

Abstract
B-Raf is the most mutated gene in melanoma; however, mechanism through which it promotes early melanomas remains uncertain. Most nevi contain activated V600EB-Raf but few develop into melanoma and expression in melanocytes is inhibitory with low protein levels present in surviving cells, suggesting unknown cooperative oncogenic events are necessary for melanoma development. Since many melanomas have V600EB-Raf and active Akt3, it is possible these proteins cooperatively facilitate melanocyte transformation. In this study, Akt3 is shown to phosphorylate V600EB-Raf to lower its activity as well as that of the downstream MAP kinase pathway to levels promoting early melanoma development. Expression of active Akt3 in early melanoma cells containing V600EB-Raf reduced MAP kinase signaling and promoted anchorage independent growth. Furthermore, expression of both V600EB-Raf and active Akt3 in melanocytes promoted a transformed phenotype. Mechanistically, aberrant Akt3 activity in early melanomas serves to phosphorylate serines 364 and 428 on V600EB-Raf in order to reduce activity of V600EB-Raf to levels that promote rather than inhibit proliferation, which aids melanocytic transformation. Inhibition of V600EB-Raf or Akt3 in advanced melanoma cells in which both pathways were active reduced anchorage independent growth and tumor development in a cooperatively acting manner. Inhibition of Akt3 alone in these cells led to increased MAP kinase signaling. In summary, these results suggest that activating B-Raf mutations initially promote nevi development but the resulting high, intense activation of the MAP kinase pathway inhibits further tumor progression requiring Akt3 activation to bypass this barrier and aid melanoma development.
INTRODUCTION
Despite being the least prevalent type of skin cancer, malignant melanoma is by far the deadliest due to its highly metastatic nature (1). PI3 and MAP kinase pathways play prominent roles in melanoma development by regulating diverse cellular processes including cellular proliferation, survival, invasion and angiogenesis (2-4). The mechanism through which these pathways interact or cooperate to promote melanoma development remains to be fully elucidated (2).
Increased activity of the PI3 kinase pathway occurs in ∼70% of sporadic melanomas due to loss of PTEN and/or increased expression of Akt3 resulting from increased gene copy number (5). Highest level of Akt3 activity occur in metastatic melanomas, deregulating apoptotic signaling and promoting chemoresistance, making it attractive therapeutic target for metastatic disease (6, 7), (8). Targeting Akt3 decreases tumorigenesis by increasing cellular apoptosis in advanced stage melanomas but it is not known whether it plays a second independent role in early melanoma development (7, 9).
The MAP kinase pathway is activated through Ras mutations in 10-15% and B-Raf mutations in ∼60% of melanomas (10-16). A single-base mutation converting T to A at nucleotide 1799 of B-Raf, substituting a valine for a glutamic acid at codon 600 (V600E) in exon 15 is the most prominent mutation (12) and is acquired but not inherited during development of sporadic melanomas (17-19). Since B-Raf is the most mutated gene in melanomas, it is an attractive therapeutic target for melanoma patients (20).
While V600EB-Raf regulates melanoma cellular proliferation (21-23), angiogenesis (21), survival (22, 23), invasion and metastasis (24, 25), it alone is not sufficient for melanoma development. Ectopic expression of Ras or Raf in primary or cancer cell lines causes cell cycle arrest or senescence (26), likely due to duration or level of MAP kinase pathway activation (27), which induces cyclin dependent kinase inhibitors (28, 29). Thus, other factors are necessary to facilitate melanoma tumor progression, likely involving loss of tumor suppressor genes or acquisition of cooperating oncogene activation (30-33). Possibly, deregulation of p53 (31, 33) and/or Rb pathway through p16INK4A loss (30, 31) might be necessary. Another unexplored possibility might involve cooperation between V600EB-Raf and Akt3 signaling cascades, which are frequently activated in the same cells (3, 4). In support of this possibility, Akt has been shown to phosphorylate B-Raf to decrease its activity in normal cells; however, it is unknown whether this process could play any role in melanoma development (34-36).
In this study, V600EB-Raf and Akt3 are shown to cooperatively promote melanoma development. Akt3 is demonstrated to phosphorylate mutant V600EB-Raf in order to reduce its and MAP kinase pathway activity to levels promoting rather than retarding melanocytic cell growth and transformation. Thus, these results suggest that activating V600EB-Raf mutation initially promotes nevi development but the resulting high, intense activation of the MAP kinase pathway inhibits further tumor progression requiring Akt3 activation to phosphorylate V600EB-Raf in order to bypass this barrier and promote melanoma development.
MATERIALS AND METHODS
Cell Lines and Culture Conditions
Normal human melanocytes and melanoma cell lines; WM35, UACC 903, and SK-MEL-24 cells were cultured as previously described (7, 37-39). Mouse melan-a melanocytes were grown in RPMI 1640 supplemented with 10% FBS, 2 mM L-glutamine, and 200 nM TPA.
In vitro Pharmacological Agent Studies
The PI3 kinase inhibitor LY-294002 (Qbiogene, Carlsbad, CA) was used to pharmacologically inhibit Akt activity. Briefly, 1×106 NHEM, WM35, SK-MEL-24 or 1.5×105 UACC 903 cells were grown in complete media supplemented with FBS or growth factors Two days later, cells were fed with serum/growth factor free media. 24-hours later, cells were treated with 50 mM LY-294002 or DMSO for 20 minutes followed by stimulation using serum or growth factors upto180 minutes. At each time point protein lysates were collected for Western blot analysis.
Western Blot Analysis
Protein lysates were collected, quantitated, and analyzed by Western blotting as detailed previously (21). Antibodies to pErk and pAkt serine-473 and caspase-3 from Cell Signaling Technologies (Danvers, MA); cyclin D1, p27 Kip1, α-enolase, anti-hemagglutinin (HA), and B-Raf from Santa Cruz Biotechnology (Santa Cruz, CA); and Akt3 from Upstate Biotechnology (Lake Placid, NY) were used.
Plasmid Constructs
Hemagglutinin tagged wild type and kinase-dead Akt3 (40) were subcloned (Eco RI and Xba I) into pcDNA3.1puro. A myristoylation tag was added by PCR to the N terminus of Akt3 and subcloned into the Eco RI and Xba I. Site directed mutagenesis (Quick Change, Stratagene, La Jolla, CA) was used to create E40K Akt3 mutant with primers TGGCTCATTCATAGGATATAAAAAGAAACCTCAAGATGTGGATTT and AAATCCACATCTTGAGGTTTCTTTTTATATCCTATGAATGAGCCA. For B-Raf constructs, HA-tagged wild type B-Raf (provided by Ann Vojtek, University of Michigan) (41) was excised (Hind III/Eco RV) and subcloned into pcDNA6/V5-HisA (Invitrogen, Carlsbad, CA). Site directed mutagenesis with primers GATTTTGGTCTAGCTACAGAGAAATCTCGATGGAGTGGG and CCCACTCCATCGAGATTTCTCTGTAGCTAGACCAAAATC generated V600EB-Raf. Similarly, primers CGAGACCGATCCTCAGCAGCTCCCAATGTGCAT and ATGCACATTGGGAGCTGCTGAGGATCGGTCTCG generated S364A V600EB-Raf, primers GCGAGAAAGGAAGTCAGCTTCATCCTCAGAAGAC and GTCTTCTGAGGATGAAGCTGACTTCCTTTCTCGC generated S428A V600EB-Raf. Construct inserts were sequence verified. Plasmids were introduced into cell lines with the Amaxa Nucleofector (Koeln, Germany). Reagent R, K17 program was used for UACC 903 cells while NHEM reagent, U20 program was used for human melanocytes, WM35 and melan-a cells. Transfection efficiencies were determined to be ∼60% using pMaxGFP as a GFP expression plasmid control.
Anchorage Independent Growth Studies
Plates were coated with PolyHEMA (12.0 mg/ml) (Sigma, St Louis, MI) in 95% ethanol to assess anchorage independent growth.
siRNA-mediated inhibition
Duplexed “Stealth” siRNA from Invitrogen (Carlsbad, CA) was used for inhibiting protein expression as described previously (7, 9, 25, 39). siRNA (100 or 200 pmoles) was nucleofected into 2×106 human melanocytes, UACC 903 or WM35 cells allowed to recover for 1.5-2 days. Cells were then trypsinized and 2×104 seeded into PolyHEMA coated 96 well plates. Protein lysates for Western blot analysis was harvested. For UACC 903, 1×104 cells/well were seeded into DMEM supplemented with 10% FBS or 2×104 cells/well in serum free conditions. Cell proliferation was quantified 3 days later by MTS assay (Promega, Madison, WI).
Ectopic plasmid expression studies
HA-tagged Akt3 constructs (1.45 pmoles pcDNA3.1puro HA wt-Akt3, E40K-Akt3, myr-Akt3, and dead-Akt3) were nucleofected alone or in combination with siRNA to PRAS40 (100 pmoles) or a scrambled siRNA into 2×106 cells, 5×104 cells/50 μl plated into 96-well PolyHEMA coated plates. Twenty-four hours later, cells were selected using 1.6 μg/ml puromycin containing media. Following 7-days of selection, anchorage independent growth was assessed by MTS assay.
Melan-a transformation assays
2×106 cells were nucleofected with 1.45 pmoles of pcDNA6 V600EB-Raf, pcDNA6 AA-V600EB-Raf alone or in combination with pcDNA3.1puro myr-Akt3 or E40K-Akt3. 4×104 cells/50 μl were added to 96-well PolyHEMA coated plates. After overnight recovery, 50 μl DMEM supplemented 10% FBS, 6 μg/ml puromycin and/or 6 μg/ml blasticidin was added to give a final concentration of 3 μg/ml. After 3 days selection, cell viability was quantified by MTS assay.
V600EB-Raf Kinase Assay
1.45 pmoles of HA-tagged pcDNA6 V600EB-Raf, S364A V600EB-Raf, S428A V600EB-Raf, AA-V600EB-Raf (S364A, S428A) or Dead B-Raf were nucleofected together with 1.45 pmoles pcDNA 3.1myc-His myr-Akt3 into melan-a cells to phosphorylate ectopically expressed B-Raf at serine residues 364 and 428. Cell lysates were harvested 3 days later using lysis buffer containing 20 mM Tris pH 8.0, 2 mM EDTA, 50 mM beta-glycerophosphate, 1mM activated sodium orthovanadate, 1% Triton X-100, 10% glycerol, 1 mM PMSF, and Protease Inhibitor Cocktail (Sigma, St Louis, MI). Various HA-tagged B-Raf proteins were immunoprecipitated from 150 μg of total lysates using 0.2 μg of antibody against HA, which was pre-coupled to Gamma Bind G sepharose (Amersham Pharmacia Biotech, Piscataway, NJ). Activity of immunoprecipitated B-Raf was measured as previously reported (42) using full length Mek protein substrate (Santa Cruz Biotechnology, Santa Cruz, CA). Samples were separated on duplicate NuPAGE gels (Invitrogen, Carlsbad, CA), with protein on one gel transferred onto PVDF membrane (Western blot analysis) and other gel fixed in 10% acetic acid/50% methanol, dried, and then exposed to HyBlot CL autoradiography film (Denville, Metuchen, NJ) for 32P Mek detection.
Phosphorylated B-Raf Analysis
HA-tagged B-Raf expressing plasmids (pcDNA6 WT B-Raf, V600EB-Raf, S364A V600EB-Raf, and AA-V600EB-Raf (S364A, S428A)) were nucleofected separately or in combination with pcDNA 3.1myc-His myr-Akt3 into melan-a cells, seeded into PolyHEMA coated plates and cell lysates harvested 3 days later. Various HA-tagged B-Raf proteins were immunoprecipitated from 20-40 μg of total lysates using 0.1 μg of antibody against HA using Gamma Bind G sepharose. Samples were separated on NuPAGE gels for Western blot analysis and probed with anti-Hemagglutinin and anti-phosphorylated-Akt substrate monoclonal antibody (Cell Signaling, Danvers, MA). Western blot images were scanned and quantified using the Image Gauge 4.0 software (Fuji Photo Film Co., Tokyo, Japan).
Animal Studies
siRNA (112.5 pmoles for Akt3, C-Raf and scrambled; 12.5 pmoles for V600EB-Raf together with 100 pmoles of scrambled siRNA; or 12.5 pmoles for V600EB-Raf together with 100 pmoles of Akt3 siRNA) was nucleofected into 1×106 UACC 903 cells. 36-hours later, 1×106 UACC 903 cells in 0.2 ml of DMEM solution were injected subcutaneously into the left and right flanks of 4-6 week old nude mice. Dimensions of developing tumors were measured on alternate days up to day 17.5.
Statistical Analysis
One-way or Two-way Analysis of Variance (ANOVA) was used for groupwise comparisons, followed by the Tukey’s or Bonferroni post hoc test. Results were considered significant at a p-value of < 0.05. Chou-Talalay method (Calcusyn software) was used for determining the combination Index (Biosoft, Cambridge, United Kingdom) (43).
RESULTS
PI3k pathway inhibition increases MAP kinase activity in melanomas but not normal melanocytes
In melanomas, the PI3k pathway is activated through increased Akt3 expression and/or PTEN loss (7, 39). The MAP kinase pathway is primarily activated through mutation of B-Raf to a constitutively active V600E form (7, 21, 22, 24, 39, 44). Whether these pathways cooperate in early melanoma development remains an unanswered question. To investigate this possibility, activation of each pathway in melanocytes, having normal levels of Akt3 activity and wild type B-Raf, was compared to UACC 903 melanoma cells having increased Akt3 activity as well as constitutively active V600EB-Raf, following growth factor starvation and stimulation. Densitometric analysis of Western blots quantified pErk and pAkt levels, which estimates MAP and PI3 kinase pathway activity respectively, and were plotted as a continuum from 0 to 180 minutes following serum stimulation (Figs. 1A & 1B). Note, profiles represent best-fit lines showing minor perturbations due to graphical software. In starved melanocytes, activity levels of both pathways were initially low (Fig. 1A), however, upon growth factor stimulation, activity of both pathways increases transiently and declined over time to basal levels due to receptor desensitization, as reported for other model systems (34). In contrast, UACC 903 cells, which had significantly higher basal activity than melanocytes showed in Fig. 1B an increase in pAkt accompanied by a corresponding decrease in MAP kinase pathway activity determined by measuring pErk (7, 14, 21). Following serum stimulation, activity of both pathways in UACC 903 cells increased; however, as observed with other cell systems, growth factor stimulation led to transient increases in activity of both pathways which declined over time (34). This decrease was not observed following Akt inhibition using LY-294002; rather, pErk plateaued at high levels. Representative Western blots showing pErk and pAkt levels 30 and 60 minutes after growth factor or serum stimulation are shown in Fig. 1C. While melanocytes showed no increase in pErk levels following inhibition of Akt using LY-294002, inhibition increased pErk levels for both UACC 903 and SK-MEL-24 cells lines (Fig. 1C). Different serum stimulation times that corresponded to the peak pAkt/Erk levels for each cell line were selected in order to easily observe changes in phosphorylation following agent manipulation (34). Thus, inhibition of PI3k pathway increased MAP kinase activity suggesting inhibitory cross-regulation between these pathways in melanoma cells but not melanocytes.
Figure 1.




Crosstalk between the Akt3 and MAP Kinase pathway occurs in melanoma cells but not normal melanocytes. Phosphorylated Akt and Erk levels of melanocytes (A.) and UACC 903 (B.) treated with or without LY-294002. Graphs represent densitometric scans of pAkt and pErk Western blots normalized to a α-enolase loading control. Cells were starved 24 hours, treated with or without LY-294002 for 20 minutes, then serum stimulated before harvesting lysates at different time points for Western blot analysis. Analysis shows LY-294002 abrogates activation of Akt (pAkt) and consequently increase Erk activation (phosphorylation) in melanoma cells but not in human melanocytes. C, Inhibition of Akt activation results in higher pErk levels in melanoma cell lines but not in melanocytes. Western blots are shown for pAkt and pErk levels in normal human melanocytes and two human metastatic melanoma cell lines, UACC 903 and SK-MEL-24. Cells were starved 24 hours, then untreated or treated with DMSO or 50 mM LY-294002 for 20 minutes, and then stimulated with growth factors or serum in media and protein lysates harvested for Western blot analysis. The two metastatic melanoma cell lines showed evidence that LY-294002 treatment increased pErk levels, which did not occur in normal human melanocytes. D, Inhibition of Akt3 increased pErk levels in melanoma cells. Normal melanocytes were transfected with scrambled siRNA, or siRNA targeting PTEN or Akt3 alone or in combination. After 48 h, cell lysates were analyzed for PTEN, Akt3, pAkt and pErk levels. Inhibiting Akt3 protein expression has no effect on pErk levels. UACC 903 cells were transfected with buffer, scrambled siRNA, or Akt3 targeting siRNA, starved 24 hours, then stimulated with media containing serum for 30 or 60 minutes and protein lysates harvested for Western blot analysis. siRNA-mediated inhibition of Akt3 increased pErk levels compared to buffer or scrambled siRNA controls. α-enolase served as a control for protein loading.
Akt3 kinase regulates MAP kinase pathway activity
To establish whether Akt3 in the PI3k signaling cascade downregulated MAP kinase pathway activity, siRNA targeting Akt3 was introduced into UACC 903 cells to inhibit protein expression and cells starved, then stimulated with serum followed by measurement of pAkt/Erk levels. Reducing Akt3 protein expression (activity) in UACC 903 cells caused a corresponding decrease in pAkt levels (Fig. 1D). In contrast, no change was observed in levels of pErk in melanocytes following siRNA-mediated knockdown of Akt3, similar to results observed following pharmacological inhibition of Akt signaling in melanocytes (Fig. 1C). Significantly, decreased Akt3 activity caused a corresponding increase in MAP kinase activity observed by measuring pErk1/2 levels. Thus, Akt3 regulated activity of the MAP kinase pathway.
Presence of V600EB-Raf and absence of constitutively activated Akt3 increased MAP kinase activity
The interactive roles of Akt3 and mutant V600EB-Raf in melanoma development were evaluated measuring in vitro anchorage independent growth of WM35 cells. Cells were derived from an early stage melanocytic lesion in the radial growth phase containing the (T1799A) B-Raf mutation (19). However, unlike UACC 903 cells, WM35 cells have low endogenous Akt3 activity due to presence of functional PTEN (7). Thus, WM35 cells are similar to ∼90% of human nevi containing V600EB-Raf and having low Akt3 activity (6, 7, 14, 15, 44), making it a suitable model for evaluating the role played by Akt3 in melanocytic tumor progression. Densitometric analysis of Western blots showed pErk/Akt levels in WM35 cells following stimulation with growth factors after 24 hours starvation (Fig. 2A). Upon growth factor stimulation, activity of both pathways increased transiently in WM35 cells and, over time, pAkt declined to basal levels due to receptor desensitization similar to that observed for melanocytes since both cell types contain PTEN (19, 34). In contrast, levels of pErk did not decline to near baseline levels, rather pErk levels remained at near maximal amounts for duration of the experiment due to V600EB-Raf constitutively activating the downstream MAP kinase pathway (Figs. 1A and 2A). Thus, presence of V600EB-Raf and absence of constitutively activated Akt3 led to high MAP kinase activity, with potential to retard tumor progression.
Figure 2.




V600EB-Raf and Akt3 regulate anchorage independent growth of early melanoma cells. A, WM35 has constitutive activation of MAP kinase pathway signaling due to presence of V600EB-Raf but normal Akt activity. Densitometric quantitation of pAkt and pErk levels from Western blots of the WM35 cell line following 24 hours serum starvation followed by stimulation, shows constitutively high levels of pErk despite serum starvation, which is caused by V600EB-Raf activating the MAP kinase signaling cascade. In contrast, the Akt activation profile is similar to that observed for melanocytes. B, Inhibition of pAkt in WM35 cells using LY-294002 resulted in higher pErk while expression of active Akt3 decreased pErk levels. Treatment with LY-294002 led to a reduction in pAkt and a corresponding increase in pErk levels. Ectopic expression of active Akt3 decreased pErk levels. WM35 cells were nucleofected with Akt3 expressing constructs and following two days recovery, cells were starved 24 hours, and then stimulated with serum containing media and protein lysates collected for Western blot analysis. Ectopic myristoylated-Akt3 expression led to decreased pErk levels. C, Inhibition of B-Raf decreases anchorage independent growth of early melanoma cells. WM35 cells were nucleofected with siRNA targeting V600EB-Raf (100 and 200 pmoles), scrambled siRNA, or a water control. Cells were then plated onto PolyHEMA coated 96 well plates to measure effect on anchorage independent growth. MTS assay, 3 days later, was used to quantify viable cell number. Western blot showed efficient knockdown of B-Raf protein expression and decreased pErk levels. α-enolase served as a control for protein loading. D, Ectopic expression of active Akt3 enhanced anchorage independent growth. Viability of WM35 cells nucleofected with Akt3 is shown along with Western blot analysis of cell lysates. WM35 cells were nucleofected with various Akt3 expressing constructs: empty vector, WT-Akt3, Dead-Akt3 and active Akt3: myristoylated-Akt3 (myr-Akt3) or E40K-Akt3 alone or in combination with a siRNA targeting PRAS40 protein. After nucleofection, cells were plated directly onto PolyHEMA coated 96 well plates and following 3 days of growth in selection media, an MTS assay was used to quantify cell viability. Myr-Akt3 significantly increased cell growth compared to less active E40K-Akt3 that had a more modest effect. Changes in cell growth corresponded directly to pAkt level, which is shown by Western blot analysis and quantified by densitometry. Knocking down PRAS40 had no significant effect on cell viability. HA probed Western blot shows equal ectopic expression of the different Akt3 proteins.
Regulating MAP kinase pathway activity by targeting Akt3
Regulation of V600EB-Raf activity by Akt3 was examined using pharmacological inhibition of Akt or expression of active myristoylated Akt3 in WM35 cells to examine effect on pErk/Akt levels. While WM35 cells have low levels of pAkt compared to UACC 903 cells, inhibition of Akt using LY-294002 decreased levels of pAkt causing a corresponding increase in pErk (Fig. 2B), similar to that observed for advanced melanoma cell lines (Fig. 1C). Since WM35 cells had levels of pAkt similar to those observed in normal melanocytes, constructs ectopically expressing constitutively active Akt3 were used to produce pAkt levels similar to those observed in more aggressive melanomas to confirm Akt3-mediated regulation of MAP kinase pathway activity. Western blot analysis showed increased pAkt accompanied by a corresponding decrease in pErk following ectopic expression of active myristoylated Akt3 but not dead Akt3 (control producing inert protein) expression (Fig. 2B). Thus, cross regulation between Akt3 and MAP kinase pathway activity occur with active Akt3.
V600EB-Raf and Akt3 promote anchorage independent growth
Effect of V600EB-Raf and Akt3 expression on melanocytic transformation was measured by regulating expression of each protein and quantifying anchorage independent growth (a phenotypic measurement of cell transformation). WM35 cells were nucleofected with siRNA against B-Raf and seeded onto PolyHEMA coated 96-well plates. A significant decrease of viable WM35 cells was observed following nucleofection with B-Raf siRNA compared to a scrambled control (Fig. 2C, p<0.05; One-way ANOVA). Western blot analysis confirmed B-Raf protein knockdown and corresponding decrease in downstream pErk (Fig. 2C). Next, myristoylated-Akt3 having high activity or the E40K-Akt3 mutant having activity in the normal physiological range was expressed in WM35 cells containing endogenous V600EB-Raf to determine whether it could enhance anchorage independent growth (45). Akt3 expression significantly enhanced anchorage independent growth compared to vector controls, which was directly proportional to level of pAkt (Fig. 2D, p<0.001; One-way ANOVA). Expression of wild type protein had lowest pAkt and least effect on anchorage independent growth. To demonstrate that Akt3 mediated anchorage independent growth of WM35 cells was not due to decreased apoptosis reported in advanced melanoma (7, 9), the downstream target of Akt3 regulating apoptosis, called PRAS40, was knocked down using siRNA while active Akt3 was expressed (9). Similar anchorage independent growth was observed irrespective of levels of PRAS40 expression (Fig. 2D). Thus, deregulated apoptosis mediated by active Akt3 was not leading to enhanced anchorage independent growth. This raised the possibility that Akt3 could be interacting with V600EB-Raf to regulate anchorage independent growth since Akt has been reported to downregulate B-Raf activity through direct phosphorylation (34-36). Thus, V600EB-Raf and Akt3 mediate anchorage independent growth in the WM35 cell model and potentially cooperate in melanocyte transformation.
Akt3 cooperates with V600EB-Raf to transform melanocytes
Since Akt3 and V600EB-Raf independently regulated anchorage independent growth of WM35 cells, effect of ectopic co-expression on melanocyte transformation was examined using melan-a mouse melanocytes to assess transformation by anchorage independent growth. This model was chosen since melan-a cells undergo transformation following low level V600EB-Raf expression (46, 47). Ectopically expressed V600EB-Raf or active-Akt3 alone, promoted anchorage independent growth compared to empty vector (p<0.001, Fig. 3A). Numerous groups have reported the oncogenic properties of myr-Akt3 (48). However, a limitation is that the activity resulting from myr-Akt3 is very high, which could be an explanation for cell transformation. Therefore, we incorporated the E40K Akt3 mutant that leads to more moderate physiological pAkt levels similar to those present in more aggressive melanoma cells. Co-expression of V600EB-Raf with active E40K- or myr-Akt3 led to statistically significant enhanced anchorage independent growth compared to single genes (Lanes 5 vs 2 or 3, p<0.05; Lanes 6 vs. 2 or 4, p<0.001; Fig. 3A). Western blot analysis of cell lysates showed that active myr-Akt3 reduced levels of pErk accompanying V600EB-Raf expression (Fig. 3A). A similar but less significant effect was seen following E40K Akt3 coexpression with V600EB-Raf (Fig. 3A). These results suggest that active Akt3 and V600EB-Raf cooperate in melanocyte transformation, promoting anchorage independent growth.
Figure 3.


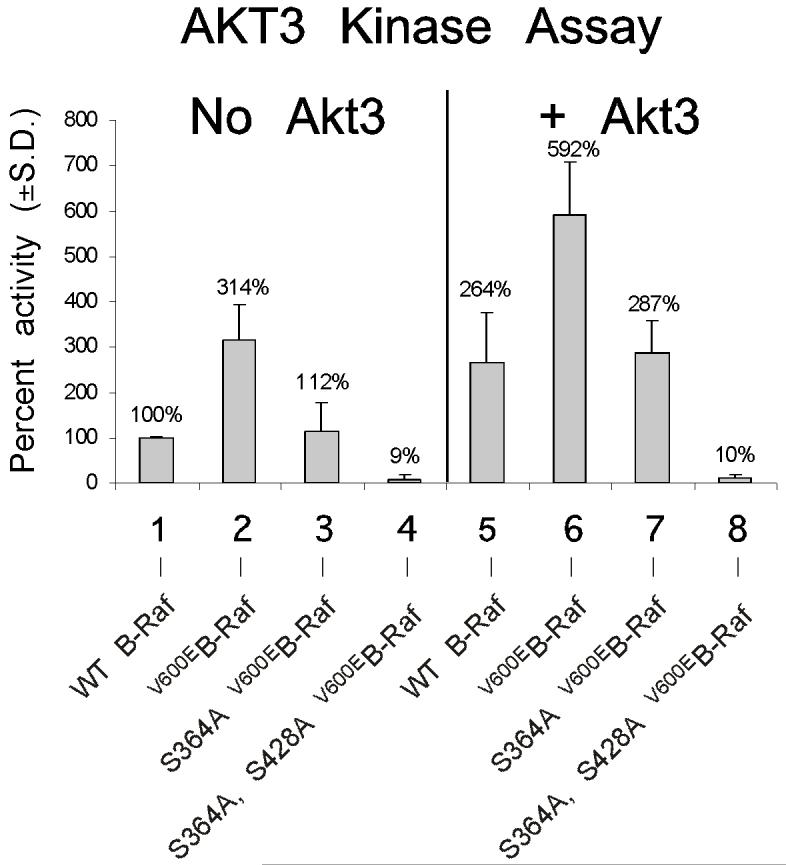
V600EB-Raf and Akt3 cooperate in melanocyte transformation. A, Melan-a transformation, following ectopic V600EB-Raf and Akt3 expression, was quantified by determined effect on anchorage independent growth. Melan-a cells were nucleofected with various combinations of V600EB-Raf and Akt3 expression plasmids (resistant to blasticidin and puromycin, respectively) and then directly plated onto PolyHEMA coated 96 well plates. Transfected cells were selected for blasticidin and puromycin resistance for three days followed by MTS assay to quantify cell viability. Results of a representative MTS assay are shown indicating effects of V600EB-Raf and Akt3 expression on anchorage independent growth. Nucleofections of either V600EB-Raf or myr-Akt3 led to increased cell viability; however, co-expression of both genes resulted in a more statistically significant effect. Western blots indicate changes in signaling pathways following ectopic expression of B-Raf and Akt3. Error bars; mean ± S.D. B, In vitro B-Raf kinase assay showing activity of V600EB-Raf compared to protein in which phosphorylation sites are converted to alanine S364A and S428A. Upper blot show autoradiography film of 32P labeled Mek substrate while lower shows a Western blot quantifying the amount of immunoprecipitated B-Raf protein used for the kinase assay. The graph represents average densitometry analysis (±S.D.) of three independent experiments, where values indicate level of labeled Mek normalized to the amount of HA B-Raf, relative to Dead B-Raf set as the background control. C, Akt3 phosphorylates V600EB-Raf on serines 364 and 428. Quantitation of B-Raf phosphorylation using densitometric scans of Western blot that were normalized to total immunoprecipitated HA B-Raf. Both wild type and V600EB-Raf were phosphorylated by Akt3. Conversion of serine 364 to alanine reduced levels of V600EB-Raf phosphorylation, while mutation of both serines 364 and 428 completely abrogated phosphorylation by Akt3. Graph is the average of three independent experiments; error bars, mean ±S.D.
Akt3 kinase phosphorylates V600EB-Raf to lower in vitro activity
To determine whether phosphorylation mediated by Akt3 regulated V600EB-Raf activity, serine residues 364 and/or 428 in V600EB-Raf were converted to alanine. Plasmids expressing these B-Raf constructs were co-nucleofected with myr-Akt3 into melan-a cells to promote phosphorylation of ectopically expressed B-Raf proteins on serines 364 and 428. V600EB-Raf constructs containing alanine at residues 364 and/or 428 could not be phosphorylated by Akt3. Various HA-tagged B-Raf proteins were then immunoprecipitated and activity measured using an in vitro kinase assay with Mek as the substrate (Fig. 3B). AA-V600EB-Raf (S364A, S428A) exhibited highest level of activity compared to V600EB-Raf, V600EB-Raf (S364A) or V600EB-Raf (S428A) (p<0.05; Fig. 3B) demonstrating that direct phosphorylation of V600EB-Raf protein by Akt3 decreased its activity. Thus, phosphorylation of V600EB-Raf has significant potential to decrease activity of the mutant protein.
Active Akt3 phosphorylates V600EB-Raf in melanocytic cells
To demonstrate that Akt3 phosphorylates mutant V600EB-Raf in melanocytic cells, melan-a melanocytes were transfected with HA-tagged V600EB-Raf, V600EB-Raf (S364A), or AA-V600EB-Raf (S364A, S428A) alone or with myr-Akt3. Western blot analysis using an antibody recognizing consensus sequences typically phosphorylated by Akt was used to quantify levels of phosphorylation of immunoprecipitated HA tagged B-Raf from cells (Fig. 3C). V600EB-Raf (S428A) was not used since the antibody does not recognize this site. Increased phosphorylation of V600EB-Raf occurred when coexpressed with active Akt3 (p<0.01; Fig. 3C). Mutation of serine 364 to alanine (V600EB-Raf (S364A)) decreased pB-Raf levels (p<0.01; Fig.3C). Furthermore, mutation of both serine 364 and serine 428 to alanines (AA-V600EB-Raf (S364A, S428A)) completely blocked phosphorylation by Akt3 (p<0.01; Fig. 3C). Thus, active Akt3 is capable of phosphorylating V600EB-Raf in melanocytic cells.
Phosphorylation of V600EB-Raf decreases MAP kinase pathway signaling overcoming inhibitory growth effects associated with high activity
Effect of phosphorylated versus unphosphorylated V600EB-Raf on melanocytic transformation was examined by expressing V600EB-Raf or AA-V600EB-Raf (S364A, S428A) alone or with active Akt3 in melan-a cells and measuring anchorage independent growth. Compared to V600EB-Raf expression alone, AA-V600EB-Raf (S364A, S428A) protein significantly decreased cell proliferation (p<0.001; Fig. 4A). Similarly, co-expression of AA-V600EB-Raf (S364A, S428A) with myristoylated or E40K Akt3 also significantly reduced proliferation when compared to cells expressing V600EB-Raf or Akt3 (Fig. 4A, P<0.001). Western blot analysis of cell lysates showed AA-V600EB-Raf (S364A, S428A) expression accompanied by higher MAP kinase pathway activity than occurred with V600EB-Raf (Fig. 4B). Furthermore, co-expression of AA-V600EB-Raf (S364A, S428A) with myristoylated-Akt3 did not lower pErk levels as observed when V600EB-Raf and myristoylated Akt3 were co-expressed (Fig. 4B). Induction of cyclin D1 was also impaired following AA-V600EB-Raf (S364A, S428A) expression (Fig. 4B). Thus, phosphorylation of V600EB-Raf by Akt3 decreased MAP kinase signaling overcoming growth inhibitory effects associated with high pathway activity.
Figure 4.

V600EB-Raf and Akt3 mediated transformation requires Akt3 phosphorylation of V600EB-Raf. A, Representative graph showing anchorage independent growth mediated by V600EB-Raf and Akt3 coexpression. Melan-a cells were nucleofected with different combinations of S364A/S428A V600EB-Raf (AA-V600EB-Raf) or V600EB-Raf and Akt3 expressing plasmids, and plated onto PolyHEMA coated 96 well plates. Transfected cells were then selected for three days for purimycin resistance followed by MTS assay to measure cell viability. Conversion of the two serine phosphorylation sites on V600EB-Raf to alanines (S364A/S428A) led to significantly decreased anchorage independent growth mediated by V600EB-Raf and Akt3 expression; error bars, mean ±S.D. of four independent experiments. B, Western blots showing changes in signaling pathways following ectopic expression of B-Raf and Akt3. Increased cyclin D1 levels are associated with coexpression of V600EB-Raf and Akt3.
Targeting V600EB-Raf and Akt3 in melanomas cooperatively inhibits tumorigenesis
To evaluate the therapeutic potential of simultaneously inhibiting Akt3 and mutant V600EB-Raf in melanoma tumorigenesis, a siRNA was used to inhibit protein expression. Effects on tumor development was studied by nucleofecting siRNA against each target into UACC 903 cells, allowing 1.5 days of recovery in culture followed by subcutaneous injection of 1 million cells into nude mice. Reduced expression of each protein was observed for as long as 9.5 days after nucleofection in cultured cells and in tumors of nude mice (data not shown) (25). Size of developing tumors was measured using calipers every other day until day 17. Initially, siRNA against Akt3 and B-Raf was titered to establish optimal concentration inhibiting tumorigenesis to a similar extent. One hundred pmoles of Akt3 siRNA was found to inhibit tumorigenesis similar to that occurring with 12.5 pmoles of siRNA against B-Raf (Fig. 5A). In comparison to controls (buffer, scrambled siRNA and C-Raf siRNA), siRNA against Akt3 or V600EB-Raf significantly decreased the tumorigenic potential of the UACC 903 cells (Fig. 5A). An even more dramatic reduction in tumorigenesis was observed following simultaneous inhibition of both Akt3 and B-Raf. Under these conditions cooperatively acting tumor inhibition was observed (Fig. 5A, p<0.001; Two-way ANOVA). Tumors removed from mice 9.5 days after siRNA nucleofection were found to have reduced Akt3 (Fig. 5B) or V600EB-Raf (Fig. 5C) protein expression demonstrating effective knockdown of these proteins. Thus, targeting both Akt3 and B-Raf led to cooperatively acting tumor inhibition.
Figure 5.

Targeting V600EB-Raf and Akt3 dramatically retards melanoma tumorigenesis. A, Nucleofection of siRNA against both B-Raf and Akt3 into UACC 903 cells led to a statistically significant reduction of tumor development in animals. The cells were nucleofected with siRNA, allowed to recover in culture for 1.5 days, and then injected subcutaneously into nude mice. Tumor size was measured on alternate days up to day 17.5. Inhibition of Akt3 or B-Raf alone led to a significant reduction in growth compared to controls; however, targeting both proteins led to an even more dramatic reduction compared to targeting Akt3 or B-Raf individually. Bars represent mean ± S.E. B, Western blot of UACC 903 tumors isolated from mice 8 days after nucleofection with siRNA targeting V600EB-Raf. siRNA against B-Raf significantly reduced level of B-Raf protein compared to control cells nucleofected with siRNA to C-Raf. C, Western blot of UACC 903 tumors isolated from mice 8 days after nucleofection with siRNA targeting Akt3. Akt3 protein level was significantly reduced with siRNA targeting Akt3 compared to cells nucleofected with a scrambled siRNA control.
Simultaneously targeting V600EB-Raf and Akt3 inhibits anchorage independent growth
Since targeting Akt3 and V600EB-Raf led to cooperative tumor inhibition, mechanistic basis for inhibition was examined next. siRNA targeting Akt3 and/or V600EB-Raf was nucleofected into UACC 903 cells and effect on in vitro anchorage independent growth examined. Simultaneous siRNA-mediated inhibition of Akt3 and B-Raf dramatically inhibited growth of cells compared to either Akt3 or B-Raf (Fig 6A, Lanes 2, 4, and 7, and Lanes 2, 5 and 8; ANOVA, p<0.001) (49). To determine whether inhibition of anchorage independent growth was additive or synergistic following siRNA-mediated inhibition of Akt3 and V600EB-Raf the Chou-Talalay method for determining the combination index (CI) using Calcusyn software was undertaken (43). Using this approach, CI values < 0.9 are synergistic, > 1.1 are antagonistic, and values 0.9-1.1 are nearly additive. Experimentally, Akt3 siRNA was kept at a constant dose of 200 pmoles while the concentration of B-Raf siRNA was titrated at doses of 3, 6, and 12 pmoles. The resulting CI for each combination was calculated to be 0.909, 1.023, and 1.107, for 3, 6, and 12 pmoles of B-Raf siRNA respectively combined with 200 pmoles of Akt3 siRNA, which suggest nearly additive cooperation. Thus, simultaneous targeting V600EB-Raf and Akt3 inhibits anchorage independent growth in a cooperatively additive manner.
Figure 6.

Simultaneously targeting V600EB-Raf and Akt3 inhibits in vitro melanoma growth in an additively cooperating manner. A, In vitro anchorage independent growth of UACC 903 cells nucleofected with siRNA targeting V600EB-Raf and Akt3 in cell culture media containing 0% serum. UACC 903 cells nucleofected with siRNA were plated onto PolyHEMA coated 96 well plates, allowed to grow for 3 days in DMEM containing no serum and cell viability quantified by MTS assay. UACC 903 cells nucleofected with no siRNA (lane 1) was set as 100% and others compared to this value. Targeting both B-Raf and Akt3 resulted in statistically significant inhibition of growth compared to cells in which B-Raf or Akt3 had been targeted individually. B, Western blots showing effects of targeting V600EB-Raf and Akt3 on cyclin D1, cleaved caspase-3 and p27Kip1 protein levels. siRNA-mediated inhibition of V600EB-Raf and Akt3 led to significant reductions in cyclin D1 and corresponding increases in cleaved caspase3 and p27Kip1 protein levels compared to cells in which proteins had been targeted individually.
Simultaneously targeting both Akt3 and V600EB-Raf enhances growth inhibition and apoptotic effects on signaling cascades
Mechanistic basis for inhibition of anchorage independent growth following V600EB-Raf and Akt3 targeting was examined using Western blotting to measure levels of protein expression involved in proliferation (cyclin D1) and apoptosis (cleaved caspase-3). Control UACC 903 cells had high cyclin D1 and low cleaved caspase-3 levels (Lane 1, Fig 6B). In contrast, siRNA-mediated inhibition of Akt3 increased cleaved caspase-3 levels indicating increased cellular apoptosis (Lane 2, Fig 6B). B-Raf siRNA did not significantly alter levels of cleaved caspase 3 but did significantly decreased cyclin D1 levels with a corresponding increase in p27 (Fig 6B, Lanes 3 to 5). Significantly, inhibition of both Akt3 and mutant V600EB-Raf dramatically increased levels of cleaved caspase 3 indicative of high apoptosis compared to cells in which Akt3 alone had been inhibited (Fig. 6B; Lanes 6, 7, and 8 versus lane 2). Furthermore, the combination approach led to more dramatic decrease in cyclin D1 than observed when V600EB-Raf was inhibited alone. Thus, simultaneously targeting both Akt3 and V600EB-Raf has a more dramatic effect on the signaling cascades of these pathways than inhibiting each individually.
DISCUSSION
Nevi or moles are benign proliferations of melanocytes that initially proliferated and then stopped growing (30). Approximately 90% harbor activating B-Raf mutations, but never progress into melanomas (12, 14, 15). This is caused by cellular senescence associated with high MAP kinase pathway activity due to presence of V600EB-Raf, which is linked to induction of cyclin dependent kinase inhibitors, such as p21Cip1, p16Ink4a, and p27Kip1, functioning as a defense mechanism of normal cells to overcome oncogene activation (26, 28, 29). In contrast, moderate levels of MAP kinase pathway activation promote cell cycle progression and cancer development (27). Therefore cells containing V600EB-Raf require additional genetic changes for senescent melanocytic cells to reenter the cell cycle (31).
Identification of proteins cooperating with V600EB-Raf in melanocytic transformation are being characterized by studying nevi development using transgenic zebrafish (30). Prolonged hyperactivation of MAP kinase signaling in zebrafish melanocytes mediated by V600EB-Raf initially cause promote cellular growth and development of structures resembling nevi followed by a senescent phase; a situation comparable to that occurring with most human moles (30). Zebrafish nevi did not develop into melanomas until p53 was deleted, suggesting deregulation of multiple pathways for melanocytic tumor progression (30). Thus, genes such as Akt3 with prominent roles development ∼70% of melanomas likely cooperate with V600EB-Raf in melanocytic transformation (7, 9, 39, 44, 50).
Akt had been shown to regulate B-Raf activity (34, 35); however, it was unknown whether Akt3 would phosphorylate mutant V600EB-Raf, and if so, whether this process played any role in melanoma development. In this report, using a combination of pharmacological and genetic approaches, Akt3 was shown to downregulate V600EB-Raf mediated activation of MAP kinase pathway signaling in order to promote tumor progression. Ectopic expression of active Akt3 in early melanoma cells, phenotypically resembling nevi cells by containing V600EB-Raf with low endogenous Akt3 activity, increased anchorage independent growth. Furthermore, expression of both proteins in melanocytes promoted anchorage independent growth, demonstrating that V600EB-Raf and active Akt3 act cooperatively together to promote melanocyte transformation; thereby, confirming the importance of these proteins in early melanoma development. Mechanistically, Akt3 was shown to directly phosphorylate B-Raf on serine residues 364 and 428 to regulate activity of mutant V600EB-Raf protein in order to promote melanocyte transformation (41). This second role for Akt in melanoma development was confirmed by inhibiting the downstream target of Akt3 regulating apoptosis, called PRAS40 (9). Constitutively active Akt3 was expressed to activate the signaling cascade, while siRNA-mediated knockdown was used to inhibit PRAS40 expression/activity. Since similar anchorage independent growth was observed irrespective of levels of PRAS40 expression, deregulated apoptosis mediated by active Akt3 was not leading to enhanced anchorage independent growth. Thus, a second important role has been identified for Akt3 in melanoma development.
Melan-a mouse melanocytes used as a model of transformation are considered to be a normal mouse melanocyte cell line and is an accepted model for studying V600EB-Raf mediated transformation (47). However, it lacks p16INK4a as well as ARF protein expression. Data presented in this study show that melan-a transformation mediated by V600EB-Raf expression in a p16INK4a /ARF null background requires expression of active Akt3 to promote anchorage independent growth. This suggests that not only was loss of p16INK4a /ARF important in bypassing V600EB-Raf induced senescence, but Akt3 activation was required to phosphorylate V600EB-Raf in order to modulate the level of MAP kinase activity.
The therapeutic potential of simultaneously targeting Akt3 and V600EB-Raf is also demonstrated in this study. Tumor inhibition was most significant in animals containing cells in which both proteins had been targeted. In vitro studies confirmed the effects of the combination approach demonstrating that targeting both led to cooperatively acting inhibition significantly increasing cellular apoptosis and decreasing proliferation. Thus, therapeutics targeting both pathways in human patients is predicted to be more effective than targeting each individually.
In conclusion, Akt3 has been shown to cooperate with V600EB-Raf in promoting melanoma development by phosphorylating the mutant protein in order to decrease its activity to levels that promote rather than inhibit melanocytic cell growth. Furthermore, simultaneously targeting both Akt3 and V600EB-Raf was more effective at inhibiting melanomas that targeting each individually, which suggest that therapies for human patients should simultaneously target these signaling pathways for maximal clinical efficacy.
Acknowledgments
Grant support: The Melanoma Research Foundation, The American Cancer Society (RSG-04-053-01-GMC), The Foreman Foundation for Melanoma Research, NIH grant CA-127892-01A1, and Elsa U. Pardee Foundation
REFERENCES
- 1.Cancer Facts and Figures. American Cancer Society; Atlanta: 2005. [Google Scholar]
- 2.Smalley KS, Herlyn M. Targeting intracellular signaling pathways as a novel strategy in melanoma therapeutics. Ann N Y Acad Sci. 2005;1059:16–25. doi: 10.1196/annals.1339.005. [DOI] [PubMed] [Google Scholar]
- 3.Hsu MY, Meier F, Herlyn M. Melanoma development and progression: a conspiracy between tumor and host. Differentiation. 2002;70:522–36. doi: 10.1046/j.1432-0436.2002.700906.x. [DOI] [PubMed] [Google Scholar]
- 4.Chudnovsky Y, Khavari PA, Adams AE. Melanoma genetics and the development of rational therapeutics. J Clin Invest. 2005;115:813–24. doi: 10.1172/JCI24808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kantrow SM, Boyd AS, Ellis DL, et al. Expression of activated Akt in benign nevi, Spitz nevi and melanomas. J Cutan Pathol. 2007;34:593–6. doi: 10.1111/j.1600-0560.2006.00675.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dai DL, Martinka M, Li G. Prognostic significance of activated Akt expression in melanoma: a clinicopathologic study of 292 cases. J Clin Oncol. 2005;23:1473–82. doi: 10.1200/JCO.2005.07.168. [DOI] [PubMed] [Google Scholar]
- 7.Stahl JM, Sharma A, Cheung M, et al. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 2004;64:7002–10. doi: 10.1158/0008-5472.CAN-04-1399. [DOI] [PubMed] [Google Scholar]
- 8.DeFeo-Jones D, Barnett SF, Fu S, et al. Tumor cell sensitization to apoptotic stimuli by selective inhibition of specific Akt/PKB family members. Mol Cancer Ther. 2005;4:271–9. [PubMed] [Google Scholar]
- 9.Madhunapantula SV, Sharma A, Robertson GP. PRAS40 deregulates apoptosis in malignant melanoma. Cancer Res. 2007;67:3626–36. doi: 10.1158/0008-5472.CAN-06-4234. [DOI] [PubMed] [Google Scholar]
- 10.Brose MS, Volpe P, Feldman M, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Research. 2002;62:6997–7000. [PubMed] [Google Scholar]
- 11.Reifenberger J, Knobbe CB, Sterzinger AA, et al. Frequent alterations of Ras signaling pathway genes in sporadic malignant melanomas. International Journal of Cancer. 2004;109:377–84. doi: 10.1002/ijc.11722. [DOI] [PubMed] [Google Scholar]
- 12.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 13.Dong J, Phelps RG, Qiao R, et al. BRAF oncogenic mutations correlate with progression rather than initiation of human melanoma. Cancer Res. 2003;63:3883–5. [PubMed] [Google Scholar]
- 14.Miller CJ, Cheung M, Sharma A, et al. Method of mutation analysis may contribute to discrepancies in reports of (V599E)BRAF mutation frequencies in melanocytic neoplasms. J Invest Dermatol. 2004;123:990–2. doi: 10.1111/j.0022-202X.2004.23468.x. [DOI] [PubMed] [Google Scholar]
- 15.Pollock PM, Harper UL, Hansen KS, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- 16.Uribe P, Wistuba II, Gonzalez S. BRAF mutation: a frequent event in benign, atypical, and malignant melanocytic lesions of the skin. Am J Dermatopathol. 2003;25:365–70. doi: 10.1097/00000372-200310000-00001. [DOI] [PubMed] [Google Scholar]
- 17.Lang J, Boxer M, MacKie R. Absence of exon 15 BRAF germline mutations in familial melanoma. Human Mutation. 2003;21:327–30. doi: 10.1002/humu.10188. [DOI] [PubMed] [Google Scholar]
- 18.Laud K, Kannengiesser C, Avril MF, et al. BRAF as a melanoma susceptibility candidate gene? Cancer Research. 2003;63:3061–5. [PubMed] [Google Scholar]
- 19.Meyer P, Klaes R, Schmitt C, Boettger MB, Garbe C. Exclusion of BRAFV599E as a melanoma susceptibility mutation. Int J Cancer. 2003;106:78–80. doi: 10.1002/ijc.11199. [DOI] [PubMed] [Google Scholar]
- 20.Tuveson DA, Weber BL, Herlyn M. BRAF as a potential therapeutic target in melanoma and other malignancies. Cancer Cell. 2003;4:95–8. doi: 10.1016/s1535-6108(03)00189-2. [DOI] [PubMed] [Google Scholar]
- 21.Sharma A, Trivedi NR, Zimmerman MA, Tuveson DA, Smith CD, Robertson GP. Mutant V599EB-Raf regulates growth and vascular development of malignant melanoma tumors. Cancer Res. 2005;65:2412–21. doi: 10.1158/0008-5472.CAN-04-2423. [DOI] [PubMed] [Google Scholar]
- 22.Hingorani SR, Jacobetz MA, Robertson GP, Herlyn M, Tuveson DA. Suppression of BRAF(V599E) in human melanoma abrogates transformation. Cancer Res. 2003;63:5198–202. [PubMed] [Google Scholar]
- 23.Karasarides M, Chiloeches A, Hayward R, et al. B-RAF is a therapeutic target in melanoma. Oncogene. 2004;23:6292–8. doi: 10.1038/sj.onc.1207785. [DOI] [PubMed] [Google Scholar]
- 24.Sumimoto H, Miyagishi M, Miyoshi H, et al. Inhibition of growth and invasive ability of melanoma by inactivation of mutated BRAF with lentivirus-mediated RNA interference. Oncogene. 2004;23:6031–9. doi: 10.1038/sj.onc.1207812. [DOI] [PubMed] [Google Scholar]
- 25.Sharma A, Tran MA, Liang S, et al. Targeting Mitogen-Activated Protein Kinase/Extracellular Signal-Regulated Kinase Kinase in the Mutant (V600E) B-Raf Signaling Cascade Effectively Inhibits Melanoma Lung Metastases. Cancer Res. 2006;66:8200–9. doi: 10.1158/0008-5472.CAN-06-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McMahon M, Woods D. Regulation of the p53 pathway by Ras, the plot thickens. Biochim Biophys Acta. 2001;1471:M63–71. doi: 10.1016/s0304-419x(00)00027-5. [DOI] [PubMed] [Google Scholar]
- 27.Woods D, Parry D, Cherwinski H, Bosch E, Lees E, McMahon M. Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol Cell Biol. 1997;17:5598–611. doi: 10.1128/mcb.17.9.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kolch W, Kotwaliwale A, Vass K, Janosch P. The role of Raf kinases in malignant transformation. Expert Rev Mol Med. 2002;2002:1–18. doi: 10.1017/S1462399402004386. [DOI] [PubMed] [Google Scholar]
- 29.Chang F, Steelman LS, Shelton JG, et al. Regulation of cell cycle progression and apoptosis by the Ras/Raf/MEK/ERK pathway (Review) Int J Oncol. 2003;22:469–80. [PubMed] [Google Scholar]
- 30.Michaloglou C, Vredeveld LC, Soengas MS, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–4. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 31.Bennett DC. Human melanocyte senescence and melanoma susceptibility genes. Oncogene. 2003;22:3063–9. doi: 10.1038/sj.onc.1206446. [DOI] [PubMed] [Google Scholar]
- 32.Bouchard C, Thieke K, Maier A, et al. Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. Embo J. 1999;18:5321–33. doi: 10.1093/emboj/18.19.5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patton EE, Widlund HR, Kutok JL, et al. BRAF mutations are sufficient to promote nevi formation and cooperate with p53 in the genesis of melanoma. Curr Biol. 2005;15:249–54. doi: 10.1016/j.cub.2005.01.031. [DOI] [PubMed] [Google Scholar]
- 34.Moelling K, Schad K, Bosse M, Zimmermann S, Schweneker M. Regulation of Raf-Akt Cross-talk. J Biol Chem. 2002;277:31099–106. doi: 10.1074/jbc.M111974200. [DOI] [PubMed] [Google Scholar]
- 35.Rommel C, Clarke BA, Zimmermann S, et al. Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science. 1999;286:1738–41. doi: 10.1126/science.286.5445.1738. [DOI] [PubMed] [Google Scholar]
- 36.Zimmermann S, Moelling K. Phosphorylation and regulation of Raf by Akt (protein kinase B) Science. 1999;286:1741–4. doi: 10.1126/science.286.5445.1741. [DOI] [PubMed] [Google Scholar]
- 37.Satyamoorthy K, DeJesus E, Linnenbach AJ, et al. Melanoma cell lines from different stages of progression and their biological and molecular analyses. Melanoma Res. 1997;7(Suppl 2):S35–42. [PubMed] [Google Scholar]
- 38.Cook AL, Donatien PD, Smith AG, et al. Human melanoblasts in culture: expression of BRN2 and synergistic regulation by fibroblast growth factor-2, stem cell factor, and endothelin-3. J Invest Dermatol. 2003;121:1150–9. doi: 10.1046/j.1523-1747.2003.12562.x. [DOI] [PubMed] [Google Scholar]
- 39.Stahl JM, Cheung M, Sharma A, Trivedi NR, Shanmugam S, Robertson GP. Loss of PTEN promotes tumor development in malignant melanoma. Cancer Res. 2003;63:2881–90. [PubMed] [Google Scholar]
- 40.Brodbeck D, Hill MM, Hemmings BA. Two splice variants of protein kinase B gamma have different regulatory capacity depending on the presence or absence of the regulatory phosphorylation site serine 472 in the carboxyl-terminal hydrophobic domain. J Biol Chem. 2001;276:29550–8. doi: 10.1074/jbc.M104633200. [DOI] [PubMed] [Google Scholar]
- 41.Guan KL, Figueroa C, Brtva TR, et al. Negative regulation of the serine/threonine kinase B-Raf by Akt. J Biol Chem. 2000;275:27354–9. doi: 10.1074/jbc.M004371200. [DOI] [PubMed] [Google Scholar]
- 42.Erhardt P, Troppmair J, Rapp UR, Cooper GM. Differential regulation of Raf-1 and B-Raf and Ras-dependent activation of mitogen-activated protein kinase by cyclic AMP in PC12 cells. Mol Cell Biol. 1995;15:5524–30. doi: 10.1128/mcb.15.10.5524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 44.Dhawan P, Singh AB, Ellis DL, Richmond A. Constitutive activation of Akt/protein kinase B in melanoma leads to up-regulation of nuclear factor-kappaB and tumor progression. Cancer Res. 2002;62:7335–42. [PubMed] [Google Scholar]
- 45.Bellacosa A, Chan TO, Ahmed NN, et al. Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene. 1998;17:313–25. doi: 10.1038/sj.onc.1201947. [DOI] [PubMed] [Google Scholar]
- 46.Wellbrock C, Ogilvie L, Hedley D, et al. V599EB-RAF is an oncogene in melanocytes. Cancer Res. 2004;64:2338–42. doi: 10.1158/0008-5472.can-03-3433. [DOI] [PubMed] [Google Scholar]
- 47.Sviderskaya EV, Hill SP, Evans-Whipp TJ, et al. p16(Ink4a) in melanocyte senescence and differentiation. J Natl Cancer Inst. 2002;94:446–54. doi: 10.1093/jnci/94.6.446. [DOI] [PubMed] [Google Scholar]
- 48.Aoki M, Batista O, Bellacosa A, Tsichlis P, Vogt PK. The akt kinase: molecular determinants of oncogenicity. Proc Natl Acad Sci U S A. 1998;95:14950–5. doi: 10.1073/pnas.95.25.14950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Christensen C, Guldberg P. Growth factors rescue cutaneous melanoma cells from apoptosis induced by knockdown of mutated (V600E) B-RAF. Oncogene. 2005 doi: 10.1038/sj.onc.1208758. [DOI] [PubMed] [Google Scholar]
- 50.Robertson GP. Functional and therapeutic significance of Akt deregulation in malignant melanoma. Cancer Metastasis Rev. 2005;24:273–85. doi: 10.1007/s10555-005-1577-9. [DOI] [PubMed] [Google Scholar]