

Abstract
We have previously reported that a subunit protein vaccine based on the receptor-binding domain (RBD) of severe acute respiratory syndrome coronavirus (SARS-CoV) spike protein and a recombinant adeno-associated virus (rAAV)-based RBD (RBD-rAAV) vaccine could induce highly potent neutralizing Ab responses in immunized animals. In this study, systemic, mucosal, and cellular immune responses and long-term protective immunity induced by RBD-rAAV were further characterized in a BALB/c mouse model, with comparison of the i.m. and intranasal (i.n.) routes of administration. Our results demonstrated that: 1) the i.n. vaccination induced a systemic humoral immune response of comparable strength and shorter duration than the i.m. vaccination, but the local humoral immune response was much stronger; 2) the i.n. vaccination elicited stronger systemic and local specific cytotoxic T cell responses than the i.m. vaccination, as evidenced by higher prevalence of IL-2 and/or IFN-γ-producing CD3+/CD8+ T cells in both lungs and spleen; 3) the i.n. vaccination induced similar protection as the i.m. vaccination against SARS-CoV challenge in mice; 4) higher titers of mucosal IgA and serum-neutralizing Ab were associated with lower viral load and less pulmonary pathological damage, while no Ab-mediated disease enhancement effect was observed; and 5) the vaccination could provide long-term protection against SARS-CoV infection. Taken together, our findings suggest that RBD-rAAV can be further developed into a vaccine candidate for prevention of SARS and that i.n. vaccination may be the preferred route of administration due to its ability to induce SARS-CoV-specific systemic and mucosal immune responses and its better safety profile.
Since the outbreak of severe acute respiratory syndrome (SARS),3 an emerging infectious disease, in November 2002, SARS has claimed the lives of 774 among 8098 affected cases (www.who.int/csr/sars/country/Table XX03_09_23/en). SARS coronavirus (SARS-CoV) is the etiological pathogen of SARS (1-3). The development of SARS vaccines must remain a high priority due to the possibility of re-emergence of the disease (4-6).
Among four structural proteins encoded by SARS-CoV, spike protein (S protein) plays an important role in SARS-CoV infection (7-9). It interacts with the cellular receptor(s) to mediate membrane fusion, allowing viral entry into host cells (10, 11). The S protein is also a major inducer of neutralizing Abs (NA) and protective immunity which prevent SARS-CoV infection (12-14). Thus, SARS-CoV S protein is a key factor for developing SARS vaccines.
Several vaccine strategies proposed for prevention of SARS include inactivated virus-based vaccines (15), DNA-based vaccines (16), recombinant subunit vaccines (17), and viral vector-based vaccines (18). These vaccine candidates are able to induce protective immune responses against SARS-CoV, including NA and T cell immune responses (14, 19-21). A majority of these vaccines are based on the SARS-CoV S protein (22-25).
Recently reported SARS vaccines are based on the full-length or fragments of SARS-CoV S protein (20, 25, 26). They can effectively induce NA, cellular, and/or protective immune responses against SARS-CoV (16, 17, 22). However, some may cause liver damage in those vaccinated animals, in which SARS-CoV infection was not prevented by the mobilized immune responses (27, 28). Thus, vaccines encoding truncated fragments of SARS-CoV S protein may prove to be more promising. In this regard, the receptor-binding domain (RBD) of SARS-CoV S protein has been shown to harbor multiple conformation-dependent epitopes that induce highly potent NA responses, and vaccines based on the RBD elicit long-term protective immunity in immunized animals (29-32), suggesting that candidates based on the RBD can be developed into safe and effective SARS vaccines.
Recombinant adeno-associated virus (rAAV) has emerged as a promising viral vector for vaccine development. As a nonpathogenic parvovirus containing a ssDNA, AAV infects a wide variety of human cell lines, with long-term transgene expression and high transduction efficiency (33-35). The rAAV encoding different pathogenic Ags can induce vaccinated animals to produce strong immune responses via various delivery methods (36, 37). These appealing qualities have promoted vectors based on the rAAV to be widely used for vaccine development (38-40). Up to now, eight serotypes of AAV vectors have been described and characterized as vectors for gene therapy, among which the most extensively studied is serotype 2 (AAV-2) (41). In light of this, we used AAV-2 as a vector for delivery of SARS-CoV immunogen.
Our previous study has demonstrated that a rAAV expressing the RBD of SARS-CoV S protein (RBD-rAAV) elicited humoral immune response with neutralizing activity in i.m.-vaccinated BALB/c mice (42). In this study, we further investigated local and systemic immune responses and long-term protective immunity that might be induced by the RBD-rAAV vaccine via i.m. and intranasal (i.n.) administration routes. These two routes were also compared for immunogenicity and protection from SARS-CoV challenge.
Materials and Methods
Cell lines and animals
HEK293T cells, for packaging of RBD-rAAV recombinant viral vector, and Vero E6 cells, for neutralization assay, were purchased from American Type Culture Collection. Female BALB/c mice at the age of 4–6 wk were used for i.m. and i.n. vaccinations. All mice for the study were purchased from the Laboratory Animal Unit (University of Hong Kong). Animals were housed in the animal facility of the Department of Microbiology (University of Hong Kong), and maintained in accordance with the animal care protocol. All of the animal studies were approved by the Department of Health (Government of Hong Kong Special Administration Region).
Construction and titration of RBD-rAAV viral vector
The rAAV encoding a 193-aa RBD domain (residues 318–510) of SARS-CoV S protein (RBD-rAAV) was produced as described previously (42). Briefly, RBD-rAAV plasmid was cotransfected with pHelper and pAAV-RC plasmids into HEK293T cells using a calcium phosphate transfection method (Stratagene) according to the manufacturer's protocol. Transfected cells and supernatant were harvested 72 h posttransfection. rAAV was purified by chloroform-NaCl-PEG8000 method and titrated by real-time quantitative PCR (Q-PCR) following protocols described in a previous study (42). RBD-rAAV vector was adjusted to 1012 viral particles (VP)/ml in PBS and used for the following vaccinations.
Mice vaccination via i.m. and i.n. routes and sample collection
As shown in Table I, six groups of mice were vaccinated with RBD-rAAV or blank AAV, respectively, via the i.m. and i.n. routes, following the protocols described previously with some modifications (42-44). For the i.m. vaccination, BALB/c mice were given with a single prime dose (i.m.P) or prime-boost doses at 1.5-mo interval (i.m.B) of RBD-rAAV (2 × 1011 VP/200 μl/dose). For the i.n. vaccination, mice were immunized with a single prime dose (i.n.P) or prime-boost doses at an interval of 0.5 mo (i.n.B) of RBD-rAAV (2 × 1010 VP/20 μl/dose). Two groups of mice i.m. or i.n. vaccinated with prime-boost doses of blank AAV were used as negative controls. Samples were collected as shown in Fig. 1. Four mice per group were challenged with SARS-CoV 1 mo after the booster vaccination (young mice), and five mice/group were boosted at the end of 12 mo postvaccination and challenged with SARS-CoV 15 days later (aged mice).
Table I.
Number of mice vaccinated with RBD-rAAV (RBD) or blank AAV (AAV)a
| Vaccinations | Total (n) |
Humoral IR (n) |
Local IR (n) |
CTL IR (n) |
SARS-CoV (#) |
||
|---|---|---|---|---|---|---|---|
| Young | Aged | ||||||
| RBD | |||||||
| im.P | 28 | 5 | 4 | 5 | 4 | 5 | |
| im.B | 28 | 5 | 4 | 5 | 4 | 5 | |
| in.P | 14 | 5 | 4 | 5 | |||
| in.B | 62 | 5 | 4 + 39 | 5 | 4 | 5 | |
| AAV | |||||||
| im.B | 28 | 5 | 4 | 5 | 4 | 5 | |
| in.B | 62 | 5 | 4 + 39 | 5 | 4 | 5 | |
Five mice per group were monitored for systemic humoral immune response (IR; serum IgG and NA). Four mice per group were tested for local IR (lung flush IgA). Mice with i.n. prime-boost vaccination of RBD-rAAV or blank AAV (in.B, 39 mice/group) were used for detection of lung flush IgA and NA at different time points at 0.5-mo intervals for up to 6 mo, and three mice for each group were tested at each time point. n, The number of mice and SARS-CoV means challenged with SARS-CoV. im.P, in.P, and im.B indicate i.m. or i.n. immunized with a single prime dose, or i.m. prime-boost doses of RBD-rAAV or blank AAV, respectively.
FIGURE 1.
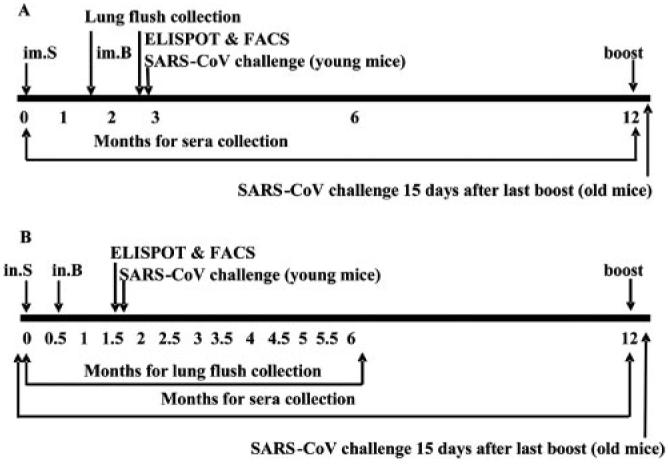
The immunization schedule of the RBD-rAAV vaccine is listed. Mice were i.m. (A) or i.n. (B) vaccinated with a single prime dose (im.P, in.P) or primer boost doses (im.B, in.B) of RBD-rAAV or blank AAV. Time points of sample collection or detection are indicated. FACS, flow cytometry analysis.
ELISA for systemic IgG and local IgA detection
Specific IgG and IgA against SARS-CoV in mouse sera and lung flush were tested by ELISA using the protocol described previously with some modifications (42). Briefly, serially diluted mouse sera were added to 96-well microtiter plates precoated with the protein mixture from SARS-CoV viral lysates. The plates were incubated at 37°C for 30 min, followed by four washes with PBS containing 0.1% Tween 20 (PBST). Bound Abs were then reacted with HRP-conjugated goat anti-mouse IgG (DakoCytomation) at 37°C for 20 min. After four washes, the substrate 3,3′,5,5′-tetramethylbenzidine (Zymed Laboratories) was added to the plates and the reaction was stopped by adding 1 N H2SO4. The absorbance at 450 nm was measured by an ELISA plate reader (Victor 1420 Multilabel Counter; PerkinElmer). In the case of IgA detection, collected mouse lung flush was added to precoated 96-well microtiter plates and incubated for 1 h. HRP-conjugated goat anti-mouse IgA Ab (Zymed Laboratories) was then added at a dilution of 1/1000 and incubated for 1 h, followed by measurement of absorbance at 450 nm.
Neutralization assay
Titers of NA in sera and lung flush of mice immunized with RBD-rAAV or blank AAV via i.m. and i.n. pathways were detected in Vero E6 cells as previously described (42). Briefly, Vero E6 cells were seeded at 104/well in 96-well culture plates and cultured at 37°C to form a monolayer. Serial 2-fold dilutions of serum samples were mixed separately with 100 TCID50 (50% tissue-culture infectious dose) of SARS-CoV strain GZ50 (GenBank accession no. AY304495), incubated at 37°C for 1 h, and added to the monolayer of Vero E6 cells in tetrad. Cells infected with 100 TCID50 SARS-CoV and without the virus were applied as positive and negative controls, respectively. The cytopathic effect (CPE) in each well was observed daily and recorded on day 3 postinfection. The neutralizing titers of mouse antisera and lung flush that completely prevented CPE in 50% of the wells were calculated by the Reed-Muench method.
IL-2 and IFN-γ ELISPOT assay
The assay was performed using an ELISPOT mouse kit (Mabtech) according to the manufacturer's protocol and our previous work (45). In brief, 96-well ELISPOT plates were coated with anti-IL-2 and -IFN-γ mAbs overnight at 4°C, and blocked by sterile RPMI 1640 containing 10% FBS for 2 h at room temperature. Single-cell suspensions prepared from the spleens of vaccinated mice were added to the wells at the concentration of 2 × 105 cells/well. Cells were incubated for 24 h in the presence or absence of an identified MHC-H-2d-restricted SARS-CoV-specific CTL peptide (N50: S365–374, KCYGVSATKL) (46) plus anti-mouse CD28 mAb (1 μg/ml; BD Pharmingen) at 37°C with 5% CO2. Plates were washed with PBS, followed by incubation with biotinylated-labeled anti-mouse IL-2 and IFN-γ mAbs at 1/1000 for 2 h at room temperature. After additional washes, wells were incubated with streptavidin-conjugated HRP for 1 h at room temperature. Wells were extensively washed again, and developed with 3,3′,5,5′-tetramethylbenzidine substrate solutions included in the kit. Spots of IL-2 and IFN-γ-producing T cells were counted by using an automated ELISPOT reader system and ImmunoSpot 3 software (Cellular Technology). Results were expressed as the number of spot-forming cells (SFC) per 106 input cells.
Cell surface markers/intracellularr cytokine staining and FACS
Single-cell suspensions (2 × 106) from spleens and lungs of vaccinated mice were stimulated with or without SARS-CoV S-specific CTL peptide (N50, 1 μg/ml) plus anti-mouse CD28 (1 μg/ml). PMA (5 ng/ml; Sigma-Aldrich) and ionomycin (250 ng/ml; Sigma-Aldrich) were used as positive controls. Cells with stimulatory agents were incubated for 5 h at 37°C with 5% CO2 in the presence of GolgiPlug containing brefeldin A (1 μl/ml; BD Pharmingen). The cells were fixed using a Cytofix/Cytoperm Plus kit in accordance with the manufacturer's protocol (BD Pharmingen), and stained directly with conjugated mAbs specific for cell surface Ags (anti-mouse-CD3 (PerCP) and anti-mouse-CD8 (allophycocyanin)) and intracellular cytokines (anti-mouse-IL-2 (PE) and anti-mouse-IFN-γ (FITC; BD Pharmingen)) for 30 min at 4°C. Appropriate isotype-matched controls for cytokines were included in each staining. The stained cells were analyzed using a flow cytometer (FACSCalibur; BD Biosciences). Lymphocyte population was gated by forward light scatter vs side light scatter, and 10,000 events for the CD3+/CD8+ lymphocyte subpopulation were acquired to determine the percentage of CD3+/CD8+ T cells positive for specific cytokines. FACS data were analyzed by CellQuest software (BD Biosciences).
SARS-CoV challenge in mice
Mice were anesthetized with isoflurane and i.n. inoculated with 50 μl of SARS-CoV strain GZ50 (100 TCID50) according to national animal care and use guidelines in an approved animal BSL-3 laboratory. The mice were sacrificed 3 days (for young mice) or 8 days (for aged mice) after virus challenge, and the lungs were removed. The lung tissues were stored at −80°C for virological tests or were fixed immediately with 10% buffered formalin for histopathological analysis.
Quantitative RT-PCR
The viral RNA copies in lung tissues of challenged mice were determined by quantitative RT-PCR according to the protocol described previously with some modifications (42, 47). Briefly, total RNA was extracted from 20 mg of lung tissue using an RNeasy Mini kit (Qiagen). Then cDNA was synthesized using random primers and the SuperScript II RT kit (Invitrogen Life Technologies). Extracted RNA (10 μl) was reverse transcribed in a 20-μl reaction mixture containing 1× first strand buffer, 100 mM DTT, 10 mM each dNTP, 50 ng of random primers, 40 U of RNaseOUT, and 200 U of SuperScript II RT at 42°C for 50 min, followed by 15 min at 70°C. The solution was incubated with RNase H (Invitrogen Life Technologies) at 37°C for 20 min. Synthesized cDNA was quantified using Power SYBR Green PCR Master Mix (Applied Biosystems) in a 20-μl mixture containing 5 μl of cDNA (1/10), 10 μl of 2 × Power SYBR Green PCR Master Mix, 3 μl of RNase-free H2O, 10 μM forward primer F (5′-GCT TAG GCC CTT TGA GAG AGA CA-3′) and reverse primer R (5′-GCC AAT GCC AGT AGT GGT GTA A-3′) in a Mx3000 QPCR System (Stratagene).
Histopathological analysis
The lung tissues of challenged mice were immediately fixed in 10% buffered formalin and embedded in paraffin wax. Sections were made of 4- to 6-μm thickness and mounted on slides. Histopathological changes caused by SARS-CoV infection were examined by H&E staining and viewed under the light microscope as described previously (48, 49).
Statistical analysis
Values were presented as mean with SE. Statistical significance among different vaccination groups was calculated by the Student t test using Stata statistical software. Values of p < 0.05 were considered significant.
Results
Intranasal vaccination induced a shorter-duration systemic humoral immune response but a stronger and prolonged mucosal IgA response than i.m. vaccination
To evaluate the long-term systemic humoral immune response to RBD-rAAV vaccination, and to compare the differences between immune responses to vaccination via i.m. and i.n. routes, serum samples collected from vaccinated mice at different time points were detected by ELISA for specific IgG Ab to SARS-CoV. As shown in Fig. 2A, a single prime dose i.m. vaccination of RBD-rAAV (RBD.im.P) induced a moderate level of specific IgG and sustained this during the 12-mo observation period, while i.m. prime-boost immunization of RBD-rAAV (RBD.im.B) induced a high level of IgG Ab response, which reached the peak within 3 mo, maintained the plateau level for 3 more months, and gradually decreased to a moderate level at 12 mo postimmunization. A single prime dose i.n. vaccination of RBD-rAAV did not induce significant Ab response (data not shown). After booster (RBD.in.B), the vaccination quickly elicited a high level of IgG Ab response, reaching the highest titer 1 mo postvaccination, which was almost the same level as that of RBD.im.B. between months 3 and 6. However, the IgG Ab level also dropped down to a low level a month later and was maintained at a similar level thereafter. NA levels in these serum samples were further detected by neutralization assay using SARS-CoV, which showed a similar pattern as that of the IgG Ab responses (Fig. 2B). These results indicated that i.n. vaccination induced a similar NA level but shorter duration systemic humoral immune response than i.m. immunization.
FIGURE 2.
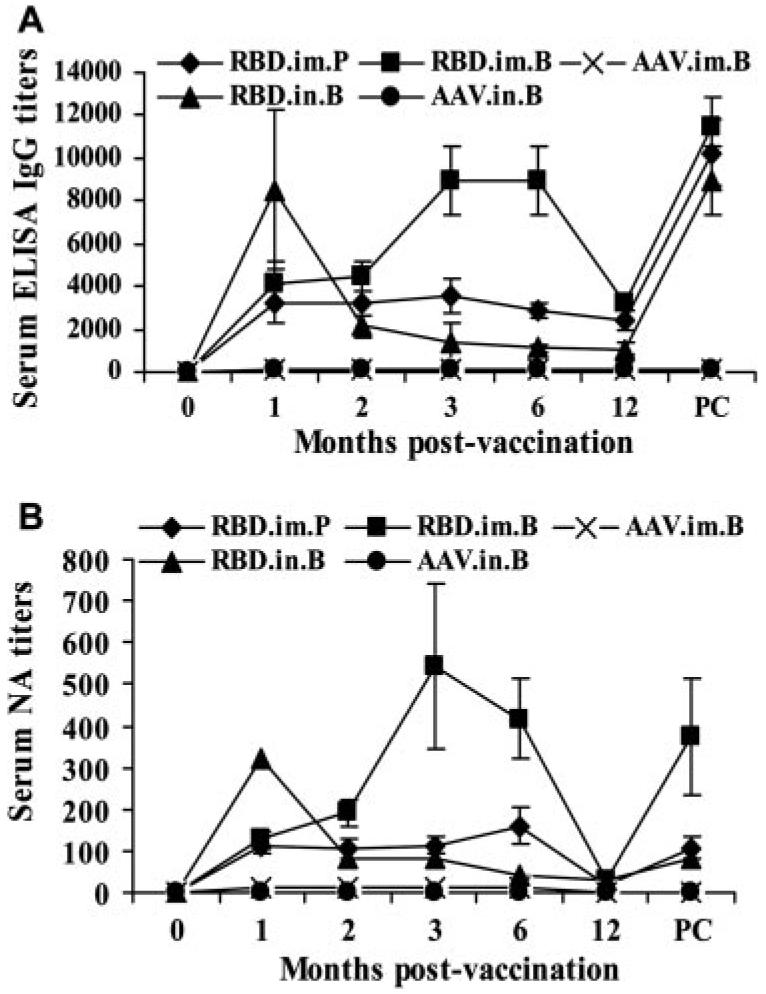
RBD-rAAV vaccinations induced long-term systemic humoral immune responses with neutralizing activity. Mice were i.m. immunized with a single prime dose of RBD-rAAV (RBD.im.P), i.m. (RBD. im.B), and i.n. prime-boost doses (RBD.in.B) of RBD-rAAV or blank AAV (AAV.im.B and AAV.in.B). The vaccinated mice were observed for a period of 12 mo. All mice were boosted again at 12 mo postvaccination. A, SARS-CoV-specific IgG in serum samples detected by ELISA. IgG were reported at 0, 1, 2, 3, 6, and 12 mo postvaccination and prechallenge (PC). The data are presented as mean ± SE of five mice per group. B, Serum NA titers measured by neutralization assay. The titers were determined as the highest dilutions of sera that could completely prevent CPE in at least 50% of the wells and presented as mean ± SE of five mice per group.
To assess the ability of i.n. vaccination to induce local immune response, mucosal IgA SARS-CoV-specific Ab was further detected by ELISA in the lung flush of vaccinated mice. As shown in Fig. 3A, RBD-rAAV i.n. prime boost (RBD.in.B) induced strong IgA Ab response, which was significantly higher than that elicited by RBD-rAAV i.m. with a single prime dose (RBD.im.P) and prime-boost doses (RBD.im.B), respectively (p = 0.004). Compared with RBD-rAAV, blank AAV (AAV.im.P, AAV.im.B) did not elicit detectable IgA Ab in lung flush (OD450 < 0.05). These data indicated that the i.n. rather than i.m. vaccination route could induce strong mucosal immune response. Titers of IgA Ab and NA induced by RBD-rAAV i.n. prime boost in mouse lung flush were further analyzed by ELISA and neutralization assay at 0.5-mo intervals. It was shown that the mucosal IgA Ab level reached its peak at 1 mo postvaccination, and gradually decreased to a low level in the following 5 mo (Fig. 3B). Nevertheless, the blank AAV control did not induce detectable IgA Ab during the detection period of 6 mo. Strikingly, the lung flush from RBD-rAAV i.n. prime-boost-vaccinated mice (RBD.in.B) contained high-level and long-lasting NA against SARS-CoV, which was highly detectable during the detection period of 6 mo, even though the sample had been diluted 1000 times in PBS during the process of sample collection (Fig. 3C). By comparison, no NA was detected from mouse lung flush samples of i.n. prime boost of blank AAV (AAV.in.B). The above data indicated that i.n. vaccination of RBD-rAAV induced a long-term mucosal immune response with neutralizing activity, implying that mucosal vaccination with RBD-rAAV should provide effective protective immune response against SARS-CoV.
FIGURE 3.
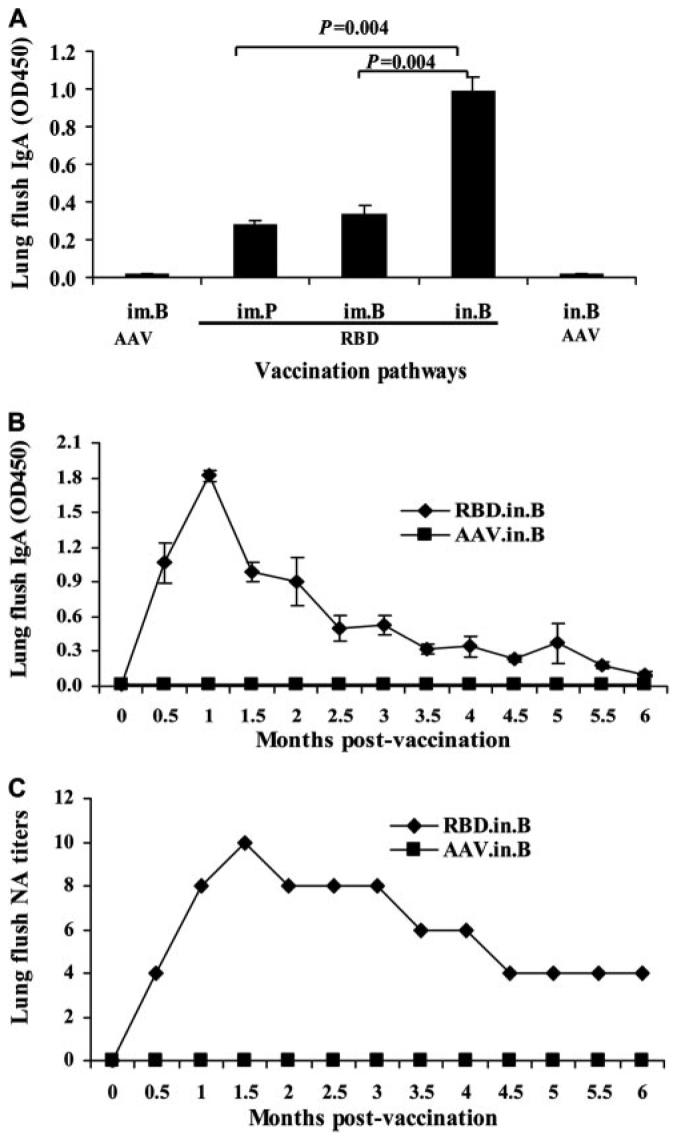
RBD-rAAV i.n. vaccination induced long-term local humoral immune responses with neutralizing activity. A, Comparison of mucosal IgA in i.m.- and i.n.-vaccinated mice. Four mice of each group were sacrificed at 1.5 mo postvaccination and lung flush was examined by ELISA for detection of IgA Ab. The data are presented as mean OD450 + SE of four mice per group. B, Monitoring of mucosal IgA Ab levels in mice i.n. vaccinated with RBD-rAAV. Three mice of each group were sacrificed at 0.5-mo intervals and lung flush was examined by ELISA for detection of IgA Ab. The data are presented as mean OD450 ± SE of three mice per group. C, Detection of NA titers in lung flush of i.n.-vaccinated mice. The NA titers were determined in pooled lung flush from three mice per group at each time point by neutralization assay.
For all vaccination groups, although IgG Abs had dropped down to low levels at 12 mo postvaccination, it rebounded quickly when the mice were reboosted (Fig. 2). These results suggested that RBD-rAAV may induce long-term memory immune responses, especially after booster immunization, by both i.m. and i.n. routes.
Intranasal vaccination induced strong CTL responses in spleen and lungs
To examine CTL responses induced by RBD-rAAV vaccination, splenocytes and lung lymphocytes were measured by ELISPOT and FACS. As shown in Fig. 4, i.n. vaccination of RBD-rAAV (RBD.in.B) induced a markedly higher level of Agspecific IL-2+ T cells but a slightly lower level of IFN-γ+ T cells in the spleen, as compared with those from the i.m. vaccination group (RBD.im.B). Nevertheless, splenocytes from mice receiving i.n. or i.m. vaccinations of blank AAV did not show Ag-specific CTL responses, resembling the negative controls that were significantly less responsive than RBD-rAAV vaccination groups (p < 0.05). In contrast, single dose i.m. or i.n. vaccination with RBD-rAAV did not induce significant IL-2+ and IFN-γ+ T cell response (data not shown), suggesting that booster immunization is necessary for inducing Ag-specific CTL response. The above data showed that i.n. vaccination could induce much stronger systemic IL-2+ CTL response than i.m. vaccination, while IFN-γ+ CTL response elicited by i.m and i.n. routes was of comparable strength. Specific CTL responses induced by RBD-rAAV vaccinations were further evaluated in the mouse splenocytes and lung lymphocytes by cell surface marker and intracellular cytokine staining followed by FACS. As shown in Fig. 5, RBD-rAAV i.n. vaccination (RBD. in.B) induced a markedly higher frequency of IL-2+ cells in the CD3+/CD8+ T cell population in both splenocytes and lung cells, as compared with RBD-rAAV i.m. vaccination (RBD. im.B). In addition, IFN-γ-producing CD3+/CD8+ T cells were significantly higher in splenocytes of RBD-rAAV i.n.-vaccinated vs i.m.-vaccinated mice, but were similar or slightly lower in lung lymphocytes of i.n.-vaccinated vs i.m.-immunized mice. However, a single prime dose i.m. and i.n. vaccinations with RBD-rAAV merely yielded low or undetectable levels of IL-2+ and IFN-γ+ CTL responses (data not shown). These results demonstrated that both i.m. and i.n. vaccination with RBD-rAAV could induce SARS-CoV specific CTL responses, and the i.n. route elicited higher systemic (in splenocytes) and local (in lungs) CTL responses than the i.m. route.
FIGURE 4.
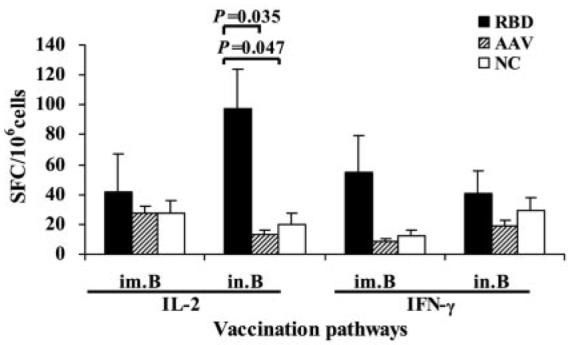
SARS-CoV-specific IL-2 and IFN-γ-producing T cells were detected by ELISPOT. Splenocytes from vaccinated mice were stimulated with SARS-CoV S-specific CTL peptide and anti-CD28 mAb for 24 h. Anti-CD28 alone was applied as the negative control (NC). Frequencies of cytokine-producing cells are expressed as mean + SE of cytokine SFC/106 cells of five mice per group.
FIGURE 5.

SARS-CoV specific CTL responses were further detected by cell surface marker and intracellular cytokine staining followed by FACS. IL-2 and IFN-γ-producing CD3+/CD8+ T lymphocytes from the spleen (A) and lung (B) were stimulated by SARS-CoV S-specific CTL peptide. Anti-CD28 alone was applied as the negative control (NC). The graphs are presented as mean value of five mice for each group. Numbers in the upper right corner of each graph represent the frequencies of IL-2 or IFN-γ-producing CD3+/CD8+ T cells. The data are determined by the isotype control and those showing significant increase are highlighted in bold.
RBD-rAAV vaccination suppressed SARS-CoV replication in mouse lungs
The protective efficacies of the vaccinations were further investigated in the mice challenged with 100 TCID50 of SARS-CoV strain GZ50. Mice were sacrificed 3 days postchallenge, and virus replication was assessed by viral load in challenged mouse lung tissue by Q-RT-PCR. Fig. 6 shows that viral loads (RNA copies/μg of lung tissues) in all mice immunized with RBD-rAAV were significantly lower than that of the corresponding control group immunized with blank AAV via i.m. and i.n. routes (p < 0.05), indicating that SARS-CoV replication was suppressed in vaccinated mice.
FIGURE 6.
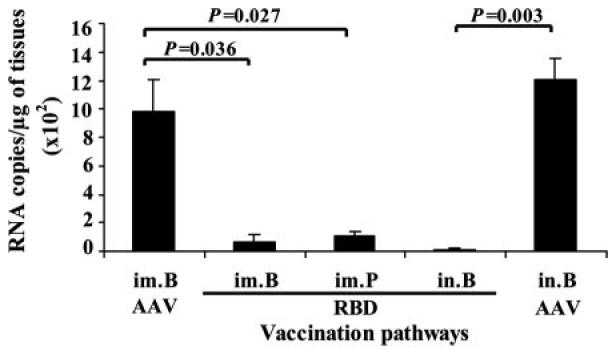
Viral load in lung tissues of challenged mice was detected by Q-RT-PCR. Viral titers of SARS-CoV in lung tissues from mice i.m. or i.n. vaccinated with a single prime dose (im.P) or prime-boost doses (im.B, in.B) of RBD-rAAV were determined. Mice i.m. and i.n. vaccinated with blank AAV were used as negative controls. The data are expressed as mean + SE of RNA copies per microgram of lung tissue from four mice for each group.
Correlation of serological data with virus protection
To understand the relationship between immune responses, vaccination pathways, and virus protection, mouse sera were collected before virus challenge to detect serum-specific IgG Ab levels and NA activities. Lung flush from corresponding mice was also collected for detecting specific IgA Ab. It was shown in Table II that there were clear correlations among the levels of SARS-CoV-specific serum IgG Ab, lung flush IgA Ab, NA, and the protection against i.n. virus challenge with live SARS-CoV. In general, a higher serum IgG titer correlated with a higher NA titer, resulting in a higher protection from virus challenge. For example, i.m. prime boost of RBD-rAAV (RBD.im.B) induced a higher serum IgG titer of 8.0 ± 1.6 × 103 and a higher NA titer of 3.7 ± 1.4 × 102 at the time of virus challenge, accompanied by a lower viral load of 0.6 ± 0.6 × 102 detected in the mouse lung tissue after challenge. In contrast, i.m. single prime dose of RBD-rAAV (RBD. im.P) elicited a lower serum IgG titer (3.2 × 103) and a lower NA titer (1.2 ± 0.4 × 102), leading to a higher virus replication (1.1 ± 0.2 × 102) in the mouse lung tissue. However, IgA produced in mouse lungs in i.n.-vaccinated mice (RBD.in.B) could also play a part in suppressing SARS-CoV replication, even though serum IgG Ab or NA levels were lower than that of the i.m.-vaccinated mice. For instance, RBD.in.B induced a much higher titer of IgA in mouse lungs, but lower serum IgG Ab and NA titers than RBD.im.B, while virus replication in i.n. prime boost (0.5 ± 0.2 × 102) was lower than in i.m. prime boost. These data indicated that both mucosal- (local) and serum- (systemic) specific Abs, especially NAs, could provide some protection for vaccinated mice from subsequent virus challenge, while mucosal immune response was indispensable for controlling SARS-CoV infection.
Table II.
Correlation of serum IgG, serum NA, mucosal IgA, and virus protectiona
| Serum IgG |
Serum NA |
Lung Flush |
Viral RNA Copies |
||
|---|---|---|---|---|---|
| Group | Vaccinations | Titer (×103) | Titer (×102) | IgA Titer (OD450) | (×102)/μg of Tissues |
| RBD | im.P | 3.2 | 1.2 ± 0.4 | 0.20 ± 0.03 | 1.1 ± 0.2 |
| im.B | 8.0 ± 1.6 | 3.7 ± 1.4 | 0.31 ± 0.05 | 0.6 ± 0.6 | |
| in.B | 4.8 ± 0.9 | 1.9 ± 0.7 | 1.00 ± 0.07 | 0.5 ± 0.2 | |
| AAV | im.B | <0.1 | <0.05 | <0.05 | 4.8 ± 1.4 |
| in.B | <0.1 | <0.05 | <0.05 | 12.0 ± 1.0 |
Mice were i.m. or i.n. vaccinated and/or boosted with RBD-rAAV. Serum IgG and NA titers of vaccinated mice (four mice per group) were recorded before SARS-CoV challenge. Corresponding lung flush was also collected for IgA detection. Results of IgG, IgA, and NA are presented as mean ± SE of four mice per group. Viral loads in lungs of challenged mice are expressed as mean ± SE of RNA copies per microgram of lung tissue of four mice per group.
RBD-rAAV vaccination provided long-term protection against SARS-CoV challenge
To detect the long-term protective effect of the candidate vaccine against SARS-CoV infection, mice (five mice per group) were boosted with RBD-rAAV 12 mo after the first RBD-rAAV immunization, and challenged with 100 TCID50 of SARS-CoV. Challenged mice were sacrificed 8 days postchallenge for examination of histopathological changes. Serious pulmonary interstitial pneumonias were observed in the lung tissues of all control mice vaccinated with blank AAV after SARS-CoV challenge (Fig. 7A). The lung showed broadening interstitial spaces, focal fusions with some alveolar compensatory expansion, pulmonary vascular dilatation and congestion, focal hemorrhage and exudation, scattered lymphocytic infiltration, especially perivascular infiltration. Focal desquamation of epithelial cells into alveolar spaces was found with scattered RBC and variable numbers of macrophages. Multinucleate giant cells were found in the alveoli and pulmonary interstitial space. Bronchial epithelial showed cytopathic effect, including necrosis and desquamation with a small amount of exudation and lymphocytic infiltration. In contrast, mice that had received RBD-rAAV vaccination showed no significant pulmonary effect after virus challenge. Three mice (three of five) in i.m. single prime dose and four mice (four of five) in i.m. or i.n. prime-boost doses presented normal lung structures or developed slightly interstitial pneumonia in the lung tissues with occasionally lymphocytic infiltration. Other mice in RBD-rAAV-vaccinated groups developed mild pulmonary interstitial pneumonia compared with those of the control AAV group (Fig. 7B). The above results demonstrated that RBD-rAAV vaccinations lessened the alveolar damage of challenged mouse lungs, and provided long-term protective immunity to prevent vaccinated mice from SARS-CoV infection.
FIGURE 7.

Mouse lung tissues infected with SARS-CoV collected 8 days postinfection were detected for histopathological changes. All sections of mouse lung tissues were stained with H&E and examined under the microscope (original magnification, ×100). A, Representative images of histopathological damage of lung tissue from the control mice administered with blank AAV in either i.m. (C1 and C2) or i.n. (C3 and C4) boost vaccinations. These control mice developed interstitial pneumonia. Predominantly infiltrating lymphocytes and mononuclear cells were identified around small blood vessels (open arrow). Pulmonary vascular peripheral lymphocyte infiltration was also shown, with bronchial epithelial cell degeneration, necrosis, desquamation (solid arrow), broadening interstitial spaces, and exudation. B, Representative images of histopathological changes of lung tissue from mice i.m. vaccinated with a single prime dose (M1) or prime-boost doses (M2), and i.n. vaccinated with prime-boost doses (M3 and M4) of RBD-rAAV. The lung tissue from a single prime dose i.m.-vaccinated mice showed mild interstitial pneumonia change with focal broadening interstitional spaces and lymphocytic infiltration (arrowhead) (M1). Mice with i.m. prime-boost vaccinations developed slightly interstitial pneumonia with normal alveolar, slightly widened pulmonary interval, and small lymphocytic infiltration (M2). Mice with i.n. prime-boost vaccinations showed almost normal vascular structure, bronchiole, alveolar, and alveolar lung spacing (M3 and M4).
Discussion
Development of effective vaccines against SARS-CoV is crucial in the prevention of recurrence of SARS. Currently reported candidate SARS vaccines are able to induce cellular, humoral immune responses and/or provide protective immunity against SARS-CoV infection, but they may have unfavorable features. Inactivated SARS-CoV-based vaccines, for example, might have the possibility of recovering virulence or causing accidental infection because of the incomplete inactivation of SARS-CoV, raising great concerns about the safety and applicability of this vaccine candidate when produced in a large quantity (50). DNA vaccines may cause toxicity to the injection sites when repeated doses were used (51). Protein-based vaccines are mainly Ab responses which have to be induced with the help of adjuvant, with weak or no cellular or mucosal immune responses (32, 52). Some viral vector-based vaccines, such as adenovirus or rMVA vectors, may have pre-existing immunity (28) or cause harmful immune responses (53) in vaccinated animals.
The rAAV vector has been recently applied for delivering vaccine Ags of various pathogens, with production of specific serum and mucosal Abs without the help of an adjuvant (38, 54). It is the only nonpathogenic viral vector now available and has been used successfully to establish long-term gene expression without toxicity in both dividing and nondividing cells (37). In addition, rAAV has the capacity of highly efficient transduction of target cell types such as muscles (33, 55) and low intrinsic adjuvant properties. These features have placed rAAV in a unique position over other contemporary candidates in SARS vaccine development.
Our previous study has shown that RBD-rAAV vaccination can elicit high humoral immune response with neutralizing activities through the i.m. route (42). This study was designed to further determine whether this candidate vaccine can provide protective immunity by different vaccination pathways, which pathway provides better protection, as well as record any side effects the vaccination may have. Thus, we compared the systemic, local humoral and cellular immune responses of BALB/c mice vaccinated with RBD-rAAV via i.m. and i.n. routes, and challenged the vaccinated animals with SARS-CoV to investigate the protective immunity and potential side effects.
Compared with i.m. vaccination, a single prime dose i.n. vaccination with RBD-rAAV could not induce detectable systemic humoral immune response (results not shown). After booster immunization, however, i.n. vaccination induced systemic Ab response of a similar level with shorter duration (Fig. 2), but much stronger and prolonged (lasting at least 5 mo) mucosal IgA Ab response with neutralizing activity (Fig. 3). One advantage of using viral vectors to deliver vaccine candidates is that the live viral vectors may induce strong mucosal humoral immune response (56), which may not be achieved by other types of vaccine candidates, such as protein and DNA vaccines (57, 58). Because the respiratory tract is the natural infection site of SARS-CoV, RBDrAAV vaccination of the lung via the i.n. route may play an important role for prevention of SARS-CoV infection by inducing a high level of IgA Ab with neutralizing activity. This has been confirmed in our study. Our results showed that the protective efficacy of RBD-rAAV vaccination against SARS-CoV infection is correlated with the Ab level, especially lung IgA Ab level (Table II). Although the i.n. vaccination induced lower systemic Ab responses than the i.m. vaccination, it provided higher protection against virus challenge (Fig. 6).
AAV-based vaccines have been shown to be able to induce both strong humoral and cell-mediated immunity (37). Our study also demonstrated that RBD-rAAV vaccination can induce not only strong humoral but also strong CTL responses. Surprisingly, the i.n. vaccination of RBD-rAAV also elicited stronger specific CTL responses, as indicated by higher frequencies of IL-2 and/or IFN-γ-producing CD3+/CD8+ cells not only in the lung but also in the spleen, than the i.m. vaccination (Fig. 5). It is well-known that cellular immune responses, especially CTL responses, play an important role in antiviral immunity (59, 60). It has been further reported that pulmonary T cell immunity is important in protecting naive natural hosts against pulmonary viral infections (61). Thus, higher frequencies of SARS-CoV-specific CTL induced by the i.n. vaccination, especially those pulmonary CTL, may also contribute to higher protective efficacy mediated by the i.n. immunization. However, potential functional differences between IL-2 and IFN-γ-producing CTL in suppressing SARS-CoV infection are unclear and should be further investigated.
Although humoral responses induced by RBD-rAAV vaccination dropped down to very low levels at 12 mo postvaccination, the Ab levels increased quickly after the animals received booster immunization, reaching the highest level (Fig. 2), which provided a potent protection against SARS-CoV challenge in the animals (Fig. 7). This may be attributed to the low antigenicity of the AAV vector itself, which does not induce significant immunity against AAV to interfere with the booster immunization, and administrated with AAV-based vaccines via the i.n. route may ward off the humoral immune response against AAV capsid proteins (62). In contrast, for other viral vectors with high antigenicity, such as adenovirus, the immune effects of vaccination may be significantly affected by the pre-existing immunity against adenovirus acquired through either natural infection or primary vaccination (53, 63).
It should be noted that some viral vector-based SARS vaccine candidates might have a harmful impact or side effects. For example, rMVA-based SARS vaccine candidate has been shown to produce strong Ab-mediated disease enhancement (ADE) effects, in which NA induced by SARS-CoV S protein did not protect ferrets from SARS-CoV challenge, but increased viral replication, or inflammatory responses (27, 28). Our study showed that both i.n. and i.m. vaccinations with RBD-rAAV did not cause ADE. In contrast, the higher IgG/IgA Ab and NA levels were associated with lower viral replication (Fig. 6, Table II) and less pathological damage (Fig. 7).
It has been reported that the wild-type AAV DNA is able to integrate into the human genome at specific sites, preferentially on chromosome 19q, and rAAV vectors may integrate randomly into nonchromosome 19q locations, although with low frequency (64). In this regard, mucosal immunization offers greater advantage in preventing potential long-term side effects which may be induced by i.m. vaccination of the rAAV vector, because the regenerated AAV-gene integrated surface mucosal cells are rapidly replaced by basal cells from mucosa.
As a vaccine vector for delivering Ags of various pathogens, rAAV has the characteristics of eliciting specific serum and mucosal Abs without the help of an adjuvant (38, 54). Likewise, rAAV vector expressing the RBD of SARS-CoV S protein was able to induce serum IgG and/or mucosal IgA immune responses as well as protection against SARS-CoV infection in the established mouse model. The major limitation of the rAAV vector is its inability to package DNA inserts >4.7 kb (65). However, for a RBD-based vaccine, only an insert of <1.0 kb is needed to be packaged in the RBD-rAAV vector. Therefore, the function of the RBD-rAAV vector is not affected by the insert size limitation. Further studies are warranted to determine the immune responses of RBD-rAAV in the presence of long-term RBD Ag expression and using different vaccination regimens, e.g., priming with DNA vaccine expressing the RBD and boosting with RBD-rAAV, as the adenovirus vector-based vaccine strategies (66).
Taken together, our study demonstrated that i.n. vaccination with RBD-rAAV can induce systemic humoral immune response of comparable strength but shorter duration, much stronger local humoral immune response, and stronger systemic and pulmonary CTL response, as compared with i.m. vaccination. The immune responses elicited by i.n. route can provide similar protection as i.m. route against SARS-CoV challenge in vaccinated mice. It is well-known that SARS is a pulmonary infection and the respiratory tract is the portal of entry for SARS-CoV. As the first line of defense to combat respiratory tract pathogens, the i.n. vaccination pathway can induce both local and systemic immune responses (54). Furthermore, it has also been demonstrated that this vacci-nation strategy did not cause ADE in the animals, and the risk of long-term side effects potentially resulting from the integration of the AAV gene into the host chromosome might also be minimized. Therefore, compared with the i.m. route, i.n. vaccination with RBD-rAAV may fulfill multiple criteria for an effective and safe SARS-CoV vaccine.
Footnotes
This work was supported by the Research Fund for the Control of Infectious Diseases, the Health, Welfare and Food Bureau of the Hong Kong Special Administrative Region government; by the National 973 Basic Research Program of China (2005CB523001); and by the National Institutes of Health of the United States (RO1 AI68002).
Abbreviations used in this paper: SARS, severe acute respiratory syndrome; SARS-CoV, SARS coronavirus; S protein, spike protein; NA, neutralizing Ab; RBD, receptor-binding domain; rAAV, recombinant adeno-associated virus; i.n., intranasal; TCID50, 50% tissue culture infectious dose; VP, viral particle; CPE, cytopathic effect; SFC, spot-forming cell; Q-RT-PCR, quantitative RT-PCR; ADE, Ab-mediated disease enhancement.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Peiris JS, Lai ST, Poon LL, Guan Y, Yam LY, Lim W, Nicholls J, Yee WK, Yan WW, Cheung MT, et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, Tong S, Urbani C, Comer JA, Lim W, et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 3.Zhong NS, Zheng BJ, Li YM, Poon, Xie ZH, Chan KH, Li PH, Tan SY, Chang Q, Xie JP, et al. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People's Republic of China, in February, 2003. Lancet. 2003;362:1353–1358. doi: 10.1016/S0140-6736(03)14630-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guan Y, Zheng BJ, He YQ, Liu XL, Zhuang ZX, Cheung CL, Luo SW, Li PH, Zhang LJ, Guan YJ, et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science. 2003;302:276–278. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- 5.Lau SK, Woo PC, Li KS, Huang Y, Tsoi HW, Wong BH, Wong SS, Leung SY, Chan KH, Yuen KY. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. USA. 2005;102:14040–14045. doi: 10.1073/pnas.0506735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, Wang H, Crameri G, Hu Z, Zhang H, et al. Bats are natural reservoirs of SARS-like coronaviruses. Science. 2005;310:676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- 7.Chan CP, Siu KL, Chin KT, Yuen KY, Zheng B, Jin DY. Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2006;80:9279–9287. doi: 10.1128/JVI.00659-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holmes KV. SARS-associated coronavirus. N. Engl. J. Med. 2003;348:1948–1951. doi: 10.1056/NEJMp030078. [DOI] [PubMed] [Google Scholar]
- 9.Du L, Kao RY, Zhou Y, He Y, Zhao G, Wong C, Jiang S, Yuen KY, Jin DY, Zheng BJ. Cleavage of spike protein of SARS coronavirus by protease factor Xa is associated with viral infectivity. Biochem. Biophys. Res. Commun. 2007;359:174–179. doi: 10.1016/j.bbrc.2007.05.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bosch BJ, van der Zee R, de Haan CA, Rottier PJ. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J. Virol. 2003;77:8801–8811. doi: 10.1128/JVI.77.16.8801-8811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buchholz UJ, Bukreyev A, Yang L, Lamirande EW, Murphy BR, Subbarao K, Collins PL. Contributions of the structural proteins of severe acute respiratory syndrome coronavirus to protective immunity. Proc. Natl. Acad. Sci. USA. 2004;101:9804–9809. doi: 10.1073/pnas.0403492101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bisht H, Roberts A, Vogel L, Bukreyev A, Collins PL, Murphy BR, Subbarao K, Moss B. Severe acute respiratory syndrome coronavirus spike protein expressed by attenuated vaccinia virus protectively immunizes mice. Proc. Natl. Acad. Sci. USA. 2004;101:6641–6646. doi: 10.1073/pnas.0401939101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang ZY, Kong WP, Huang Y, Roberts A, Murphy BR, Subbarao K, Nabel GJ. A DNA vaccine induces SARS coronavirus neutralization and protective immunity in mice. Nature. 2004;428:561–564. doi: 10.1038/nature02463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang L, Zhu Q, Qin E, Yu M, Ding Z, Shi H, Cheng X, Wang C, Chang G, Zhu Q, et al. Inactivated SARS-CoV vaccine prepared from whole virus induces a high level of neutralizing antibodies in BALB/c mice. DNA Cell Biol. 2004;23:391–394. doi: 10.1089/104454904323145272. [DOI] [PubMed] [Google Scholar]
- 16.Zhao P, Ke JS, Qin ZL, Ren H, Zhao LJ, Yu JG, Gao J, Zhu SY, Qi ZT. DNA vaccine of SARS-Cov S gene induces antibody response in mice. Acta Biochim. Biophys. Sin. 2004;36:37–41. doi: 10.1093/abbs/36.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou Z, Post P, Chubet R, Holtz K, McPherson C, Petric M, Cox M. A recombinant baculovirus-expressed S glycoprotein vaccine elicits high titers of SARS-associated coronavirus (SARS-CoV) neutralizing antibodies in mice. Vaccine. 2006;24:3624–3631. doi: 10.1016/j.vaccine.2006.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu RY, Wu LZ, Huang BJ, Huang JL, Zhang YL, Ke ML, Wang JM, Tan WP, Zhang RH, Chen HK, et al. Adenoviral expression of a truncated S1 subunit of SARS-CoV spike protein results in specific humoral immune responses against SARS-CoV in rats. Virus Res. 2005;112:24–31. doi: 10.1016/j.virusres.2005.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spruth M, Kistner O, Savidis-Dacho H, Hitter E, Crowe B, Gerencer M, Bruhl P, Grillberger L, Reiter M, Tauer C, et al. A double-inactivated whole virus candidate SARS coronavirus vaccine stimulates neutralising and protective antibody responses. Vaccine. 2006;24:652–661. doi: 10.1016/j.vaccine.2005.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao B, Jin NY, Wang RL, Zhang LS, Zhang YJ. Immunization of mice with a DNA vaccine based on severe acute respiratory syndrome coronavirus spike protein fragment 1. Viral Immunol. 2006;19:518–524. doi: 10.1089/vim.2006.19.518. [DOI] [PubMed] [Google Scholar]
- 21.Bukreyev A, Lamirande EW, Buchholz UJ, Vogel LN, Elkins WR, St. Claire M, Murphy BR, Subbarao K, Collins PL. Mucosal immunisation of African green monkeys (Cercopithecus aethiops) with an attenuated parainfluenza virus expressing the SARS coronavirus spike protein for the prevention of SARS. Lancet. 2004;363:2122–2127. doi: 10.1016/S0140-6736(04)16501-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Z, Zhang L, Qin C, Ba L, Yi CE, Zhang F, Wei Q, He T, Yu W, Yu J, et al. Recombinant modified vaccinia virus Ankara expressing the spike glycoprotein of severe acute respiratory syndrome coronavirus induces protective neutralizing antibodies primarily targeting the receptor binding region. J. Virol. 2005;79:2678–2688. doi: 10.1128/JVI.79.5.2678-2688.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faber M, Lamirande EW, Roberts A, Rice AB, Koprowski H, Dietzschold B, Schnell MJ. A single immunization with a rhabdovirus-based vector expressing severe acute respiratory syndrome coronavirus (SARS-CoV) S protein results in the production of high levels of SARS-CoV-neutralizing antibodies. J. Gen. Virol. 2005;86:1435–1440. doi: 10.1099/vir.0.80844-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao W, Tamin A, Soloff A, D'Aiuto L, Nwanegbo E, Robbins PD, Bellini WJ, Barratt-Boyes S, Gambotto A. Effects of a SARS-associated coronavirus vaccine in monkeys. Lancet. 2003;362:1895–1896. doi: 10.1016/S0140-6736(03)14962-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kapadia SU, Rose JK, Lamirande E, Vogel L, Subbarao K, Roberts A. Long-term protection from SARS coronavirus infection conferred by a single immunization with an attenuated VSV-based vaccine. Virology. 2005;340:174–182. doi: 10.1016/j.virol.2005.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee JS, Poo H, Han DP, Hong SP, Kim K, Cho MW, Kim E, Sung MH, Kim CJ. Mucosal immunization with surface-displayed severe acute respiratory syndrome coronavirus spike protein on Lactobacillus casei induces neutralizing antibodies in mice. J. Virol. 2006;80:4079–4087. doi: 10.1128/JVI.80.8.4079-4087.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weingartl H, Czub M, Czub S, Neufeld J, Marszal P, Gren J, Smith G, Jones S, Proulx R, Deschambault Y, et al. Immunization with modified vaccinia virus Ankara-based recombinant vaccine against severe acute respiratory syndrome is associated with enhanced hepatitis in ferrets. J. Virol. 2004;78:12672–12676. doi: 10.1128/JVI.78.22.12672-12676.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Czub M, Weingartl H, Czub S, He R, Cao J. Evaluation of modified vaccinia virus Ankara based recombinant SARS vaccine in ferrets. Vaccine. 2005;23:2273–2279. doi: 10.1016/j.vaccine.2005.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Du L, Zhao G, He Y, Guo Y, Zheng BJ, Jiang S, Zhou Y. Receptor-binding domain of SARS-CoV spike protein induces long-term protective immunity in an animal model. Vaccine. 2007;25:2832–2838. doi: 10.1016/j.vaccine.2006.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He Y, Lu H, Siddiqui P, Zhou Y, Jiang S. Receptor-binding domain of severe acute respiratory syndrome coronavirus spike protein contains multiple conformation-dependent epitopes that induce highly potent neutralizing antibodies. J. Immunol. 2005;174:4908–4915. doi: 10.4049/jimmunol.174.8.4908. [DOI] [PubMed] [Google Scholar]
- 31.He Y, Zhou Y, Liu S, Kou Z, Li W, Farzan M, Jiang S. Receptor-binding domain of SARS-CoV spike protein induces highly potent neutralizing antibodies: implication for developing subunit vaccine. Biochem. Biophys. Res. Commun. 2004;324:773–781. doi: 10.1016/j.bbrc.2004.09.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zakhartchouk AN, Sharon C, Satkunarajah M, Auperin T, Viswanathan S, Mutwiri G, Petric M, See RH, Brunham RC, Finlay BB, et al. Immunogenicity of a receptor-binding domain of SARS coronavirus spike protein in mice: implications for a subunit vaccine. Vaccine. 2007;25:136–143. doi: 10.1016/j.vaccine.2006.06.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blankinship MJ, Gregorevic P, Allen JM, Harper SQ, Harper H, Halbert CL, Miller DA, Chamberlain JS. Efficient transduction of skeletal muscle using vectors based on adeno-associated virus serotype 6. Mol. Ther. 2004;10:671–678. doi: 10.1016/j.ymthe.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 34.Bouchard S, MacKenzie TC, Radu AP, Hayashi S, Peranteau WH, Chirmule N, Flake AW. Long-term transgene expression in cardiac and skeletal muscle following fetal administration of adenoviral or adeno-associated viral vectors in mice. J. Gene Med. 2003;5:941–950. doi: 10.1002/jgm.421. [DOI] [PubMed] [Google Scholar]
- 35.Amiss TJ, McCarty DM, Skulimowski A, Samulski RJ. Identification and characterization of an adeno-associated virus integration site in CV-1 cells from the African green monkey. J. Virol. 2003;77:1904–1915. doi: 10.1128/JVI.77.3.1904-1915.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xin KQ, Ooki T, Mizukami H, Hamajima K, Okudela K, Hashimoto K, Kojima Y, Jounai N, Kumamoto Y, Sasaki S, et al. Oral administration of recombinant adeno-associated virus elicits human immunodeficiency virus-specific immune responses. Hum. Gene Ther. 2002;13:1571–1581. doi: 10.1089/10430340260201662. [DOI] [PubMed] [Google Scholar]
- 37.Xin KQ, Urabe M, Yang J, Nomiyama K, Mizukami H, Hamajima K, Nomiyama H, Saito T, Imai M, Monahan J, et al. A novel recombinant adeno-associated virus vaccine induces a long-term humoral immune response to human immunodeficiency virus. Hum. Gene Ther. 2001;12:1047–1061. doi: 10.1089/104303401750214276. [DOI] [PubMed] [Google Scholar]
- 38.Hara H, Monsonego A, Yuasa K, Adachi K, Xiao X, Takeda S, Takahashi K, Weiner HL, Tabira T. Development of a safe oral Abeta vaccine using recombinant adeno-associated virus vector for Alzheimer's disease. J. Alzheimers Dis. 2004;6:483–488. doi: 10.3233/jad-2004-6504. [DOI] [PubMed] [Google Scholar]
- 39.Wendtner CM, Kofler DM, Theiss HD, Kurzeder C, Buhmann R, Schweighofer C, Perabo L, Danhauser-Riedl S, Baumert J, Hiddemann W, et al. Efficient gene transfer of CD40 ligand into primary B-CLL cells using recombinant adeno-associated virus (rAAV) vectors. Blood. 2002;100:1655–1661. [PubMed] [Google Scholar]
- 40.Ponnazhagan S, Mahendra G, Curiel DT, Shaw DR. Adeno-associated virus type 2-mediated transduction of human monocyte-derived dendritic cells: implications for ex vivo immunotherapy. J. Virol. 2001;75:9493–9501. doi: 10.1128/JVI.75.19.9493-9501.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zaiss AK, Muruve DA. Immune responses to adeno-associated virus vectors. Curr. Gene Ther. 2005;5:323–331. doi: 10.2174/1566523054065039. [DOI] [PubMed] [Google Scholar]
- 42.Du L, He Y, Wang Y, Zhang H, Ma S, Wong CK, Wu SH, Ng F, Huang JD, Yuen KY, et al. Recombinant adeno-associated virus expressing the receptor-binding domain of severe acute respiratory syndrome coronavirus S protein elicits neutralizing antibodies: implication for developing SARS vaccines. Virology. 2006;353:6–16. doi: 10.1016/j.virol.2006.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mancheno-Corvo P, Martin-Duque P. Viral gene therapy. Clin. Transl. Oncol. 2006;8:858–867. doi: 10.1007/s12094-006-0149-y. [DOI] [PubMed] [Google Scholar]
- 44.Qu D, Zheng B, Yao X, Guan Y, Yuan ZH, Zhong NS, Lu LW, Xie JP, Wen YM. Intranasal immunization with inactivated SARS-CoV (SARS-associated coronavirus) induced local and serum antibodies in mice. Vaccine. 2005;23:924–931. doi: 10.1016/j.vaccine.2004.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yao X, Zheng B, Zhou J, Xu DZ, Zhao K, Sun SH, Yuan ZH, Wen YM. Therapeutic effect of hepatitis B surface antigen-antibody complex is associated with cytolytic and non-cytolytic immune responses in hepatitis B patients. Vaccine. 2007;25:1771–1779. doi: 10.1016/j.vaccine.2006.11.019. [DOI] [PubMed] [Google Scholar]
- 46.Huang J, Cao YN, Du JL, Bu XZ, Ma R, Wu CY. Priming with SARS CoV S DNA and boosting with SARS CoV S epitopes specific for CD4+ and CD8+ T cells promote cellular immune responses. Vaccine. 2007;25:6981–6991. doi: 10.1016/j.vaccine.2007.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zheng BJ, Guan Y, Tang Q, Du C, Xie FY, He ML, Chan KW, Wong KL, Lader E, Woodle MC, et al. Prophylactic and therapeutic effects of small interfering RNA targeting SARS-coronavirus. Antivir. Ther. 2004;9:365–374. [PubMed] [Google Scholar]
- 48.Zheng BJ, Ng MH, Chan KW, Tam S, Woo PC, Ng SP, Yuen KY. A single dose of oral DNA immunization delivered by attenuated Salmonella typhimurium down-regulates transgene expression in HBsAg transgenic mice. Eur. J. Immunol. 2002;32:3294–3304. doi: 10.1002/1521-4141(200211)32:11<3294::AID-IMMU3294>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 49.Zheng BJ, Ng MH, He LF, Yao X, Chan KW, Yuen KY, Wen YM. Therapeutic efficacy of hepatitis B surface antigen-antibodies-recombinant DNA composite in HBsAg transgenic mice. Vaccine. 2001;19:4219–4225. doi: 10.1016/s0264-410x(01)00158-x. [DOI] [PubMed] [Google Scholar]
- 50.Marshall E, Enserink M. Medicine. Caution urged on SARS vaccines. Science. 2004;303:944–946. doi: 10.1126/science.303.5660.944. [DOI] [PubMed] [Google Scholar]
- 51.Sheets RL, Stein J, Manetz TS, Andrews C, Bailer R, Rathmann J, Gomez PL. Toxicological safety evaluation of DNA plasmid vaccines against HIV-1, Ebola, severe acute respiratory syndrome, or West Nile virus is similar despite differing plasmid backbones or gene-inserts. Toxicol. Sci. 2006;91:620–630. doi: 10.1093/toxsci/kfj170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bisht H, Roberts A, Vogel L, Subbarao K, Moss B. Neutralizing antibody and protective immunity to SARS coronavirus infection of mice induced by a soluble recombinant polypeptide containing an N-terminal segment of the spike glycoprotein. Virology. 2005;334:160–165. doi: 10.1016/j.virol.2005.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhi Y, Figueredo J, Kobinger GP, Hagan H, Calcedo R, Miller JR, Gao G, Wilson JM. Efficacy of severe acute respiratory syndrome vaccine based on a nonhuman primate adenovirus in the presence of immunity against human adenovirus. Hum. Gene Ther. 2006;17:500–506. doi: 10.1089/hum.2006.17.500. [DOI] [PubMed] [Google Scholar]
- 54.Kuck D, Lau T, Leuchs B, Kern A, Muller M, Gissmann L, Kleinschmidt JA. Intranasal vaccination with recombinant adeno-associated virus type 5 against human papillomavirus type 16 L1. J. Virol. 2006;80:2621–2630. doi: 10.1128/JVI.80.6.2621-2630.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chao H, Liu Y, Rabinowitz J, Li C, Samulski RJ, Walsh CE. Several log increase in therapeutic transgene delivery by distinct adeno-associated viral serotype vectors. Mol. Ther. 2000;2:619–623. doi: 10.1006/mthe.2000.0219. [DOI] [PubMed] [Google Scholar]
- 56.Crotty S, Lohman BL, Lu FX, Tang S, Miller CJ, Andino R. Mucosal immunization of cynomolgus macaques with two serotypes of live poliovirus vectors expressing simian immunodeficiency virus antigens: stimulation of humoral, mucosal, and cellular immunity. J. Virol. 1999;73:9485–9495. doi: 10.1128/jvi.73.11.9485-9495.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen AY, Fry SR, Forbes-Faulkner J, Daggard G, Mukkur TK. Evaluation of the immunogenicity of the P97R1 adhesin of Mycoplasma hyopneumoniae as a mucosal vaccine in mice. J. Med. Microbiol. 2006;55:923–929. doi: 10.1099/jmm.0.46088-0. [DOI] [PubMed] [Google Scholar]
- 58.Wang D, Kandimalla ER, Yu D, Tang JX, Agrawal S. Oral administration of second-generation immunomodulatory oligonucleotides induces mucosal Th1 immune responses and adjuvant activity. Vaccine. 2005;23:2614–2622. doi: 10.1016/j.vaccine.2004.11.028. [DOI] [PubMed] [Google Scholar]
- 59.Fu TM, Dubey SA, Mehrotra DV, Freed DC, Trigona WL, Adams-Muhler L, Clair JH, Evans TG, Steigbigel R, Jacobson JM, et al. Evaluation of cellular immune responses in subjects chronically infected with HIV type 1. AIDS Res. Hum. Retrovir. 2007;23:67–76. doi: 10.1089/aid.2006.0114. [DOI] [PubMed] [Google Scholar]
- 60.Nikolova MH, Muhtarova MN, Taskov HB, Kostov K, Vezenkov L, Mihova A, Boumsell L, Bensussan A. The CD160+ CD8high cytotoxic T cell subset correlates with response to HAART in HIV-1+ patients. Cell. Immunol. 2005;237:96–105. doi: 10.1016/j.cellimm.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 61.Charley B, Riffault S, Van Reeth K. Porcine innate and adaptative immune responses to influenza and coronavirus infections. Ann. NY Acad. Sci. 2006;1081:130–136. doi: 10.1196/annals.1373.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hernandez YJ, Wang J, Kearns WG, Loiler S, Poirier A, Flotte TR. Latent adeno-associated virus infection elicits humoral but not cell-mediated immune responses in a nonhuman primate model. J. Virol. 1999;73:8549–8558. doi: 10.1128/jvi.73.10.8549-8558.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xiang Z, Gao G, Reyes-Sandoval A, Cohen CJ, Li Y, Bergelson JM, Wilson JM, Ertl HC. Novel, chimpanzee serotype 68-based adenoviral vaccine carrier for induction of antibodies to a transgene product. J. Virol. 2002;76:2667–2675. doi: 10.1128/JVI.76.6.2667-2675.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tenenbaum L, Lehtonen E, Monahan PE. Evaluation of risks related to the use of adeno-associated virus-based vectors. Curr. Gene Ther. 2003;3:545–565. doi: 10.2174/1566523034578131. [DOI] [PubMed] [Google Scholar]
- 65.Owens RA. Second generation adeno-associated virus type 2-based gene therapy systems with the potential for preferential integration into AAVS1. Curr. Gene Ther. 2002;2:145–159. doi: 10.2174/1566523024605627. [DOI] [PubMed] [Google Scholar]
- 66.Chunling M, Kun Y, Jian X, Jian Q, Hua S, Minsheng Z. Enhanced induction of SARS-CoV nucleocapsid protein-specific immune response using DNA vaccination followed by adenovirus boosting in BALB/c mice. Intervirology. 2006;49:307–318. doi: 10.1159/000094247. [DOI] [PMC free article] [PubMed] [Google Scholar]