

Introduction
The signal transducers and activators of transcription (STATs) are a class of transcription factor proteins that regulate cell growth and survival by modulating the expression of specific target genes.[1] A total of seven different STAT isoforms, encoded in distinct genes, have been identified in mammalian cells. Stat3, a member of the STAT family, has been identified in an increasing number of tumor cell lines. Stat3 drives malignant progression through the misregulation of key proteins, including cell survival proteins such as Bcl-xL and Mcl-1, cell cycle regulators such as cyclin D1/D2 and c-myc, and inducers of angiogenesis such as vascular endothelial growth factor (VEGF).[2] In contrast to normal cells, where Stat3 activation is rapid and transient, neoplastic cells are found to display constitutive Stat3 activation that, once inhibited, correlates with suppression of both cell transformation and growth, and induction of apoptosis.[3-8] While STAT signaling is just one of many pathways compromised in oncogenesis, interruption of this pathway is sufficient to block cell transformation; this suggests that these cells have an irreversible dependence on constitutively active Stat3 for survival. Numerous reports highlight the relevance of persistent Stat3 activation in human cancers; abnormal levels of Stat3 activation have been observed in breast,[9,10] ovarian,[9] prostate,[11] haematological,[12] and head and neck cancer cell lines.[13] These investigations suggest that agents designed to disrupt Stat3 signaling hold considerable promise for the prevention and treatment of human cancers.
Junctures for Stat3 Molecular Intervention
STAT activation and transcriptional function relies on an intricate series of intracellular protein complexation events mediated by several phosphorylative processes. Structurally, the STATs incorporate a conserved N terminus, a DNA binding domain, and a Src-homology 2 (SH2) domain that is involved in both receptor recruitment and dimerization. The Stat3 signaling pathway is composed of multiple, distinct steps, affording several junctures for molecular intervention (Figure 1). Stat3 signaling involves the following steps: 1) cell stimulation by growth factors or cytokines, resulting in receptor dimerization and activation; 2) phosphorylation of the receptor’s cytoplasmic tail, providing docking sites for the recruitment of monomeric, non-phosphorylated STAT proteins via their SH2 domains; 3) activated tyrosine kinases (JAK) phosphorylate recruited STAT proteins at a specific tyrosine near the C terminus; 4) phosphorylated STAT proteins are released from the receptor, and homodimerization occurs through a reciprocal phosphotyrosine-SH2 interaction; 5) STAT dimers translocate to the nucleus, and form STAT-DNA complexes. Each of these five steps towards the activation of functional Stat3 offers a unique opportunity to inhibit aberrant Stat3 activity.
Figure 1.

JAK/STAT signaling pathway. STAT activity, arrowheaded lines; inhibitory events, barred lines. Abbreviations: Ub, ubiquitin; pY, pTyr; PTP, phosphotyrosine phosphatase; SOCS, suppressors of cytokine signaling; PIAS, protein inhibitors of activated STAT; SUMO, small ubiquitin-related modifier.
In 1998, Müller and colleagues solved the crystal structure of the Stat3β homodimer bound to DNA.[14] Their work revealed the structural composition of the key binding “hot spots” of Stat3 proteins, including the SH2 domain, allowing the rational design and screening of potential inhibitors of Stat3. Indeed, since the disclosure of this crystal structure, significant research efforts have focused on the design of compounds that disrupt the transcriptionally active Stat3 dimeric complex. Accordingly, a plethora of small molecules targeting the reciprocal phosphotyrosine-SH2 domain interaction, which effectively compete with the native binding sequence, have been reported.
The crystal structure of the Stat3β:Stat3β-DNA complex (Figure 2) highlights the important interactions between the SH2 domain of Stat3 and the native phosphotyrosine binding sequence, including the contacts made by the key phosphotyrosine pTyr 705 and its flanking residues 702-711 (Ala-Ala-Pro-pTyr-Leu-Lys-Thr-Lys-Phe-Ile). pTyr 705 makes crucial interactions with the side chains of Lys 591, Arg 609, Ser 611 and Ser 613, as well as with the backbone NH group of Glu 612. In addition, the α-NH of Leu 706 hydrogen bonds to the main chain C=O of Ser 636 (dotted line) (Figure 2).[15]
Figure 2.
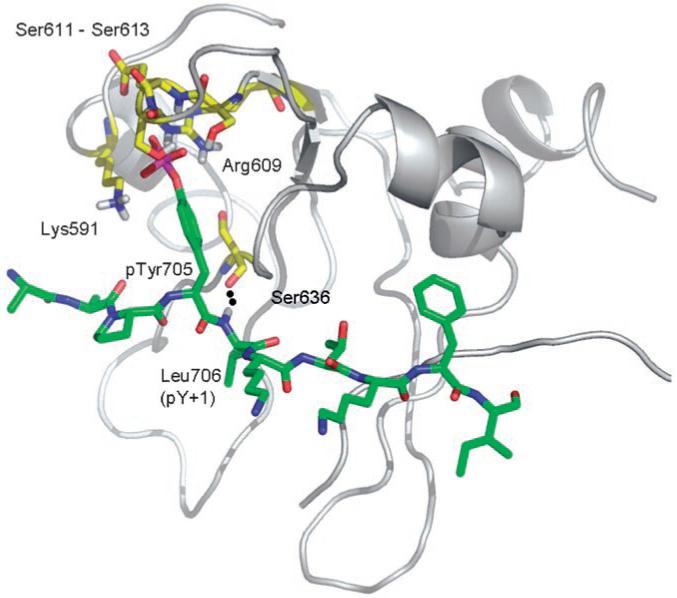
The crystal structure of the Stat3β:Stat3β-DNA complex (PDB: 1BG1).[14] Depicted is the phosphopeptide segment Ala-Ala-Pro-pTyr-Leu-Lys-Thr-Lys-Phe-Ile (residues 702-711) (green) of one Stat3 molecule and its interactions with the SH2 domain of the other Stat3 molecule (gray), in particular with Lys 591, Arg 609, Ser 611 and Ser 613, and Glu 612 (residues in yellow). The image was generated with PyMol and was reproduced with permission.[15]
Herein, we discuss the progress made towards developing isoform-specific Stat3 inhibitors that operate by one or more of the following mechanisms: suppression of Stat3 recruitment to activated tyrosine kinases (step 3 above); prevention of phosphorylated Stat3 dimerization (step 4 above); or prevention of Stat3-DNA binding and aberrant transcriptional activity (step 5 above). Direct inhibitors of Stat3 can be categorized into four different classes of compounds: peptides, peptidomimetics, small molecules, and platinum complexes. This review considers each of these classes of inhibitors in turn, highlighting their modes of inhibition, describing the pertinent structural facets that elicit their activities, and commenting on their selectivities for the Stat3 isoform.
Peptidic inhibitory sequences
In early work, Turkson et al. provided preliminary evidence that Stat3:Stat3 dimer disruption was a valid target for molecular intervention.[16,17] Effective disruption of Stat3:Stat3-DNA binding in vitro was achieved with the native peptide binding sequence Pro-pTyr-Leu-Lys-Thr-Lys (1a), corresponding to the core of the native C-terminal Stat3-SH2 domain binding sequence Gly-Ser-Ala-Ala-Pro-pTyr-Leu-Lys-Thr-Lys-Phe-Ile-Cys. Treating nuclear extracts from cells containing constitutively activated Stat3 (NIH3T3/v-Src transformed mouse fibroblasts) with 1a, and subsequently measuring the Stat3:Stat3-DNA binding in vitro using a radiolabeled probe and electrophoretic mobility shift assay (EMSA) showed significant levels of induced dimer disruption.[17] The tyrosine phosphorylated sequence 1a suppressed Stat3:Stat3-DNA binding with a DB50 (50 % decrease in binding) value of 235 μm. The corresponding unphosphorylated sequence induced no inhibitory effects, highlighting the importance of the phosphate group to SH2 domain recognition.
Alanine scanning analysis of phosphopeptide 1a identified the pTyr-Leu residues to be critical, with simple tripeptides Xxx-pTyr-Leu (e.g. Pro-pTyr-Leu (2), DB50 = 182 μm; Ala-pTyr-Leu (3), DB50 = 217 μm) sufficient to block Stat3 homodimerization.[17] While the disruption of a protein-protein interaction is considered taxing,[18] disruption of a pre-formed Stat3:Stat3 dimer in a protein-protein-DNA ternary complex, as measured by this assay, is even more challenging, and so it is important to note these triple-digit DB50 values of very promising leads. Cellular studies with a membrane-permeable conjugate of phosphopeptide 1a, Pro-pTyr-Leu-Lys-Thr-Lys-mts (membrane translocation sequence), induced a 28 % inhibition of transformed cell lines containing persistent Stat3 activation (NIH3T3/vSrc) at 1 mm concentration. Significantly, this work validated the rationale for disrupting oncogenic Stat3 activity through inhibition of the functional Stat3 dimer complex, and furthermore, identified Stat3 as an important and worthy therapeutic target.
considered taxing,[18] disruption of a pre-formed Stat3:Stat3 dimer in a protein-protein-DNA ternary complex, as measured by this assay, is even more challenging, and so it is important to note these triple-digit DB50 values of very promising leads. Cellular studies with a membrane-permeable conjugate of phosphopeptide 1a, Pro-pTyr-Leu-Lys-Thr-Lys-mts (membrane translocation sequence), induced a 28 % inhibition of transformed cell lines containing persistent Stat3 activation (NIH3T3/vSrc) at 1 mm concentration. Significantly, this work validated the rationale for disrupting oncogenic Stat3 activity through inhibition of the functional Stat3 dimer complex, and furthermore, identified Stat3 as an important and worthy therapeutic target.
In addition to the native Stat3 sequence, McMurray and coworkers identified a high-affinity phosphopeptide inhibitor of Stat3 protein derived from known Stat3-binding receptor sequences.[19] Binding through its SH2 domain, Stat3 is recruited by phosphotyrosine residues on glycoprotein 130 (gp130),[20] leukemia inhibitory factor receptor (LIFR),[21] epidermal growth factor receptor (EGFR),[22] interleukin 10 receptor (IL-10R),[23] and granulocyte colony stimulating factor receptor (G-CSFR).[24] Using solid-phase, Fmoc chemistry, a series of tyrosine-phosphorylated hexapeptides were synthesized based on the known Stat3 docking sites of these receptor proteins. Generally, hexapeptides containing the pTyr-Xxx-Xxx-Gln-Xxx-Xxx sequence were the most active inhibitors of Stat3-DNA binding, as determined by EMSAs. Moreover, the peptides in which the third residue was proline, i.e. Ac-pTyr-Xxx-Pro-Gln-Xxx-Xxx-NH2, were even more potent, with their most effective phosphopeptide Ac-pTyr-Leu-Pro-Gln-Thr-Val-NH2 (4), derived from the gp130-Stat3 binding sequence, disrupting the Stat3-DNA interaction with an IC50 value of 150 nm. This represents a 133-fold improvement in inhibitory activity over the Stat3-derived peptide Ac-pTyr-Leu-Lys-Thr-Lys-Phe-NH2 (1b; IC50 = 20 μm).
The authors studied 4 further, and, by conducting truncation experiments, determined that at least four amino acid residues are required for significant inhibition. Furthermore, alanine scanning confirmed the importance of leucine at position pTyr+1, proline at pTyr+2 and glutamine at pTyr+3, whilst removal of the phosphoryl group from pTyr was not tolerated, giving the authors additional confidence that their phosphopeptides target the SH2 domain of Stat3. Altogether, their data showed that each residue from pTyr to pTyr + 3 contributes to the binding energy of their best phosphopeptide Ac-pTyr-Leu-Pro-Gln-Thr-Val-NH2 (4), providing a strong starting point for the design of more “drug-like” peptidomimetics.
Peptidomimetic Stat3 inhibitors
Peptidic agents commonly suffer from limited cell permeability, a property that has restricted their practical application in vivo at therapeutic doses. The Stat3 peptide leads have, however, provided an excellent starting point from which more cell-permeable peptidomimetics have been subsequently developed.
As mentioned earlier, structure-activity relationship (SAR) studies conducted by Turkson et al. on the Stat3 dimerization-disrupting phosphopeptide Pro-pTyr-Leu-Lys-Thr-Lys (1a) revealed the importance of the Pro-pTyr-Leu (2) (or Ala-pTyr-Leu (3)) tripeptide sequence for inhibitor activity. Structural diversification of this tripeptide was first achieved by substitution of the N-terminal proline with a range of groups of varying size, polarity and orientation, a selection of which is depicted in Figure 3.[25] The replacement of the pTyr-1 proline (or alanine) with 4-cyanobenzoyl (ISS610 (5)), or 2,6-dimethoxybenzoyl (ISS637 (6)), led to a fivefold increase in inhibition of Stat3-DNA binding activity relative to the lead tripeptide, Pro-pTyr-Leu (2); as determined by EMSA analysis (Figure 3). Moreover, these peptidomimetics exhibited very good selectivity for the disruption of Stat3:Stat3 dimers over Stat1:Stat1 and Stat5:-Stat5 (e.g. ISS610 (5): Stat3:Stat3 IC50 = 42 μm; Stat1:Stat1, IC50 = 310 μm; Stat5:Stat5, IC50 = 285 μm).
Figure 3.

N-terminal structural diversification of pTyr-Leu. IC50 values for Stat3:Stat3 dimer disruption were determined in vitro by EMSA analysis.[25]
In general, phenyl-(e.g. ISS610 (5)), pyridyl- (e.g. ISS221 (7)) and pyrazinyl-based (ISS493 (8)) carboxylic acids were tolerated as alternatives to proline (or alanine) in Pro-pTyr-Leu peptidomimetic inhibitors of Stat3, although it should be noted that these inhibitors were particularly sensitive to substitution on the aromatic ring. However, the incorporation of bulky carboxylic acids; such as those derived from quinoline (ISS363 (9)), naphthalene, and biphenyl (e.g. ISS355 (10)), furnished peptidomimetic inhibitors with little or no activity relative to the tripeptide leads. Taken together, these data suggest there may be a small hydrophobic domain that is just amino-terminal to the pTyr binding pocket that can be exploited to improve inhibitor binding. Importantly, the dephosphorylated form of ISS610 (ISS610NP) did not disrupt Stat3-DNA binding, supporting the hypothesis that it is the pTyr-SH2 interaction that is disrupted by the peptidomimetics. The lowest energy GOLD[26] flexible ligand docking conformation indicated that the 4-cya-nophenyl group in ISS610 (5) has access to a hydrophobic domain and any available hydrogen bonding interactions on the protein surface in addition to those hydrophilic and hydrophobic interactions that are achieved by the lead Pro-pTyr-Leu peptide 2. It is likely that these additional interactions explain the improved activities of many of the authors’ peptidomimetics over the lead tripeptides.
Studies on the effects of ISS610 (5) in NIH3T3/v-Src cells demonstrated selective blockage of Stat3-DNA binding and transcriptional activities. These studies were extended to human tumor cell lines, including breast (MDA-MB 231, MDA-MB 435, MDA-MB 468) and lung (A549) carcinoma;[27] significant suppression of Stat3 activation was again observed. This research represents the first report of a successful peptidomimetic approach to the inhibition of Stat3 dimerization, and provides proof-of-principle that peptidomimetic inhibitors of Stat3, such as ISS610 (5), represent potential novel anticancer therapeutics.
Hamilton, Gunning, et al. further developed the ISS610 (5) peptidomimetic by functionalization of the C terminus to generate a library of 11 compounds. Initially designed to further enhance Stat3 inhibitory potency, one member of this library, ISS840 (11), an m-methoxyaniline amide derivative of ISS610 (5), was serendipitously identified as a potent Stat1 inhibitor, as shown in Figure 4.[28] As determined by EMSA analysis, ISS840 (11) displayed a 20-fold selectivity in the disruption of the Stat1 homodimer (IC50 = 31 μm) over the Stat3 homodimer (IC50 = 560 μm). Notably, ISS840 (11) was more than 13 times less active towards Stat3 inhibition compared with the lead, ISS610 (5). Indeed, all eleven of the library members were poorer inhibitors of Stat3. However, despite these disappointing results for Stat3 inhibition, it is interesting to note that condensation of the C-terminal carboxylic acid in ISS610 (5) with a hydrophobic amine generally led to increased inhibition of the Stat1 isoform, possibly due to additional hydrophobic contacts with Tyr 603 and Ile 616.
Figure 4.
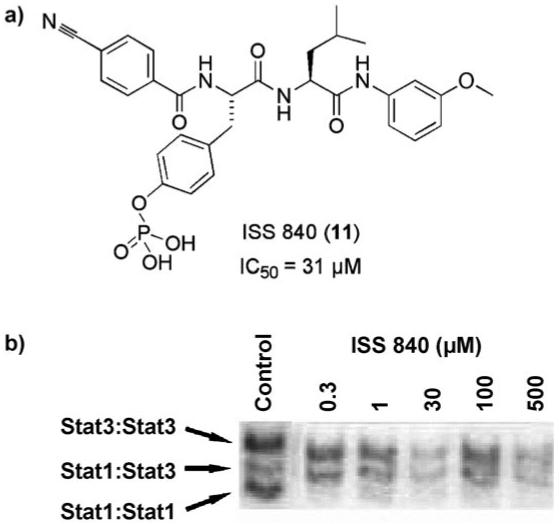
a) The chemical structure of ISS840 (11) and its IC50 value for the disruption of the Stat1:Stat1 interaction; b) EMSA analysis showing selective disruption of STAT family members by ISS840 (11).[28]
Meanwhile, McMurray and co-workers embarked on their own course of peptidomimetic research, which is summarized in Figure 5.[15] Performing extensive SAR studies on their phosphopeptide Ac-pTyr-Leu-Pro-Gln-Thr-Val-NH2 (4), the authors created a library of approximately 50 synthetic compounds that enabled them to draw a number of conclusions about the importance of backbone and side chain interactions between 4 and the Stat3-SH2 domain. First, using the simpler phosphopeptide Ac-pTyr-Leu-Pro-Gln-Thr-NH2 (12) as their reference, the researchers identified a hydrophobic patch on the surface of the SH2 domain where the pTyr residue is predicted to bind, confirming the earlier findings of Turkson et al.[25] As a specific example, replacement of the acetyl group with the more lipophilic hydrocinnamoyl group led to an approximate threefold improvement in inhibition of Stat3 (Ac-pTyr-Leu-Pro-Gln-Thr-NH2 (12) IC50 = 739 nm; PhCH2CH2CO-pTyr-Leu-Pro-Gln-Thr-NH2 (13) IC50 = 276 nm) as determined by fluorescence polarization (FP) assays.[29]
Figure 5.

Structural evolution of gp130 peptidomimetics (IC50 values determined by FP analysis[29]).[15]
Further SAR analysis confirmed the importance of the NH group of the pTyr-Leu amide bond. This is in agreement with the crystal structure of Stat3b dimer bound to DNA that reveals a hydrogen bond between the backbone NH of Leu 706 at the pTyr+1 position of pTyr 705-Leu-Lys-Thr-Lys-Phe and the backbone C=O of Ser 636. Indeed, the backbone NH of Leu proved critical to inhibitor activity; replacement of the Leu NH in 4 with NMe to give phosphopeptide 14 led to a decrease in inhibitor IC50 from 0.739 μm to > 100 μm. Constraining the peptide backbone by replacement of proline with cis-3,4-meth-anoproline at the pTyr+2 position led to a threefold increase in activity, whilst removal of the side chain amide protons of the glutamine at the pTyr+3 residue was poorly tolerated with a > 100-fold drop in activity from Ac-pTyr-Leu-Pro-Gln-Thr-NH2 (12) to Ac-pTyr-Leu-Pro-Gln(Me)2-Thr-NH2 (15). It is not clear if these protons are required for intra- or intermolecular interactions, or if the methyl groups cause unfavorable steric clashes, but further modifications with the glutamine isostere methionine sulfoxide suggested the entire carboxamide CONH2 side chain may be involved in key interactions. Subsequent analysis revealed that the C-terminal Thr-Val-NH2 dipeptide unit could be replaced with benzylamine with minimal loss of activity (Ac-pTyr-Leu-Pro-Gln-Thr-Val-NH2 (4), IC50 = 290 nm vs. Ac-pTyr-Leu-Pro-Gln-NHBn (16), IC50 = 409 nm), a successful step towards achieving their desired peptide mimicry. Combining the findings from substitutions at the N and C termini, and from the structural requirements at the pTyr+1 to pTyr+3 positions, McMurray’s work peaked with the identification of peptidomimetic PhCH2CH2CO-Tyr-Leu-(cis-3,4-methanoPro)-Gln-NHBn (17) with an IC50 value of 125 nm. More recently, the authors have developed a hypothesis for the binding of Ac-pTyr-Leu-Pro-Gln-NHBn (16) to the SH2 domain of Stat3 using computer modeling;[30] as well as depicting the established salt bridge interactions of the pTyr residue, the docked pose of phosphopeptide 16 accounts for the importance of Glu at pTyr+3, which is engaged in a network of hydrogen bonds.
In a subsequent investigation, McMurry and colleagues achieved a similarly potent, yet simpler, less peptidic, small molecule by replacing the N-terminal Ac-pTyr unit with 4-phosphorylcinnamoyl (pCin) to give pCin-Leu-Pro-Gln-NHBn (18), which inhibited Stat3 with an IC50 value of 135 nm (FP assay).[31] In order to engineer Stat3 inhibitors with improved potency in physiological environments, replacement of the glutamine in phosphopeptide 18 with residues that may impart resistance to proteases and glutaminases was next investigated. Substitution of glutamine in 18 with the known glutamine mimetic O-carbamolyserine (Ser(CONH2)) to give pCin-Leu-Pro-Ser-(CONH2)-NHBn (19) was not well tolerated, leading to an approximate threefold loss in activity (IC50 = 379±49 vs. 135±8 nm) (Figure 6); this could be due to a number of factors, such as a loss of hydrogen bonding.[31] More informative, however, was the alternative substitution of glutamine with O-carbamoylthreonine (Thr(CONH2)) to furnish pCin-Leu-Pro-Thr-(CONH2)-NHBn (20), which led to an even greater loss in activity of around sixfold relative to the parent phosphopeptide 18. This latter result suggests that the region of the Stat3 protein that binds the glutamine side chain in the peptide 18 is a tight binding pocket or cleft, unable to accommodate the additional β-methyl group present in Thr(CONH2).
Figure 6.

Structural evolution of gp130 peptidomimetics with glutamine mimetics.[31]
Returning to their C-terminal truncated lead phosphopeptide Ac-pTyr-Leu-Pro-Gln-Thr-NH2 (12), McMurry and colleagues examined the geometry of the Leu-Pro peptide bond—Xxx-Pro peptide bonds are known to undergo cis/trans isomerization— that is adopted when 12 is bound to the SH2 domain of Stat3.[32] 2,2-Dimethyloxazolidine pseudoproline analogues (ΨPro) induce predominantly cis conformation in Xxx-Pro peptide bonds. Hence, by replacing proline in phosphopeptide 12 with pseudoproline analogues 2,2-dimethyl serine-, and threonine-derived oxazolidines, Ser(ΨMe,Mepro) and Thr(ΨMe,Mepro) respectively, the authors created phosphopeptides 21 and 22 with enhanced cis conformation of Leu-ΨPro peptide bonds (Figure 7). 1H NMR studies in aqueous solution revealed that substitution of Pro with Ser(ΨMe,Mepro) led to an increased percentage of cis over trans, from 2 to 69 %, of the Leu-Pro peptide bond, whilst incorporation of Thr(ΨMe,Mepro) lead to 63 % cis Leu-ΨPro peptide bond. These phosphopeptides containing the cis-constraining pseudoproline analogues proved weaker inhibitors of Stat3. Indeed, the greater the cis content, the poorer the inhibitor (e.g. Ac-pTyr-Leu-Pro-Gln-Thr-NH2 (12): 2% cis Leu-Pro peptide bond, IC50 = 739 nm vs. Ac-pTyr-Leu-Ser(ΨMe,Mepro)-Gln-Thr-NH2 (21): 69 % cis Leu-ΨPro peptide bond, IC50 = 3830 nm (IC50s determined by FP assays)). Additional data implied that the ΨPro side chain points away from the protein surface, indicating that the decreased IC50 values are not due to steric conflict of the gem-dimethyl groups. Altogether, their research provides considerable evidence that the Leu-Pro peptide bond in Stat3 phosphopeptide inhibitors is trans when bound to the Stat3-SH2 domain.
Figure 7.

Pseudoproline analogues of Ac-pTyr-Leu-Pro-Gln-Thr-NH2 (12) to investigate the geometry of the inhibitor Leu-Pro peptide bond upon binding Stat3.[32]
In a study that complements McMurray’s peptidomimetic research, scientists in the Garbay research group investigated the replacement of the critical phosphotyrosine residue in the phosphopeptide Ac-pTyr-Leu-Pro-Gln-Thr-Val-OH (23 a) with alternative pTyr mimetics (Figure 8).[33] Substitution of the phosphate group with an O-methylenecarboxylic acid (23b) or tetrazole (23c or 23d), a more lipophilic bioisostere of a carboxylic acid, was poorly tolerated. It is suggested that mono-anionic pTyr mimetics are incapable of forming as many hydrogen bonds and salt bridges with the two positively charged amino acid residues in the pTyr binding pocket, specifically Lys 591 and Arg 609, compared with pTyr. In agreement with this hypothesis, activity was substantially improved by the introduction of a di-anionic species, as with phosphonate 23e and O-malonate 23f, although these derivatives were still weaker Stat3 inhibitors than the parent phosphate 23a by at least an order of magnitude. Nonetheless, due to the higher pKa values of carboxylic acids, malonate derivative 23f may be anticipated to exhibit improved whole-cell activity over phosphate derivative 23a, through enhanced cell penetration.
Figure 8.
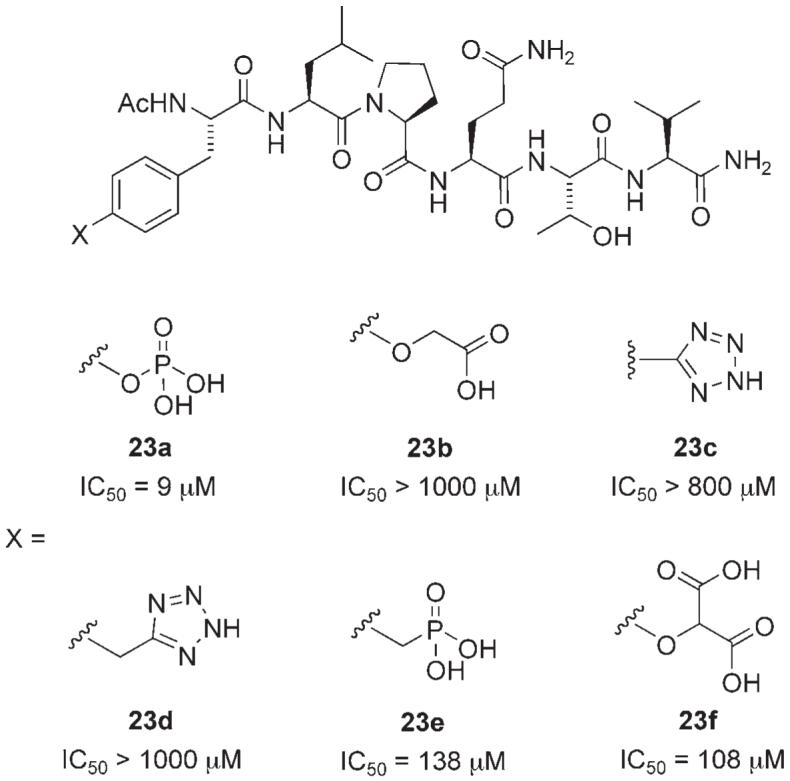
Stat3 inhibitors incorporating pTyr mimetics. IC50 values determined by ELISA assay.[33]
Wang and co-workers designed and synthesized a novel, conformationally constrained, macrocyclic peptidomimetic inhibitor of Stat3 based on the phosphopeptide Ac-pTyr-Leu-Lys-Thr-Lys-Phe-NH2 (1b), which had been found to inhibit Stat3 dimerization.[34] Using the previously discussed crystal structure of the peptide segment pTyr-Leu-Lys-Thr-Lys-Phe bound to Stat3 (as the Stat3 dimer) to guide their investigations, Wang and colleagues discovered that the two lysine side chain amino groups are solvent exposed, and so do not contribute to inhibitor binding, providing an opportunity to introduce conformational constraint through cyclization of these two side chains. Cyclic peptidomimetics can be more resistant to digestion by proteases than their linear counterparts and hence may show improved biological activity in whole-cell assays. Moreover, higher binding affinity may be observed with such cyclic compounds owing to a reduced entropic cost upon binding to the target protein. Wang and colleagues prepared macrocycle 24 in which the ring cyclization was accomplished using “click chemistry” between a terminal alkyne and an azide (Figure 9).[34] Employing a modified version of the FP assay reported by Schust and Berg,[29] it was found that 1b and its cyclic analogue 24 inhibited recombinant Stat3 with Ki values of 25.9±2.5 and 7.3±0.9 μm, respectively. These data suggest that the introduction of conformational constraint in Ac-pTyr-Leu-Lys-Thr-Lys-Phe-NH2 (1b) has furnished a threefold improvement in Stat3 inhibition.
Figure 9.
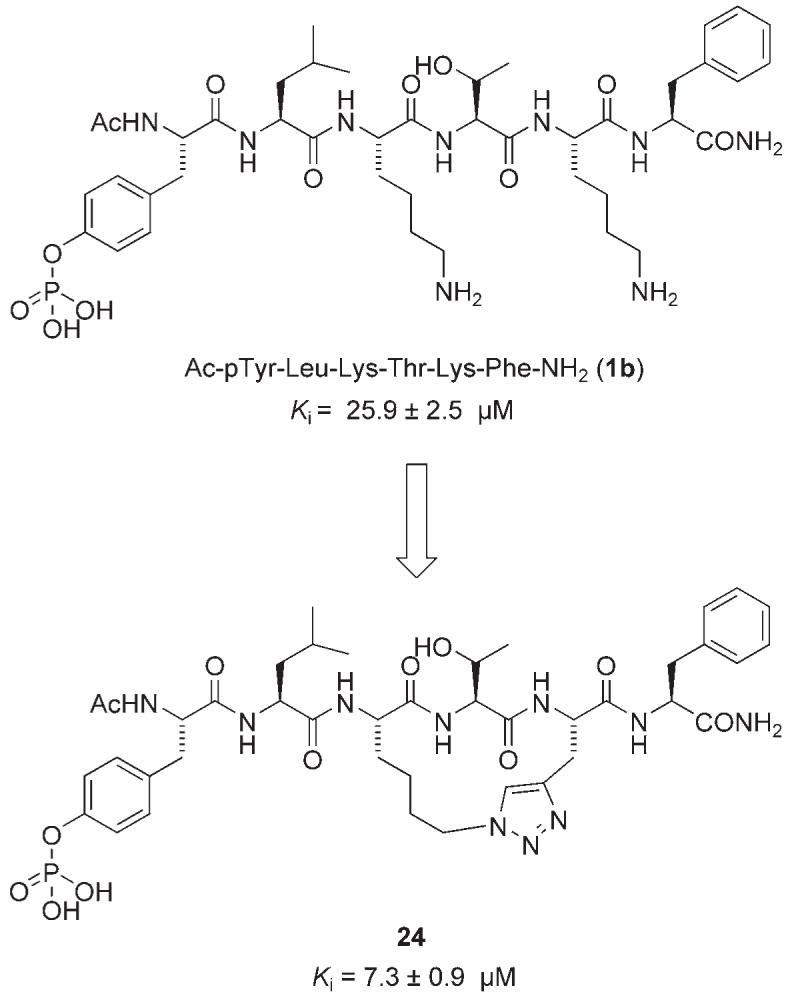
A conformationally constrained macrocyclic peptidomimetic inhibitor.[34]
Small-molecule inhibitors of Stat3 function
The ultimate goal of peptidomimetic research is to achieve a peptide mimic devoid of all peptidic characteristics. Alternatively, Stat3 inhibition has been achieved by small molecules identified by rational design or through the screening of chemical libraries.
Hamilton and co-workers recently designed a library of oxazole-based, small-molecule inhibitors of Stat3, structurally inspired by the peptidomimetic inhibitor ISS610 (5) (Figure 3).[35] In conjunction with previous ISS610-QSAR studies, the authors’ rational design approach utilized computationally aided GOLD docking studies to identify a number of low-micromolar inhibitors of Stat3, the most potent of which was S3I-M2001 (25). The Stat3-SH2 domain is composed of three sub-domains: a hydrophilic cleft formed by Arg 609, Ser 611, Ser 613, and Lys 591; a partially hydrophobic pocket formed by Ile 597, Ile 634, the tetramethylene portion of the Lys 592 side chain, and the trimethylene portion of the Arg 595 side chain; and finally a hydrophobic channel predominantly formed by Phe 716 and Trp 623. The authors state that the trigonal arrangement of the oxazole core of 25 gave optimal interactions with all three of the SH2 sub-domains.
According to GOLD[26] docking studies, the phosphate moiety of S3I-M2001 (25) appears to bind tightly within the Arg 609 hydrophilic cleft. This is in agreement with the crystal structure in Figure 2. Turkson, Hamilton, et al. argue that the orientation of this small molecule is likely to be predetermined by this critical interaction; earlier we commented on the fact that removal of the phosphoryl group of ISS610 (5) to give ISS610NP completely abolished Stat3 inhibitory activity.[25] The hydrophobic naphthyl group makes significant contact with the Ile 634 side chain, and the lipophilic hexyl substituent is partially encased within the Phe 716/Trp 623 hydrophobic channel. Altogether, the three sets of proposed binding interactions of S3I-M2001 (25) with the Stat3-SH2 domain are predicted to confer potent inhibitory activity on this small molecule. Indeed, EMSA analyses of S3I-M2001 (25) gave similar in vitro results to its peptidomimetic predecessor ISS610 (5) (25, IC50 = 79 μm vs. 5, IC50 = 42 μm). Importantly, although S3I-M2001 (25) displayed marginally decreased potency in the Stat3-DNA binding assay, it inhibited Stat3 activation in a whole-cell assay at 100 μm. This is in contrast to the 10-fold higher concentration (1 mm) of ISS610 (5) that was required to inhibit intracellular Stat3 activity. In addition to preventing Stat3-dependant malignant transformation, survival, migration, and invasion of mouse and human cancer cells containing constitutive Stat3 activity, S3I-M2001 (25) also inhibited the growth of human breast cancer xenographs. Interestingly, the inhibition of aberrant Stat3 in malignant cells by this oxazole induced early Stat3 aggregation in the perinuclear aggresomes, and a latephase proteasome-mediated degradation.[35]
Several prominent inhibitors of Stat3 have been identified through the screening of chemical libraries. Using their FP assay, Berg and co-workers surveyed >17 000 small molecules for their affinity to the Stat3-SH2 domain, which culminated in the identification of Stattic (26), a benzothiophene derivative.[36] Stattic (26) was found to inhibit Stat3 with an IC50 value of 5.1±0.8 μm. Interestingly, the simple benzothiophene structure does not incorporate, nor rely upon, a phosphate, or phosphate mimetic, to confer binding potency to the Stat3-SH2 domain, negating the cell permeability problems invariably associated with phosphate-containing therapeutics.
Testing similar benzothiophene derivatives revealed that both the nitro group and the conjugated double bond of Stattic (26) are important to its inhibitory activity. Schust et al. noted that the binding potency, displayed both in vitro and in vivo, may be due to Michael addition of Stattic (26) to nucleophilic residues; this would correlate with the decreased activity of the benzothiophene lacking the α,β-unsaturation, although conclusive evidence has yet to be reported for covalent attachment of Stattic (26) to the Stat3-SH2 protein surface. Nevertheless, their data showed that Stattic (26) compromised the Stat3-SH2 domain in vitro, and, in addition to the effective disruption of Stat3 in cancer cell lines, inhibited Stat3 translocation and homodimerization. Furthermore, Stattic (26) displayed good selectivity for Stat3 over isoforms Stat1 and Stat5b.
Structure-based virtual screening has yielded two notable small-molecule inhibitors of Stat3 function in cancer, specifically STA-21 (27) and S3I-201 (28). STA-21 (27), a deoxytetrangomycin natural product, was identified from an in silico screen of 429 000 compounds from the National Cancer Institute (NCI), Merck, Sigma-Aldrich and Ryan databases.[37] Small molecules were ranked according to their calculated binding affinities for the Stat3-SH2 domain (Stat3β homodimer); a second round of screening, utilizing DOCK[38] and X-SCORE,[39] reduced the number of compounds expected to inhibit SH2 domain activity to 100. Subsequently, a whole-cell luciferase assay was employed to screen these 100 compounds for Stat3-dependent luciferase activity in cancer cells, leading to the identification of the promising small molecule STA-21 (27). Computational visualization of STA-21 (27) docking showed that the inhibitor binds in the same site as the native phosphopeptide, forming critical contacts with key basic residues Arg 595 and Arg 609, presumably through its phenolic and carbonyl functional groups. In addition, the predominantly hydrophobic and planar STA-21 (27) is predicted to make beneficial interactions with Ile 634, mirroring the positioning of the naphthyl substituent of S3I-M2001 (25). STA-21 (27) inhibited Stat3 nuclear translocation, and DNA binding and transcriptional activities, and induced apoptosis in human breast tumor cell lines, expressing aberrant Stat3, at 20-30 μm. Whilst no evidence was presented to show direct disruption of phosphopeptide binding to the protein, encouragingly, no appreciable suppression of non-Stat3-dependant luciferase activity was observed.
More recently, Turkson and colleagues identified S3I-201 (28), a low-molecular-weight 4-aminosalicylic acid-based SH2-binding agent from the NCI chemical libraries, as a Stat3 inhibitor.[40] In a similar approach to the discovery of STA-21 (27), small molecule S3I-201 (28) was discovered using structure-based virtual screening with a computer model of the Stat3-SH2 domain bound to its Stat3 phosphotyrosine peptide, derived from the X-ray crystal structure of the Stat3β homodimer.[14] Structurally, the salicylic acid group is a known phosphate mimetic;[41] Turkson cites its incorporation as being vital to Stat3 binding. Docking studies using GLIDE[42] (Grid-based Ligand Docking from Energetics) software suggested that the carboxylic acid forms hydrogen bonds with Arg 609, Ser 611, and Ser 613, whilst the phenol is hydrogen-bonded to Lys 591. Furthermore, S3I-201 (28) contains a hydrophobic tolyl group that is predicted to access the second of the three main subpockets of the SH2-domain, forming van der Waals interactions with the side chains of Ile 597 and Ile 634, as well as with the hydrophobic portions of the side chains of Arg 595 and Lys 592. In silico docking highlighted some interesting differences in structural occupation that exist between S3I-M2001 (25) and S3I-201 (28) (Figure 10). Whilst both successfully access the phosphate binding pocket and the adjacent hydrophobic pocket (Ile 634 side chain), S3I-201 (28) is structurally incapable of projecting into the third sub-domain composed primarily of Phe 716 and Trp 623. This may explain the lower inhibition of Stat3-DNA binding activity by S3I-201 (28), with a mean IC50 value of 86 μm (EMSA). S3I-201 (28) exhibited good isoform selectivity for inhibition of Stat3 over both Stat1 and Stat5 (Stat1:Stat1, IC50 > 300 μm and Stat5:Stat5, IC50 = 166 μm). Turkson and co-workers showed that S3I-201 (28) repressed the expression of the Stat3-regulated genes encoding cyclin D1, Bcl-xL, and survivin, and preferentially induced apoptosis of malignant cells harboring constitutively activated Stat3. Furthermore, S3I-201 (28) induced regression of human breast tumor xenografts.
Figure 10.
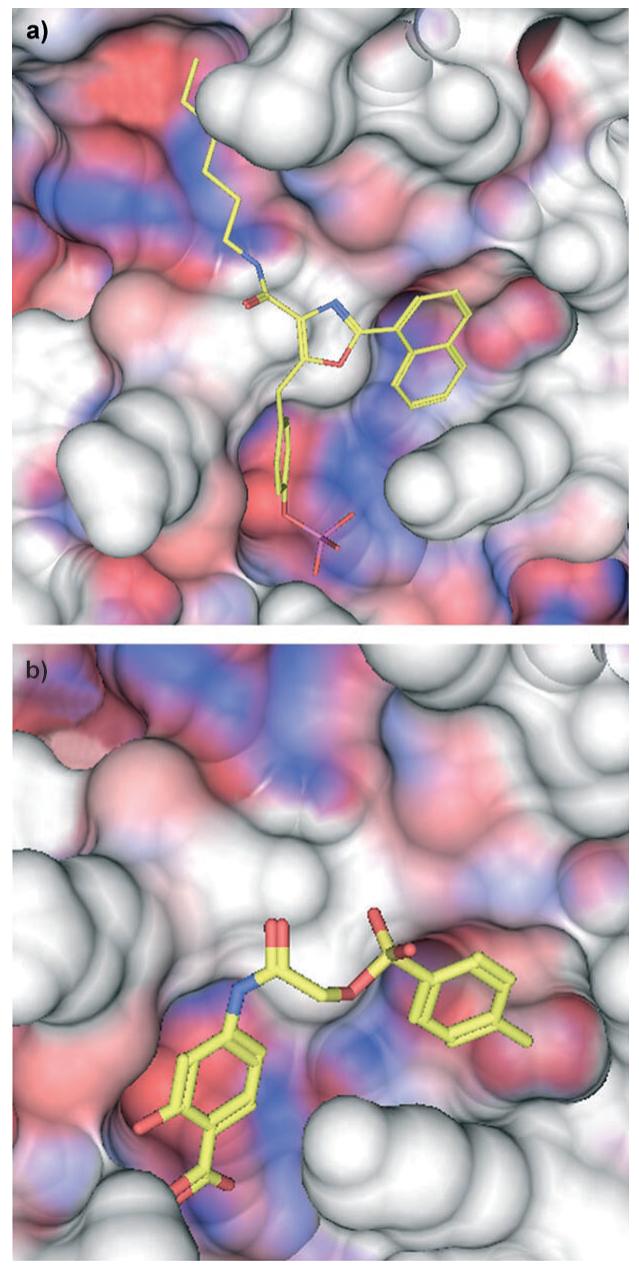
Lowest energy GOLD[26] docking results for inhibitors (colored by atom type). a) S31-2001 (25) and b) S31-201 (28) bound in the SH2 domain (red, hydrophobic; blue, hydrophilic) of the Stat3β dimer (PDB: 1BG1[14]).
Platinum-based inhibitors of Stat3
Whilst the majority of Stat3 inhibitors take the form of peptides, peptidomimetics, or small molecules, there have been a few reports on the successful inhibition of Stat3 by metal complexes. For example, Turkson et al. have described the inhibition of Stat3 with a variety of platinum complexes.[43] There is much evidence to suggest that these agents, such as the platinum(II) drug cisplatin (29), exert their anticancer activity through cross-linking DNA.[44] However, very little had been published on the effects of platinum complexes on signal transduction events, until now.[43] Starting with a series of novel platinum complexes, the authors identified potent, and selective with respect to other signaling pathways, disruptors of STAT activity, with Stat3 being the most inhibited protein of the family of STATs investigated (Stat1, Stat3 and, Stat5). Each of the platinum(IV) compounds CPA-1 (30), CPA-7 (32), and PtCl4 (33), and the platinum(II) compound CPA-3 (31), were all  found to inhibit Stat3 activity in vitro at low micromolar concentrations; CPA-7 (32) was the most potent inhibitor of Stat3 (IC50 = 1.5 μm, determined by EMSA analysis). In addition, activities in whole cells were also observed at 10-20 μm. Upon administration of CPA-1 (30) or CPA-7 (32) to malignant cells harboring constitutively activated Stat3, cell growth was inhibited and apoptosis was initiated. Furthermore, CPA-7 (32) induced regression of colon CT26 tumors in mouse models, which correlated with Stat3 inhibition.
found to inhibit Stat3 activity in vitro at low micromolar concentrations; CPA-7 (32) was the most potent inhibitor of Stat3 (IC50 = 1.5 μm, determined by EMSA analysis). In addition, activities in whole cells were also observed at 10-20 μm. Upon administration of CPA-1 (30) or CPA-7 (32) to malignant cells harboring constitutively activated Stat3, cell growth was inhibited and apoptosis was initiated. Furthermore, CPA-7 (32) induced regression of colon CT26 tumors in mouse models, which correlated with Stat3 inhibition.
Continuing their exploration of platinum(IV) compounds as inhibitors of STATs, Turkson, Jove, et al. identified IS3295 (34) from the NCI 2000 diversity set as another novel platinum compound that selectively inhibits constitutive Stat3 signaling over Stat1 (Stat3, IC50 = 1.4 μm vs. Stat1, IC50 = 4.1 μm), and induces cell-cycle arrest and apoptosis of malignant cells,[45] consistent with previous findings of other platinum(IV) complexes.[43] Additional data suggested IS3295 (34) behaves as a Stat3 inhibitor by binding directly to the protein, both to the Stat3 monomer and to the activated Stat3:Stat3 dimer, leading to decreased Stat3 phosphotyrosine levels, DNA binding activity, and transcriptional regulation. Furthermore, the kinetics of IS3295 (34)-mediated inhibition of Stat3-DNA binding activity were found to be noncompetitive, and given the known interactions of other platinum complexes with thiol-containing biological molecules, the authors speculate that IS3295 (34) imparts its inhibitory activity by irreversibly binding Stat3 through covalent attachment to a cysteine residue. Once again, treatment of human and mouse tumor cells harboring constitutively activated Stat3 with the platinum(IV) compound IS3295 (34) led to cell-cycle arrest and apoptosis, events that were induced by the inhibition of aberrant Stat3 signaling.
Outlook
To date,[46-48] the majority of Stat3 inhibitors have disrupted the oncogenic protein indirectly by exerting their effects through the intervention of tyrosine kinases, or their receptors. The direct disruption of Stat3 with molecular inhibitors represents an extremely challenging goal due to the many formidable obstacles associated with the disruption of protein-protein interactions, events that feature extensively in Stat3 signaling. Nevertheless, in less than a decade, there has been much progress towards this goal with a number of prominent small-molecule inhibitors being discovered through several avenues of research, including peptidomimicry, de novo rational design, and screening chemical libraries. Given the significant role of Stat3 in the resistance of cancer to current chemotherapeutic strategies, it is likely that these inhibitors will play a significant role in the future of cancer and adjuvant cancer therapies.
Acknowledgements
We thank the Canadian Foundation for Innovation and the University of Toronto (Connaught Award) for financial support of our research in this area.
References
- [1].Darnell JE., Jr. Recent Prog. Horm. Res. 1996;51:391–403. [PubMed] [Google Scholar]
- [2].Darnell JE., Jr. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- [3].Buettner R, Mora LB, Jove R. Clin. Cancer Res. 2002;8:945–954. [PubMed] [Google Scholar]
- [4].Bowman T, Garcia R, Turkson J, Jove R. Oncogene. 2000;19:2474–2488. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- [5].Darnell JE., Jr. Nat. Med. 2005;11:595–596. doi: 10.1038/nm0605-595. [DOI] [PubMed] [Google Scholar]
- [6].Yu H, Jove R. Nat. Rev. Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- [7].Turkson J. Expert Opin. Ther. Targets. 2004;8:409–422. doi: 10.1517/14728222.8.5.409. [DOI] [PubMed] [Google Scholar]
- [8].Haura EB, Turkson J, Jove R. Nat. Clin. Pract. Oncol. 2005;2:315–324. doi: 10.1038/ncponc0195. [DOI] [PubMed] [Google Scholar]
- [9].Burke WM, Jin X, Lin H-J, Huang M, Liu R, Reynolds KR, Lin J. Oncogene. 2001;20:7925–7934. doi: 10.1038/sj.onc.1204990. [DOI] [PubMed] [Google Scholar]
- [10].Berishaj M, Gao SP, Ahmed S, Leslie K, Al-Ahmadie H, Gerald WL, Bornmann W, Bromberg JF. Breast Cancer Res. 2007;9:R32. doi: 10.1186/bcr1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Barton BE, Karras JG, Murphy TF, Barton A, Huang HF-S. Mol. Cancer Ther. 2004;3:11–20. [PubMed] [Google Scholar]
- [12].a) Kruger A, Anderson SM. Oncogene. 1991;6:245–256. [PubMed] [Google Scholar]; b) Chaturvedi P, Sharma S, Reddy EP. Mol. Cell. Biol. 1997;17:3295–3304. doi: 10.1128/mcb.17.6.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, Ciliberto G, Moscinski L, FernRndez-Luna JL, Nunez G, Dalton WS, Jove R. Immunity. 1999;10:105–115. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- [13].Song JL, Grandis JR. Oncogene. 2000;19:2489–2495. doi: 10.1038/sj.onc.1203483. [DOI] [PubMed] [Google Scholar]
- [14].Becker S, Groner B, Müller CW. Nature. 1998;394:145–151. doi: 10.1038/28101. [DOI] [PubMed] [Google Scholar]
- [15].Coleman DR, IV, Ren R, Mandal PK, Cameron AG, Dyer GA, Muranjan S, Chen X, McMurray JS. J. Med. Chem. 2005;48:6661–6670. doi: 10.1021/jm050513m. [DOI] [PubMed] [Google Scholar]
- [16].Turkson J, Bowman T, Garcia R, Caldenhoven E, De Groot RP, Jove R. Mol. Cell. Biol. 1998;18:2545–2552. doi: 10.1128/mcb.18.5.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Turkson J, Ryan D, Kim JS, Zhang Y, Chen Z, Haura E, Laudano A, Sebti SM, Hamilton AD, Jove R. J. Biol. Chem. 2001;276:45 443–45 455. doi: 10.1074/jbc.M107527200. [DOI] [PubMed] [Google Scholar]
- [18].a) Fletcher S, Hamilton AD. Curr. Top. Med. Chem. 2007;7:922–927. doi: 10.2174/156802607780906735. [DOI] [PubMed] [Google Scholar]; b) Fletcher S, Hamilton AD. J. R. Soc. Interface. 2006;3:215–233. doi: 10.1098/rsif.2006.0115. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Fletcher S, Hamilton AD. Curr. Opin. Chem. Biol. 2005;9:632–638. doi: 10.1016/j.cbpa.2005.10.006. [DOI] [PubMed] [Google Scholar]
- [19].Ren Z, Cabell LA, Schaefer TS, McMurray JS. Bioorg. Med. Chem. Lett. 2003;13:633–636. doi: 10.1016/s0960-894x(02)01050-8. [DOI] [PubMed] [Google Scholar]
- [20].Stahl N, Farruggella TJ, Boulton TJ, Zhong Z, Darnell JE, Yancopoulos GD. Science. 1995;267:1349–1353. doi: 10.1126/science.7871433. [DOI] [PubMed] [Google Scholar]
- [21].Coffer P, Kruijer W. Biochem. Biophys. Res. Commun. 1995;210:74–81. doi: 10.1006/bbrc.1995.1629. [DOI] [PubMed] [Google Scholar]
- [22].Weber-Nordt RM, Riley JK, Greenlund AC, Moore KW, Darnell JE, Schreiber RD. J. Biol. Chem. 1996;271:27 954–27 961. doi: 10.1074/jbc.271.44.27954. [DOI] [PubMed] [Google Scholar]
- [23].Chakraborty A, Dyer KF, Cascio M, Mietzner TA, Tweardy DJ. Blood. 1999;93:15–24. [PubMed] [Google Scholar]
- [24].Park OK, Schaefer LK, Wang W, Schaefer TS. J. Biol. Chem. 2000;275:32 244–32 249. doi: 10.1074/jbc.M005082200. [DOI] [PubMed] [Google Scholar]
- [25].Turkson J, Kim JS, Zhang S, Yaun J, Haung M, Glenn M, Haura E, Sebti S, Hamilton AD, Jove R. Mol. Cancer Ther. 2004;3:261–269. [PubMed] [Google Scholar]
- [26].Jones G, Willett P, Glen RC, Leach AC, Taylor R. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- [27].Cailleau R, Olive M, Crueiger QVJ. In Vitro. 1978;14:911–915. doi: 10.1007/BF02616120. [DOI] [PubMed] [Google Scholar]
- [28].Gunning PT, Katt WP, Glenn MP, Siddiquee KAZ, Kim JS, Jove R, Sebti SM, Turkson J, Hamilton AD. Bioorg. Med. Chem. Lett. 2007;17:1875–1878. doi: 10.1016/j.bmcl.2007.01.077. [DOI] [PubMed] [Google Scholar]
- [29].Schust J, Berg T. Anal. Biochem. 2004;330:114–118. doi: 10.1016/j.ab.2004.03.024. [DOI] [PubMed] [Google Scholar]
- [30].McMurray JS. Biopolymers. 2008;90:69–79. doi: 10.1002/bip.20901. [DOI] [PubMed] [Google Scholar]
- [31].Mandal PK, Heard PA, Ren Z, Chen X, McMurray JS. Bioorg. Med. Chem. Lett. 2007;17:654–656. doi: 10.1016/j.bmcl.2006.10.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Coleman DR, IV, Kaluarachchi K, Ren Z, Chen X, McMurray JS. Int. J. Pept. Res. Ther. 2008;14:1–9. doi: 10.1007/s10989-012-9313-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Dourlat J, Valentin B, Liu W-C, Garbay C. Bioorg. Med. Chem. Lett. 2007;17:3939–3942. doi: 10.1016/j.bmcl.2007.04.107. [DOI] [PubMed] [Google Scholar]
- [34].Chen J, Nikolovska-Coleska Z, Yang C-Y, Gomez C, Gao W, Krajewski K, Jiang S, Roller P, Wang S. Bioorg. Med. Chem. Lett. 2007;17:3939–3942. doi: 10.1016/j.bmcl.2007.04.096. [DOI] [PubMed] [Google Scholar]
- [35].a) Siddiquee K, Gunning PT, Glenn M, Glenn WP, Sebti SM, Jove R, Hamilton AD, Turkson J. ACS Chem. Biol. 2007;2:787–796. doi: 10.1021/cb7001973. [DOI] [PubMed] [Google Scholar]; b) Gunning PT, Glenn M, Siddiquee KAZ, Katt WP, Masson E, Sebti SM, Turkson J, Hamilton AD. ChemBioChem. 2008 doi: 10.1002/cbic.200800291. DOI: 10.1002/cbic.200800291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Chem. Biol. 2006;13:1235–1242. doi: 10.1016/j.chembiol.2006.09.018. [DOI] [PubMed] [Google Scholar]
- [37].Song H, Wang R, Wang S, Lin J. Proc. Natl. Acad. Sci. USA. 2005;102:4700–4705. doi: 10.1073/pnas.0409894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ewing T, Makino S, Skillman A, Kuntz I. J. Comput.-Aided Mol. Des. 2001;15:411–428. doi: 10.1023/a:1011115820450. [DOI] [PubMed] [Google Scholar]
- [39].Wang R, Lai L, Wang S. J. Comput.-Aided Mol. Des. 2002;16:11–26. doi: 10.1023/a:1016357811882. [DOI] [PubMed] [Google Scholar]
- [40].Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, Yip MLR, Jove R, Laughlin MM, Lawrence NJ, Sebti SM, Turkson J. Proc. Natl. Acad. Sci. USA. 2007;104:7391–7396. doi: 10.1073/pnas.0609757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zhang S, Zhang Z-Y. Drug Discovery Today. 2007;12:373–381. doi: 10.1016/j.drudis.2007.03.011. [DOI] [PubMed] [Google Scholar]
- [42].Halgren T, Murphy R, Friesner R, Beard H, Frye L, Pollard W, Banks J. J. Med. Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- [43].Turkson J, Zhang S, Palmer J, Kay H, Stanko J, Mora LB, Sebti S, Yu H, Jove R. Mol. Cancer Ther. 2004;3:1533–1542. [PubMed] [Google Scholar]
- [44].Bose RN. Mini-Rev. Med. Chem. 2002;2:103–111. doi: 10.2174/1389557024605500. [DOI] [PubMed] [Google Scholar]
- [45].Turkson J, Zhang S, Mor LB, Sebti S, Jove R. J. Biol. Chem. 2005;280:32 979–32 988. doi: 10.1074/jbc.M502694200. [DOI] [PubMed] [Google Scholar]
- [46].Aggarwal BB, Sethi G, Ahn KS, Sandur SK, Pandey MK, Kun-numakkara AB, Sung B, Ichikawa H. Ann. N. Y. Acad. Sci. 2006;1091(Part B):151–169. doi: 10.1196/annals.1378.063. Signal Transduction Pathways. [DOI] [PubMed] [Google Scholar]
- [47].Deng J, Grande F, Neamati N. Curr. Cancer Drug Targets. 2007;7:91–107. doi: 10.2174/156800907780006922. [DOI] [PubMed] [Google Scholar]
- [48].Berg T. ChemBioChem. 2008 DOI: 10.1002/cbic.200800274. [Google Scholar]