

Abstract
Approximately 5% of patients with neurofibromatosis type 1 (NF1) have deletions of the entire NF1 gene. The phenotype usually includes early onset, large number of neurofibromas, presence of congenital anomalies, cognitive deficiency, and variable dysmorphic features and growth abnormalities. Connective tissue abnormalities are not generally recognised as a part of NF1 microdeletion syndrome, but mitral valve prolapse, joint laxity, and/or soft skin on the palms have been reported in a few patients. We describe clinical findings in six newly diagnosed patients with NF1 microdeletions, five of whom presented with connective tissue abnormalities. A literature review of the clinical findings associated with NF1 microdeletion was also performed. Our report confirms that connective tissue dysplasia is common in patients with NF1 microdeletions. Given the potential for associated cardiac manifestation, screening by echocardiogram may be warranted. Despite the large number (>150) of patients with known NF1 microdeletions, the clinical phenotype remains incompletely defined. Additional reports of patients with NF1 microdeletions, including comprehensive clinical and molecular information, are needed to elucidate possible genotype–phenotype correlation.
Keywords: neurofibromatosis type 1, microdeletion, connective tissue dysplasia
Neurofibromatosis type 1 (NF1) is a common autosomal dominant disorder with a prevalence of 1:4000.1 Large deletions involving the entire gene have been found in at least 4.4% patients with NF1,2 although earlier reports suggested a higher frequency of 5–10%.3,4,5,6 The first patient with NF1 microdeletion was reported in 1992,7 and since then, 151 patients have been reported in the literature.3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,35,36,37,38,39
Patients with NF1 with microdeletions that encompass the entire coding region exhibit a more severe clinical phenotype (early onset, large number of neurofibromas, cognitive deficiency, dysmorphic features). Frequently noted dysmorphic features include: prominent forehead, ptosis, downslanted palpebral fissures, hypertelorism, short nose, low set ears, micrognathia, and a general appearance of facial coarsening. In addition, previous articles suggest that patients with NF1 microdeletions are at increased risk for developing malignant tumours, both NF1 associated (malignant peripheral nerve sheath tumours; MPNST) and those not known to be associated with NF1 (cerebellar medulloblastoma, retroperitoneal fibrosarcoma, cerebellar neuroblastoma).8,9,10,11,15,16,17,18,19,20,21,23,24,25,26,27,30,31 Less persistent clinical features, such as short stature, large hands and feet, microcephaly/macrocephaly, scoliosis, iris coloboma, and pulmonary stenosis have also been described. Although increased joint mobility, mitral valve prolapse, and aortic dilatation have been reported in a few patients,6,17,20,22,26,29 the association of connective tissue abnormalities and NF1 microdeletion has not been clearly established.
We describe six new patients with microdeletions involving the entire NF1 gene, five of whom presented with marked signs of connective tissue dysplasia.
MATERIALS AND METHODS
All patients signed a release giving consent for anonymised publication of clinical information and photographs. Approval was given by the institutional review board of Mayo College of Medicine.
Cytogenetic studies
Probe design and validation
To detect the deletion of the NF1 gene region associated with del (17q11.2), direct labelled fluorescent in situ hybridisation (FISH) probes were designed from bacterial artificial chromosomes (BACs) covering the gene of interest. The search for BAC clones on 17q11.2 was accomplished using the University of California Santa Cruz (UCSC) Genome Browser (www.uscs.edu) and the website of the National Center for Biotechnology Information (NCBI; www.ncbi.nlm.nih.gov/). The four clones used were: RP11‐848P1 (210 kb), RP11‐142O6 (145 kb), CTD‐2370N5 (113 kb), and CTD‐2538H15 (160 kb). The RP11‐848P1 and CTD‐2538H15 clones flanked the NF1 gene to increase the size of the probe. Glycerol stocks of the BAC clones (supplied by ResGen Invitrogen Corp.) were plated and propagated immediately upon arrival. Isolation and purification of DNA was performed using the Qiagen Plasmid Maxi kit (25) according to manufacturer's instructions. The sequence for each BAC was obtained from the NCBI website (fig 1).

Figure 1 A direct labelled FISH probe, made in our laboratory, was designed to span the NF1 gene region. The probe schematic indicates the four BACs covering this gene region (approximately 550 kb) and genes within close proximity to NF1.
Nick translation was performed using the Vysis nick translation kit (Vysis Inc., Downers Grove, IL, USA) to produce the fluorescence labelled DNA probe for FISH. Each NF1 clone was labelled with Spectrum Orange dUTP (Vysis Inc.). Following probe labelling, the probes were precipitated and concentrated in sterile water. Probe working solutions were made by mixing three parts of each labelled BAC with seven parts LSI/WCP hybridisation buffer (Vysis Inc.) Signal validation of each labelled BAC was verified by observing both interphase and metaphase cells in normal blood samples. After individual BAC signal validation, all four NF1 probes were combined and signal validation was repeated. After signal validation, the combined probes were stored in the dark at −20°C. The NF1 probe working solution was prepared by mixing three parts combined NF1 probe with seven parts LSI/WCP hybridisation buffer. CEP 17 (D1721) Spectrum Green dUTP (Vysis Inc.) probe working solution was made by mixin one part concentrated probe with nine parts LSI/WCP hybridisation buffer and added to the NF1 working solution as a control. The NF1 and CEP 17 (D1721) probe working solutions were mixed together in a 1:1 ratio. Specimens were set up following standard FISH pretreatment and hybridisation protocols for tissue.
Primers were designed for each BAC clone using either the NF1 gene sequence or sequence tagged sites (STS) within and flanking the NF1 gene region. To verify the presence of the gene or region of interest in each BAC clone, PCR and gel electrophoresis were performed both before and after DNA isolation.
Molecular testing
Deletion mapping
Clones RP11‐848P1, CTC‐499I20, CTD‐2370N5, and CTD‐2538H15 comprised the NF1 probe and served as the basis of our deletion map (fig 1). To determine the extent of the deletion, we created direct labelled FISH probes from BAC and P1 derived artificial chromosome (PAC) clones flanking the NF1 gene region both centromeric and telomeric. The search for BACs and PACs on 17q11.2 was accomplished using the UCSC and NCBI websites. Clones selected centromeric to NF1 were: RP11‐271K11 (199 kb), CTD‐2349P21 (119 kb), and RP13‐753N3 (153 kb) (fig 2). The clones selected telomeric to NF1 were: RP1‐41C23 (208 kb), RP11‐805L22 (108 kb), CTC‐542B22 (130 kb), RP11‐164M10 (170 kb), CTD‐2095E4 (104 kb), RP11‐443G13 (156 kb), and 227G15 (183 kb) (fig 2). Each clone was PCR validated using STS markers. Flanking clones were labelled in opposite colours (Spectrum Orange or Spectrum Green), and applied to cytogenetically normal metaphases to verify their chromosomal location. Probes in opposite colours were applied in groups of two sequentially away from the NF1 gene region until the deletion end point was determined (fig 2).

Figure 2 Direct labelled FISH probes were designed to span and flank the NFI gene region. The probe schematic demonstrates the 12 BAC/PAC probes that were deleted, indicated by the dashed lines. The BACs represented with a solid line indicate the probes that were present. The total size of the deletion was determined to be 1.94 Mb. Genes located at the deletion end points are also indicated on the schematic.
RESULTS
FISH
FISH test validation
The NF1 probe was applied to 40 blood specimens, the identity of which had been blinded. The NF1 probe (fig 1) was used to analyse 20 blood samples from patients with previously defined NF1 and 20 blood samples from healthy controls (negative controls). With a cut off of 4.5%, 20 normal specimens yielded normal results. One specimen from the 20 patients with known NF1 showed a deletion of the NF1 gene region, while the other 19 specimens were found to have two NF1 signals.
Molecular testing
Additional FISH probes were created to determine the extent of the deletion in each patient. These probes were applied to samples from each of the six patients with confirmed NF1 gene deletion. For each sample, probes centromeric to NF1, RP13‐753N3, and CTD‐2349P21 were found to be deleted and probe RP11‐271K11 was found to be present (fig 2). The probes telomeric to NF1, RP1‐41C23, RP11‐805L22, CTC‐542B22, RP11‐164M10, CTD‐2095E4 and RP11‐443G13 were deleted, while probe RP11‐227G15 was present (fig 2). Although two signals were present for probe RP11‐227G15, one signal was of much weaker intensity and smaller in size, indicating that the deletion junction probably resides within this particular BAC clone. Results from the FISH mapping suggest a similar deletion size of 1.94 Mb in each of the six patients.
Clinical reports
Patient 1
Patient 1 (fig 3) was first evaluated at 1 day of age because of dysmorphic features, redundant skin, and polydactyly. Pregnancy and delivery were normal. Birth weight and length were at the 50th percentile, while the child's head circumference was between the 5th and 10th percentiles. Neonatal examination revealed shallow orbits, bilateral inverted epicanthic folds, long philtrum, thin upper lip, small mandible, and C shaped overfolded helices with upper level 5 mm above the canthal line. There was a pedunculated postaxial polydactyly from the middle phalanx of the left little finger. The patient had prominent hypermobility of the wrist and small joints in both hands. The skin over the proximal arms, legs, and hands was notably redundant and soft. He had bilateral inguinal hernia.

Figure 3 Patient 1: (A) at 9 months, (B) 18 months, (C) 5 years, showing dysmorphic features, including epicanthic folds, long philtrum, thin upper lip, small mandible, and C shaped overfolded helices. Informed consent for publication of photographs was obtained.
At 2 weeks of age, the patient presented with new finding of 2/6 systolic ejection murmur, and echocardiogram disclosed severe, asymmetrical ventricular septal hypertrophy. Treatment was initiated and there was complete resolution of the septal hypertrophy by age of 13 months. Follow up echocardiograms remained normal. Interestingly, the asymptomatic mother was found to have hypertrophic cardiomyopathy. The unusual cardiac presentation in this family has been reported previously.32
Impressive skin laxity persisted in the patient. Electron microscopy of skin showed decreased amount of elastin, but elastin protein expression and RNA expression were normal. Urine test to evaluate elastin breakdown was also normal.
At the age of 7 months, a few hyperpigmented macules over the abdomen (one >10 mm) were noted. Cognitive and speech development showed delay, but physical growth was excellent.
Several diagnoses (cutis laxa, NF1, and Langer‐Giedion, Noonan, Costello, and cardiofaciocutaneous syndromes) were considered. Chromosome analysis from blood and skin was normal (46,XY). Molecular analysis of NF1 by protein truncation, performed at Roche Laboratories, was normal.
The patient was again examined at 3 years of age. Significant clinical findings included: >10 café au lait macules >10 mm, several smaller hyperpigmented spots, and unilateral inguinal freckling. The diagnosis of NF1 (Watson type) was made.
Over the course of the following 5 years (age 3–8 years), the patient experienced rapid development of NF1 with associated manifestations (more than 50 café au lait macules, intertriginous freckling, one Lisch nodule, multiple cutaneous and subcutaneous neurofibromas, and a large plexiform neurofibroma of the brachial plexus). Cognitive and speech development remained delayed, requiring special education.
FISH performed on skin sample showed a deletion of approximately 1.94 Mb covering the entire NF1 gene. Neither parent had a deletion. In addition, aside from hypertrophic cardiomyopathy in the mother, parents were both generally healthy with no dysmorphism or skin/joint laxity.
Patient 2
Patient 2 (fig 4) was diagnosed with NF1 at 17 years of age when he presented with several café au lait macules and interriginous freckling. Numerous subcutaneous neurofibromas developed during the second decade of life. At 41 years of age, magnetic resonance imaging (MRI) of the cervical spine revealed a plexiform neurofibroma in the left prevertebral region extending from the mid‐cervical to upper thoracic levels.

Figure 4 Patient 2. (A) Long narrow face, hypertelorism, downslanted palpebral fissures, prominent glabella with deeply set eyes, and right exotropia. Patient has a high narrow palate and a prominent cross bite. Ears are normally set, nose is prominent with fleshy nasal tip. (B) Macrocephaly and marfanoid body build. (C) Skin of palms was wrinkled and redundant. Informed consent for publication of photographs was obtained.
Pregnancy, delivery, and neonatal histories were unremarkable. Childhood medical history was significant for poor coordination, mild learning disability, strabismus, a deviated nasal septum, and unilateral clubfoot. Bacterial endocarditis was diagnosed at 38 years of age, secondary to previously undiagnosed mitral valve prolapse with regurgitation.
The patient was examined when he was 41 years old. He was tall and slim, with marfanoid body build and obvious facial dysmorphism. Weight was 76.3 kg (75–90%), and height was 1.84 m (90%). Head was large, with a circumference of 625 mm (97%). Extremities were disproportionally long and thin, with large hands and feet. Joints were hypermobile. Skin on the palms and soles was soft, loose, and redundant (fig 4). Skin elsewhere was soft and hyperextensible. No unusual scarring or striae were noted. Cognitive abilities were age appropriate. Mild speech articulation difficulty and hypernasal speech were observed. There was prominent cervical and upper thoracic kyphosis. Chest was mildly asymmetric, but there was no pectus excavatum deformity. Heart rhythm was regular, with a 2–3/6 systolic murmur. There were innumerable cutaneous and subcutaneous neurofibromas. Except for mild muscular hypotonia, neurological examination was normal.
High resolution karyotype was normal (46,XY), and molecular testing for mutations in the fibrillin‐1 gene was negative. FISH performed on a peripheral blood sample showed a deletion of approximately 1.94 Mb covering the entire NF1 gene. Family history is negative for NF1, and neither parent had a deletion.
Patient 3
Patient 3 (fig 5) was aged 5 years and 3 months at the time of his initial evaluation. Pregnancy, delivery, and neonatal histories were unremarkable. Birth weight was average.

Figure 5 Patient 3, at 9 years of age. Prominent forehead, bulbous nose, and shortened upper lip are apparent. Informed consent for publication of photographs was obtained.
A few darkly pigmented skin macules were observed at birth. Over time, skin changes became more noticeable, and new lesions appeared. At 5 years of age, inguinal freckling was observed and NF1 diagnosed. On examination at this time, the patient was mildly dysmorphic, had hypernasal speech, and showed global developmental delay. There was mild hypotonia. Weight was 24 kg and height 1.18 m (both 95th percentile), and head circumference was 520 mm (50th percentile).
The head was brachycephalic with a prominent forehead and widely spaced eyes. The nose was bulbous, and the upper lip short. The thorax had mild anterior prominence, and there was minimal thoracic scoliosis. Heart rate was regular with normal heart sounds and no murmurs. Hands were large (over the 97th percentile for age) with short metatarsals, and the carrying angle of the elbows was increased. The great toes were broad.
Head MRI was normal except for the presence of multiple foci of increased T2 signal in the brainstem and basal ganglia.
FISH performed on a peripheral blood sample showed a deletion of approximately 1.94 Mb covering the entire NF1 gene. Family history was negative. The parents were not tested, but neither showed clinical features of NF1.
Patient 4
Patient 4 (fig 6) was diagnosed with NF1 at 4 years of age because of multiple café au lait macules and cutaneous neurofibromas.

Figure 6 Patient 4. (A) Macrocephaly, prominent forehead, broad fleshy nasal tip, broad neck with sloping shoulders, and multiple cutaneous neurofibromas; (B) large hands. Informed consent for publication of photographs was obtained.
Pregnancy and delivery were unremarkable. Birth weight was average. Unilateral cleft lip and cleft of the right ear lobulus were noted upon delivery. Surgical correction for the cleft lip was performed at 3 months of age, with subsequent revision at 8 years. Psychomotor development was reported to be normal. Café au lait macules were present in infancy, and an increased number of cutaneous neurofibromas became present in the early teens, resulting in a large tumour burden before the age of 20 years.
At 19 years of age, medical genetics evaluation was significant for a prominent forehead, downslanting palpebral fissures, broad fleshy nasal tip, broad neck with sloping shoulders, macrocephaly, large hands and feet, soft fleshy palms and soles, and deep palmar/plantar creases. "Constitutional connective dysplasia" was manifested as joint laxity, skin laxity, and mitral valve prolapse. There was moderate thoracolumbar scoliosis and a somewhat marfanoid body build. Lisch nodules and mild high frequency deafness were also noted.
The patient remained generally healthy except for chronic depression during the subsequent 20 years. During that time, numerous cutaneous and subcutaneous neurofibromas continued to develop over the entire body and face, resulting in disfigurement and severe deformation of the eyelids.
FISH performed on a peripheral blood sample showed a deletion of approximately 1.94 Mb covering the entire NF1 gene. Family history is negative. The parents were not tested but neither showed clinical features of NF1.
Patient 5
Patient 5 (fig 7) was diagnosed with NF1 at 3 years of age because of multiple café au lait macules and intertriginous freckling.

Figure 7 Patient 5. (A) macrocephaly, downslanted palpebral fissures, bulbous nose, and facial coarsening; (B) numberous cutaneous neurofibromas on the back. Informed consent for publication of photographs was obtained.
Pregnancy, delivery, and neonatal course were unremarkable. Childhood medical history is significant only for mild strabismus and migraine headaches. Computed tomography of the head performed at 10 years of age did not disclose any significant intracranial abnormalities. Aside from mild gross motor delay, development was age appropriate. Psychometric testing showed average ability.
The patient was examined at 25 years of age. She was macrocephalic and mildly dysmorphic. Downturned palpebral fissures, wide and bulbous nasal tip, and low set ears were observed. There was a general appearance of facial coarsening. There was no scoliosis. The skin was generally very soft and hyperextensible. Scars were wide and had a keloid appearance. Diffuse cutaneous and subcutaneous neurofibromas (>100) were noted over the head, trunk, and extremities. No plexiform neurofibromas were noted. There was marked joint laxity. Echocardiogram revealed mild mitral valve prolapse with regurgitation.
FISH performed on a peripheral blood sample showed a deletion of approximately 1.94 Mb. Family history is negative. The parents were not tested but neither showed clinical features of NF1.
Patient 6
Patient 6 (fig 8) was diagnosed with NF1 at 16 years of age because of axillary freckling and cutaneous neurofibromas.
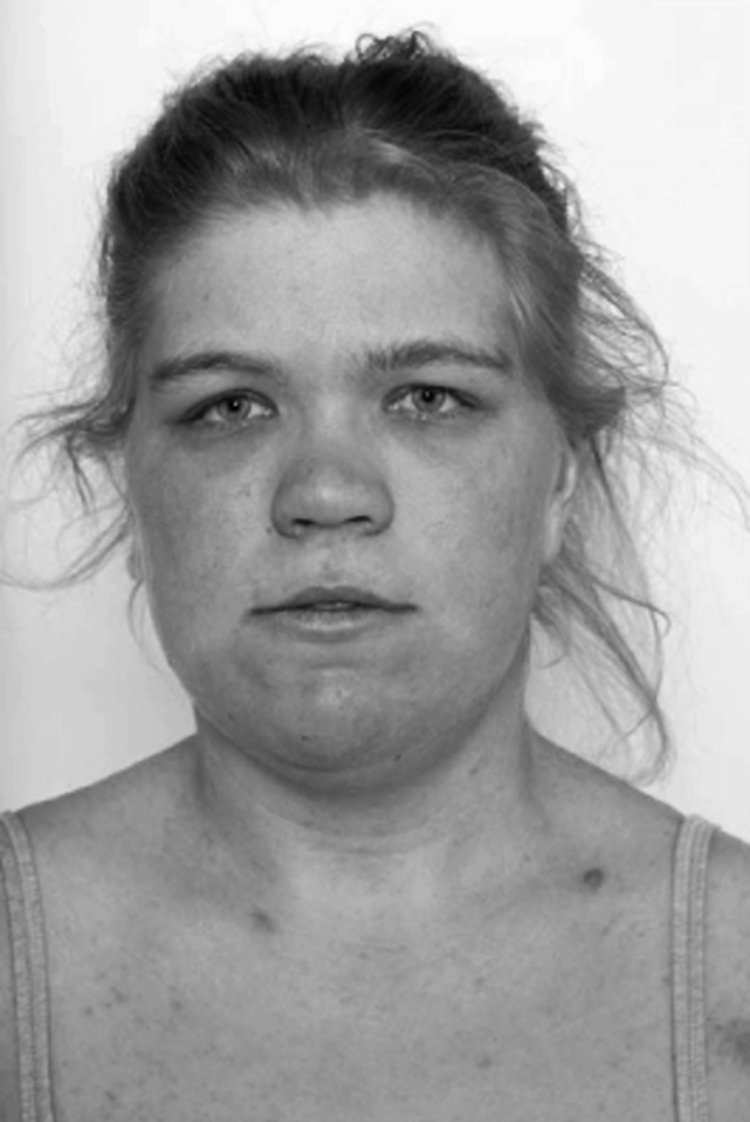
Figure 8 Patient 6. Broad nasal bridge, hypertelorism, downslanted palpebral fissures, and “bulbous” nose. Informed consent for publication of photographs was obtained.
Pregnancy, delivery, and neonatal histories were unremarkable. Childhood medical history was significant for multiple injuries, including broken bones and several sprains, as a consequence of poor motor coordination. Developmental history was otherwise normal. At 16 years of age, the patient underwent surgery for a cartilage chip in the left knee joint.
The patient was examined at 27 years of age. There was mild dysmorphism including hypertelorism, downslanted palpebral fissures, and broad nasal bridge. There was minimal thoracic scoliosis and significant joint laxity. Hands were large with long digits. She had numerous cutaneous and subcutaneous neurofibromas. No plexiform neurofibromas were noted. The skin on the palms was very soft and somewhat redundant. Scars were thin and soft. No plexiform neurofibromas were noted.
Family history is negative for NF1. FISH performed on a peripheral blood sample showed a deletion of approximately 1.94 Mb covering the entire NF1 gene.
Review of the literature
We performed an extensive literature review using OVID and PubMed databases. Our search identified 39 references, providing a variable amount of clinical information for 151 patients with NF1 microdeletions. We report six additional patients. Of this total 157 patients, cognitive and/or developmental status was recorded for 118, of whom 70% (83/118) showed cognitive deficiency and/or developmental delay of various degree, ranging from learning disability to severe mental retardation. Of these 83 patients, 53 had mental retardation, based on IQ <83, 12 had developmental delay, and 16 had learning disability. Our review confirms that the proportion of patients with NF1 microdeletions and some degree of cognitive deficiency, learning disability, or developmental delay is greater than the 30–65%33 expected in the general NF1 population.
The presence and description of dysmorphic features was reported for 118 of the 157 patients, and 75% showed one or more dysmorphic features. Those most commonly noted were: hypertelorism, coarse face, broad, fleshy nose +/− broad nasal bridge, ptosis, downslanting of the palpebral fissures, and micrognathia (table 1). The proportion of patients with dysmorphic features and large deletion of the NF1 gene (75%) exceeded that expected from the general NF1 population (5–15%).27
Table 1 Dysmorphic characteristics in patients with NF1 microdeletions.
| No. of patients* | Facial dysmorphism | Prominent forehead | Coarse face | Broad, fleshy nose +/− broad nasal bridge | Hyper‐telorism | Tele‐canthus | Down‐slanted palpebral fissures | Ptosis | Micro‐gnathia | Large ears | Facial asymmetry | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Kayes et al7,13† | 5 | 5 | 1 | 1 | 4 | 2 | 4 | 5 | 1 | ||||||||||||||
| Wu et al28 | 4 | 4 | 1 | 1 | 2 | 1 | 2 | 2 | |||||||||||||||
| Leppig et al16 | 2 | 2 | 1 | 1 | 1 | 1 | |||||||||||||||||
| Colley et al9 | 2 | 2 | 1 | 2 | |||||||||||||||||||
| Upadhyaya et al6 | 1 | 1 | 1 | 1 | 1 | ||||||||||||||||||
| Leppig et al17 | 6 | 6 | 1 | 5 | 1 | 6 | 6 | 1 | 4 | ||||||||||||||
| Tonsgard et al3 | 5 | 3 | 1 | 1 | 1 | 1 | 1 | 1 | |||||||||||||||
| Wu et al29 | 2 | 2 | 2 | 2 | 1 | ||||||||||||||||||
| Cnossen et al4; Van Asperen et al26 | 5 | 5 | 1 | 5 | 3 | 1 | 2 | 4 | 2 | 2 | |||||||||||||
| Valero et al25 | 6 | 3 | 1 | 1 | |||||||||||||||||||
| Upadhyaya et al24‡ | 18 | 10 | |||||||||||||||||||||
| Ruggieri et al22 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |||||||||||||||
| Riva et al38‡; Riva et al23 | 7 | 4 | 1 | 3 | 4 | 1 | 1 | 3 | 1 | ||||||||||||||
| Lopez‐Correa et al18 | 7 | 6 | 6 | 6 | 6 | ||||||||||||||||||
| Dorschner et al10 | 9 | 9 | 4 | 1 | 3 | 1 | 1 | 4 | |||||||||||||||
| Jenne et al12 | 1 | 1 | 1 | ||||||||||||||||||||
| Oktenli et al20 | 1 | 1 | 1 | 1 | 1 | 1 | |||||||||||||||||
| Venturin et al27 | 14 | 10 | 2 | 1 | 5 | 6 | 2 | 1 | 1 | 2 | |||||||||||||
| Kehrer‐Sawatzki et al14 | 16 | 7 | 7 | 7 | 7 | 7 | |||||||||||||||||
| Our patients | 6 | 6 | 3 | 3 | 5 | 4 | 5 | 2 | 1 | 2 | |||||||||||||
| Total | 118 | 88 | 12 | 37 | 37 | 40 | 8 | 31 | 25 | 19 | 10 | 5 | |||||||||||
Includes four patients with small intragenic deletion from report by Upadhyaya et al 1998. Thirteen patients are mosaic (Lazaro et al,5 Wu et al,28 Colman et al,37 Ainsworth et al,8 Tonsgard et al,3 Rasmussen et al 1997,21 Riva et al,23 Kehrer‐Sawatzki et al,14). *Some reports provided only limited information about phenotype of patients; †four patients reported in Kayes et al,7,13 later also reported in Dorschner et al,10 and in Jenne et al,11,12 excluded from later references; ‡includes only 16 of 22 reported patients for whom information about dysmorphism was available.
Patients with a microdeletion are often described with unusual body habitus and disproportionate body build, sometimes having a Noonan, Weaver, or Watson syndrome phenotype. In addition, many patients show non‐specific findings, most commonly large hands and/or feet (25/114), macrocephaly (22/114), and broad neck with sloping shoulders (21/114) (table 2). Five patients, including two of our cases, had marfanoid body build.
Table 2 Body habitus in patients with NF1 microdeletions§.
| No. of reported patients* | Broad neck/sloping shoulders | Macro‐cephaly | Micro‐cephaly | Short stature | Tall stature/over‐growth | Hemi‐hypertrophy | Large hands +/− large feet | Noonan‐like | Weaver‐like | Watson‐like | Marfanoid | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Kayes et al7,13 | 5 | 4 | 3 | 2 | 1 | |||||||||||||||||||
| Wu et al28 | 4 | 3 | 2 | |||||||||||||||||||||
| Leppig et al17 | 2 | 1 | 1 | 1 | ||||||||||||||||||||
| Colley et al9 | 2 | 1 | 2 | 2 | ||||||||||||||||||||
| Upadhyaya et al6 | 1 | 1 | 1 | |||||||||||||||||||||
| Ainsworth et al8 | 3 | 1 | 1 | |||||||||||||||||||||
| Leppig et al16 | 6 | 6 | 2 | 5 | 1 | |||||||||||||||||||
| Tonsgard et al3 | 5 | 1 | 1 | 1 | 1 | |||||||||||||||||||
| Valero et al25 | 6 | 1 | 1 | |||||||||||||||||||||
| Wu et al29 | 2 | 2 | 2 | |||||||||||||||||||||
| Cnossen et al4; Van Asperen et al26 | 5 | 1 | 2 | 2 | 2 | |||||||||||||||||||
| Rasmussen et al21 | 5 | 2 | 1 | |||||||||||||||||||||
| Ruggieri et al22 | 1 | 1 | 1 | 1 | ||||||||||||||||||||
| Riva et al23,28 | 7 | 1 | 2 | 2 | ||||||||||||||||||||
| Lopez‐Correa et al18 | 7 | 2 | 5 | |||||||||||||||||||||
| Dorschner et al10 | 9 | 2 | 2 | 3 | 1 | 5 | 1 | 1 | ||||||||||||||||
| Jenne et al12 | 1 | 1 | 1 | |||||||||||||||||||||
| Oktenli et al20 | 1 | 1 | 1 | |||||||||||||||||||||
| Venturin et al27 | 14 | 1 | 1 | 1 | 3 | 2 | 1 | |||||||||||||||||
| Kehrer‐Sawatzki et al14 | 22 | 1 | 1 | |||||||||||||||||||||
| Our patients | 6 | 2 | 3 | 1 | 3 | 4 | 1 | 2 | ||||||||||||||||
| Totals | 114 | 21 | 22 | 4 | 15 | 19 | 2 | 25 | 5 | 2 | 2 | 5 | ||||||||||||
*Only those patients with some description of body habitus.
A summary of associated internal anomalies, skeletal anomalies, tumours, and connective tissue abnormalities in the literature is presented in tables 3, 4, 5, and 6, respectively. Interestingly, we noted that four patients were reported with gynaecomastia, previously unknown to be associated with NF1.
Table 3 Reported internal and other anomalies in patients with NF1 microdeletions.
| Brain anomalies | Eyes | CHD | Hearing loss | Other | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Kayes et al13 | ASD | Urticaria pigmentosa; hirsutism | ||||||||
| Wu et al28 | Dilated ventricles; Chiari I with hydrocephalus; septum cavum pellucidum | Iris coloboma | Heart murmur | |||||||
| Colley et al9 | FTT; generalised oedema | |||||||||
| Upadhyaya et al6 | PDA | Pelviureteric obstruction; undescended testes | ||||||||
| Leppig et al16 | Lower lid eversion | Aortic dissection | Shawl scrotum; GCM | |||||||
| Tonsgard et al3 | Agenesis of corpus callosum | PS | Accessory nipple; single palmar crease; moles; GCM | |||||||
| Wu et al29 | Callosal dysgenesis | MVP | HT; facial nerve palsy | |||||||
| Valero et al25 | Cerebral chronic inflammatory tumour | Acromegaly | ||||||||
| Van Asperen et al26 | Unilateral hernia | |||||||||
| Rasmussen et al21 | Bilateral sensorineural | Precocious puberty | ||||||||
| Korf et al15 | Chiari I; septum cavum pellucidum and vergae; callosum dysgenesis | |||||||||
| Riva et al23 | Strabismus | PS | Facial nerve palsy; GCM | |||||||
| Lopez‐Correa et al18 | Unilateral deafness | Hypertrichosis; epilepsy | ||||||||
| Dorschner et al10 | ASD; PS | |||||||||
| Oktenli et al20 | Retinal detachment; optic disk drusen | MVP | No facial hair; GCM | |||||||
| Kehrer‐Sawatzki et al14 | Ataxia; hypogonadism | |||||||||
| Venturin et al27 | Strabismus; amblyopia | VSD; mitral insufficiency; HCM | Hearing loss | Umbilical hernia | ||||||
| Our patients | UBO | Exotropia; strabismus | HCM; MVP | Mild hearing loss | GCM |
ASD, atrial septal defect; HT, hypertension; PS, pulmonary stenosis; FTT, failure to thrive; UBO, unidentified bright object; GCM, gynaecomastia; MVP, mitral valve prolapse; VSD, ventricular septal defect; HCM, hypertophic cardiomyopathy; PDA, patent ductus arteriosus.
Table 4 Skeletal anomalies in NF1 microdeletion patients.
| No. of patients* | Pectus excavatum | Scoliosis | Brachy‐dactyly | Brachy‐cephaly | Cubitus valgus | Metaphyseal anomalies | Poly‐dactyly | Other | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Kayes et al7,13 | 3 | 2 | 1 | 2 | Absent coccyx; shield chest | |||||||||||||
| Wu et al28 | 1 | 1 | Metatarsus adductus | |||||||||||||||
| Leppig et al17 | 2 | 1 | Spinal stenosis; Madelung deformity | |||||||||||||||
| Colley et al9 | 2 | 2 | ||||||||||||||||
| Leppig et al16 | 6 | 2 | 2 | 2 | Dislocation of radii | |||||||||||||
| Tonsgard et al3 | 1 | Tibial torsion | ||||||||||||||||
| Cnossen et al4 | 1 | 1 | 1 | 1 | ||||||||||||||
| Valero et al25 | 1 | 1 | ||||||||||||||||
| Rasmussen et al21 | 3 | 2 | Fibular pseudoarthrosis | |||||||||||||||
| Ruggieri et al22 | 1 | 1 | ||||||||||||||||
| Riva et al23,28 | 2 | 1 | 1 | Broad diaphyses; flared rib cage; congenital hip dislocation; leg anomalies | ||||||||||||||
| Lopez‐Correa et al18 | 4 | 3 | 1 | |||||||||||||||
| Oktenli et al20 | 1 | 1 | 1 | 1 | Osteoporosis; spina bifida | |||||||||||||
| Venturin et al27 | 5 | 1 | 2 | 4 | 1 | Syndactyly; pes planus | ||||||||||||
| Kehrer‐Sawatzki et al14 | 3 | 2 | 1 | Camptodactyly | ||||||||||||||
| Our patients | 6 | 1 | 4 | 1 | 1 | 1 | Club foot; increased carrying angle; pectus carinatum | |||||||||||
| Total | 42 | 14 | 18 | 9 | 3 | 4 | 1 | 1 |
*Only those patients with some description of skeletal phenotype.
Table 5 Tumours in patients with NF1 microdeletions.
| No. of patients | Multiple SC neurofibromas | Plexiform neurofibromas | MPNST | Paraspinal neurofibromas | Optic glioma | Other | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Kayes et al7,13 | 5 | 5 | 5 | 1 | 1 | |||||||||
| Wu et al28 | 4 | 3 | 1 | 2 | Cerebellar neuroblastoma | |||||||||
| Leppig et al17* | 2 | 2 | 1 | 1 | ||||||||||
| Leppig et al16 | 6 | 5 | 2 | Peritoneal neurofibrosarcoma; cerebellar medulloblastoma | ||||||||||
| Tonsgard et al3 | 5 | 5 | 3 | |||||||||||
| Wu et al29 | 2 | 2 | 1 | |||||||||||
| Ainsworth et al8 | 3 | 2 | 1 | |||||||||||
| Van Asperen et al26; Cnossen et al4 | 5 | 4 | 1 | 1 | 1 | |||||||||
| Valero et al25 | 6 | 5 | 5‡ | 1 | 1 | Astrocytoma type I; malignant mediastinal neurilemmoma | ||||||||
| Upadhyaya et al24 | 18 | 3 | 2 | 1 | 1 | 1 | ||||||||
| Rasmussen et al21 | 5 | 4 | Hypothalamic glioma | |||||||||||
| Riva et al23 | 7 | 4 | 2 | |||||||||||
| Ruggieri et al 2000 | 1 | 1 | 1 | 1 | ||||||||||
| Lopez‐Correa et al18,19 | 20 | 14 | 2 | 3 | ||||||||||
| Dorschner et al10 | 9 | 8 | 5 | 1 | 1 | 2 | ||||||||
| Jenne et al12 | 1 | 1 | ||||||||||||
| Oktenli et al20 | 1 | 1 | ||||||||||||
| Venturin et al27 | 14 | 2 | 1 | Thalamic hamartoma | ||||||||||
| DeRaedt et al31† | 9 | 9 | ||||||||||||
| Kehrer‐Sawatzki et al14 | 16 | 13 | 2 | 2 | 1 | Astrocytoma | ||||||||
| Our patients | 6 | 5 | 1 | 1 | ||||||||||
| Total | 145 | 89 | 34 | 19 | 8 | 8 |
Of 157 patients, 42 (28%) (including our six newly reported patients) were reported to have significant skeletal anomaly, the most common of which were scoliosis (43%) and pectus excavatum (33%) (table 4). This is higher than the general NF1 population, in which scoliosis is seen in approximately 10% and pectus excavatum in approximately 20% of patients.34
Quantification of tumours was recorded for 145 of the 157 described patients with NF1 microdeletions. Of these, 89/145 (61%) showed multiple subcutaneous neurofibromas, while 34/141 (24%) had plexiform neurofibromas, which is similar to the 25–30%27 observed in the general population of patients with NF1. Age of development of neurofibromas was provided for only a few patients, thus we were unable to assess number of tumours corrected for age. However, the impression is that tumour load in these patients is very large and that tumours develop early. Development of MPNST was reported in 19/145 of patients, which slightly exceeds the 5–10% expected for the general NF1 population (table 5).
Finally, connective tissue abnormalities were described in 19 of the 157 patients (12%) (table 6). Six patients (including three from our group) had mitral valve prolapse.20,27,29 It is difficult to make a comparison between connective tissue phenotype observed in the general population of patients with NF1 and those patients with NF1 with microdeletions. Connective tissue involvement is indeed part of the classic NF1 phenotype, as scoliosis, bone dysplasia, and vascular anomalies are common features. The connective tissue phenotype observed in patients with NF1 microdeletions appears to be more pronounced such that it also includes joint laxity, hyperextensible skin, and mitral valve prolapse.
Table 6 Connective tissue dysplasia in patients with NF1 microdeletions.
| No. of patients* | Hyper‐extensible skin | Excessive wrinkled skin on palms | Deep palmar/plantar creases | Soft, fleshy palms/soles | Loose joints | MVP | Aortic dissection | Umbilical hernia | Inguinal hernia | Other | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Wu et al28 | 1 | Soft tissue hypertrophy (arm, back) | ||||||||||||||||||||
| Leppig et al17 | 1 | 1 | ||||||||||||||||||||
| Upadhyaya et al6 | 1 | 1 | ||||||||||||||||||||
| Leppig et al16 | 4 | 1 | 1 | 3 | 3 | 1 | Friable aorta; dislocation of radii | |||||||||||||||
| Wu et al29 | 1 | 1 | ||||||||||||||||||||
| Van Asperen et al26 | 2 | 1 | 1 | |||||||||||||||||||
| Ruggieri et al 2000 | 1 | Multiple coronary artery aneurysms | ||||||||||||||||||||
| Oktenli et al20 | 1 | 1 | 1 | |||||||||||||||||||
| Venturin et al27 | 2 | 1 | 1 | |||||||||||||||||||
| Our patients | 5 | 5 | 3 | 1 | 4 | 5 | 3 | 1 | ||||||||||||||
| Total | 19 | 6 | 4 | 2 | 7 | 11 | 6 | 1 | 2 | 1 |
*Number of patients with described connective tissue abnormality.
DISCUSSION
We present six newly diagnosed patients with NF1 and microdeletions encompassing the entire NF1 gene. All represent de novo cases of NF1. Five of these patients presented with symptoms of connective tissue abnormalities. Mitral valve prolapse +/− marfanoid body habitus was noted in three of these five patients. Two sets of parents were available for cytogenetic studies, and tested negative.
Despite a large number of patients with known NF1 microdeletions, the clinical phenotype and the correlation between size of deletion and clinical phenotype remain incompletely defined. This is partly due to a discrepancy between the amount of known clinical information and the molecular work performed on the same patients. Many reports with extensive molecular data contain very limited clinical information, and vice versa, making analysis of phenotype–genotype correlation difficult. Additionally, the review of the literature was complicated by description of the same patient in multiple reports. For example, the same patient is identified as UWA 1131,12 ULM,11 and 11311.10
The known clinical phenotype of patients with NF1 microdeletions includes early onset and large number of neurofibromas, presence of congenital anomalies, cognitive deficiency, variable dysmorphic characteristics, and growth abnormalities. Joint laxity, soft skin on the palms, and mitral valve prolapse have been reported previously in a few patients, but connective tissue abnormalities are not generally recognised as a part of NF1 microdeletion syndrome. Marked connective tissue abnormalities in our five patients confirms that soft tissue involvement is part of the NF1 microdeletion phenotype and raises the suspicion that this may be a more common association than previously thought. One of our patients developed the potentially life threatening complication of bacterial endocarditis secondary to mitral valve insufficiency. This is a preventable complication when the defect is appropriately diagnosed and treated.
Venturin et al27 recently provided phenotypic descriptions of 14 new patients and reviewed 78 previously reported patients. They showed that cardiac anomalies, learning disability with or without developmental delay, mental retardation, and dysmorphism are more frequent in patients with microdeletion than in patients with classic NF1. Based on deletion characterisation of 44 genetically and clinically diagnosed patients (from the literature and their 14 new patients), they proposed that haploinsufficiency of the OMG and/or CDK5R1 genes may be implicated in learning disability, and that the JJAZ1 and CENTA2 genes may be implicated in development of cardiovascular anomalies.
Our review of the literature describing the clinical phenotype of NF1 microdeletion revealed skeletal anomalies to be a relatively common clinical finding. The prior report by Venturin et al showed no statistical difference in frequency of scoliosis between microdeleted patients and patients with classic NF1. Oktenli et al20 recently described a patient with unusual ocular findings and multiple skeletal abnormalities including osteoid osteoma in ethmoid sinus, spina bifida, wide metaphyses, delayed bone age, osteoporosis, clinodactyly and abnormal body proportions with marfanoid body habitus. We found that 42 of 157 patients (including our six newly reported patients) with some phenotypic description reported, were found to have significant skeletal anomalies. A few patients had multiple skeletal anomalies.3,4,7,9,13,16,19,21,23,27,28 The most common bony anomalies in this group were scoliosis (18/42), pectus excavatum (14/42), and brachydactyly (9/42). Owing to inconsistency in clinical reports, one should interpret these numbers with caution. However, these clinical findings, when seen, may raise suspicion for NF1 microdeletion.
Like Venturin et al, we identified a higher prevalence of cognitive deficiency, learning disability, dysmorphic features, and skeletal anomalies in patients with microdeletions as compared with the general NF1 population. However, this comparison is inadequate, as statistics from the general NF1 population include patients with NF1 with microdeletions, and phenotypic information specific to the population of patients with NF1 with without such mutations is not available. Therefore, further studies of the clinical phenotype in conjunction with a clearly defined genotype will be required to define this correlation.
Even though most patients with NF1 microdeletion were found to have more complex clinical symptoms (“NF1 plus”), there is no clear genotype–phenotype correlation, even in cases with identical deletions.11,23 It could be expected that patients with mosaicism for NF1 microdeletion have an attenuated phenotype, as suggested by Kehrer et al.14 However, some reported mosaic patients have more severe phenotype including mental retardation,3,21 which may represent variability in degree and tissue distribution of NF1 microdeletion. New deletions are more commonly seen on the maternal allele.11,24 Some patients share common deletion breakpoints, resulting in the loss of at least 11 functional genes.
All six of the patients we report here had a similar de novo NF1 microdeletion, estimated to be 1.94 Mb in size. Five of these patients had prominent connective tissue symptoms, while one did not have any of these clinical findings. This observation may indicate that factors other than size of deletion have a role in phenotypic expression.
The available literature is deficient in both clinical and detailed molecular information of patients with large deletions, thus extracting specific information from the literature is difficult. Additional reports of patients with NF1 microdeletions, which include comprehensive clinical and molecular information, may shed light on possible genotype–phenotype correlation, point to genes responsible for connective tissue disorder in proximity to the NF1 gene, or lead to identification of other factors, perhaps modifier genes, which influence NF1 phenotype.
ACKNOWLEDGEMENTS
We thank D Johnson for assistance in preparation of the manuscript. We also extend deep appreciation to our patients for sharing their medical histories and photographs for medical publication.
Abbreviations
BAC - bacterial artificial chromosome
FISH - fluorescent in situ hybridisation
MPNST - malignant peripheral nerve sheath tumours
MRI - magnetic resonance imaging
NCBI - National Center for Biotechnology Information
NF1 - neurofibromatosis 1
PAC - P1 derived artificial chromosome
STS - sequence tagged site
UCSC - University of California Santa Cruz
Footnotes
Competing interests: there are no competing interests.
All photographs used in this manuscript were obtained and used only after written informed consent of patients authorising use for medical education and publication.
References
- 1.Friedman J M. Epidemiology of neurofibromatosis type 1. Am J Med Genet 1999891–6. [PubMed] [Google Scholar]
- 2.Kluwe L, Siebert R, Gesk S, Friedrich R E, Tinschert S, Kehrer‐Sawatzki H, Mautner V F. Screening 500 unselected neurofibromatosis 1 patients for deletions of the NF1 gene. Hum Mutat 200423111–116. [DOI] [PubMed] [Google Scholar]
- 3.Tonsgard J H, Yelavarthi K K, Cushner S, Short M P, Lindgren V. Do NF1 gene deletions result in a characteristic phenotype? Am J Med Genet 19977380–86. [DOI] [PubMed] [Google Scholar]
- 4.Cnossen M H, van der Est M N, Breuning M H, van Asperen C J, Breslau‐Siderius E J, van der Ploeg A T, de Goede‐Bolder A, van den Ouweland A M, Halley D J, Niermeijer M F. Deletions spanning the neurofibromatosis type 1 gene: Implications for genotype‐phenotype correlations in neurofibromatosis type 1? Hum Mutat 19979458–464. [DOI] [PubMed] [Google Scholar]
- 5.Lazaro C, Gaona A, Lynch M, Kruyer H, Ravella A, Estivill X. Molecular characterization of the breakpoints of a 12‐kb deletion in the NF1 gene in a family showing germ‐line mosaicism. Am J Hum Genet 1995571044–1049. [PMC free article] [PubMed] [Google Scholar]
- 6.Upadhyaya M, Roberts S H, Maynard J, Sorour E, Thompson P W, Vaughan M, Wilkie A O, Hughes H E. A cytogenetic deletion, del(17)(q11.22q21.1), in a patient with sporadic neurofibromatosis type 1 associated with dysmorphism and developmental delay. J Med Genet 199633148–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kayes L M, Riccardi V M, Burke W, Bennett R L, Stephens K. Large de novo DNA deletion in a patient with sporadic neurofibromatosis 1, mental retardation, and dysmorphism. J Med Genet 199229686–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ainsworth P J, Chakraborty P K, Weksberg R. Example of somatic mosaicism in a series of de novo neurofibromatosis type 1 cases due to a maternally derived deletion. Hum Mutat 19979452–457. [DOI] [PubMed] [Google Scholar]
- 9.Colley A, Donnai D, Evans D G. Neurofibromatosis/Noonan phenotype: A variable feature of type 1 neurofibromatosis. Clin Genet 19964959–64. [DOI] [PubMed] [Google Scholar]
- 10.Dorschner M O, Sybert V P, Weaver M, Pletcher B A, Stephens K. NF1 microdeletion breakpoints are clustered at flanking repetitive sequences. Hum Mol Genet 2000935–46. [DOI] [PubMed] [Google Scholar]
- 11.Jenne D E, Tinschert S, Stegmann E, Reimann H, Nurnberg P, Horn D, Naumann I, Buske A, Thiel G. A common set of at least 11 functional genes is lost in the majority of NF1 patients with gross deletions. Genomics 20006693–97. [DOI] [PubMed] [Google Scholar]
- 12.Jenne D E, Tinschert S, Dorschner M O, Hameister H, Stephens K, Kehrer‐Sawatzki H. Complete physical map and gene content of the human NF1 tumor suppressor region in human and mouse. Genes Chromosomes Cancer 200337111–120. [DOI] [PubMed] [Google Scholar]
- 13.Kayes L M, Burke W, Riccardi V M, Bennett R, Ehrlich P, Rubenstein A, Stephens K. Deletions spanning the neurofibromatosis 1 gene: Identification and phenotype of five patients. Am J Hum Genet 199454424–436. [PMC free article] [PubMed] [Google Scholar]
- 14.Kehrer‐Sawatzki H, Kluwe L, Sandig C, Kohn M, Wimmer K, Krammer U, Peyrl A, Jenne D E, Hansmann I, Mautner V F. High frequency of mosaicism among patients with neurofibromatosis type 1 with microdeletions caused by somatic recombination of the JJAZ1 gene. Am J Hum Genet 200475410–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korf B R, Schneider G, Poussaint T Y. Structural anomalies revealed by neuroimaging studies in the brains of patients with neurofibromatosis type 1 and large deletions. Genet Med 19991136–140. [DOI] [PubMed] [Google Scholar]
- 16.Leppig K A, Viskochil D, Neil S, Rubenstein A, Johnson V P, Zhu X L, Brothman A R, Stephens K. The detection of contiguous gene deletions at the neurofibromatosis 1 locus with fluorescence in situ hybridization. Cytogenet Cell Genet 19967295–98. [DOI] [PubMed] [Google Scholar]
- 17.Leppig K A, Kaplan P, Viskochil D, Weaver M, Ortenberg J, Stephens K. Familial neurofibromatosis 1 microdeletions: Cosegregation with distinct facial phenotype and early onset of cutaneous neurofibromata. Am J Med Genet 199773197–204. [DOI] [PubMed] [Google Scholar]
- 18.Lopez‐Correa C, Brems H, Lazaro C, Estivill X, Clementi M, Mason S, Rutkowski J L, Marynen P, Legius E. Molecular studies in 20 submicroscopic neurofibromatosis type 1 gene deletions. Hum Mutat 199914387–393. [DOI] [PubMed] [Google Scholar]
- 19.Lopez Correa C, Brems H, Lazaro C, Marynen P, Legius E. Unequal meiotic crossover: A frequent cause of NF1 microdeletions. Am J Hum Genet 2000661969–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oktenli C, Saglam M, Demirbas S, Thompson P, Upadhyaya M, Consoli C, Ulucan H, Koz C, Durukan A H, Bozkurt A, Koc B, Kocar I H, Gul D. A large deletion (1.5 Mb) encompassing the neurofibromatosis type 1 gene in a patient with sporadic NF1 associated with dysmorphism, mental retardation, and unusual ocular and skeletal features. Clin Dysmorphol 200312199–201. [DOI] [PubMed] [Google Scholar]
- 21.Rasmussen S A, Colman S D, Ho V T, Abernathy C R, Arn P H, Weiss L, Schwartz C, Saul R A, Wallace M R. Constitutional and mosaic large NF1 gene deletions in neurofibromatosis type 1. J Med Genet 199835468–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruggieri M, D'Arrigo G, Abbate M, Distefano A, Upadhyaya M. Multiple coronary artery aneurysms in a child with neurofibromatosis type 1. Eur J Pediatr 2000159477–480. [DOI] [PubMed] [Google Scholar]
- 23.Riva P, Corrado L, Natacci F, Castorina P, Wu B L, Schneider G H, Clementi M, Tenconi R, Korf B R, Larizza L. NF1 microdeletion syndrome: Refined FISH characterization of sporadic and familial deletions with locus‐specific probes. Am J Hum Genet 200066100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Upadhyaya M, Ruggieri M, Maynard J, Osborn M, Hartog C, Mudd S, Penttinen M, Cordeiro I, Ponder M, Ponder B A, Krawczak M, Cooper D N. Gross deletions of the neurofibromatosis type 1 gene are predominantly of maternal origin and commonly associated with a learning disability, dysmorphic features and developmental delay. Hum Genet 1998102591–597. [DOI] [PubMed] [Google Scholar]
- 25.Valero M C, Pascual‐Castroviejo I, Velasco E, Moreno F, Hernandez‐Chico C. Identification of de novo deletions at the NF1 gene: No preferential paternal origin and phenotypic analysis of patients. Hum Genet 199799720–726. [DOI] [PubMed] [Google Scholar]
- 26.Van Asperen C J, Overweg‐Plandsoen W C, Cnossen M H, van Tijn D A, Hennekam R C. Familial neurofibromatosis type 1 associated with an overgrowth syndrome resembling Weaver syndrome. J Med Genet 199835323–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Venturin M, Guarnieri P, Natacci F, Stabile M, Tenconi R, Clementi M, Hernandez C, Thompson P, Upadhyaya M, Larizza L, Riva P. Mental retardation and cardiovascular malformations in NF1 microdeleted patients point to candidate genes in 17q11.2. J Med Genet 20044135–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu B L, Austin M A, Schneider G H, Boles R G, Korf B R. Deletion of the entire NF1 gene detected by the FISH: Four deletion patients associated with severe manifestations. Am J Med Genet 199559528–535. [DOI] [PubMed] [Google Scholar]
- 29.Wu B L, Schneider G H, Korf B R. Deletion of the entire NF1 gene causing distinct manifestations in a family. Am J Med Genet 19976998–101. [PubMed] [Google Scholar]
- 30.Wu R, Lopez‐Correa C, Rutkowski J L, Baumbach L L, Glover T W, Legius E. Germline mutations in NF1 patients with malignancies. Genes Chromosomes Cancer 199926376–380. [PubMed] [Google Scholar]
- 31.De Raedt T, Brems H, Wolkenstein P, Vidaud D, Pilotti S, Perrone F, Mautner V, Frahm S, Sciot R, Legius E. Elevated risk for MPNST in NF1 microdeletion patients. Am J Hum Genet 2003721288–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eidem B W, Lindor N M, Driscoll D J. Resolution of neonatal hypertrophic cardiomyopathy in an infant with an affected mother. Pediatr Cardiol 199920208–211. [DOI] [PubMed] [Google Scholar]
- 33.North K. Neurofibromatosis type 1. Am J Med Genet 200097119–127. [DOI] [PubMed] [Google Scholar]
- 34.Friedman J, Guttman, MacCollin Neurofibromatosis: phenotype, natural history, and pathogenesis. 3rd ed. Baltimore: Johns Hopkins University Press, 1999
- 35.Leroy K, Dumas V, Martin‐Garcia N, Falzone M ‐ C, Voisin M ‐ C, Wechsler J, Revuz J, Creange A, Levy E, Lantieri L, Zeller J, Wolkenstein P. Malignant peripheral nerve sheath tumors associated with neurofibromatosis type I. Arch Dermatol 2001137908–913. [PubMed] [Google Scholar]
- 36.Kaplan P, Rosenblatt B. A distinctive facial appearance in neurofibromatosis von Recklinghausen. Am J Med Genet 198521463–470. [DOI] [PubMed] [Google Scholar]
- 37.Colman S D, Rasmussen S A, Ho V T, Abernathy C R, Wallace M R. Somatic mosaicism in a patient with neurofibramatosis type I. Am J Med Genet 199658484–490. [PMC free article] [PubMed] [Google Scholar]
- 38.Riva P, Castorina P, Manoukian S, Dalpra L, Doneda L, marini G, den Dunnen J, Larizza L. Characterization of a cytogenetic 17q11.2 deletion in an NF1 patient with a contiguous gene syndrome. Hum Genet 199698646–650. [DOI] [PubMed] [Google Scholar]
- 39.Wu B L, Boles R G, Yaari H, Weremowicz S, Schneider G H, Korf B R. Somatic mosaicism for deletion of the entire NF1 gene identified by FISH. Hum Genet 199799209–213. [DOI] [PubMed] [Google Scholar]