.4 (br, m, 1H), 3.3 - 3.15 (br, m, 1H), 3.15 - 3.0 (br, m, 1H), 2.1 - 1.8 (br, m, 6H), 1.35 - 1.15 (m, 1H), 1.1 - 0.9 (m, 1H), 0.79 (t, 3H); 13C NMR δ 195.7, 143.3, 136.9, 136.1, 131.8, 131.3, 128.0, 95.7, 67.5, 53.8, 51.9, 31.5, 22.8, 17.2, 13.6; APCI MS m/z 358 (M + 1); Anal. (C15H21ClINO) C, H, N, Cl.
4-(2-Pyrrolidin-1-yl-pentanoyl)benzonitrile hydrochloride (4i)
4-(2-Bromopentanoyl)benzonitrile, 3i: 1H NMR δ 8.11 (d, 2H), 7.80 (d, 2H), 5.07 (dd, 1H), 2.25 - 2.05 (m, 2H), 1.7 - 1.35 (m, 2H), 1.00 (t, 3H) was prepared (General Procedure B) from 4-cyanovalerophenone 2i35 and converted to 4i as described in General Procedure C (70% yield); Mp 197 – 199 °C (dec.); 1H NMR δ 10.9 - 10.7 (br, 1H), 8.24 (d, 2H), 8.14 (d, 2H), 5.7 - 5.55 (br, m, 1H), 3.7 - 3.6 (br, m, 1H), 3.6 - 3.5 (br, m, 1H), 3.3 - 3.1 (br, m, 2H), 2.1 - 1.8 (m, 6H), 1.4 - 1.2 (m, 1H), 1.1 - 0.9 (m, 1H), 0.77 (t, 3H); 13C NMR δ 196.2, 137.5, 133.2, 129.4, 117.9, 116.6, 67.8, 53.7, 51.9, 31.3, 22.9, 17.2, 13.7; APCI MS m/z 257 (M + 1); Anal. (C16H21ClN2O.1/4H2O) C, H, N, Cl.
1-(4-Hydroxymethylphenyl)-2-pyrrolidin-1-yl-pentan-1-one hydrochloride (4j)
2-Bromo-1-(4-hydroxymethylphenyl)-pentan-1-one 3j: 1H NMR δ 8.01 (d, 2H), 7.48 (d, 2H), 5.15 (dd, 1H), 4.79 (br, d, 2H), 2.25 - 2.05 (m, 2H), 2.05 - 1.95 (br, 1H), 1.65 - 1.4 (m, 2H), 0.99 (t, 3H) was prepared (General Procedure B) from 1-(4-hydroxymethylphenyl)pentan-1-one 2j35 and converted to 4j as described in General Procedure C (79% yield); Mp 186 – 187 °C (dec.); 1H NMR δ 10.6 - 10.4 (br, 1H), 8.05 (d, 2H), 7.56 (d, 2H), 5.7 - 5.4 (br, m, 2H), 4.62 (s, 2H), 3.7 - 3.55 (m, 1H), 3.55 - 3.3 (m, 1H), 3.35 - 3.15 (m, 1H), 3.1 - 3.0 (m, 1H), 2.1 - 1.8 (m, 6H), 1.3 - 1.15 (m, 1H), 1.15 - 0.95 (m, 1H), 0.78 (t, 3H); 13C NMR δ 196.2, 150.4, 132.8, 128.8, 126.7, 67.4, 62.2, 53.8, 51.9, 31.8, 22.8, 17.3, 13.7; APCI MS m/z 262 (M + 1); Anal. (C16H24ClNO2.1/4H2O) C, H, N, Cl.
1-(4-Hydroxyphenyl)-2-pyrrolidin-1-yl-pentan-1-one hydrochloride (4k)
1-(4-Methoxyphenyl)-2-pyrrolidin-1-yl-pentan-1-one 4m (9.00 g, 30.3 mmol) was freed from its hydrochloride salt by basification to pH 8–9 with 20% aqueous Na2CO3 and extraction into CH2Cl2. The free base was dissolved in CH2Cl2 (50 mL) and cooled to −78 °C. BBr3 (90 mL, 1.0 M solution in CH2Cl2, 90 mmol) was added to the solution over 0.5 h. The mixture was stirred for a further 1 h before warming gradually to room temperature. The gummy mixture, which became difficult to stir, was quenched after 2 h with saturated aqueous NaHCO3 and the neutral organics were extracted into CH2Cl2. A white solid precipitated from the aqueous layer and was collected on a frit (2.8 g). This material was dissolved in Et2O and treated with 2 M ethereal HCl. The solid obtained was collected by filtration then recrystallized from ethanol to give pure 1-(4-hydroxyphenyl)-2-pyrrolidin-1-yl-pentan-1-one as its hydrochloride 4k (2.9 g, 34%). Mp 235 °C (dec.); 1H NMR (CD3OD) δ 7.99 (d, 2H), 6.93 (d, 2H), 5.26 (t, J = 5.5 Hz, 1H), 3.7 - 3.0 (br, 4H), 2.2 - 1.9 (br, m, 6H), 1.4 - 1.1 (m, 2H), 0.89 (t, 3H); 13C NMR δ 195.0, 156.8, 132.9, 127.3, 117.0, 69.8, 33.9, 24.1, 18.6, 14.2; APCI MS m/z 248 (M + 1); Anal. (C15H22ClNO2) C, H, N, Cl.
1-(4-Nitrophenyl)-2-pyrrolidin-1-yl-pentan-1-one hydrochloride (4l)
A 50% w/w aqueous solution of H2O2 (7 mL, 0.12 mol) was added to CH2Cl2 (50 mL) which had been cooled on an ice bath. Trifluoroacetic anhydride (23 mL, 0.14 mol) was added slowly via syringe. The solution was then warmed to room temperature. N-[4-(2-Pyrrolidin-1-ylpentanoyl)phenyl]acetamide hydrochloride 4o (4.5 g, 18 mmol) was added over 20 min, and the mixture was heated to reflux for 1 h. The solution was cooled, then quenched cautiously with aqueous Na2SO3 (100 mL of a 1.6 M solution, 0.16 mol). The organics were separated and extracted into Et2O, then back-extracted into 1 M aqueous HCl. The acidic extracts were basified with 20% aqueous Na2CO3 to pH 8–9 and extracted into Et2O. The organic extracts were dried (MgSO4), filtered, and then treated with 2 M ethereal HCl. The resulting white precipitate was collected on a frit, dissolved in water, and then freeze-dried to give pure 1-(4-nitrophenyl)-2-pyrrolidin-1-yl-pentan-1-one hydrochloride 4l (290 mg, 5%). Mp 189 °C (dec.); 1H NMR δ 10.8 - 10.6 (br, 1H), 8.45 (d, 2H), 8.32 (d, 2H), 5.65 (m, 1H), 3.7 - 3.3 (br, m, 2H), 3.3 - 3.1 (br, m, 2H), 2.1 - 1.8 (br, m, 6H), 1.4 - 1.2 (m, 1H), 1.1 - 0.9 (m, 1H), 0.78 (t, 3H); 13C NMR δ 196.0, 150.8, 138.7, 130.4, 124.3, 68.1, 53.9, 52.0, 31.2, 22.9, 17.2, 13.7; APCI MS m/z 277 (M + 1); Anal. (C15H21ClN2O3.1/2H2O.1/10HCl) C, H, N, Cl.
1-(4-Methoxyphenyl)-2-pyrrolidin-1-yl-pentan-1-one hydrochloride (4m)
1-(4-Methoxyphenyl)pentan-1-one 2m, obtained by methylation of commercially available 1-(4-hydroxyphenyl)pentan-1-one, 2k, with MeI/K2CO3 in refluxing acetone, was brominated (General Procedure B) to afford 2-bromo-1-(4-methoxyphenyl)pentan-1-one 3m: 1H NMR δ 8.01 (d, 2H), 6.96 (d, 2H), 5.12 (dd, 1H), 3.89 (s, 3H), 2.25 - 2.05 (m, 2H), 1.65 - 1.35 (m, 2H), 0.98 (t, 3H). Compound 4m was obtained as a colorless solid from 3m (General Procedure C) (68% yield); 1H NMR δ 10.8 - 10.6 (br, 1H), 8.10 (d, 2H), 7.15 (d, 2H), 5.55 (m, 1H), 3.89 (s, 3H), 3.7 - 3.55 (br, m, 1H), 3.55 - 3.4 (br, m, 1H), 3.3 - 3.15 (br, m, 1H), 3.1 - 2.95 (br, m, 1H), 2.15 - 1.85 (br, m, 6H), 1.34 - 1.15 (m, 1H), 1.15 - 1.0 (m, 1H), 0.79 (t, 3H); 13C NMR δ 194.7, 164.5, 131.4, 127.4, 114.5, 66.7, 55.8, 53.4, 51.8, 32.0, 22.9, 17.5, 13.7; APCI MS m/z 262 (M + 1); Anal. (C16H24ClNO2.1/2H2O.1/2HCl) C, H, N, Cl.
4-(2-Pyrrolidin-1-yl-pentanoyl)benzoic acid methyl ester hydrochloride (4n)
4-(2-Bromopentanoyl)benzoic acid methyl ester 3n: 1H NMR δ 8.14 (d, 2H), 8.06 (d, 2H), 5.13 (t, 1H), 3.96 (s, 3H), 2.2 - 2.05 (m, 2H), 1.65 - 1.35 (m, 2H), 1.00 (t, 3H) was prepared (General Procedure B) from 2n35 and converted to 4n as described in General Procedure C (77% yield); Mp 202 °C (dec.); 1H NMR δ 10.7 - 10.5 (br, 1H), 8.3 - 8.1 (m, 4H), 5.58 (m, 1H), 3.91 (s, 3H), 3.7 - 3.5 (br, m, 2H), 3.3 - 3.05 (br, m, 2H), 2.15 - 2.85 (br, m, 6H), 1.4 - 1.2 (m, 1H), 1.15 - 0.95 (m, 1H), 0.77 (t, 3H); 13C NMR δ 196.5, 165.3, 137.6, 134.6, 129.8, 129.2, 67.9, 53.9, 52.7, 51.9, 31.4, 22.9, 17.2, 13.7; APCI MS m/z: 290 ((M + 1), 100%), 275; Anal. (C17H24ClNO3) C, H, N, Cl.
N-[4-(2-Pyrrolidin-1-yl-pentanoyl)phenyl]acetamide hydrochloride (4o)
N-(4-Pentanoylphenyl)acetamide, 2o, prepared in 60% yield by Friedel-Crafts acylation of acetanilide in CS2, and purified by recrystallization from hot MeOH: 1H NMR δ 7.94 (d, 2H), 7.61 (d, 2H), 7.41 (br, s, 1H), 2.94 (t, 2H), 2.22 (s, 3H), 1.8 - 1.65 (m, 2H), 1.45 - 1.35 (m, 2H), 0.95 (t, 3H); 13C NMR δ 168.4, 142.0, 132.9, 129.5, 118.8, 38.2, 26.6, 24.8, 22.5, 14.0, was brominated (General Procedure B) to provide N-[4-(2-bromopentanoyl)phenyl]acetamide, 3o: 1H NMR δ 8.00 (d, 2H), 7.65 (br, m, 3H), 5.12 (dd, 1H), 2.23 (s, 3H), 2.2 - 2.05 (m, 2H), 1.7 - 1.35 (m, 2H), 0.98 (t, 3H). Compound 4o was prepared from 3o as described in General Procedure C (56% yield); Mp 195 °C (dec.); 1H NMR δ 10.76 (s, 1H), 10.55 - 10.35 (br, 1H), 8.05 (d, 2H), 7.85 (d, 2H), 5.5 - 5.4 (br, m, 1H), 3.7 - 3.55 (br, 1H), 3.5 - 3.3 (br, 1H), 3.3 - 3.15 (br, m, 1H), 3.15 - 3.0 (br, m, 1H), 2.13 (s, 3H), 2.1 - 1.8 (br, m, 6H), 1.3 - 1.15 (m, 1H), 1.15 - 1.0 (m, 1H), 0.79 (t, 3H); 13C NMR δ 194.8, 169.4, 145.4, 130.4, 128.8, 118.4, 67.0, 53.6, 51.9, 32.0, 24.2, 22.8, 17.4, 13.7; APCI MS m/z 289 (M + 1); Anal. (C17H25ClN2O2.1/2H2O) C, H, N, Cl.
2-Pyrrolidin-1-yl-1-(4-trifluoromethylphenyl)pentan-1-one hydrochloride (4p)
1-(4-Trifluoromethylphenyl)pentan-1-one 2p, prepared in 95% yield from 4-trifluoromethylbenzonitrile (General Procedure A): 1H NMR δ 8.06 (d, 2H), 7.43 (d, 2H), 3.00 (t, 2H), 1.74 (m, 2H), 1.41 (m, 2H), 0.96 (t, 3H) was brominated (General Procedure B) to provide 2-bromo-1-(4-trifluoromethylphenyl)pentan-1-one, 3p: 1H NMR δ 8.13 (d, 2H), 7.76 (d, 2H), 5.11 (dd, 1H), 2.25 - 2.1 (m, 2H), 1.7 - 1.4 (m, 2H), 1.00 (t, 3H). Compound 4p was prepared from 3p as described in General Procedure C (44% yield); Mp 228 °C (dec.); 1H NMR δ 10.8 - 10.6 (br, 1H), 8.28 (d, 2H), 8.03 (d, 2H), 5.62 (m, 1H), 3.7 - 3.4 (br, m, 2H), 3.3 - 3.05 (br, m, 2H), 2.1 - 1.8 (br, m, 6H), 1.4 - 1.2 (m, 1H), 1.1 - 0.9 (m, 1H), 0.78 (t, 3H); 13C NMR δ 196.2, 137.4, 129.7, 126.3, 67.8, 53.8, 51.9, 31.3, 22.9, 17.2, 13.7; APCI MS m/z 300 (M + 1); Anal. (C16H21ClF3NO) C, H, N, Cl.
1-(4-Propynylphenyl)-2-pyrrolidin-1-yl-pentan-1-one hydrochloride (4q)
1-(4-Iodophenyl)-2-pyrrolidin-1-yl-pentan-1-one hydrochloride (500 mg, 1.27 mmol) 4g, was taken up in Et2NH (10 mL) and degassed by purging with N2. [PdCl2(PPh3)2] (18 mg, 2.5 × 10−5 mol) and CuI (2.4 mg, 1.3 × 10−5 mol) were added to the stirring solution at room temperature. Propyne was then bubbled through the resulting yellow mixture for 7 h. The mixture was filtered and reduced to an oil in vacuo. The oil was taken up in Et2O and extracted into 1M aqueous HCl, then back-extracted into Et2O by treatment with 20% aqueous Na2CO3 until pH 8–9. The organic extracts were dried (MgSO4), filtered, and reduced in vacuo to a pale yellow oil. The hydrochloride was prepared from 2M ethereal HCl and recrystallized twice from EtOH/Et2O to give pure 1-(4-propynylphenyl)-2-pyrrolidin-1-yl-pentan-1-one 4q as a colorless crystalline solid (260 mg, 67%). Mp 231 °C (dec.); 1H NMR δ 10.6 - 10.4 (br, 1H), 8.04 (d, 2H), 7.62 (d, 2H), 5.55 - 5.4 (br, m, 1H), 3.7 - 3.55 (br, 1H), 3.55 - 3.4 (br, 1H), 3.3 - 3.1 (br, m, 1H), 3.1 - 2.95 (br, m, 1H), 2.12 (s, 3H), 2.1 - 1.8 (br, m, 6H), 1.3 - 1.15 (m, 1H), 1.15 - 0.95 (m, 1H), 0.78 (t, 3H); 13C NMR δ 195.9, 133.1, 131.9, 129.9, 129.1, 92.1, 79.0, 67.5, 53.8, 51.9, 31.7, 22.8, 17.2, 13.7, 4.1; APCI MS m/z 270 (M + 1); Anal. (C18H24ClNO) C, H, N, Cl.
1-(2-Methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one hydrochloride (4r)
1-(2-Methylphenyl)pentan-1-one 2r obtained in 75% yield from 2-methylbenzonitrile (General Procedure A) and purified by distillation (Bp 58 – 60°C, 0.05 mm Hg): 1H NMR δ 7.62 (m, 1H), 7.36 (m, 1H), 7.26 (m, 2H), 2.89 (t, 2H), 2.48 (s, 3H), 1.68 (m, 2H), 1.39 (m, 2H), 0.94 (t, 3H) was brominated (General Procedure B) to afford 2-bromo-1-(2-tolyl)pentan-1-one 3r: 1H NMR δ 7.63 (d, 1H), 7.42 (m, 1H), 7.27 (m, 2H), 5.05 (dd, 1H), 2.50 (s, 3H), 2.25 - 2.0 (m, 2H), 1.65 - 1.35 (m, 2H), 0.99 (t, 3H). Compound 4r was prepared from 3r as described in General Procedure C (39% yield); 1H NMR δ 10.9 - 10.7 (br, 1H), 8.12 (d, 1H), 7.58 (t, 1H), 7.44 (t, 2H), 5.56 (m, 1H), 3.7 - 3.5 (br, 2H), 3.35 - 3.1 (br, m, 2H), 2.46 (s, 3H), 2.1 - 1.7 (br, m, 6H), 1.4 - 1.2 (m, 1H), 1.1 - 0.9 (m, 1H), 0.76 (t, 3H); 13C NMR δ 199.1, 138.8, 134.4, 133.2, 132.3, 130.0, 126.2, 68.9, 53.5, 51.8, 31.4, 23.0, 20.7, 17.5, 13.7; APCI MS m/z 246 (M + 1); Anal. (C16H24ClNO.H2O) C, H, N, Cl.
1-(3-Methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one hydrochloride (4s)
1-(3-Methylphenyl)pentan-1-one 2s, obtained in 98% yield from 3-methylbenzonitrile (General Procedure A) and purified by distillation (Bp 64 – 68°C, 0.1 mm Hg): 1H NMR δ 7.86 (d, 2H), 7.26 (d, 2H), 2.94 (t, 2H), 2.41 (s, 3H), 1.71 (m, 2H), 1.41 (m, 2H), 0.95 (t, 3H), was brominated (General Procedure B) to provide 2-bromo-1-(3-methylphenyl)pentan-1-one, 3s: 1H NMR δ 7.81 (m, 2H), 7.40 (m, 2H), 5.15 (dd, 1H), 2.43 (s, 3H), 2.25 - 2.05 (m, 2H), 1.7 - 1.35 (m, 2H), 0.99 (t, 3H). Compound 4s was prepared from 3s as described in General Procedure C (53% yield); Mp 166 °C (dec.); 1H NMR δ 10.8 - 10.6 (br, 1H), 7.90 (d, 2H), 7.65 - 7.5 (m, 2H), 5.57 (m, 1H), 3.7 - 3.55 (br, 1H), 3.55 - 3.4 (br, 1H), 3.3 - 3.15 (br, m, 1H), 3.15 - 3.0 (br, m, 1H), 2.42 (s, 3H), 2.1 - 1.8 (br, m, 6H), 1.35 - 1.15 (m, 1H), 1.15 - 0.95 (m, 1H), 0.78 (t, 3H); 13C NMR δ 196.7, 138.8, 135.6, 134.5, 129.1, 126.1, 67.4, 53.6, 51.9, 31.7, 22.9, 20.8, 17.3, 13.7; APCI MS m/z 246 (M + 1); Anal. (C16H24ClNO) C, H, N, Cl.
1-Naphthalen-2-yl-2-pyrrolidin-1-yl-pentan-1-one hydrochloride (4t)
1-Naphthalen-2-yl-pentan-1-one 2t prepared in 95 % yield from naphthalene-2-carbonitrile (General Procedure A): 1H NMR δ 8.48 (s, 1H), 8.04 (dd, 1H), 7.97 (d, 1H), 7.90 (m, 2H), 7.57 (m, 2H), 3.11 (t, 2H), 1.79 (m, 2H), 1.44 (m, 2H), 0.98 (t, 3H) was brominated (General Procedure B) to afford 2-bromo-1-naphthalen-2-yl-pentan-1-one 3t: 1H NMR δ 8.55 (s, 1H), 8.1 - 7.85 (m, 4H), 7.60 (m, 2H), 5.33 (dd, 1H), 2.3 - 2.1 (m, 2H), 1.7 - 1.4 (m, 2H), 1.01 (t, 3H). Compound 4t was prepared from 3t as described in General Procedure C (51% yield); Mp 221 – 223 °C (dec.); 1H NMR δ 10.8 - 10.6 (br, 1H), 8.92 (s, 1H), 8.2 - 8.0 (m, 4H), 7.75 (dt, 2H), 5.73 (m, 1H), 3.75 - 3.6 (br, 1H), 3.6 - 3.4 (br, m, 1H), 3.35 - 3.1 (br, m, 2H), 2.2 - 1.8 (m, 6H), 1.4 - 1.2 (m, 1H), 1.2 - 1.0 (m, 1H), 0.78 (t, 3H); 13C NMR δ 196.6, 135.7, 132.0, 131.8, 131.7, 129.9, 129.7, 129.0, 127.8, 127.5, 123.4, 67.3, 53.6, 52.0. 31.9, 22.9, 17.4, 13.7; APCI MS m/z 282 (M + 1); Anal. (C19H24ClNO) C, H, N, Cl.
1-(3,4-Dichlorophenyl)-2-pyrrolidin-1-yl-pentan-1-one hydrochloride (4u)
1-(3,4-Dichlorophenyl)pentan-1-one 2 u prepared in 93% yield from 3,4-dichlorobenzonitrile (General Procedure A) and used crude in the next step of the reaction: 1H NMR δ 8.03 (d, 1H), 7.78 (dd, 1H), 7.54 (d, 1H), 2.92 (t, 2H), 1.71 (m, 2H), 1.39 (m, 2H), 0.94 (t, 3H) was brominated (General Procedure B) to afford 2-bromo-1-(3,4-dichlorophenyl)pentan-1-one 3u: 1H NMR δ 8.09 (d, 1H), 7.84 (dd, 1H), 7.55 (d, 1H), 5.02 (dd, 1H), 2.25 - 2.05 (m, 2H), 1.65 - 1.35 (m, 2H), 0.99 (t, 3H). Compound 4u was prepared from 3u as described in General Procedure C (32% yield); Mp 195 °C (dec.); 1H NMR δ 10.8 - 10.6 (br, 1H), 8.35 (d, 1H), 8.04 (dd, 1H), 7.94 (d, 1H), 5.58 (m, 1H), 3.7 - 3.6 (br, 1H), 3.6 - 3.45 (br, m, 1H), 3.3 - 3.05 (br, m, 2H), 2.15 - 2.85 (br, m, 6H), 1.35 - 1.15 (m, 1H), 1.15 - 0.95 (m, 1H), 0.79 (t, 3H); 13C NMR δ 195.0, 137.8, 134.5, 132.3, 131.6, 130.8, 128.8, 67.5, 53.7, 51.9, 31.4, 22.9, 17.2, 13.6; APCI MS m/z 300, 302, 304 (M + 1); Anal. (C15H20Cl3NO) C, H, N, Cl.
1-(3,4-Dihydroxyphenyl)-2-pyrrolidin-1-yl-pentan-1-one hydrobromide (4v)
1-(3,4-Dimethoxyphenyl)-2-pyrrolidin-1-yl-pentan-1-one 4w (1.50 g, 4.6 mmol) was freed from its hydrochloride salt by treatment with aqueous Na2CO3 and extraction into CH2Cl2. The organics were dried (MgSO4), filtered, and reduced to a pale yellow oil in vacuo. The oil was taken up in CH2Cl2 (10 mL) and cooled to −78 °C, whereon BBr3 (46 mL, 1.0 M solution in CH2Cl2, 46 mmol) was added dropwise over 0.5 h. The resulting yellow mixture was warmed slowly to room temperature and stirred for 3 h. The yellow solution was hydrolyzed cautiously with aq. Na2CO3 (20% solution) until the pH was 8, then water (50 mL) was added and the solution was allowed to stand overnight. Neutral organics were extracted from the mixture by separation of the CH2Cl2 layer, which was then discarded. The aqueous layer was acidified to pH 3 with 1 M HCl, most of the water was removed by rotary evaporation, and the remaining volume of ca. 10 mL was allowed to cool in the refrigerator. After 3 d, a white solid separated from the solution and was collected by filtration. Recrystallization (EtOH/Et2O) afforded pure 1-(3,4-dihydroxyphenyl)-2-pyrrolidin-1-yl-pentan-1-one 4v as its hydrobromide, an off-white solid (0.60 g, 44%); Mp 181 – 182 °C; 1H NMR δ 10.42 (s, 1H), 10.1 - 9.9 (br, 1H), 9.59 (s, 1H), 7.51 (dd, 1H), 7.43 (d, 1H), 6.91 (d, 1H), 5.35 - 5.25 (br, 1H), 3.75 - 3.5 (br, 1H). 3.5 - 3.3 (br, 1H), 3.3 - 3.15 (br, 1H), 3.0 - 2.85 (br, 1H), 2.1 - 1.8 (m, 6H), 1.3 - 1.0 (m, 2H), 0.80 (t, 3H); 13C NMR δ 194.8, 153.4, 146.4, 126.7, 123.5, 116.0, 115.9, 67.5, 54.5, 52.3, 32.8, 23.2, 17.9, 14.3; APCI MS m/z 264 (M + 1); Anal. (C15H22BrNO3) C, H, N, Br.
1-(3,4-Dimethoxyphenyl)-2-pyrrolidin-1-yl-pentan-1-one hydrochloride (4w)
2-Bromo-1-(3,4-dimethoxyphenyl)pentan-1-one 3w was obtained together with 2-bromo-1-(2-bromo-4,5-dimethoxyphenyl)pentan-1-one by General Procedure B. The compounds were separated by flash column chromatography (10% EtOAc/hexane) to provide 2-bromo-1-(3,4-dimethoxyphenyl)-pentan-1-one 3w: 1H NMR δ 7.66 (dd, 1H), 7.58 (d, 1H), 6.91 (d, 1H), 5.15 (dd, 1H), 3.97 (s, 3H), 3.95 (s, 3H), 2.25 - 2.05 (m, 2H), 1.7 - 1.35 (m, 2H), 1.01 (t, 3H), and 2-bromo-1-(2-bromo-4,5-dimethoxyphenyl)pentan-1-one: 1H NMR δ 7.07 (s, 1H), 7.04 (s, 1H), 5.28 (dd, 1H), 3.92 (s, 3H), 3.90 (s, 3H), 2.3 - 2.0 (m, 2H), 1.7 - 1.4 (m, 2H), 1.00 (t, 3H). Compound 4w was then prepared from 3w as described in General Procedure C to provide a solid (74% yield); Mp 177 °C (dec.); 1H NMR δ 10.5 - 10.3 (br, 1H), 7.78 (d, 1H), 7.53 (d, 1H), 7.18 (d, 1H), 5.55 - 5.4 (br, m, 1H), 3.90 (s, 3H), 3.86 (s, 3H), 3.7 - 3.55 (br, m, 1H), 3.5 - 3.3 (br, m, 1H), 3.3 - 3.15 (br, m, 1H), 3.05 - 2.9 (br, m, 1H), 2.1 - 1.8 (m, 6H), 1.3 - 1.0 (m, 2H), 0.80 (t, 3H); 13C NMR δ 194.7, 154.7, 149.0, 127.2, 124.6, 111.2, 110.5, 66.7, 56.0, 55.7, 53.7, 51.8, 32.1, 22.8, 17.4, 13.7; APCI MS m/z 292 (M + 1); Anal. (C17H26ClNO3) C, H, N, Cl.
1-(4-Furan-2-ylphenyl)-2-pyrrolidin-1-yl-pentan-1-one hydrochloride (4x)
This compound was prepared using a procedure analogous to that described later for the preparation of 4z, except that commercially available 2-tributylstannyl furan was employed as a starting material, and chromatography was not performed on the crude free base. The crude hydrochloride was recrystallized from hot EtOH to give pure 4x as a colorless crystalline solid: (59% yield); Mp 236 °C (dec.); 1H NMR (DMSO-d6 + 6 drops CD3OD) δ 8.14 (d, 2H), 7.95 (d, 2H), 7.90 (d, 1H), 7.29 (d, 1H), 6.71 (dd, 1H), 5.51 (m, 1H), 3.7 - 3.6 (br, m, 1H), 3.6 - 3.45 (br, m, 1H), 3.35 - 3.2 (br, m, 1H), 3.15 - 3.0 (br, m, 1H), 2.15 - 1.85 (br, m, 6H), 1.35 - 1.15 (m, 1H), 1.15 - 1.0 (m, 1H), 0.81 (t, 3H); 13C NMR δ 195.7, 151.8, 145.1, 136.0, 132.6, 130.0, 123.8, 112.9, 109.9, 67.8, 54.2, 52.0, 32.0, 22.9, 17.3, 13.7; APCI MS m/z 298 (M + 1); Anal. (C19H24ClNO2) C, H, N, Cl.
2-Pyrrolidin-1-yl-1-(4-thiophen-2-yl-phenyl)pentan-1-one hydrochloride (4y)
This compound was prepared using a procedure analogous to that described later for the preparation of 4z, except that commercially available 2-tributylstannyl thiophene was employed as a starting material, and chromatography was not performed on the crude free base. The crude hydrochloride was readily obtained by treatment of the crude free base with 2M ethereal HCl. Recrystallization from hot EtOH gave pure 4v as a colorless crystalline solid (61% yield). Mp 220 °C (dec.); 1H NMR (DMSO-d6 + 12 drops CD3OD) δ 8.12 (d, 2H), 7.93 (d, 2H), 7.77 (dd, 1H), 7.72 (dd, 1H), 7.23 (dd, 1H), 5.5 - 5.4 (br, 1H), 3.7 - 3.45 (br, m, 2H), 3.3 - 3.2 (br, m, 1H), 3.1 - 3.0 (br, m, 1H), 2.2 - 1.9 (br, m, 6H), 1.35 - 1.2 (m, 1H), 1.2 - 1.0 (m, 1H), 0.83 (t, 3H); 13C NMR δ 195.9, 141.8, 140.3, 132.9, 130.3, 129.3, 128.6, 126.6, 126.0, 68.1, 54.5, 52.1, 32.2, 23.1, 17.4, 13.8; APCI MS m/z 314 (M + 1); Anal. (C19H24ClNOS) C, H, N, Cl.
1-(4-N-Methylpyrrolephenyl)-2-pyrrolidin-1-yl-pentan-1-one hydrochloride (4z)
To a cooled (−78 °C) solution of N-methylpyrrole (1.14 g, 14 mmol) in THF (10 mL), tBuLi (9.1 mL of a 1.7M solution in pentane, 15 mmol) was added drop-wise. The mixture was then warmed to room temperature for 2 h, then cooled to −78 °C. Chlorotributylstannane (5.0 g, 15 mmol) was added to the mixture dropwise. On completion of addition, the mixture was warmed to room temperature and stirred for 1 h. The mixture was filtered and reduced to an oil in vacuo. This oil (crude 2-tributylstannyl-(N-methylpyrrole)) was added to a solution of 2-pyrrolidin-1-yl-1-(4-bromophenyl)-pentan-1-one (which had been freed from its hydrochloride 4f by treatment with 20% aqueous Na2CO3 and extraction into Et2O) in dioxane (30 mL). The resulting solution was degassed by purging with N2. [Pd(PPh3)4] (264 mg, 0.22 mmol) was added and the mixture was heated to 95 – 100 °C (oil bath temperature) for a period of 10 h. The solvent was removed in vacuo. The pure free base was obtained by column chromatography (5% MeOH/CH2Cl2) as a yellow oil. The hydrochloride was prepared by treatment with 2M ethereal HCl. Lyophilization of an aqueous solution of the salt afforded 1-(4-N-methylpyrrolephenyl)-2-pyrrolidin-1-yl-pentan-1-one hydrochloride as a pale green solid 4z (1.4 g, 36%). Mp 185 °C; 1H NMR δ 10.6 - 10.45 (br, 1H), 8.11 (d, 2H), 7.72 (d, 2H), 7.00 (dd, 1H), 6.45 (dd, 1H), 6.15 (dd, 1H), 5.54 (m, 1H), 3.77 (s, 3H), 3.7 - 3.55 (br, 1H), 3.55 - 3.4 (br, 1H), 3.35 - 3.15 (br, m, 1H), 3.15 - 3.0 (br, m, 1H), 2.1 - 1.85 (br, m, 6H), 1.35 - 1.2 (m, 1H), 1.2 - 1.0 (m, 1H), 0.82 (t, 3H); 13C NMR δ 195.6, 139.1, 131.9, 131.5, 129.4, 127.4, 127.1, 111.1, 108.2, 67.2, 53.7, 51.9, 35.6, 31.9, 22.9, 17.4, 13.7; APCI MS m/z 311 (M + 1); Anal. (C20H27ClN2O.2/3H2O) C, H, N, Cl.
1-(4-Methylphenyl)pent-2-en-1-one (5a)
This compound was prepared as described below for 5b employing 2-bromo-1-(4-methylphenyl)pentan-1-one 3a as starting material (82% yield); 1H NMR δ 7.85 (d, 2H), 7.25 (d, 2H), 7.10 (dt, 1H), 6.88 (dt, 1H), 2.39 (s, 3H), 2.32 (m, 2H), 1.13 (t, 3H); 13C NMR δ 190.3, 150.6, 143.2, 135.3, 129.0, 128.5, 124.7, 25.7, 21.5, 12.2.
1-(3,4-Dichlorophenyl)pent-2-en-1-one (5b)
2-Bromo-1-(3,4-dichlorophenyl) pentan-1-one, 3u, (3.36 g, 10.9 mmol) was dissolved in DMF (60 mL). Li2CO3 (1.28 g, 17 mmol) and LiBr (0.99 g, 11.5 mmol) were added to the solution, which was then heated with stirring to 110 – 120 °C (oil bath temperature) for 1.5 h. The mixture was diluted with H2O (100 mL) and the organics were extracted into EtOAc (3 × 50 mL). The ethyl acetate layer was collected and washed with saturated brine (2 × 50 mL), dried (MgSO4), filtered, and reduced to an oil in vacuo. Flash column chromatography (1% EtOAc/hexane to 2.5% EtOAc/hexane) furnished pure 5b as a colorless solid (1.5 g, 60%). 1H NMR δ 8.01 (d, 1H), 7.76 (dd, 1H), 7.55 (d, 1H), 7.15 (dt, 1H), 6.80 (dt, 1H), 2.37 (m, 2H), 1.15 (t, 3H); 13C NMR δ 188.5, 152.8, 137.6, 137.1, 133.2, 130.6, 130.5, 127.5, 124.1, 26.0, 12.2.
1-(3,4-Dichlorophenyl)-3-pyrrolidin-1-yl-pentan-1-one hydrochloride (6b)
1-(3,4-Dichlorophenyl)pent-2-en-1-one 5b (1.29 g, 5.63 mmol) was taken up in EtOH (10 mL), cooled on an ice bath, and degassed by purging with N2. Pyrrolidine (0.80 g, 11 mmol) was added dropwise over 2 min. After 0.5 h, the ethanolic solution was separated between 1M aqueous HCl and Et2O. The HCl extracts were collected and back-extracted into Et2O by treatment with 20% aqueous Na2CO3. The ethereal extracts were dried (MgSO4), filtered, and treated with 2M ethereal HCl. Trituration afforded 1-(3,4-dichlorophenyl)-2-pyrrolidin-1-yl-methylpentan-1-one hydrochloride 6b as a white powder which was filtered and washed copiously with Et2O (0.99 g, 50%); Mp 104 – 107 °C (dec.); 1H NMR δ 11.1 - 10.9 (br, 1H), 8.27 (d, 1H), 7.98 (dd, 1H), 7.87 (d, 1H), 3.9 - 3.35 (br, m, 5H), 3.15 - 2.95 (br, 2H), 2.05 - 1.8 (br, m, 5H), 1.8 - 1.6 (m, 1H), 0.90 (t, 3H); 13C NMR δ 195.0, 136.4, 136.1, 131.8, 131.1, 130.3, 128.1, 59.2, 50.7, 50.1, 38.2, 23.8, 22.9, 10.0; APCI MS m/z 300, 302, 304 (M + 1); Anal. (C15H20Cl3NO.1/3H2O) C, H, N, Cl.
1-(4-Methylphenyl)-3-pyrrolidin-1-yl-pentan-1-one hydrochloride (6a)
This compound was prepared from 1-(4-methylphenyl)-2-en-1-one 5a using the same procedure as that described for 6b. Mp 97 °C (dec.); 1H NMR δ 11.1 - 10.9 (br, 1H), 7.94 (d, 2H), 7.38 (d, 2H), 3.9 - 3.75 (br, 1H), 3.7 - 3.6 (m, 1H), 3.6 - 3.3 (m, 3H), 3.15 - 2.95 (br, m, 2H), 1.96 (s, 3H), 2.0 - 1.8 (br, m, 5H), 1.8 - 1.6 (m, 1H), 0.88 (t, 3H); 13C NMR δ 196.2, 144.3, 133.5, 129.3, 128.3, 59.7, 50.7, 50.4, 37.9, 23.8, 22.9, 22.8, 21.2, 9.9; APCI MS m/z 246 (M + 1); Anal. (C16H24ClNO) C, H, N, Cl.
1-(3,4-Dichlorophenyl)-2-pyrrolidin-1-yl-methylpentan-1-one hydrochloride (7b)
2-Bromo-1-(3,4-dichlorophenyl)pentan-1-one 3u (3.5 g, 15 mmol), pyrrolidine.HCl (2.4 g, 23 mmol) and paraformaldehyde (1.35 g, 45 mmol) were taken up in iPrOH (25 mL) containing concentrated HCl (0.2 mL). The mixture was brought to reflux for 16 h. The solvent was removed by rotary evaporation and the residue was separated between 1 M aqueous HCl and Et2O. The aqueous extracts were basified with 20% aqueous Na2CO3 to pH 8–9 and the organics were extracted into Et2O. The organics were dried (MgSO4), filtered, and reduced to an oil in vacuo. Column chromatography (10% MeOH/CH2Cl2) gave the pure free base. Reaction with 2 M ethereal HCl and filtration of the resulting white precipitate provided 1-(3,4-dichlorophenyl)-2-pyrrolidin-1-yl-methylpentan-1-one hydrochloride, 7b (0.61 g, 12%). Mp 168 °C (dec.); 1H NMR δ 10.7 - 10.5 (br, 1H), 8.29 (d, 1H), 8.05 (dd, 1H), 7.88 (d, 1H), 4.3 - 4.1 (br, 1H), 3.7 - 3.5 (br, m, 2H), 3.5 - 3.25 (br, m, 2H), 3.15 - 2.85 (br, m, 2H), 2.1 - 1.75 (br, m, 4H), 1.75 - 1.4 (m, 2H), 1.35 - 1.05 (m, 2H), 0.81 (t, 3H); 13C NMR δ 198.9, 136.6, 135.9, 132.1, 131.4, 131.2, 130.5, 130.3, 128.7, 128.5, 54.1, 53.4, 42.3, 42.2, 33.1, 22.7, 22.4, 18.8, 13.8; APCI MS m/z 314, 312, 310 (M + 1); Anal. (C16H22Cl3NO) C, H, N, Cl.
1-(4-Methylphenyl)-2-pyrrolidin-1-yl-methylpentan-1-one hydrochloride (7a)
This compound was prepared from 1-(2-methylphenyl)pentan-1-one (3.5 g, 20 mmol) using the same method as described for 7b with the following modifications. No chromatography was performed. The hydrochloride salt of the crude free base was isolated after extraction of the crude reaction mixture into 1 M aqueous HCl, and back extraction (with 20% aqueous Na2CO3) into Et2O, followed by acidification with 2M HCl in Et2O. The product was recrystallized from EtOH/Et2O to give pure crystalline 1-(4-methylphenyl)-2-pyrrolidin-1-yl-methylpentan-1-one hydrochloride 7a (2.6 g, 44%). Mp 176 °C (dec.); 1H NMR δ 10.8 - 10.6 (br, 1H), 7.98 (d, 2H), 7.39 (d, 2H), 4.25 - 4.15 (br, m, 1H), 3.65 - 3.5 (m, 2H), 3.5 - 3.25 (m, 2H), 3.1 - 2.95 (br, m, 1H), 2.95 - 2.8 (br, m, 1H), 2.40 (s, 3H), 2.0 - 1.75 (m, 4H), 1.7 - 1.4 (m, 2H), 1.3 - 1.1 (m, 2H), 0.81 (t, 3H); 13C NMR δ 200.4, 144.4, 135.2, 129.7, 129.5, 128.7, 128.5, 54.0, 53.7, 53.3, 41.9, 33.5, 22.8, 22.3, 21.1, 19.0, 13.8; APCI MS m/z 260 (M + 1); Anal. (C17H26ClNO) C, H, N, Cl.
1-(3,4-Dichlorophenyl)-2-pyrrolidin-1-yl-butan-1-one hydrochloride (9a)
1-(3,4-Dichlorophenyl)butan-1-one, prepared in quantitative yield from 3,4-dichlorobenzonitrile and n-PrMgCl (General Procedure A); 1H NMR δ 8.01 (d, 1H), 7.78 (dd, 1H), 7.54 (d, 1H), 2.91 (t, 2H), 1.77 (sextet, 2H), 1.01 (t, 3H), was brominated according to General Procedure B to give 2-bromo-1-(3,4-dichlorophenyl)butan-1-one; 1H NMR δ 8.09 (d, 1H), 7.84 (dd, 1H), 7.57 (d, 1H), 4.95 (dd, 1H), 2.35 - 2.05 (m, 2H), 1.09 (t, 3H). Compound 9a was prepared according to General Procedure C (71% yield); Mp 211 °C (dec.); 1H NMR δ 10.95 - 10.75 (br, 1H), 8.35 (d, 1H), 8.06 (dd, 1H), 7.92 (d, 1H), 5.75 - 5.65 (br, m, 1H), 3.65 - 3.35 (br, m, 2H), 3.3 - 3.1 (br, m, 2H), 2.15 - 1.9 (br, m, 6H), 0.78 (t, 3H); 13C NMR δ 194.7, 137.7, 134.5, 132.3, 131.6, 130.7, 128.8, 68.5, 53.7, 51.8, 23.0, 22.6, 8.4; APCI MS m/z 286, 288, 290 (M + 1); Anal. (C14H18Cl3NO) C, H, N.
4-Methyl-2-pyrrolidin-1-yl-1-(4-methylphenyl)pentan-1-one hydrochloride (9b)
4-Methyl-1-(4-methylphenyl)pentan-1-one, prepared in quantitative yield by Friedel-Crafts acylation of toluene with 4-methylvaleroyl chloride: 1H NMR δ 7.86 (d, 2H), 7.26 (d, 2H), 3.94 (t, 2H), 2.41 (s, 3H), 1.62 (m, 3H), 0.94 (d, 6H) was converted to 2-bromo-4-methyl-1-(4-methylphenyl)pentan-1-one, as described in General Procedure B: 1H NMR δ 7.92 (d, 2H), 7.29 (d, 2H), 5.21 (dd, 1H), 2.43 (s, 3H), 2.15 - 1.95 (m, 2H), 1.95 - 1.75 (m, 1H), 0.96 (d, 6H). 4-Methyl-2-pyrrolidin-1-yl-1-(4-methylphenyl)pentan-1-one hydrochloride 9b was then prepared as described in General Procedure C (68% yield); Mp 218 °C (dec.); 1H NMR δ 10.9 - 10.75 (br, 1H), 8.06 (d, 2H), 7.45 (d, 2H), 5.46 (m, 1H), 3.75 - 3.6 (br, 1H), 3.6 - 3.4 (br, 1H), 3.3 - 3.0 (br, m, 2H), 2.42 (s, 3H), 2.1 - 1.7 (m, 6H), 1.45 - 1.3 (m, 1H), 0.82 (dd, J = 2, 6 Hz, 6H); 13C NMR δ 197.2, 164.0, 132.9, 129.9, 129.0, 64.4, 52.7, 51.2, 24.2, 23.3, 22.8, 21.5, 21.3; APCI MS m/z 260 (M + 1); Anal. (C17H26ClNO) C, H, N, Cl.
1-(4-Methylphenyl)-2-pyrrolidin-1-yl-pent-4-ene-1-one hydrochloride (9c)
This compound was prepared as described previously.29 Mp 196 °C (dec.); 1H NMR δ 10.8 - 10.6 (br, 1H), 7.96 (d, 2H), 7.43 (d, 2H), 5.8 - 5.6 (m, 2H), 5.03 (s, 1H), 5.00 (m, 1H), 3.75 - 3.6 (br, 1H), 3.6 - 3.4 (br, 1H), 3.4 - 3.2 (br, m, 1H), 3.15 - 3.0 (br, m, 1H), 3.85 - 3.65 (br, m, 2H), 2.42 (s, 3H), 2.2 - 1.85 (br, m, 4H); 13C NMR δ 195.2, 145.8, 131.8, 130.6, 129.7, 129.0, 120.1, 66.9, 53.8, 52.0, 34.2, 22.9, 21.3; APCI MS m/z 244 (M + 1); Anal. (C16H22ClNO) C, H, N, Cl.
1-(3,4-Dichlorophenyl)-2-pyrrolidin-1-yl-pent-4-ene-1-one hydrochloride (9d)
This compound was prepared as described for 9c 29 Mp 176 °C (dec.); 1H NMR δ 10.8 - 10.6 (br, 1H), 8.29 (d, 1H), 8.00 (dd, 1H), 7.94 (d, 1H), 5.8 - 5.6 (m, 2H), 5.07 (s, 1H), 5.02 (m, 1H), 3.75 - 3.6 (br, m, 1H), 3.6 - 3.3 (br, m, 1H), 3.3 - 3.1 (br, m, 2H), 2.77 (m, 2H), 2.2 - 1.8 (br, m, 4H); 13C NMR δ 194.2, 137.8, 134.4, 132.2, 131.6, 130.8, 130.3, 128.8, 120.6, 67.2, 53.9, 52.1, 33.8, 22.9; APCI MS m/z 302 ((M + 1), 100%), 300, 298; Anal. (C15H18Cl3NO) C, H, N, Cl.
1-(4-Methylphenyl)-2-pyrrolidin-1-yl-pent-4-yn-1-one hydrochloride (9e)
1-(4-Methylphenyl)-2-pyrrolidin-1-ylethanone 829 (25 g, 104 mmol) was freed from its hydrochloride by treatment with aqueous Na2CO3 and extraction into Et2O. The organics were dried (MgSO4), filtered and reduced in vacuo to a yellow oil. This oil was taken up in toluene (200 mL), and NaNH2 was added to the stirring solution, which was then heated to approximately 120 °C (oil bath temperature) for 0.5 h. The solution was allowed to cool to about 100 °C and propargyl bromide (13 mL, 80% w/w solution in toluene, 14 g, 115 mmol) was added to the orange mixture at such a rate that steady reflux was maintained with concomitant NH3 evolution. Upon complete addition (0.5 h), the mixture was allowed to cool to room temperature and was then hydrolyzed cautiously by addition of water (100 mL). The toluene layer was separated and the aqueous layer was extracted with toluene (2 × 50 mL). The combined organics were dried (MgSO4), filtered and reduced in vacuo to a brown oil that was taken up in Et2O (50 mL). 2 M HCl in Et2O was added to the ethereal solution of the oil. Trituration afforded a brown solid that could not be crystallized from EtOH/Et2O. The solvents were removed in vacuo and the free base was prepared by addition of 2 M NaOH solution until pH 8–9. The organics were extracted into Et2O (3 × 100 mL) to give a light brown solution. Back-extraction into 1 M HCl (3 × 50 mL) gave a light yellow solution. The water was removed by rotary evaporation; lyophilization than gave a light brown gum (5.3 g). Recrystallization from EtOH/Et2O afforded pure 1-(4-methylphenyl)-2-pyrrolidin-1-yl-pent-4-yn-1-one hydrochloride 9e (3.15 g, 11%): Mp 178 °C (dec.); 1H NMR δ 10.6 - 10.4 (br, 1H), 7.97 (d, 2H), 7.45 (d, 2H), 5.66 (m, 1H), 3.7 - 3.2 (m, 3H), 3.2 - 2.9 (m, 4H), 2.43 (s, 3H), 2.1 - 1.8 (m, 4H); 13C NMR δ 193.9, 146.0, 131.1, 129.7, 129.2, 76.8, 76.6, 65.2, 54.0, 52.0, 22,9, 22.9, 21.3, 20.0; APCI MS m/z 242 (M + 1); Anal. (C16H20ClNO) C, H, N, Cl.
2-Butylamin-1-yl-1-(3,4-dichlorophenyl)pentan-1-one hydrochloride (9f)
Compound 9f (an off-white solid) was obtained from 3u (described above) and n-butylamine, according to General Procedure C (71% yield); Mp 185 °C (dec.); 1H NMR δ 9.8 - 9.6 (br, 1H), 9.3 - 9.1 (br, 1H), 8.35 (d, 1H), 8.04 (dd, 1H), 7.91 (d, 1H), 5.4 - 5.25 (br, 1H), 3.05 - 2.75 (br, m, 2H), 2.05 - 1.8 (br, m, 2H), 1.8 - 1.6 (br, m, 2H), 1.4 - 1.2 (m, 3H), 1.2 - 1.0 (m, 1H), 0.88 (t, 3H), 0.78 (t, 3H); 13C NMR δ 194.8, 137.6, 134.3, 132.3, 131.5, 130.6, 128.7, 60.8, 45.7, 31.5, 27.4, 19.3, 17.2, 13.6, 13.5; APCI MS m/z 302, 304, 306 (M + 1); Anal. (C15H22Cl3NO) C, H, N, Cl.
1-(3,4-Dichlorophenyl)-2-piperidin-1-yl-pentan-1-one hydrochloride (9g)
Compound 9g was prepared from 3u (described above) and piperidine, as described in General Procedure C (35% yield); Mp 202 °C (dec.); 1H NMR δ 10.5 - 10.3 (br, 1H), 8.40 (d, 1H), 8.10 (dd, 1H), 7.94 (d, 1H), 5.45 - 5.35 (br, m, 1H), 3.7 - 3.55 (br, m, 1H), 3.45 - 3.3 (br, m, 1H), 3.2 - 1.95 (br, m, 2H), 2.1 - 1.65 (br, m, 7H), 1.5 - 1.3 (br, 1H), 1.2 - 1.0 (br, m, 2H), 0.81 (t, 3H); 13C NMR δ 195.3, 138.0, 135.3, 132.4, 131.6, 130.7, 128.8, 65.8, 52.0, 50.2, 29.3, 22.3, 22.0, 21.5, 17.8, 13.7; APCI MS m/z 314, 316, 318 (M + 1); Anal. (C16H22Cl3NO) C, H, N, Cl.
1-(4-Methylphenyl)-2-pyrrolidin-1-yl-pentan-1-ol hydrochloride (diastereoisomers 9h and 9j)
1-(4-Methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one hydrochloride 4a (1.50 g, 5.32 mmol) was suspended in THF (20 mL). LiAlH4 (0.20 g, 5.3 mmol) was added in several small portions at room temperature to the stirring mixture with slight heat evolution. The resulting clear solution was hydrolyzed cautiously with H2O, then made acidic by addition of 1M aqueous HCl. The aqueous extracts were collected and basified to pH 8–9 with 20% aqueous Na2CO3. The organics were extracted into Et2O, dried (MgSO4), filtered, and reduced to an oil in vacuo. Chromatography (5% NEt3/15% EtOAc/80% hexane) gave the two diastereoisomers 9h and 9j. The hydrochlorides were prepared from 2M ethereal HCl and recrystallized from EtOH/Et2O to afford 1-(4-methylphenyl)-2-pyrrolidin-1-yl-pentan-1-ol hydrochloride 9h, a colorless crystalline solid (0.57 g, 37%); Mp 140 – 142 °C; 1H NMR δ 10.15 - 10.0 (br, 1H), 7.32 (d, 2H), 7.19 (d, 2H), 6.20 (d, J = 5 Hz, 1H), 5.24 (s, 1H), 3.75 - 3.65 (br, m, 1H), 3.65 - 3.5 (br, m, 1H), 3.4 - 3.3 (br, 2H), 3.2 - 3.05 (br, m, 1H), 2.30 (s, 3H), 2.1 - 1.8 (br, m, 4H), 1.75 - 1.6 (m, 1H), 1.4 - 1.25 (br, m, 1H), 1.1 - 0.95 (m, 1H), 0.8 - 0.6 (m, 1H), 0.57 (t, 3H); 13C NMR δ 138.3, 136.2, 128.6, 125.5, 69.3, 68.1, 51.5, 26.5, 22.7, 22.5, 20.7, 20.3, 13.7; APCI MS m/z 248 (M + 1); Anal. (C16H26ClNO) C, H, N, Cl. and 1-(4-methylphenyl)-2-pyrrolidin-1-yl-pentan-1-ol hydrochloride 9j, a colorless microcrystalline solid (159 mg, 10%), this was the more polar material; Mp 219 °C (dec.); 1H NMR δ 9.8 - 9.65 (br, 1H), 7.33 (d, 2H), 7.20 (d, 2H), 6.53 (d, J = 4 Hz, 1H), 4.65 (dd J = 4, 9 Hz, 1H), 3.55 - 3.3 (m, 3H), 3.3 - 3.15 (br, m, 1H), 3.15 - 2.95 (br, m, 1H), 2.31 (s, 3H), 2.0 - 1.85 (br, 4H), 1.55 - 1.35 (br, m, 2H), 1.05 - 0.85 (m, 1H), 1.75 - 1.6 (m, 4H); 13C NMR δ 138.4, 137.3, 128.9, 127.1, 72.1, 67.0, 40.3, 40.1, 27.6, 23.3, 23.0, 20.8, 20.0, 13.6; APCI MS m/z 248 (M + 1); Anal. (C16H26ClNO) C, H, N, Cl.
Biological Procedures
(Provided by NIDA from Oregon Health & Science University and SRI International). Unknowns were weighed and dissolved in DMSO to make a 10 mM stock solution. An initial dilution to 50 µM in assay buffer for binding, or to 1 mM in assay buffer for uptake, was made. Subsequent dilutions were made with assay buffer supplemented with DMSO, maintaining a final concentration of 0.1% DMSO. Pipetting was conducted using a Biomek 2000 robotic workstation.
Inhibition of Radioligand Binding of [125I]RTI 55 to hDAT, hSERT or hNET in Clonal Cells
Cell preparation: HEK293 cells expressing hDAT, hSERT or hNET inserts are grown to 80% confluence on 150 mm diameter tissue culture dishes and serve as the tissue source. Cell membranes are prepared as follows. Medium is poured off the plate, and the plate is washed with 10 ml of calcium- and magnesium-free phosphate-buffered saline. Lysis buffer (10 ml; 2 mM HEPES with 1 mM EDTA) is added. After 10 min, cells are scraped from plates, poured into centrifuge tubes, and centrifuged 30,000 × g for 20 min. The supernatant fluid is removed, and the pellet is resuspended in 12–32 ml of 0.32 M sucrose using a Polytron at setting 7 for 10 sec. The resuspension volume depends on the density of binding sites within a cell line and is chosen to reflect binding of 10% or less of the total radioactivity. Assay conditions: Each assay tube contains 50 µl of membrane preparation (about 10–15 µg of protein), 25 µl of unknown, compound used to define non-specific binding, or buffer (Krebs-HEPES, pH 7.4; 122 mM NaCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 10 µM pargyline, 100 µM tropolone, 0.2% glucose and 0.02% ascorbic acid, buffered with 25 mM HEPES), 25 µl of [125I]RTI-55 (40–80 pM final concentration) and additional buffer sufficient to bring up the final volume to 250 µl. Membranes are preincubated with unknowns for 10 min prior to the addition of the [125I]RTI-55. The assay tubes are incubated at 25°C for 90 min. Binding is terminated by filtration over GF/C filters using a Tomtec 96-well cell harvester. Filters are washed for six seconds with ice-cold saline. Scintillation fluid is added to each square and radioactivity remaining on the filter is determined using a Wallac µ- or beta-plate reader. Specific binding is defined as the difference in binding observed in the presence and absence of 5 µM mazindol (HEK-hDAT and HEK-hNET) or 5 µM imipramine (HEKhSERT). Two or three independent competition experiments are conducted with duplicate determinations. GraphPAD Prism is used to analyze the ensuing data, with IC50 values converted to Ki values using the Cheng-Prusoff equation (Ki=IC50/(1+([RTI-55]/Kd RTI-55))).
Filtration Assay for Inhibition of [3H]Neurotransmitter Uptake in HEK293 Cells Expressing Recombinant Biogenic Amine Transporters
Cell preparation: Cells are grown to confluence as described above. The medium is removed, and cells are washed twice with phosphate buffered saline (PBS) at room temperature. Following the addition of 3 ml Krebs-HEPES buffer, the plates are warmed in a 25°C water bath for 5 min. The cells are gently scraped and then triturated with a pipette. Cells from multiple plates are combined. One plate provides enough cells for 48 wells, which is required to generate data on two complete curves for the unknowns.
Uptake inhibition assay conditions: The assay is conducted in 96 1-ml vials. Krebs- HEPES (350 µl) and unknowns, compounds used to define non-specific uptake, or buffer (50 µl) are added to vials and placed in a 25°C water bath. Specific uptake is defined as the difference in uptake observed in the presence and absence of 5 µM mazindol (HEK-hDAT and HEK-hNET) or 5 µM imipramine (HEK-hSERT). Cells (50 µl) are added and preincubated with the unknowns for 10 min The assay is initiated by the addition of [3H]dopamine, [3H]serotonin, or [3H]norepinephrine (50 µl, 20 nM final concentration). Filtration through Whatman GF/C filters presoaked in 0.05% polyethylenimine is used to terminate uptake after 10 min. The IC50S are calculated applying the GraphPAD Prism program to triplicate curves made up of 6 drug concentrations each. Two or three independent determinations of each curve are made.
Supplementary Material
X-ray crystallographic data. CCDC 280869-280870 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Acknowledgments
This work was supported by the National Institute on Drug Abuse: DA1-8825 (PM), DA11542 (PM), DA06303 (BKM), DA11558 (BKM), DA15305 (BKM) RR00168 (BKM). The authors would like to thank the National Institute on Drug Abuse for providing the biological procedures and transporter inhibition binding data (DA 0007; Oregon Health & Science University); receptor binding and functional data (DA 1-8816; SRI International) and locomotor activity (DA 2-8822; University of North Texas Health Sciences). The authors thank Richard Laura for help with manuscript preparation.
References
- 1.Schildkraut JJ. The catecholamine hypothesis of affective disorders: A review of supporting evidence. J. Psychiatry. 1965;122:509–522. doi: 10.1176/ajp.122.5.509. [DOI] [PubMed] [Google Scholar]
- 2.Madras BK, Pristupa ZB, Niznik HB, Liang AY, Blundell P, Gonzalez MD, Meltzer PC. Nitrogen-based drugs are not essential for blockade of monoamine transporters. Synapse. 1996;24:340–348. doi: 10.1002/(SICI)1098-2396(199612)24:4<340::AID-SYN4>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 3.Biederman J, Spencer T. Attention-deficit/hyperactivity disorder (ADHD) as a noradrenergic disorder. Biol. Psychiatr. 1999;46:1234–1242. doi: 10.1016/s0006-3223(99)00192-4. [DOI] [PubMed] [Google Scholar]
- 4.Popper CW. Pharmacologic alternatives to psychostimulants for the treatment of attention-deficit/hyperactivity disorder. Child Adolesc. Psychiatr. Clin. N Am. 2000;9:605–646. [PubMed] [Google Scholar]
- 5.Fleckenstein AE, Gibb JW, Hanson GR. Differential effects of stimulants on monoaminergic transporters: pharmacological consequences and implications for neurotoxicity. Eur. J. Pharmacol. 2000;406:1–13. doi: 10.1016/s0014-2999(00)00639-7. [DOI] [PubMed] [Google Scholar]
- 6.Gorelick DA, Gardner EL, Xi ZX. Agents in development for the management of cocaine abuse. Drugs. 2004;64:1547–1573. doi: 10.2165/00003495-200464140-00004. [DOI] [PubMed] [Google Scholar]
- 7.Gainetdinov RR, Caron MG. Monoamine transporters: from genes to behavior. Ann. Rev. Pharm. and Tox. 2002;43:261–284. doi: 10.1146/annurev.pharmtox.43.050802.112309. [DOI] [PubMed] [Google Scholar]
- 8.Vocci FJ, Acri J, Elkashef A. Medications development for addictive disorders: the state of the science. Am. J. Psychiatry. 2005;162:1432–1440. doi: 10.1176/appi.ajp.162.8.1432. [DOI] [PubMed] [Google Scholar]
- 9.Coyle JT, Snyder SH. Antiparkinsonian drugs: inhibition of dopamine uptake in the corpus striatum as a possible mechanism of action. Science. 1969;166:899. doi: 10.1126/science.166.3907.899. [DOI] [PubMed] [Google Scholar]
- 10.Iversen LL. Uptake process of biogenic amines. New York: Plenum; 1975. pp. 381–442. [Google Scholar]
- 11.Meltzer PC, Blundell P, Madras BK. Structure activity relationships of inhibition of the dopamine transporter by 3-arylbicyclo[3.2.1]octanes. Med. Chem. Res. 1998;8:12–34. [Google Scholar]
- 12.Meltzer PC, Wang B, Chen Z, Blundell P, Jayaraman M, Gonzalez MD, George C, Madras BK. Synthesis of 6- and 7- hydroxy-8-azabicyclo[3.2.1]octanes and their binding affinity for the dopamine and serotonin transporters. J. Med. Chem. 2001;44:2619–2635. doi: 10.1021/jm0101242. [DOI] [PubMed] [Google Scholar]
- 13.Meltzer PC, Liu S, Blanchette HS, Blundell P, Madras BK. Design and synthesis of an irreversible dopamine-sparing cocaine antagonist. Bioorg.& Med. Chem. 2002;10:3583–3591. doi: 10.1016/s0968-0896(02)00244-4. [DOI] [PubMed] [Google Scholar]
- 14.Madras BK, Fahey MA, Miller GM, De La Garza R, Goulet M, Spealman RD, Meltzer PC, George SR, O'Dowd BF, Bonab E, Livni E, Fischman AJ. Non-amine-based dopamine transporter (reuptake) inhibitors retain properties of amine-based progenitors. Eur J Pharmacol. 2003;479:41–51. doi: 10.1016/j.ejphar.2003.08.055. [DOI] [PubMed] [Google Scholar]
- 15.Kennedy LT, Hanbauer I. Sodium sensitive cocaine binding to rat striatal membrane: possible relationship to dopamine uptake sites. J. Neurochem. 1983;34:1137–1144. doi: 10.1111/j.1471-4159.1983.tb13666.x. [DOI] [PubMed] [Google Scholar]
- 16.Schoemaker H, Pimoule C, Arbilla S, Scatton B, Javoy-Agid F, Langer SZ. Sodium dependent [3H]cocaine binding associated with dopamine uptake sites in the rat striatum and human putamen decrease after dopaminergic denervation and in Parkinson's disease. Naunyn-Schmiedeberg's Arch. Pharmacol. 1985;329:227–235. doi: 10.1007/BF00501873. [DOI] [PubMed] [Google Scholar]
- 17.Reith MEA, Meisler BE, Sershen H, Lajtha A. Structural requirements for cocaine congeners to interact with dopamine and serotonin uptake sites in mouse brain and to induce stereotyped behavior. Biochem. Pharmacol. 1986;35:1123–1129. doi: 10.1016/0006-2952(86)90148-6. [DOI] [PubMed] [Google Scholar]
- 18.Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science. 1987;237:1219–1223. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- 19.Madras BK, Fahey MA, Bergman J, Canfield DR, Spealman RD. Effects of cocaine and related drugs in nonhuman primates: I. [3H]Cocaine binding sites in caudate-putamen. J. Pharmacol. Exp. Ther. 1989;251:131–141. [PubMed] [Google Scholar]
- 20.Bergman J, Madras BK, Johnson SE, Spealman RD. Effects of cocaine and related drugs in nonhuman primates. III. Self-administration by squirrel monkeys. J. Pharmacol. Exp. Ther. 1989;251:150–155. [PubMed] [Google Scholar]
- 21.Canfield DR, Spealman RD, Kaufman MJ, Madras BK. Autoradiographic localization of cocaine binding sites by [3H]CFT ([3H]WIN 35,428) in the monkey brain. Synapse. 1990;6:189–195. doi: 10.1002/syn.890060211. [DOI] [PubMed] [Google Scholar]
- 22.Madras BK, Kamien JB, Fahey M, Canfield D, Milius RA, Saha JK, Neumeyer JL, Spealman RD. N-Modified fluorophenyltropane analogs of cocaine with high affinity for [3H]cocaine receptors. Pharmacol Biochem. Behav. 1990;35:949–953. doi: 10.1016/0091-3057(90)90384-t. [DOI] [PubMed] [Google Scholar]
- 23.Kaufman MJ, Madras BK. Severe depletion of cocaine recognition sites associated with the dopamine transporter in Parkinson's diseased striatum. Synapse. 1991;9:43–49. doi: 10.1002/syn.890090107. [DOI] [PubMed] [Google Scholar]
- 24.Kuhar MJ, Ritz MC, Boja JW. The dopamine hypothesis of the reinforcing properties of cocaine. Trends Neurosci. 1991;14:299–302. doi: 10.1016/0166-2236(91)90141-g. [DOI] [PubMed] [Google Scholar]
- 25.Servin A, Jacquot CN, Rapin JR. Effects of pyrovalerone on peripheral noradrenergic mechanisms. Biochem. Pharmacol. 1978;27:1693–1694. doi: 10.1016/0006-2952(78)90180-6. [DOI] [PubMed] [Google Scholar]
- 26.Perrine DM, Ross JT, Nervi SJ, Zimmerman RH. A short, one-pot synthesis of bupropion. J. Chem. Ed. 2000;77:1479–1480. [Google Scholar]
- 27.Musso DL, Mehta NB, Soroko FE, Ferris RM, Hollingsworth EB, Kenney BT. Synthesis and evaluation of the antidepressant activity of the enantiomers of bupropion. Chirality. 1993;5:495–500. doi: 10.1002/chir.530050704. [DOI] [PubMed] [Google Scholar]
- 28.Lancelot JC, Robba M, Bonnet JJ, Vaugeois JM, Costentin J. Synthesis and preliminary study of the activity of thiophene analogues of pyrovalerone on the neuronal uptake of the monoamines. Eur. J. Med. Chem. 1992;27:297–300. [Google Scholar]
- 29.Heffe W. Die Stevens-umlagerung von allyl-phenacyl-ammoniumsalzen. Helv. Chim. Acta. 1964;47:1289–1292. [Google Scholar]
- 30.Stille G, Ackermann H, Eichenberger E, Lauener H. Vergleiehende pharmakologische untersuching eines sentralem stimulans 1-p-tolyl-1-oxo-2-pyrrolidino-n-pentan-HCl. Arzneimittle-Forsch. 1963;13:871–877. [PubMed] [Google Scholar]
- 31.Holliday AR, Morris RB, Sharpley RP. Compound 84/F 1983 compare with D-amphetamine and placebo in regard to effects on human performance. Psychopharmacologia. 1964;6:192–200. doi: 10.1007/BF00404009. [DOI] [PubMed] [Google Scholar]
- 32.Gardos G, Cole JO. Evaluation of pyrovalerone in chronically fatigued volunteers. Current Therap. Res. 1971;13:631–635. [PubMed] [Google Scholar]
- 33.Fauquet J-P, Morel E, Demarty C, Rapin JR. Role des catecholamines centrales dans l'activite psychostimulante de la pyrovalerone. Arch. Int. Pharmacodyn. 1976;224:325–337. [PubMed] [Google Scholar]
- 34.Vaugeois J-M, Bonnet J-J, Duterte-Boucher D, Costentin J. In vivo occupancy of the striatal dopamine uptake complex by various inhibitors does not predict their effects on locomotion. Eur. J. Pharm. 1993;230:195–201. doi: 10.1016/0014-2999(93)90802-o. [DOI] [PubMed] [Google Scholar]
- 35.Michaelis W, Russel JH, Schindler O. The metabolism of pyrovalerone hydrochloride. J. Med. Chem. 1970;13:497–503. doi: 10.1021/jm00297a036. [DOI] [PubMed] [Google Scholar]
- 36.Eshleman AJ, Carmolli M, Cumbay M, Martens CR, Neve KA, Janowsky A. Characteristics of drug interactions with recombinant biogenic amine transporters expressed in the same cell type. J Pharmacol Exp Ther. 1999;289:877–885. [PubMed] [Google Scholar]
- 37.Calligaro DO, Eldefrawi ME. High affinity stereospecific binding of [3H]cocaine in striatum and its relationship to the dopamine transporter. Membr. Biochem. 1987;7:87–106. doi: 10.3109/09687688709039986. [DOI] [PubMed] [Google Scholar]
- 38.Glennon RA, Young R, Martin BR, Dal Cason TA. Methcathione ("cat"): an enantiomeric potency comparison. Pharmacol. Biochem. Behav. 1995;50:601–606. doi: 10.1016/0091-3057(94)00348-3. [DOI] [PubMed] [Google Scholar]
- 39.Carroll FI, Gao Y, Rahman MA, Abraham P, Parham K, Lewin AH, Boja JW, Kuhar MJ. Synthesis, ligand binding, QSAR, and CoMFA study of 3β-(p-substituted phenyl)tropane-2β-carboxylic acid methyl esters. J. Med. Chem. 1991;34:2719–2725. doi: 10.1021/jm00113a008. [DOI] [PubMed] [Google Scholar]
- 40.Newman AH, Izenwasser S, Robarge MJ, Kline RH. CoMFA study of novel phenyl ring-substituted 3α-(diphenylmethoxy)tropane analogues at the dopamine transporter. J. Med. Chem. 1999;42:3502–3350. doi: 10.1021/jm980701v. [DOI] [PubMed] [Google Scholar]
- 41.Deutsch HM. Structure-activity relationships for methylphenidate analogs and comparisons to cocaine and tropanes. Med. Chem. Res. 1998;8:91–99. [Google Scholar]
- 42.Meltzer PC, Wang P, Blundell P, Madras BK. Synthesis and evaluation of dopamine and serotonin transporter inhibition by oxacyclic and carbacyclic analogues of methylphenidate. J. Med. Chem. 2003;46:1538–1545. doi: 10.1021/jm0205292. [DOI] [PubMed] [Google Scholar]
- 43.Newman AH. Novel dopamine transporter ligands: The state of the art. Med. Chem. Res. 1998;8:1–11. [Google Scholar]
- 44.Carroll FI, Howell LL, Kuhar MJ. Pharmacotherapies for treatment of cocaine abuse: preclinical aspects. J. Med. Chem. 1999;42:2721–2736. doi: 10.1021/jm9706729. [DOI] [PubMed] [Google Scholar]
- 45.Carroll FI. 2002 medicinal chemsitry division award address: Monoamine transporters and opioid receptors. Targets for addiction therapy. J. Med. Chem. 2003;46:1775–1794. doi: 10.1021/jm030092d. [DOI] [PubMed] [Google Scholar]
- 46.Deutsch HM, Collard DM, Zhang L, Burnham KS, Deshpande AK, Holtzman SG, Schweri MM. Synthesis and pharmacology of site-specific cocaine abuse treatment agents: 2-(aminomethyl)-3-phenylbicyclo[2.2.2]- and -[2.2.1]alkane dopamine uptake inhibitors. J. Med. Chem. 1999;42:882–895. doi: 10.1021/jm980566m. [DOI] [PubMed] [Google Scholar]
- 47.Carroll FI, Pawlush N, Kuhar MJ, Pollard GT, Howard JL. Synthesis, monoamine transporter binding properties, and behavioral pharmacology of a series of 3β-(substituted phenyl)-2β-(3'-substituted isoxazol-5-yl)tropanes. J. Med. Chem. 2004;47:296–302. doi: 10.1021/jm030453p. [DOI] [PubMed] [Google Scholar]
- 48.Meltzer PC, Madras BK. Unpublished data. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
X-ray crystallographic data. CCDC 280869-280870 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.