

Abstract
Dysregulation of the endocannabinoid and dopamine systems have been implicated in schizophrenia. The purpose of this study was to examine the effects of sub-chronic treatment with two antipsychotics on CB1 receptor-mediated in vitro and in vivo effects. Adult and adolescent male and female rats were injected twice daily with haloperidol (0.3 mg/kg), clozapine (10 mg/kg), or saline for 10 days. Subsequently, CB1 receptor number and function were assessed by [3H]SR141716 and WIN55,212-2-stimulated [35S]GTPγS binding, respectively. The effects of sub-chronic antipsychotic treatment on the in vivo actions of Δ9-tetrahydrocannabinol (Δ9-THC) were also evaluated. In adult female rats, antipsychotic treatment attenuated maximal stimulation of CB1 receptor-mediated G-protein activity in the striatum (clozapine) and prefrontal cortex (both antipsychotics), but not in the ventral midbrain. Associated changes in CB1 receptor number were not observed, suggesting that this attenuation was not due to downregulation. In vivo, sub-chronic treatment with clozapine, but not haloperidol, attenuated Δ9-THC-induced suppression of activity in adult females, whereas neither drug altered hypothermia or catalepsy. In contrast, antipsychotic treatment did not change CB1 receptor-mediated G-protein activation in any brain region in adult male rats and in adolescents of either sex. In vivo, haloperidol, but not clozapine, enhanced Δ9-THC-mediated suppression of activity and hypothermia in adult male rats whereas neither antipsychotic affected Δ9-THC-induced in vivo effects in adolescent rats. These findings suggest that modulation of the endocannabinoid system might contribute in a sex- and age-selective manner to differences in motor side effects of clozapine versus haloperidol.
Keywords: antipsychotics, cannabinoids, CB1 receptors, clozapine, haloperidol, sex differences
Schizophrenia is characterized by disturbances in cognitive, emotional and social functioning. Although its underlying neural basis remains incompletely described, neurotransmitter systems that may play a role include dopamine, glutamate and serotonin (Horacek et al. 2006). Several lines of evidence suggest that alterations in the endocannabinoid system also may contribute. This system is comprised of CB1 and CB2 cannabinoid receptors found primarily in the brain and periphery, respectively, as well as at least two endogenous ligands, 2-arachidonylglycerol (2-AG) and anandamide [for a review, see (Pertwee 2006)]. Postmortem examination of brains from schizophrenic patients has revealed increased levels of CB1 receptors in the anterior cingulate (Zavitsanou et al. 2004) and dorsal lateral prefrontal (Dean et al. 2001) cortices. In patients with acute schizophrenia, elevated anandamide levels have been found in cerebrospinal fluid (Giuffrida et al. 2004) and blood (De Marchi et al. 2003), with decreases to baseline levels observed during remission. Psychiatric genetics studies have found that individuals with a polymorphism of the gene for the dopamine metabolic enzyme catechol-O-methyltransferase were more susceptible to development of psychosis if they smoked cannabis during adolescence (Caspi et al. 2005) and that a polymorphism in the CNR1 gene, which encodes the CB1 receptor, is associated with susceptibility to schizophrenia (Ujike et al. 2002). Finally, case reports have demonstrated similarities between certain symptoms of psychosis and cannabis intoxication (Grech et al. 2005).
The most frequently prescribed pharmacotherapy for schizophrenia is antipsychotics. While drugs within this class have varied profiles of receptor effects, the preponderance of research findings suggests that their primary therapeutic action results from antagonism of dopamine and/or serotonin receptors (Seeman 2002b). Antipsychotic blockade of dopamine receptors in three major pathways, mesocortical, mesolimbic and nigrostriatal, may mediate the effects of these drugs on cognition, affect and motor function, respectively. In addition to dopamine receptors, however, these areas also contain high levels of CB1 receptors and endocannabinoids (Herkenham et al. 1991). In forebrain regions, CB1 receptors are also co-localized with serotonin receptors (Hermann et al. 2002), suggesting that these monoamine neurotransmitter systems and the endocannabinoid system may interact. Further, this interaction may be bi-directional. For example, peripherally administered Δ9-tetrahydrocannabinol (Δ9-THC), the principal psychoactive substituent of marijuana, directly increased dopamine levels and preferentially increased burst activity of dopamine neurons in the nucleus accumbens and ventral tegmental area, two regions within the mesolimbic pathway (Chen et al. 1990a; Chen et al. 1990b; Diana et al. 1998; French 1997). Similarly, dopamine receptors modulate endocannabinoid release in the dorsal striatum (Giuffrida et al. 1999; Patel et al. 2003), and D1 and D2 agonists reduced cannabinoid-induced catalepsy in mice (Meschler et al. 2000). There is also evidence that dopamine activity affects endocannabinoid levels (anandamide and/or 2-AG) in limbic forebrain and prefrontal areas through intermediate glutamatergic mechanisms (Melis et al. 2004). Chronic treatment with the antipsychotic haloperidol increases D2 receptor levels and produces supersensitivity to D2 -mediated responses (Muller and Seeman 1977), with possible indirect impact on the endocannabinoid system through a similar mechanism. Given these interactions between endocannabinoid and monoamine systems, it would seem logical to propose that drugs that affect either system might have indirect effects on the other.
The major objective of the study was to determine the effects of sub-chronic antipsychotic treatment on CB1 receptors in selected brain areas and on subsequent in vivo responses to Δ9-THC in adolescent and adult rats of both sexes. Evaluation of age differences in antipsychotic interaction with the endocannabinoid system is particularly important, given that the onset of schizophrenia frequently occurs during late adolescence. [In rats, adolescence is characterized by a pattern of behaviors, including increased risk taking, novelty seeking, and increased orientation toward peers, and occurs between postnatal (PN) days 28 and 42 (Spear 2000)]. Haloperidol and clozapine were chosen as representative typical and atypical antipsychotics, respectively. Haloperidol acts primarily as a D2 dopamine receptor antagonist (Zhang and Bymaster 1999) and has a high liability for producing extrapyramidal motor side effects. In contrast, clozapine is an antagonist at numerous receptors, including 5-HT2, D2, D1, noradrenergic and muscarinic receptors (Zhang and Bymaster 1999), and is not associated with extrapyramidal motor effects. Brain areas included the ventral midbrain, which contains dopamine cell bodies, and the striatum and prefrontal cortex, which are dopaminergic terminal regions implicated in antipsychotic effects on motor function and symptoms of schizophrenia, respectively. Results from this study may help to elucidate additional (non-monoamine) mechanisms that may contribute to the therapeutic or side effect profile of these two antipsychotics. Further, these results may provide support for development of modulators of endocannabinoid functioning as pharmacotherapy for schizophrenia.
Methods
Subjects
All rats were born to adult female Long-Evans rats (Harlan, Dublin, VA), impregnated by adult male Long-Evans rats (Harlan) in our animal room facility. The dams were individually housed in clear plastic cages in a temperature-controlled (20-22°C) environment with a 12-h light-dark cycle (lights on at 7 a.m.) and were left undisturbed until they gave birth (PN0). On PN21, pups were weaned and were pair-housed with a same-sex rat. Rats in different treatment conditions were randomly chosen from different litters, with the exception that one male and one female from each litter may have received the same treatment. These studies were carried out in accordance with federal guidelines (National Research Council 1996) and were approved by our Institutional Animal Care and Use Committee.
Apparatus
Each activity chamber consisted of a clear plastic cage (22.5 cm × 44 cm × 20 cm), surrounded by 4 × 8 equally spaced photocell beams on the X- and Y-axes (Lafayette Instrument, Lafayette, IN) and housed in a sound-attenuated cabinet. A Traceable7 digital thermometer (Control Company, Friendsville, TX) was used to measure rectal temperature. The catalepsy bar apparatus consisted of a 280 mm bolt (10 mm diameter), attached to a frame by eyebolts at a height of 98 mm for adolescents and 130 mm for adults.
Procedures
Sub-Chronic Drug Administration
Rats were injected twice daily with clozapine (10 mg/kg), haloperidol (0.3 mg/kg), or saline for 10 days (in vitro study) or 9.5 days (in vivo study). Adolescent rats were aged PN30 on the first injection day whereas adults were PN60-PN70. Rats used in vitro were sacrificed by decapitation 18 h after the final injection. Prefrontal cortex, striatum (caudate putamen and nucleus accumbens plus rostral globus pallidus) and ventral midbrain (substantia nigra, ventral tegmental area) were dissected on ice. Periaqueductal gray (PAG) was also collected as a control region to compare with the dopamine containing regions. Tissue was stored at -80°C until use. For rats used in vivo, cumulative dosing with Δ9-THC and testing was initiated 24 h after last injection (PN40 for adolescents). Antipsychotic doses were selected from a range of doses that suppressed activity when administered acutely and were based upon our prior work with these drugs.
Agonist-Stimulated [35S]GTPγS Binding
Tissue samples were thawed on the day of assay, placed in 20 volumes of cold Membrane Buffer (50 mM Tris-HCl, 3 mM MgCl2, 1 mM EGTA, pH 7.4), homogenized with a Polytron and centrifuged at 48,000 × g at 4°C for 10 min. The supernatants were discarded and the pellets were re-homogenized in Membrane Buffer, centrifuged at 48,000 × g and resuspended in Assay Buffer (50 mM Tris-HCl, 3mM MgCl2, 0.2 mM EGTA, 100 mM NaCl, pH 7.4). Adenosine deaminase (final concentration=0.004 units/ml) was added to the membrane homogenates, which were then preincubated for 10 min at 30°C. Total membrane protein was measured according to (Bradford 1976). Concentration-effect curves were generated by incubating membrane protein (3.5-6 μg) in Assay Buffer with 1.25g/L bovine serum albumin (BSA) (Assay Buffer + BSA) with 0.1-30μM WIN55,212-2 in the presence of 30μM GDP and 0.1nM [35S]GTPγS in 0.5mL total volume for 2 hours at 30°C. Basal binding was measured in the absence of agonist and non-specific binding was measured in the presence of 20μM unlabeled GTPγS. The reaction was terminated by vacuum filtration though Whatman GF/B glass fiber filters, followed by three washes with 4°C Tris buffer (50 mM Tris-HCl, pH 7.4). Bound radioactivity was determined by liquid scintillation spectrophotometry at 95% efficiency after 10-hour extraction in ScintiSafe Econo 1 scintillation fluid.
[3H]SR141716A Binding
Membranes were prepared as described above. Saturation analysis was performed by incubating 8-12μg membrane protein with 0.2-3nM [3H]SR141716A in Assay Buffer + BSA in the presence or absence of 5 μM unlabeled SR141716A (to determine non-specific and total binding, respectively) for 90min at 30°C. The reaction was terminated by vacuum filtration though Whatman GF/B glass fiber filter that was pre-soaked in Tris buffer containing 5g/L BSA (Tris-BSA), followed by 3 washes with 4°C Tris-BSA. Bound radioactivity was determined by liquid scintillation spectrophotometry at 45% efficiency after extraction in ScinitSafe Econo 1 scintillation fluid.
Cell Culture
Cell lines stably expressing mouse CB1 receptors (CB1-CHO cells) were cultured in a humidified atmosphere of 5% CO2/95% air at 37°C, in a 1:1 mixture of DMEM and Nutrient Mixture F12 containing 5% fetal bovine serum, 100 units/ml each of penicillin and streptomycin and 0.4 mg/ml hygromycin B. Cells were harvested by replacing the media with PBS + 0.4% EDTA and collected by centrifugation at 1,000 × g for 15 min at 4°C. Cells were homogenized in 20 vol. ice-cold membrane buffer. The homogenate was centrifuged at 48,000 × g for 10 min at 4°C, the supernatant discarded and the pellet resuspended in assay buffer A, centrifuged, and the final pellet resuspended in assay buffer. CB1-CHO cell membranes (12 μg) were incubated at 37°C for 90 min with 0.8 μM [3H]SR141716A and varying concentrations of clozapine or haloperidol in assay buffer containing 0.5% BSA.
In Vivo Evaluation
Rats were transported in their home cages to the laboratory at least one h before the start of testing. Immediately after measurement of baseline rectal temperature, rats were injected with vehicle. Twenty min later each rat was placed in a locomotor chamber for 5-min. Upon removal, temperature was measured again. The front paws of the rat were placed on the bar apparatus 30 min post-injection. The total amount of time (in s) that both paws remained in contact with the bar during a 5-min session was recorded. If the rat voluntarily removed its paws from the bar 10 times, the session was stopped and amount of time on bar was recorded as 0. After the bar test (35 min after vehicle injection), each rat was injected with 3 mg/kg Δ9-THC. Twenty min later the testing regimen described above was repeated. Subsequently, each rat received additional doses of 7, 20, and 70 mg/kg Δ9-THC (cumulative doses of 10, 30, and 100 mg/kg) and was re-tested following an identical procedure. Inter-dose interval was 35 min and completion of the entire cumulative dose-effect curve required 175 min (20 min pre-session injection interval and 15 min for testing = 35 min per dose × 5 doses = 175 min).
Chemicals and Drugs
R(+)-[2,3-dihydro-5-methyl-3-{[(morpholinyl)methyl]pyrrolo-[1,2,3-de]-1,4-benzoxazinyl}-(1-naphthalenyl)methanone mesylate (WIN55,212), guanosine 5' diphosphate (GDP), GTPγS and bovine serum albumin (BSA) were purchased from Sigma Chemical Company (St. Louis, MO). Cell culture reagents were purchased from Gibco/BRL (Grand Island, NY). [35S]GTPγS (1150-1300 Ci/mmol) was obtained from Perkin Elmer Life Sciences (Boston, MA). [3H]SR141716A (44.0 Ci/mmol) was purchased from Amersham Pharmacia (Piscataway, NJ). The chemical name of SR141716A (rimonabant) is N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide HCl. Haloperidol (McNeil Pharmaceutical, Spring House, PA) was prepared by adding saline to a commercially available 5 mg/ml stock solution containing 1.8 mg methyl paraben, 0.2 mg propylparaben, and lactic acid. Clozapine (NIMH Chemical Synthesis and Drug Supply Program, Bethesda, MD) was prepared in purified distilled water. Δ9-THC (NIDA, Bethesda, MD) was mixed in a vehicle of absolute ethanol, Emulphor-620 (Rhone-Poulenc, Inc., Princeton, NJ), and saline in a ratio of 1:1:18. All injections were administered intraperitoneally (i.p.) at a volume of 1 ml/kg, with the exception that the 70 mg/kg dose of Δ9-THC was volume-adjusted from a 50 mg/ml solution.
Data Analysis
In vitro data are reported as mean (±SEM) of at least five experiments, each performed in triplicate. Non-specific binding was subtracted from each sample. Net stimulated [35S]GTPγS binding was defined as agonist-stimulated minus basal [35S]GTPγS binding, and percent stimulation was defined as (net-stimulated/basal [35S]GTPγS binding) × 100%. Nonlinear iterative regression analyses of agonist concentration-effect and saturation binding curves were performed with JMP 5.0 (SAS Institute, Cary, NC). Statistical significance was determined by ANOVA followed by post-hoc analysis with Dunnett's test (α=0.05).
For the in vivo evaluation, activity was measured as total number of photocell beam interruptions during the 5-min session. Rectal temperature values were expressed as the difference between control temperature (before injection) and temperatures following each drug dose (Δ°C). Catalepsy was assessed as the percentage of time that the rat had both front feet in contact with the bar during the 5-min session. For each age, sex, and antipsychotic, a separate split-plot Δ9-THC dose (within subject) X treatment (between subjects; antipsychotic vs. saline) ANOVA was performed for each dependent measure. Only a single group of rats of each age and sex received sub-chronic saline during the dosing regimen and served as the control group for both antipsychotics. When any ANOVA was significant, Tukey-Kramer post hoctests (α=0.05) were used to compare individual means.
Results
CB1 Receptor Binding and Function
Basal [35S]GTPγS binding did not differ between brains from vehicle and drug-treated male or female rats in any region (data not shown). Similarly, concentration-effect curves of WIN55,212-stimulated [35S]GTPγS binding did not differ between saline- and antipsychotic-treated adult male rats or adolescents of either sex in any brain region (Table 1). In contrast, significant decreases in WIN55,212-stimulated [35S]GTPγS binding were observed in the prefrontal cortex of adult female rats treated with either antipsychotic, and in the striatum, of those treated with clozapine (Fig. 1). Emax values for WIN 55,212 were reduced by 31% and 36% in the prefrontal cortex of haloperidol- or clozapine-treated adult female rats, respectively, and by 36% in the striatum of clozapine-treated adult female rats (Table 1). WIN55,212-2 EC50 values were significantly increased in the prefrontal cortex (approximately 1.7 and 3-fold for haloperidol and clozapine, respectively), but were not altered in the striatum. In the ventral midbrain, WIN55,212-stimulated [35S]GTPγS binding did not differ between saline- and antipsychotic-treated adult female rats (Fig. 1 and Table 1). CB1 receptor binding sites were not altered in any region, as indicated by similar Bmax and KD values for [3H]SR141716A binding in saline- and antipsychotic-treated adult rats of either sex (Table 2). The PAG was examined for comparison with the dopamine-containing regions assessed above. WIN55,212-stimulated [35S]GTPγS binding was similar in the PAG of vehicle- (Emax = 131% ± 7.2%), haloperidol- (Emax = 128% ± 6.3%) and clozapine- (Emax = 125% ± 8.5%) treated rats.
Table 1.
Effects of Sub-Chronic Dosing with Clozapine and Haloperidol on WIN 55,212-2-Stimulated [35S]GTPγS Binding
| Data are expressed as means ± S.E.M. obtained from non-linear regression analyses of WIN55,212-2-stimulated [35S]GTPγS binding. | ||||||
| Female Adults | ||||||
| Prefrontal Ctx | Striatum | Midbrain | ||||
| Drug | Emax (% Stim) |
EC50 (μM) |
Emax (% Stim) |
EC50 (μM) |
Emax (% Stim) |
EC50 (μM) |
| Saline | 205 ± 10.0 | 0.08 ± 0.02 | 194 ± 5.1 | 0.13 ± 0.02 | 137 ± 8.5 | 0.08 ± 0.03 |
| Clozapine | 130 ± 5.5* | 0.32 ± 0.06* | 123 ± 2.9* | 0.14 ± 0.02 | 126 ± 6.8 | 0.04 ± 0.01 |
| Haloperidol | 141 ± 8.7* | 0.11 ± 0.03* | 166 ± 8.3 | 0.16 ± 0.04 | 112 ± 9.9 | 0.10 ± 0.05 |
| Male Adults | ||||||
| Prefrontal Ctx | Striatum | Midbrain | ||||
| Drug | Emax(% Stim) |
EC50 (μM) |
Emax (% Stim) |
EC50 (μM) |
Emax (% Stim) |
EC50 (μM) |
| Saline | 135 ± 2.9 | 0.88 ± 0.38 | 100 ± 2.6 | 1.46 ± 0.45 | 91 ± 5.1 | 0.41 ± 0.17 |
| Clozapine | 134 ± 7.8 | 1.12 ± 0.91 | 102 ± 10.8 | 1.34 ± 0.65 | 104 ± 1.0 | 0.45 ± 0.23 |
| Haloperidol | 135 ± 13.2 | 0.96 ± 0.69 | 103 ± 7.8 | 1.10 ± 0.48 | 93 ± 8.8 | 0.74 ± 0.57 |
| Female Adolescents | ||||||
| Prefrontal Ctx | Striatum | Midbrain | ||||
| Drug | Emax (% Stim) |
EC50 (μM) |
Emax (% Stim) |
EC50 (μM) |
Emax (% Stim) |
EC50 (μM) |
| Saline | 119 ± 1.5 | 0.04 ± 0.01 | 102 ± 1.8 | 0.06 ± 0.03 | 87 ± 10.6 | 0.19 ± 0.02 |
| Clozapine | 125 ± 2.8 | 0.06 ± 0.01 | 94 ± 2.6 | 0.06 ± 0.02 | 92 ± 4.8 | 0.22 ± 0.05 |
| Haloperidol | 134 ± 3.5 | 0.08 ± 0.01 | 88 ± 2.3 | 0.06 ± 0.01 | 87 ± 5.5 | 0.20 ± 0.06 |
| Male Adolescents | ||||||
| Prefrontal Ctx | Striatum | Midbrain | ||||
| Drug | Emax (% Stim) |
EC50 (μM) |
Emax (% Stim) |
EC50 (μM) |
Emax (% Stim) |
EC50 (μM) |
| Saline | 112 ± 5.4 | 0.16 ± 0.01 | 96 ± 5.1 | 0.14 ± 0.02 | 105 ± 5.8 | 0.14 ± 0.01 |
| Clozapine | 133 ± 17.6 | 0.24 ± 0.03 | 103 ± 7.8 | 0.19 ± 0.03 | 96 ± 7.6 | 0.17 ± 0.02 |
| Haloperidol | 123 ± 12.4 | 0.24 ± 0.01 | 106 ± 16.1 | 0.23 ± 0.03 | 112 ± 6.8 | 0.16 ± 0.02 |
p<0.05 different from saline-treated rats.
Figure 1.
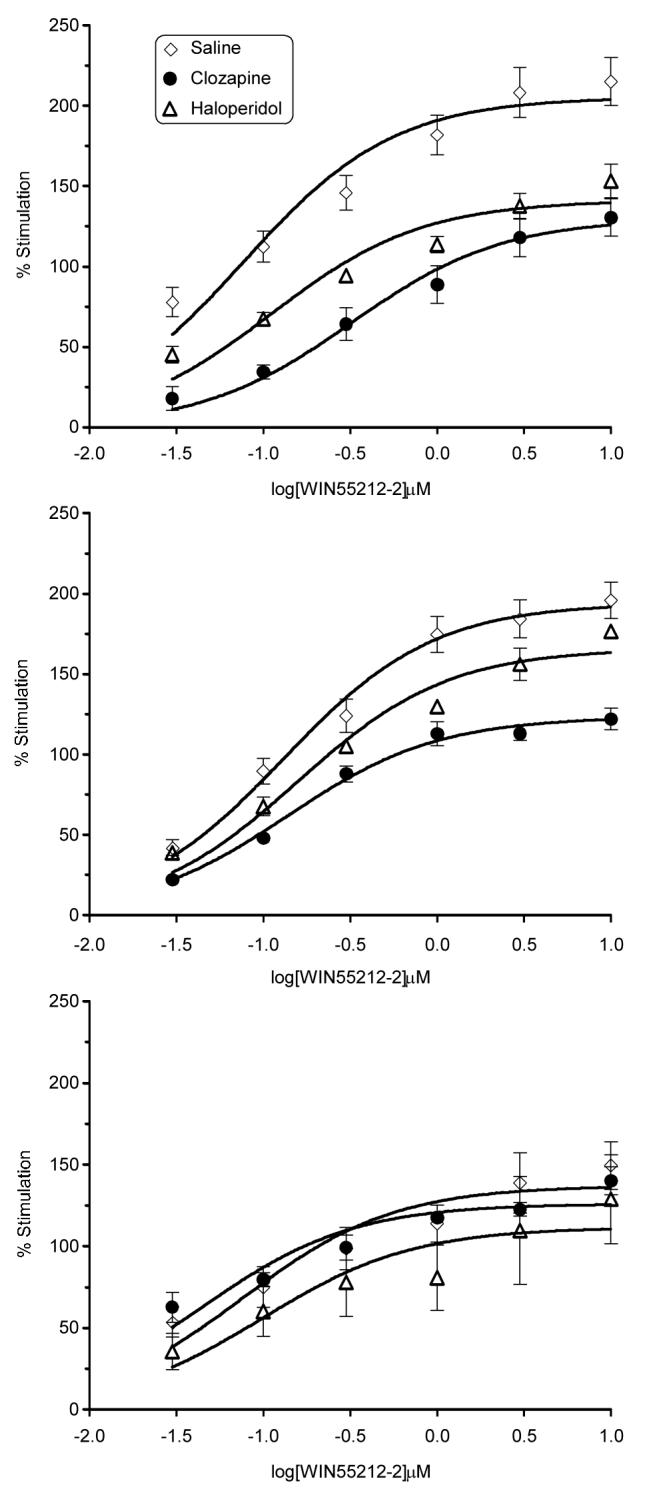
Regional effects of sub-chronic saline, clozapine and haloperidol treatment on stimulation of [35S]GTPγS binding by WIN55,212-2 in the brains of adult female rats. Regions include prefrontal cortex (top panel), striatum (middle panel) and midbrain (bottom panel). Data are expressed as mean percent stimulation values ± SEM. Associated Emax and EC50 values are provided in Table 1.
Table 2.
Effects of Sub-Chronic Dosing with Clozapine and Haloperidol on [3H]SR141716A Binding
| Data are expressed as means ± S.E.M. obtained from non-linear regression analyses of [3H]SR141716A binding. | ||||
| Female Adults | ||||
| Prefrontal Ctx | Striatum | |||
| Drug | Bmax (nM) |
KD (nM) |
Bmax (nM) |
KD (nM) |
| Saline | 3.63 ± 0.37 | 0.73 ± 0.06 | 2.53 ± 0.14 | 0.67 ± 0.10 |
| Clozapine | 3.13 ± 0.28 | 0.73 ± 0.11 | 2.65 ± 0.28 | 1.02 ± 0.24 |
| Haloperidol | 3.83 ± 0.13 | 0.78 ± 0.06 | 3.08 ± 0.33 | 0.72 ± 0.14 |
| Male Adults | ||||
| Prefrontal Ctx | Striatum | |||
| Drug | Bmax (nM) |
KD (nM) |
Bmax (nM) |
KD (nM) |
| Saline | 3.24 ± 0.33 | 0.63 ± 0.06 | 3.22 ± 0.41 | 1.57 ± 0.37 |
| Clozapine | 3.88 ± 0.46 | 0.72 ± 0.06 | 2.90 ± 0.43 | 1.32 ± 0.52 |
| Haloperidol | 3.71 ± 0.58 | 0.62 ± 0.05 | 3.64 ± 0.81 | 1.88 ± 0.85 |
To determine whether these antipsychotic drugs bound directly to CB1 receptor, competition binding experiments were conducted using [3H]SR141716A in CHO-K1 cells stably expressing the mouse CB1 receptor ([3H]SR141716A KD = 0.9 ± 0.2 nM; Bmax = 5.2 ± 0.7 pmol/mg membrane protein). Neither antipsychotic inhibited [3H]SR141617A binding in CB1 receptor-expressing CHO cells at concentrations ranging from 3 nM to 3 μM (data not shown), suggesting that the effects of these drugs on CB1 receptor function were not due to direct effects on the receptor. In contrast, the cannabinoid agonist 3 μM WIN55,212 inhibited >70% of [3H]SR141617A binding.
In Vivo Pharmacological Effects
In vivo evaluation showed that vehicle injection produced similar effects on all measures in rats treated with saline versus antipsychotic (Figs. 2 & 3, see points over “veh” to the left of each figure panel), suggesting that the sub-chronic antipsychotic dosing regimen did not produce a carry-over effect 24 h after termination of treatment. In rats of both ages and sexes that were treated sub-chronically with saline, Δ9-THC produced significant dose-dependent motor activity suppression (left panels), hypothermia (middle panels), and catalepsy (right panels) [Fig. 2, females and Fig. 3, males]. Antipsychotic treatment did not alter any of the three Δ9-THC effects in adolescent rats of either sex nor did it affect Δ9-THC-induced catalepsy in male or female adult rats (i.e., main effects of antipsychotic treatment and Δ9-THC dose X treatment interactions were not significant). In contrast, the activity suppression induced by Δ9-THC in adult female rats was attenuated (but not eliminated) by clozapine [main effect of clozapine treatment: F(1,18)=4.5, p<0.05], but not by haloperidol. In adult male rats, however, Δ9-THC's suppressive effect on activity was not affected by clozapine, but was enhanced by haloperidol [treatment condition × Δ9-THC dose interaction: F(4,72)=4.9, p<0.01]. Post hoc analysis of the latter effect revealed that male adult rats treated with haloperidol (versus saline) showed greater suppression of activity after receiving 3 or 10 mg/kg Δ9-THC. Although higher doses of Δ9-THC (30 and 100 mg/kg) also decreased activity, the magnitude of decrease was similar for rats treated with haloperidol or saline, suggesting a possible floor effect. Male adult rats also showed a significantly greater degree of Δ9-THC-induced hypothermia when treated with haloperidol (versus saline) [main effect of treatment: F(1,18)=8.4, p<0.05], but not with clozapine whereas Δ9-THC produced similar degrees of hypothermia in adult female rats regardless of sub-chronic treatment condition.
Figure 2.

Effects of cumulative doses of Δ9-THC on locomotor counts (left panels), change in rectal temperature (middle panels), and immobility in the bar test (right panels) following sub-chronic treatment with saline, haloperidol, and clozapine in adult (top panels) and adolescent (bottom panels) female rats. Points above VEH show effects of 1:1:18 (ethanol:emulphor:saline) vehicle for rats in each treatment group. Each value represents the mean ± SEM of data from adolescent rats (n=6, saline; n=8, haloperidol; n=7 clozapine) and adult rats (n=10 all treatments). h$ and c$ indicate significant main effect of Δ9-THC dose (p<0.05), as compared to vehicle (at left in each panel), in haloperidol- and clozapine-treated rats, respectively. @ (in symbol legend on figure) indicates a significant main effect of clozapine treatment (p<0.05), as compared to saline treatment, on locomotor activity.
Figure 3.

Effects of cumulative doses of Δ9-THC on locomotor counts (left panels), change in rectal temperature (middle panels), and immobility in the bar test (right panels) following sub-chronic treatment with saline, haloperidol, and clozapine in adult (top panels) and adolescent (bottom panels) male rats. Points above VEH show effects of 1:1:18 (ethanol:emulphor:saline) vehicle for rats in each treatment group. Each value represents the mean ± SEM of data from adolescent rats (n=7, saline; n=9, haloperidol; n=7, clozapine) and adult rats (n=10 all treatments). h$ and c$ indicate significant main effect of Δ9-THC dose (p<0.05), as compared to vehicle (at left in each panel), in haloperidol- and clozapine-treated rats, respectively. @ (in symbol legend on figure) indicates a significant main effect of haloperidol treatment (p<0.05), as compared to saline treatment, on change in rectal temperature. h* indicates significant haloperidol treatment X Δ9-THC dose interaction and post hoc determination of difference between haloperidol vs. saline treatment at indicated Δ9-THC dose (p<0.05).
Discussion
Sub-chronic treatment with either haloperidol or clozapine decreased CB1 receptor-mediated G-protein activation in the prefrontal cortex of adult female rats. Clozapine, but not haloperidol, also decreased activation in the striatum. Neither drug altered activation in the ventral midbrain. Similar changes were not seen in the brains of drug-treated male rats or adolescent rats of either sex. Further, antipsychotic administration did not affect [3H]SR141716 binding in any brain region tested, suggesting that the alterations resulted from changes in receptor signaling rather than from downregulation.
Two previous studies also examined the effects of repeated administration of haloperidol or clozapine on CB1 receptor binding or activation. Consistent with the results of the present study, Sundram and colleagues (2005) reported that chronic dosing with haloperidol did not affect [3H]CP55,940 binding in any of the tested brain regions of male Sprague-Dawley rats. In contrast, they found that clozapine decreased [3H]CP55,940 binding in the nucleus accumbens. While these latter results are at variance with those obtained in the present study, differences in several experimental parameters may have contributed. In the Sundram et al study, rats were treated with an antipsychotic for an extended duration (1-3 months) as compared to the 10-day period used here. Further, dosing started when the rats were 6-weeks old (i.e., during periadolescence) and continued into the early adult period. Whether or not the effects of antipsychotic administration over the entire time period of adolescence and into adulthood differ from those seen during administration only during adolescence (as in the present study) is unknown; however, given the extensive developmental changes in brain organization and dopamine receptor pruning that occur during adolescence [for a review, see (Spear 2000)], it would not be surprising. Moreover, the present study utilized whole striatum (e.g., caudate-putamen and nucleus accumbens), whereas previous results were obtained in isolated nucleus accumbens. Another group reported increased [3H]CP55,940 and [35S]GTPγS binding in striatum and substantia nigra following haloperidol in male Sprague-Dawley rats (Andersson et al. 2005). Again, however, there was substantial variation between experimental parameters used in this study and those used here. For example, the Andersson et al study utilized a higher haloperidol dose and a longer (3 day) washout period. Because CB1 receptors exhibit a high level of adaptation, this time interval could have resulted in a rebound overshoot effect in CB1 receptors. In addition, both of the studies described above used Sprague-Dawley rats (vs. Long-Evans strain in present study). Differences in findings among these studies emphasize the complex nature of the interaction between monoamine and endocannabinoid systems.
A number of possible mechanisms might mediate the observed decrease in CB1 receptor-mediated G-protein activity in the brains of adult female rats that was observed in the present study. The effects of haloperidol and clozapine on CB1 receptors are indirect, as neither antipsychotic has significant affinity for these receptors (data not shown). Desensitization, an uncoupling of the receptor from G-protein activation, can occur in the absence of receptor downregulation (Sim-Selley et al. 2000). For example, previous research has shown that chronic anandamide treatment produced CB1 receptor desensitization without downregulation (Rubino et al. 2000). Antipsychotic-induced increases in endocannabinoid levels could potentially produce similar effects. Another possibility is that antipsychotic treatment reduced the level of G-proteins. Previous studies have produced mixed results, with the majority of studies reporting no change or a decrease in Gαi or Gαs, and no change in Gαo, in striatum (Gupta and Mishra 1992; Kaplan et al. 1999; Meller and Bohmaker 1996; See et al. 1993; Shin et al. 1995) and no change or a decrease in Gαi expression in prefrontal cortex after haloperidol or clozapine treatment (Kaplan et al. 1999; Meller and Bohmaker 1996; See et al. 1993). Only one of these studies, however, examined G-protein expression in female (vs. male) rats (See et al. 1993). Consistent with the results of the present study, this study found no effect of chronic haloperidol treatment on Gαi, Gαo or Gαs expression in striatum. These results suggest alterations in G-protein expression cannot fully account for the results obtained here.
Decreased CB1-mediated G-protein activity might also result from reduced availability of G-proteins for activation by CB1 receptors. CB1 and D2 receptors have been shown to converge on the same pools of G-proteins in the striatum (Meschler and Howlett 2001) and both D2 and CB1 receptors can sequester G-proteins (Jarrahian et al. 2004; Vasquez and Lewis 1999). Increased levels of D2 or other G-protein coupled receptors that occur after antipsychotic treatment could decrease the available G-protein pool for CB1 receptors thereby reducing the level of CB1 receptor-mediated activity. Recent studies have also demonstrated that CB1 and D2 receptors can dimerize, thereby forming novel signaling complexes (Kearn et al. 2005). Under these conditions, CB1 receptors appear to switch their G-protein preference and activate Gαs. Consequently, the apparent level of receptor-mediated G-protein activity measured using agonist-stimulated [35S]GTPγS binding would be reduced because this assay primarily detects activation of inhibitory Gi/o subfamily of G-proteins in the brain.
Since CB1 and D2 receptors are not co-localized in all brain areas, the formation of these novel signaling complexes would be expected to exhibit regional differences. In the midbrain, for example, anatomical studies have shown that CB1 receptors are not co-localized with dopamine receptors (Herkenham et al. 1991). Hence, CB1 receptor functioning in this area would not be expected to change with antipsychotic treatment, as is consistent with the present results.
As noted above, only clozapine produced desensitization of CB1 receptors in the striatum. This regionally distinct effect might contribute to its low liability for extrapyramidal motor effects, as it is not shared by haloperidol. While haloperidol's motor effects are related to its action as a D2 antagonist, the degree to which the endocannabinoid system might also contribute is unknown. Cannabinoids, including anandamide, produce motor inhibition and catalepsy (Adams et al. 1995). Because CB1 receptor desensitization did not occur in striatum after haloperidol treatment (present study), increases in 2-AG levels induced by antagonism of D2 receptors (Patel et al. 2003) and consequent effects on brain pathways controlling movement, could contribute to motor deficits seen in patients treated with haloperidol. In contrast, attenuated CB1 receptor-mediated G-protein activation was found in the striatum of clozapine-treated female rats, which might mitigate adverse motor effects. Clozapine also has lower affinity for D2 receptors than haloperidol and acts at a variety of nondopamine receptors (Zhang and Bymaster 1999), both of which may contribute further to its lack of motor side effects.
One of the most intriguing findings is the sex- and age-dependent nature of antipsychotic-mediated CB1 receptor adaptation. In contrast to the striking effects found in adult female rats, CB1 receptor-mediated G-protein activity was unchanged in brains of antipsychotic-treated adult male or adolescent rats. Consistent with these findings, clinical reports have shown sex differences in the course of schizophrenia or in the efficacy and side effect profiles of antipsychotics (Seeman 1985; 2002a). Premenopausal females with schizophrenia tend to have a later onset and smoother clinical course than males of similar ages (Hafner 2003); however, they also tend to be more sensitive to the therapeutic, motor, and neuroendocrine effects of antipsychotics (Seeman 1985). These differences might be related to underlying sex differences in dopaminergic function, as confirmed by studies which have found larger increases in striatal dopamine release and catalepsy after haloperidol administration in female rats as compared to male rats (Campbell et al. 1988; Walker et al. 2006a). Hormonal modulation of dopamine signaling has also been reported (Becker 1999). Interestingly, pre-pubertal female rats (present study) and ovariectomized rats (unpublished data from our laboratory) did not exhibit antipsychotic-induced alterations in CB1 receptor activation. Since this study assessed the effects of only a single sub-chronic dose of each antipsychotic, however, the possibility that similar changes in CB1 receptor activation may have been observed in male adults and/or adolescents of both sexes with a different antipsychotic dosing regimen cannot be entirely eliminated. For example, previous research has shown that haloperidol's effect on regulation of dopamine neurotransmission is sex-dependent (Walker et al. 2006b). Since dopamine and endocannabinoid systems exhibit bidirectional interaction, it is possible that these sex differences in antipsychotic effects on dopamine functioning may indirectly result in differential activation of endocannabinoid system and consequent sex differences in CB1 receptor functioning.
In order to determine whether antipsychotic-induced changes in CB1 receptor activity were associated with functional alterations, the in vivo effects of Δ9-THC were assessed following antipsychotic treatment in tests in which cannabinoids produce a characteristic profile of actions, including suppression of activity, antinociception, hypothermia and catalepsy (Martin et al. 1991). Consistent with in vitro data, adolescent rats of both sexes showed equal sensitivity to Δ9-THC's in vivo effects regardless of their treatment status. In contrast, clozapine and haloperidol produced different patterns of effects in adult female rats across brain regions in vitro and across tasks in vivo. Whereas clozapine treatment attenuated Δ9-THC-induced motor suppression in these rats, neither antipsychotic affected Δ9-THC-mediated hypothermia or catalepsy. The striatum represents the most probable localization of the effects of clozapine on CB1 receptor desensitization (in vitro) and sensitivity to Δ9-THC (in vivo), because it is one of the many brain areas involved in modulation of locomotion. Moreover, haloperidol neither altered CB1 receptors in this brain area nor changed Δ9-THC-mediated effects on locomotion. In contrast with results in female rats, adult male rats treated with haloperidol exhibited enhanced9-THC-induced motor suppression and hypothermia that were not associated with alterations of CB1 receptor activation, suggesting that this potentiation may be related to alterations in another receptor system affected by haloperidol (e.g., dopamine). Increased catalepsy and hypothermia have also been observed in adult male rodents after acute co-administration of Δ9-THC and haloperidol, but not with acute Δ9-THC and clozapine (Marchese et al. 2003).
In summary, sub-chronic antipsychotic treatment with haloperidol or clozapine decreased CB1 receptor-mediated G-protein activity in specific forebrain regions in adult female rats without affecting CB1 receptor densities. In contrast, alterations in CB1 receptor signaling were not observed in adult male rats or in adolescent rats of either sex (with the caveat that only a single dosing regimen for each antipsychotic was evaluated). These sex and age differences were associated to some extent with differences in sensitivity to the in vivo effects of Δ9-THC, although other receptor mechanisms may also be involved. The results are also consistent with previous studies that have reported prominent sex differences in dopaminergic function, schizophrenia, and antipsychotic response. Although it is not clear whether antipsychotic modulation of CB1 receptor-mediated G-protein activity contributes to the therapeutic efficacy of haloperidol or clozapine, reports that CB1 receptors and endocannabinoids are increased in schizophrenia suggest that it might be one component of their actions, at least in adult females. Moreover, differences in the regulation of CB1 receptor-mediated G-protein activity in the striatum produced by treatment with haloperidol and clozapine might be involved in the different motor side effects of these drugs.
Acknowledgements
This research was supported by National Institutes of Health grants MH-64771 (JLW), DA-16644 (JLW), DA-05274 (DES), and DA-14277 (LJS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams IB, Ryan W, Singer M, Thomas BF, Compton DR, Razdan RK, Martin BR. Evaluation of cannabinoid receptor binding and in vivo activities for anandamide analogs. J Pharmacol Exp Ther. 1995;273:1172–81. [PubMed] [Google Scholar]
- Andersson M, Terasmaa A, Fuxe K, Stromberg I. Subchronic haloperidol increases CB(1) receptor binding and G protein coupling in discrete regions of the basal ganglia. Journal of Neuroscience Research. 2005;82:264–272. doi: 10.1002/jnr.20630. [DOI] [PubMed] [Google Scholar]
- Becker JB. Gender differences in dopaminergic function in striatum and nucleus accumbens. Pharmacol Biochem Behav. 1999;64:803–12. doi: 10.1016/s0091-3057(99)00168-9. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Campbell A, Baldessarini RJ, Cremens MC. Dose-catalepsy response to haloperidol in rat: effects of strain and sex. Neuropharmacology. 1988;27:1197–1199. doi: 10.1016/0028-3908(88)90018-4. [DOI] [PubMed] [Google Scholar]
- Caspi A, Moffitt TE, Cannon M, McClay J, Murray R, Harrington H, Taylor A, Arseneault L, Williams B, Braithwaite A, Poulton R, Craig IW. Moderation of the effect of adolescent-onset cannabis use on adult psychosis by a functional polymorphism in the catechol-O-methyltransferase gene: longitudinal evidence of a gene X environment interaction. Biol Psychiatry. 2005;57:1117–27. doi: 10.1016/j.biopsych.2005.01.026. [DOI] [PubMed] [Google Scholar]
- Chen J, Paredes W, Li J, Smith D, Lowinson J, Gardner E. Δ9 Tetrahydrocannabinol produces naloxone-blockable enhancement of presynaptic basal dopamine efflux in nucleus accumbens of conscious, freely-moving rats as measured by intracerebral microdialysis. Psychopharm. 1990a;102:156–162. doi: 10.1007/BF02245916. [DOI] [PubMed] [Google Scholar]
- Chen J, Paredes W, Lowinson JH, Gardner EL. Δ9-Tetrahydrocannabinol enhances presynaptic dopamine efflux in medial prefrontal cortex. Eur. J. Pharmacol. 1990b;190:259–262. doi: 10.1016/0014-2999(90)94136-l. [DOI] [PubMed] [Google Scholar]
- De Marchi N, De Petrocellis L, Orlando P, Daniele F, Fezza F, Di Marzo V. Endocannabinoid signalling in the blood of patients with schizophrenia. Lipids Health Dis. 2003;2:5. doi: 10.1186/1476-511X-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean B, Sundram S, Bradbury R, Scarr E, Copolov D. Studies on [3H]CP-55940 binding in the human central nervous system: regional specific changes in density of cannabinoid-1 receptors associated with schizophrenia and cannabis use. Neuroscience. 2001;103:9–15. doi: 10.1016/s0306-4522(00)00552-2. [DOI] [PubMed] [Google Scholar]
- Diana M, Melis M, Gessa GL. Increase in meso-prefrontal dopaminergic activity after stimulation of CB1 receptors by cannabinoids. Eur J Neurosci. 1998;10:2825–30. doi: 10.1111/j.1460-9568.1998.00292.x. [DOI] [PubMed] [Google Scholar]
- French ED. delta9-Tetrahydrocannabinol excites rat VTA dopamine neurons through activation of cannabinoid CB1 but not opioid receptors. Neurosci Lett. 1997;226:159–62. doi: 10.1016/s0304-3940(97)00278-4. [DOI] [PubMed] [Google Scholar]
- Giuffrida A, Leweke FM, Gerth CW, Schreiber D, Koethe D, Faulhaber J, Klosterkotter J, Piomelli D. Cerebrospinal anandamide levels are elevated in acute schizophrenia and are inversely correlated with psychotic symptoms. Neuropsychopharmacology. 2004;29:2108–14. doi: 10.1038/sj.npp.1300558. [DOI] [PubMed] [Google Scholar]
- Giuffrida A, Parsons LH, Kerr TM, Rodriguez de Fonseca F, Navarro M, Piomelli D. Dopamine activation of endogenous cannabinoid signaling in dorsal striatum [see comments] Nat Neurosci. 1999;2:358–63. doi: 10.1038/7268. [DOI] [PubMed] [Google Scholar]
- Grech A, Van Os J, Jones PB, Lewis SW, Murray RM. Cannabis use and outcome of recent onset psychosis. Eur Psychiatry. 2005;20:349–53. doi: 10.1016/j.eurpsy.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Gupta SK, Mishra RK. Effects of chronic treatment of haloperidol and clozapine on levels of G-protein subunits in rat brain. Journal of Molecular Neuroscience. 1992;3:197–201. doi: 10.1007/BF03380139. [DOI] [PubMed] [Google Scholar]
- Hafner H. Gender differences in schizophrenia. Psychoneuroendocrinology. 2003;28(Suppl 2):17–54. doi: 10.1016/s0306-4530(02)00125-7. [DOI] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, DeCosta BR, Richfield EK. Neuronal localization of cannabinoid receptors in the basal ganglia of the rat. Brain Res. 1991;547:267–274. doi: 10.1016/0006-8993(91)90970-7. [DOI] [PubMed] [Google Scholar]
- Hermann H, Marsicano G, Lutz B. Coexpression of the cannabinoid receptor type 1 with dopamine and serotonin receptors in distinct neuronal subpopulations of the adult mouse forebrain. Neuroscience. 2002;109:451–60. doi: 10.1016/s0306-4522(01)00509-7. [DOI] [PubMed] [Google Scholar]
- Horacek J, Bubenikova-Valesova V, Kopecek M, Palenicek T, Dockery C, Mohr P, Hoschl C. Mechanism of action of atypical antipsychotic drugs and the neurobiology of schizophrenia. CNS Drugs. 2006;20:389–409. doi: 10.2165/00023210-200620050-00004. [DOI] [PubMed] [Google Scholar]
- Jarrahian A, Watts VJ, Barker EL. D2 dopamine receptors modulate Galpha-subunit coupling of the CB1 cannabinoid receptor. Journal of Pharmacology and Experimental Therapeutics. 2004;308:880–886. doi: 10.1124/jpet.103.057620. [DOI] [PubMed] [Google Scholar]
- Kaplan GB, Leite-Morris KA, Keith DJ. Differential effects of treatment with typical and atypical antipsychotic drugs on adenylyl cyclase and G proteins. Neuroscience Letters. 1999;273:147–150. doi: 10.1016/s0304-3940(99)00610-2. [DOI] [PubMed] [Google Scholar]
- Kearn CS, Blake-Palmer K, Daniel E, Mackie K, Glass M. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors enhances heterodimer formation: a mechanism for receptor cross-talk? Molecular Pharmacology. 2005;67:1697–1704. doi: 10.1124/mol.104.006882. [DOI] [PubMed] [Google Scholar]
- Marchese G, Casti P, Ruiu S, Saba P, Sanna A, Casu G, Pani L. Haloperidol, but not clozapine, produces dramatic catalepsy in delta9-THC-treated rats: possible clinical implications. Br J Pharmacol. 2003;140:520–6. doi: 10.1038/sj.bjp.0705478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BR, Compton DR, Thomas BF, Prescott WR, Little PJ, Razdan RK, Johnson MR, Melvin LS, Mechoulam R, Ward SJ. Behavioral, biochemical, and molecular modeling evaluations of cannabinoid analogs. Pharmacol Biochem Behav. 1991;40:471–478. doi: 10.1016/0091-3057(91)90349-7. [DOI] [PubMed] [Google Scholar]
- Melis M, Pistis M, Perra S, Muntoni AL, Pillolla G, Gessa GL. Endocannabinoids mediate presynaptic inhibition of glutamatergic transmission in rat ventral tegmental area dopamine neurons through activation of CB1 receptors. J Neurosci. 2004;24:53–62. doi: 10.1523/JNEUROSCI.4503-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller E, Bohmaker K. Chronic treatment with antipsychotic drugs does not alter G protein alpha or beta subunit levels in rat brain. Neuropharmacology. 1996;35:1785–1791. doi: 10.1016/s0028-3908(96)00119-0. [DOI] [PubMed] [Google Scholar]
- Meschler JP, Conley TJ, Howlett AC. Cannabinoid and dopamine interaction in rodent brain: effects on locomotor activity. Pharmacol. Biochem. Behav. 2000;67:567–573. doi: 10.1016/s0091-3057(00)00390-7. [DOI] [PubMed] [Google Scholar]
- Meschler JP, Howlett AC. Signal transduction interactions between CB1 cannabinoid and dopamine receptors in the rat and monkey striatum. Neuropharmacology. 2001;40:918–926. doi: 10.1016/s0028-3908(01)00012-0. [DOI] [PubMed] [Google Scholar]
- Muller P, Seeman P. Brain neurotransmitter receptors after long-term haloperidol: dopamine, acetylcholine, serotonin, alpha-noradrenergic and naloxone receptors. Life Sciences. 1977;21:1751–1758. doi: 10.1016/0024-3205(77)90155-2. [DOI] [PubMed] [Google Scholar]
- National Research Council . Guide for the Care and Use of Laboratory Animals. National Academy Press, National Academy Press; 1996. [Google Scholar]
- Patel S, Rademacher DJ, Hillard CJ. Differential regulation of the endocannabinoids anandamide and 2-arachidonylglycerol within the limbic forebrain by dopamine receptor activity. J Pharmacol Exp Ther. 2003;306:880–8. doi: 10.1124/jpet.103.054270. [DOI] [PubMed] [Google Scholar]
- Pertwee RG. The pharmacology of cannabinoid receptors and their ligands: an overview. Int J Obes (Lond) 2006;30(Suppl 1):S13–8. doi: 10.1038/sj.ijo.0803272. [DOI] [PubMed] [Google Scholar]
- Rubino T, Vigano D, Costa B, Colleoni M, Parolaro D. Loss of cannabinoid-stimulated guanosine 5'-O-(3-[(35)S]Thiotriphosphate) binding without receptor down-regulation in brain regions of anandamide-tolerant rats. Journal of Neurochemistry. 2000;75:2478–2484. doi: 10.1046/j.1471-4159.2000.0752478.x. [DOI] [PubMed] [Google Scholar]
- See RE, Striplin C, Kalivas PW. Chronic haloperidol does not alter G protein alpha-subunit levels in rats. Molecular Brain Research. 1993;19:219–221. doi: 10.1016/0169-328x(93)90030-s. [DOI] [PubMed] [Google Scholar]
- Seeman MV. Sex and schizophrenia. Canadian Journal of Psychiatry. 1985;30:313–315. doi: 10.1177/070674378503000502. [DOI] [PubMed] [Google Scholar]
- Seeman MV. The role of sex hormones in psychopathology: focus on schizophrenia. Primary Care. 2002a;29:171–182. doi: 10.1016/s0095-4543(03)00080-0. [DOI] [PubMed] [Google Scholar]
- Seeman P. Atypical antipsychotics: mechanism of action. Can J Psychiatry. 2002b;47:27–38. [PubMed] [Google Scholar]
- Shin CJ, Kim YS, Park JB, Juhnn YS. Changes in G protein levels in the hippocampus and the striatum of rat brain after chronic treatment with haloperidol and sulpiride. Neuropharmacology. 1995;34:1335–1338. doi: 10.1016/0028-3908(95)00117-o. [DOI] [PubMed] [Google Scholar]
- Sim-Selley LJ, Selley DE, Vogt LJ, Childers SR, Martin TJ. Chronic heroin self-administration desensitizes mu opioid receptor-activated G-proteins in specific regions of rat brain. Journal of Neuroscience. 2000;20:4555–4562. doi: 10.1523/JNEUROSCI.20-12-04555.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear LP. The adolescent brain and age-related behavioral manifestations. Neurosci Biobehav Rev. 2000;24:417–63. doi: 10.1016/s0149-7634(00)00014-2. [DOI] [PubMed] [Google Scholar]
- Sundram S, Copolov D, Dean B. Clozapine decreases [3H] CP 55940 binding to the cannabinoid 1 receptor in the rat nucleus accumbens. Naunyn Schmiedebergs Archives of Pharmacology. 2005;371:428–433. doi: 10.1007/s00210-005-1074-2. [DOI] [PubMed] [Google Scholar]
- Ujike H, Takaki M, Nakata K, Tanaka Y, Takeda T, Kodama M, Fujiwara Y, Sakai A, Kuroda S. CNR1, central cannabinoid receptor gene, associated with susceptibility to hebephrenic schizophrenia. Mol Psychiatry. 2002;7:515–8. doi: 10.1038/sj.mp.4001029. [DOI] [PubMed] [Google Scholar]
- Vasquez C, Lewis DL. The CB1 cannabinoid receptor can sequester G-proteins, making them unavailable to couple to other receptors. Journal of Neuroscience. 1999;19:9271–9280. doi: 10.1523/JNEUROSCI.19-21-09271.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker QD, Ray R, Kuhn CM. Sex differences in neurochemical effects of dopaminergic drugs in rat striatum. Neuropsychopharmacology. 2006a;31:1193–1202. doi: 10.1038/sj.npp.1300915. [DOI] [PubMed] [Google Scholar]
- Walker QD, Ray R, Kuhn CM. Sex differences in neurochemical effects of dopaminergic drugs in rat striatum. Neuropsychopharmacology. 2006b;31:1193–202. doi: 10.1038/sj.npp.1300915. [DOI] [PubMed] [Google Scholar]
- Zavitsanou K, Garrick T, Huang XF. Selective antagonist [3H]SR141716A binding to cannabinoid CB1 receptors is increased in the anterior cingulate cortex in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28:355–60. doi: 10.1016/j.pnpbp.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Zhang W, Bymaster FP. The in vivo effects of olanzapine and other antipsychotic agents on receptor occupancy and antagonism of dopamine D1, D2, D3, 5HT2A and muscarinic receptors. Psychopharmacology (Berl) 1999;141:267–78. doi: 10.1007/s002130050834. [DOI] [PubMed] [Google Scholar]