

Abstract
Paclitaxel is a microtubule-targeting antineoplastic drug widely used in human cancers. Even when tumors are initially responsive, progression of disease despite continued taxane therapy is all too common in the treatment of many of the most common epithelial cancers, including breast cancer. However, the mechanisms underlying paclitaxel resistance in cancer cells are not completely understood. Our hypothesis is that changes in the intrinsic (or mitochondrial) cell death pathway controlled by the BCL-2 family are key to the development of acquired paclitaxel resistance. Here we show that paclitaxel activates the mitochondrial apoptosis pathway, which can be blocked by BCL-2 overexpression. Treatment with ABT-737, a small molecule BCL-2 antagonist, restores sensitivity to paclitaxel in BCL-2 overexpressing cells. To investigate the importance of changes in the intrinsic apoptotic pathway in the absence of enforced BCL-2 expression, we generated two independent breast cancer cell lines with acquired resistance to apoptosis induced by paclitaxel. In these lines, acquired resistance to paclitaxel is mediated either by increased antiapoptotic BCL-2 proteins or decreased proapoptotic BCL-2 proteins. In both cases, ABT-737 can engage the mitochondrial apoptosis pathway to restore sensitivity to paclitaxel to cell lines with acquired paclitaxel resistance. In summary, these findings suggest that alterations in the intrinsic apoptotic pathway controlled by BCL-2 protein family members may be crucial to causing paclitaxel resistance. Furthermore, our results suggest that combining small molecule BCL-2 antagonists with paclitaxel may offer benefit to patients with paclitaxel-resistant tumors, an oncologic problem of great prevalence.
Introduction
Resistance to chemotherapy is a major obstacle for the success of cancer therapy. Resistance may be present at the onset of therapeutic intervention and patients initially fail to respond (intrinsic or innate chemoresistance) or it may emerge over time during chemotherapy (acquired chemoresistance), even after a dramatic initial response (1). In epithelial tumors including breast, ovarian and lung cancers, resistance to chemotherapy is frequently observed and is associated with poor clinical outcome (2-5).
Paclitaxel is a diterpenoid compound initially isolated from Taxus brevifolia and is widely used against malignant epithelial tumors. Paclitaxel mainly targets β-tubulin at the molecular level and stabilizes microtubule dynamics to induce microtubule polymerization (6). There are a variety of mechanisms responsible for the development of chemoresistance in cancer cells; reduced drug uptake or increased drug efflux, increased repair of drug targets, alteration of drug targets that prevent drug binding or action and blocks in biochemical pathways that mediate drug-induced cytotoxicity. Development of acquired paclitaxel-resistance has been attributed to such mechanisms, including overexpression of P-glycoprotein, alterations in tubulin composition and mutations in β-tubulin (7-10). Nearly every patient with advanced or metastatic breast cancer in the United States receives a taxane as part of their chemotherapy. Therefore, those who die of their disease have generally ceased to respond to taxanes.
One can greatly simplify the pathway of chemotherapy response into three main steps. In the first step, drug must accumulate in the cell in sufficient concentration. In the second step, the drug must interact with target. In the third step, death signals generated from drug interaction with target must interact with a receptive apoptotic pathway. Acquired resistance due to blocking of the first two steps has been examined for paclitaxel. The third step, involving alteration of the intrinsic apoptotic pathway, has not been well explored as a mechanism of acquired paclitaxel resistance.
Apoptosis, a type of programmed cell death, is an essential mechanism for cell death following many types of chemotherapy. The mitochondrial (or intrinsic) apoptosis pathway is mainly regulated by the interplay between members of the BCL-2 protein family. Following apoptotic stimuli, including many kinds of chemotherapy, cytochrome c is released into the cytosol from mitochondria as a result of mitochondrial outer membrane permeabilization, which is followed by the formation of the apoptosome complex and caspase activation (11). The key, essentially irreversible step in the commitment to apoptotic death is the permeabilization of the mitochondrial outer membrane. It is this step that is controlled by the interaction of BCL-2 family proteins.
Proapoptotic BCL-2 proteins can be classified into two main groups: multidomain proapoptotic proteins, BAX and BAK; and BH3-only proteins, BID, BIM, BAD, NOXA, PUMA, BMF, BIK and HRK. In response to death stimuli, BH3-only proteins are upregulated by numerous means, including transcriptional upregulation, posttranslational modification and subcellular localization. Certain BH3-only proteins, the so-called activators, which include BID and BIM, induce the activation of BAX and BAK. Activated BAX and BAK undergo a conformational change, which can be recognized by conformation–specific antibodies, and oligomerize. Oligomerized BAX and BAK, perhaps in a complex with other proteins, induce permeabilization of the outer mitochondrial membrane which allows diffusion of proapoptotic proteins, including cytochrome c from the intermembrane space. Antiapoptotic BCL-2 proteins BCL-2, BCL-XL, MCL-1, BCL-w, and BFL-1 control the mitochondrial release of cytochrome c by sequestering proapoptotic BCL-2 family proteins. BAD, NOXA, PUMA, BMF, BIK and HRK, known as sensitizers, act as inhibitors of the inhibitors of apoptosis, displacing activators from antiapoptotic BCL-2 proteins, which are then freed to trigger activation and oligomerization of BAX and BAK (12, 13). Adding to this complexity, antiapoptotic BCL-2 proteins exhibit selective binding affinities to sensitizer BH3-only proteins and the dynamic nature of these interactions frequently dictate the fate of the cell in response to apoptotic stimuli, either death or survival.
Here we systematically investigate, for the first time, whether alterations in the apoptotic pathway are responsible for resistance to paclitaxel. We find that breast cancer cell lines that acquire resistance increase antiapoptotic protein expression or decrease proapoptotic protein expression. ABT-737 is a small molecule inhibitor of BCL-2, BCL-XL and BCL-w that binds to the BH3-binding cleft with very high affinity (14). ABT-737 was reported to elicit apoptosis either as a single agent, or in combination with chemotherapeutics against several cancer cell types, including non-Hodgkins lymphoma, non-small cell lung cancer, and various leukemias, (13, 15-27). An orally available derivative of ABT-737, ABT-263, is currently in study in clinical trials for non-Hodgkins lymphoma, chronic lymphocytic leukemia, and small-cell lung cancer. We find that we can reverse acquired resistance to paclitaxel using ABT-737. We suggest that combination therapy of taxanes with ABT-737 may be a useful strategy to attack the serious clinical problem of acquired taxane resistance in breast cancer.
Material and Methods
Cell lines
MCF-7 and MDA-MB-468 cells were grown in DMEM/F12 (Invitrogen) supplemented with 2 mM L-Glutamine, 10% heat-inactivated fetal bovine serum (Sigma), 100 IU/ml penicillin and 100 μg/ml streptomycin (Invitrogen) in a humidifed incubator at 37°C and 5% CO2. 5 μg/ml insulin (Sigma) was added to DMEM/F12 medium used for the culture of MCF-7 and T47D cells.
The paclitaxel-resistant MCF-7 and MDA-MB-468 cells were derived as described before (28). Following selection of paclitaxel-resistant clonal lines, MCF-7 TaxR30 and MCF-7 TaxR50 were maintained in the presence of 30 nM and 50 nM paclitaxel, MDA-MB-468 TaxR was maintained in the presence of 15 nM paclitaxel, respectively. All paclitaxel-resistant cell lines were grown in the presence of 5 μg/ml verapamil.
MCF-7 and T47D cells were stably transfected with either pCI-Neo.FlagBCL-2 (MCF-7 BCL-2, T47D BCL-2) or pCI-Neo.FLAG plasmid (MCF-7 Mock, T47D Mock) using Fugene 6 (Roche) and clonal selection was carried out in the presence of G418 (1.2 mg/mL; Sigma). The resulting clones were assayed by detection of BCL-2 or FLAG-tag using immunoblot analysis. Selected clones were maintained in growth medium with 0.2 mg/mL G418.
Chemicals
Paclitaxel was purchased from Sigma and dissolved in DMSO. ABT-737 and ABT-737 enantiomer were provided by Abbott Laboratories (Abbott Park, IL). Caspase-9 inhibitor (z-LEHD-FMK), pancaspase inhibitor (z-VAD-FMK) and caspase-8 inhibitor (z-IETD-FMK) were obtained from BD Biosciences Pharmingen.
Cell viability and apoptotic assays
Apoptosis was evaluated by Annexin-V-FITC (BioVision) staining according to the manufacturer's protocols. Apoptosis was quantified by flow cytometry on a FACSCalibur (BD Biosciences), followed by analysis using WinMDI 2.9 software (Scripps Institute, La Jolla, CA). For determination of EC50 values for paclitaxel in parental and resistant cells lines, cell viability was determined using MTT assay kit according to manufacturer's instructions following treatment with various concentrations of paclitaxel. The results were from three experiments in triplicate. The EC50 values for paclitaxel in parental and resistant cells were determined by nonlinear regression of sigmoidal dose-response curves using GraphPad Prism 3.0 software (GraphPad, San Diego, CA).
Colony-forming assay
Colony-forming assay was performed as described previously (29). Briefly, paclitaxel resistant MCF-7 and MDA-MB-468 cells were plated in 24-well plates and treated with drugs for 16 h. Cells were counted and replated into 60-mm tissue culture dishes (200 cells/plate). Following 10 days of incubation, tissue culture plates were stained with crystal violet [0.5% crystal violet in a 3:1 (v/v) mixture of distilled water:methanol], and colonies were counted. Results were expressed as the percentage of colony formation by untreated control cells.
Immunoprecipitation and immunoblotting
Total cell lysates were prepared in 1% Chaps buffer [5 mM MgCl2, 137 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Chaps, 20 mM Tris-HCl (pH 7.5), and protease inhibitors (Complete, Roche)] as described previously (30). To monitor the activation of BAX and BAK, proteins (800 μg) were immunoprecipitated with anti-BAX (6A7; BD Pharmingen) and anti-BAK (Ab-2, Oncogene Research) at 4°C for 2 h. Immunoprecipitates were captured by 50% slurry of protein G-Sepharose in lysis buffer (GE Healthcare) at 4°C for 2 h. Immunoprecipitates were then recovered by centrifugation and washed three times in 1% Chaps buffer. Immunoprecipitates, total cell extracts (40 μg) and subfractionation lysates (30 μg) were separated on NuPage 10% Bis-Tris gels. Following SDS-PAGE, proteins were transferred onto PVDF membranes (Immobilon, Millipore) and then blocked with 5% dried milk in PBS-Tween20. Membranes were incubated with primary and secondary antibodies (GE Healthcare) in a buffer containing 10% milk diluent blocking concentrate (KPL), detected with Western Lightning Chemiluminescence Reagent Plus (PerkinElmer) and exposed to Biomax MR film (Kodak). For detection of some immunoprecipitates, protein A-HRP (GE Healthcare) was employed as a secondary detection agent. The following antibodies were used for immunoblotting: anti-FLAG (Sigma), anti-BCL-2, anti-BCL-XL, anti-BMF, anti-BAD, anti-PUMA, anti-β-Actin, anti-caspase-3, anti-caspase-9, anti-caspase-8, (Cell Signaling), anti-BAX (N20; Santa Cruz), anti-BAK (G-23; Santa Cruz), anti-BIM (22-40; Calbiochem) anti-cytochrome c (6H2.B4; BD Biosciences), and anti-CoxIV (Molecular Probes).
Caspase activation assays
The activity of Caspase-3, -9 and -8 was determined by ApoAlert Caspase Profiling Plate (Clontech) according to manufacturer's protocol. The release of fluorochrome AMC was analyzed at 380 nm excitation and 460 nm emission using a multiplate fluorescence spectrophotometer. Data shown are mean ± SE of three independent experiments in duplicate and expressed in arbitrary fluorescence units (AFU)/mg of protein.
Subcellular fractionation
Subcellular fractionation was performed as described before (31). Briefly, cells were harvested and washed in ice-cold PBS and then resuspended in an isotonic buffer [250 mM sucrose, 20 mM HEPES (pH 7.5), 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM phenylmethylsulphonyl fluoride, and protease inhibitors (Complete, Roche)] on ice for 20 min. Following incubation, cells were homogenized with Dounce homogenizer and centrifuged at 800g for 10 min at 4°C. The resulting supernatant was centrifuged at 8,000g for 20 min at 4°C to obtain mitochondrial and cytosolic fractions. These fractions were used to monitor cytochrome c release from mitochondria. Mitochondrial fractions were lysed in 1% Chaps buffer for immunoblot analysis.
Statistical analysis
Statistical significance of the results was analyzed using Student's t-tail test using GraphPad Prism 3.0 software. P < 0.05 and P < 0.01 were considered significant.
Results
ABT-737 treatment overcomes BCL-2-mediated resistance to paclitaxel-induced apoptosis
If a type of cell death operates via the mitochondrial apoptotic pathway, we would expect that BCL-2 overexpression would block the death. We first tested whether BCL-2 overexpression could block apoptosis due to paclitaxel treatment. MCF-7 and T47D cells were stably transfected with either pCI-Neo.FlagBCL-2 (MCF-7 BCL-2, T47D BCL-2) or control vector (MCF-7 Mock, T47D Mock). The overexpression of BCL-2 was verified by immunoblotting of total cell extracts with anti-BCL-2 or FLAG antibody (Fig. 1A and C). Both parental and transfected cells were treated with 100 nM paclitaxel for 48 h and analyzed for apoptotic response by Annexin V staining. The results clearly demonstrated the protection against paclitaxel-induced apoptosis by BCL-2 overexpression in MCF-7 and T47D cells (Fig. 1B and D). These results imply that the mitochondrial, or intrinsic, apoptotic pathway, which is controlled by the BCL-2 family of proteins, is necessary for apoptosis following paclitaxel treatment.
Figure 1.
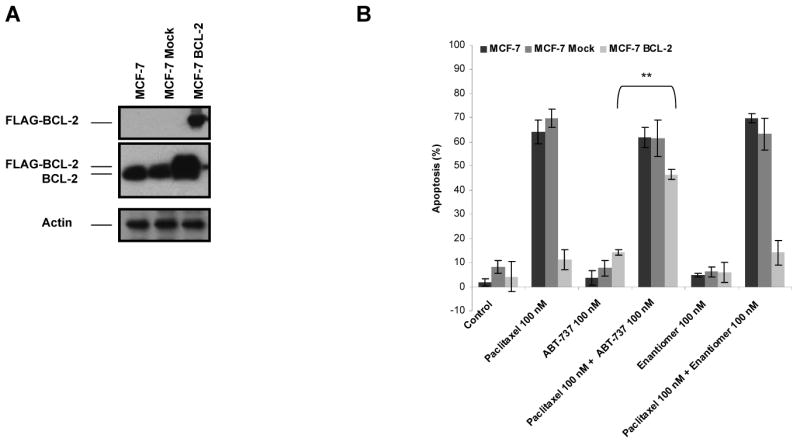
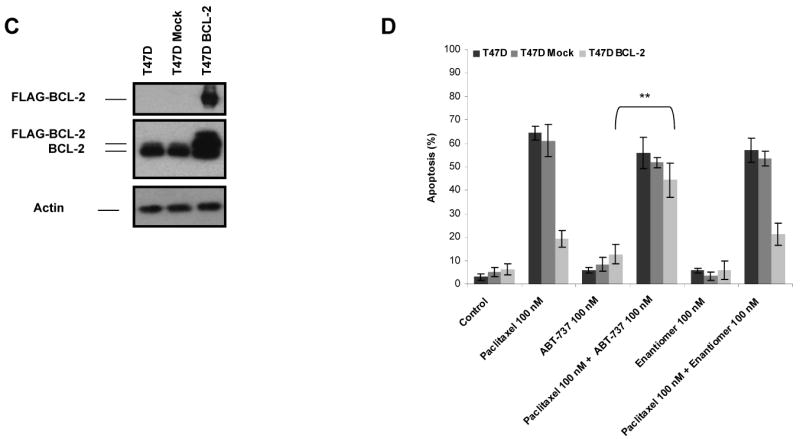
BCL-2 protects against paclitaxel-induced apoptosis and ABT-737 might overcome BCL-2-mediated paclitaxel resistance. A, MCF-7 cells were stably transfected with control vector (MCF-7 Mock) or pCI.Neo.FlagBCL-2 (MCF-7 BCL-2). The expression of FLAG-BCL-2 in transfected cells were verified by immunoblotting with anti-BCL-2 or anti-FLAG antibody. β-Actin was probed as a loading control. B, cells were treated with treated with paclitaxel (100 nM), ABT-737 (100 nM), combination of paclitaxel (100 nM) and ABT-737 (100 nM), enantiomer of ABT-737 (100 nM) or combination of enantiomer of ABT-737 (100 nM) and paclitaxel (100 nM) for 48 h. The percent apoptotic response was evaluated by Annexin V staining. Columns, mean of three independent experiments; bars, SE. **, P < 0.01, paclitaxel plus ABT-737 treated with respect to paclitaxel-only treated. C, T47-D cells were stably transfected with control vector (T47D Mock) or pCI.Neo.FlagBCL-2 (T47D BCL-2). The expression of FLAG-BCL-2 in transfected cells were verified by immunoblotting with anti-BCL-2 or anti-FLAG antibody. β-Actin was probed as a loading control. D, cells were treated with treated with paclitaxel (100 nM), ABT-737 (100 nM), combination of paclitaxel (100 nM) and ABT-737 (100 nM), enantiomer of ABT-737 (100 nM) or combination of enantiomer of ABT-737 (100 nM) and paclitaxel (100 nM) for 48 h. The percent apoptotic response was evaluated by Annexin V staining. Columns, mean of three independent experiments; bars, SE. **, P < 0.01, paclitaxel plus ABT-737 treated with respect to paclitaxel-only treated.
ABT-737 is a cell-permeable BCL-2, BCL-XL and BCL-w inhibitor, which was shown to induce apoptosis in various cancer cell types as a single agent and in combination with chemotherapeutics (18-20, 23-26). Therefore, we evaluated whether treatment with ABT-737 could reverse BCL-2-mediated protection against paclitaxel-induced apoptosis in MCF-7 BCL-2 and T47D BCL-2 cells. As shown in Fig. 1B and D, ABT-737 efficiently augmented paclitaxel-induced apoptosis in MCF-7 BCL-2 and T47D BCL-2 cells, even though its negative control enantiomer did not exert such an effect. Notably, ABT-737 or negative control enantiomer did not elicit a significant effect when utilized alone. Moreover combining ABT-737 or negative control enantiomer with paclitaxel did not enhance paclitaxel-induced apoptosis in parental or mock-transfected MCF-7 or T47D cells. Of interest, co-treatment of parental MCF-7 and T47D cells with ABT-737 and 0-100 nM paclitaxel did not elicit a major effect on apoptotic response, except a slight, but statistically insignificant, increase in Annexin V-positive cells when 100 nM ABT-737 was combined with 50 nM paclitaxel in MCF-7 cells (results not shown).
Since BCL-2 family proteins control the mitochondrial apoptotic pathway, our expectation was that the sensitization effect of ABT-737 on BCL-2 overexpressing breast cancer cells also operated through the mitochondrial apoptosis pathway. In support of this expectation, we found that co-treatment with paclitaxel and ABT-737 induced mitochondrial outer membrane permeabilization, as measured by cytochrome c release from the mitochondria (Fig 2A). The conformational change in BAX and BAK that accompanies apoptosis via the mitochondrial pathway was also observed following co-treatment (Fig. 2B). Caspase 9 activation accompanies apoptosis via the mitochondrial apoptotic pathway, while activation of caspase 8 accompanies activation via the extrinsic apoptotic pathway. Caspase assays demonstrated the activation of caspase 9, but not caspase 8, in MCF-7 BCL-2 cells treated with the combination of ABT-737 and paclitaxel (Fig. 2C). Demonstrating that caspase 9 activation was required for cell death and not simply a synchronous phenomenon, pretreatment with pancaspase inhibitor z-VAD-FMK and caspase-9 inhibitor z-LEHD-FMK, but not caspase 8 inhibitor z-IETD-FMK, attenuated ABT-737-mediated sensitization of MCF-7 BCL-2 cells to paclitaxel-induced apoptosis (Fig. 2D). These findings suggest that the apoptotic block against paclitaxel treatment imparted by BCL-2 overexpression could be overcome by co-treatment with the BCL-2 inhibitor, ABT-737 in breast cancer cells, resulting in a restoration of death via the mitochondrial, or intrinsic, apoptotic pathway.
Figure 2.
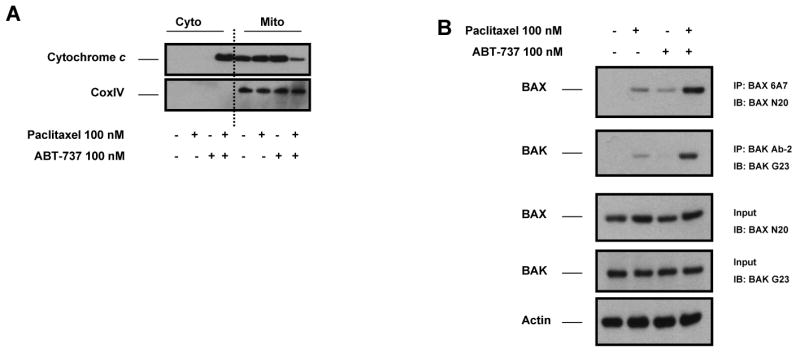

ABT-737-mediated sensitization of MCF-7 BCL-2 cells to paclitaxel-induced apoptosis via mitochondrial apoptosis pathway. A, MCF-7 BCL-2 cells were treated with paclitaxel (100 nM), ABT-737 (100 nM), or combination of paclitaxel (100 nM) and ABT-737 (100 nM) for 48 h. Cytosolic and mitochondrial fractions from MCF-7 BCL-2 cells were immunoblotted for cytochrome c. CoxIV was probed as a loading control for mitochondrial fractions. B, MCF-7 BCL-2 cells were treated with paclitaxel (100 nM), ABT-737 (100 nM), or the combination of paclitaxel (100 nM) and ABT-737 (100 nM) for 12 h. Activation of BAX and BAK was analyzed by immunoprecipitation with active conformation-specific anti BAX (6A7) and anti-BAK (Ab-2) antibodies followed by immunoblot analysis of BAX and BAK. 5% of the input for immunoprecipitation was also subjected to immunoblot analysis. β-Actin was probed as a loading control. C, MCF-7 BCL-2 cells were treated with paclitaxel (100 nM), ABT-737 (100 nM), or combination of paclitaxel (100 nM) and ABT-737 (100 nM) for 48 h. Activation of caspase-9 and caspase-8 by the combination of paclitaxel and ABT-737 treatment was evaluated by fluorometric caspase activation assays as described in Materials and methods. Data were expressed as relative fluorescence units (RFU). Columns, mean of three independent experiments; bars, SE. D, MCF-7 BCL-2 cells were pretreated with 20 μM pancaspase inhibitor (Z-VAD-FMK), 20 μM caspase-9 inhibitor (Z-LEHD-FMK) and 20 μM caspase-8 inhibitor (Z-IETD-FMK) before treatment with paclitaxel plus ABT-737 for 48 h. The percent apoptotic response was evaluated by Annexin V staining. Columns, mean of three independent experiments; bars, SE.
Acquired resistance to paclitaxel is mediated by alterations in BCL-2 protein family members
The above experiments established that enforced changes in the mitochondrial apoptotic pathway could mediate resistance to paclitaxel. We next asked whether changes in the mitochondrial apoptotic pathway were selected for in models of paclitaxel resistance in which resistance was passively derived by long-term exposure to drug. To establish paclitaxel-resistant breast cancer cell lines, we exposed MCF-7 and MDA-MB-468 cells to increasing concentrations of paclitaxel for short times. MCF-7 cells are ER+, p53 wt and MDA-MB-468 cells are ER-, p53 mutated breast cancer cell lines, which were chosen to represent different genetic properties of primary breast cancer tumors. After resistant cell pools were developed, we selected individual resistant clones by continuous exposure to paclitaxel. Two different clones of MCF-7 with different levels of resistance against paclitaxel were generated and designated as MCF-7 TaxR30 and MCF-7 TaxR50. Additionally, paclitaxel-resistant clone of MDA-MB-468 was established and designated as MDA-MB-468 TaxR. EC50 values for paclitaxel in parental and resistant cell lines were determined by nonlinear regression analysis of dose-response curves (Fig. 3A). MCF-7 TaxR30 and MCF-7 TaxR50 cells tolerated ∼100 and ∼150 fold greater doses of paclitaxel treatment in comparison to parental MCF-7 cells, respectively. Similarly, MDA-MB-468 TaxR cells tolerated ∼15 fold greater doses of paclitaxel when compared with parental MDA-MB-468 cells.
Figure 3.

Acquired resistance to paclitaxel is mediated by BCL-2 protein family members. A, EC50 values of parental (MCF-7 and MDA-MB-468) and paclitaxel-resistant cell lines (MCF-7 TaxR30, MCF-7 TaxR50 and MDA-MB-468 TaxR, respectively) were evaluated with MTT assay after treatment with various doses of paclitaxel for 48 h. EC50 values were determined by nonlinear regression analysis using GraphPad Prism software. Bars, SE. B and C, Western blot analysis of BCL-2 protein family members in parental and paclitaxel-resistant cell lines. Paclitaxel-resistant cell lines marked with “+” were grown in continuous exposure to paclitaxel (30 nM for MCF-7 TaxR30, 50 nM for MCF-7 TaxR50, 15 nM for MDA-MB-468 TaxR) or paclitaxel was withdrawn from growth media for 24 hours to reduce the possibility of acute paclitaxel-mediated variations in protein levels (marked with “-”). Total cellular proteins were isolated either in the presence or in the absence of paclitaxel in growth media and analyzed in parallel. β-Actin was probed as a loading control.
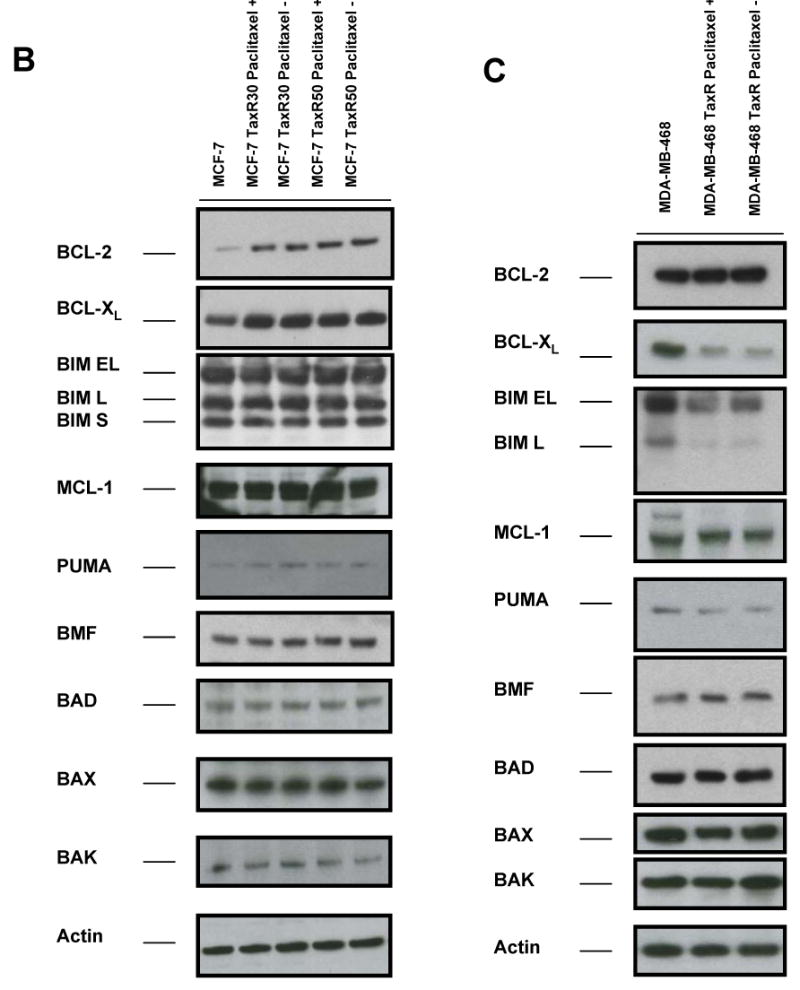
The expression of BCL-2 family proteins in parental and paclitaxel-resistant clones was compared. Since we were interested in permanent changes established by selection rather than changes due to presence of drug, we compared continuous paclitaxel to withdrawing paclitaxel from media for 24 h before isolation of total proteins, though in practice this had little effect on our findings. We then analyzed proteins isolated from resistant clones grown in media in which paclitaxel was either restored or absent. As demonstrated in Fig. 3B, BCL-2 and BCL-XL levels were increased in MCF-7 TaxR30 and MCF-7 TaxR50 cells compared with parental cells. We could not detect any change in expression levels of other BCL-2 proteins. Correspondingly, there were decreased levels of both BCL-XL and BIM in paclitaxel-resistant MDA-MB-468 TaxR cells in comparison to parental MDA-MB-468 cells (Fig. 3C). Moreover, no significant alterations in other BCL-2 protein family members were found in MDA-MB-468 TaxR cells. These results indicate that the acquired resistance to paclitaxel may be promoted either by increased levels of antiapoptotic BCL-2 proteins (BCL-2, BCL-XL) or by decreased levels of proapoptotic BH3-only proteins (BIM) in breast cancer cells.
BCL-2 inhibitor ABT-737 restores the sensitivity to paclitaxel in paclitaxel-resistant breast cancer cells
ABT-737 antagonizes BCL-2 function by acting as a mimetic of the BH3 domain of pro-apoptotic BH3-only proteins. Thus, it seemed possible that treatment with ABT-737 might reverse resistance induced by either increased expression of antiapoptotic BCL-2 and BCL-XL or decreased expression of the proapoptotic BH3-only proteins BIM. As shown in Fig. 4A, treatment of paclitaxel-resistant MCF-7 TaxR30 and MCF-7 TaxR50 cells with ABT-737 resulted in restoration of paclitaxel sensitivity in these cells as determined by Annexin V staining. To verify that the observed effects were occurring via the mitochondrial pathway, we monitored cytochrome c release from mitochondria. Paclitaxel treatment induced mitochondrial cytochrome c release in parental MCF-7 cells, and as seen in Fig. 4B, increased sensitivity of MCF-7 TaxR50 to paclitaxel when combined with ABT-737 was also accompanied by increased translocation of cytochrome c into cytosol. To further delineate the molecular mechanisms of ABT-737-mediated sensitization to paclitaxel-induced apoptosis, MCF-7 TaxR50 cells were treated with paclitaxel, ABT-737, or both and activation of BAX and BAK were evaluated by immunoprecipitation using conformation-specific antibodies. When paclitaxel-resistant cells were treated with the combination of paclitaxel and ABT-737, a significantly increased activation of BAX and BAK was detected in MCF-7 TaxR50 cells (Fig. 4C). No such effect was observed upon exposure to either paclitaxel or ABT-737 alone. Thus, as expected, ABT-737 targets a pathway upstream of BAX and BAK activation in MCF-7 TaxR50 cells to promote sensitization to paclitaxel-induced apoptosis.
Figure 4.



ABT-737 restores sensitivity to paclitaxel in paclitaxel-resistant MCF-7 cells. A, MCF-7 TaxR30 and MCF-7 TaxR50 cells were treated with paclitaxel (300 nM), ABT-737 (300 nM), or combination of paclitaxel (300 nM) and ABT-737 (300 nM) for 48 h. The percent apoptotic response was evaluated by Annexin V staining. Columns, mean of three independent experiments; bars, SE. *, P < 0.05; **, P < 0.01, paclitaxel plus ABT-737 treated with respect to paclitaxel-only treated. B, MCF-7 cells were treated with paclitaxel (100 nM) for 0-48 h, MCF-7 TaxR50 cells were treated as in A, cytosolic and mitochondrial fractions were blotted for cytochrome c. CoxIV was probed as a loading control for mitochondrial fractions. C, MCF-7 TaxR50 cells were treated with paclitaxel (300 nM), ABT-737 (300 nM) or combination of paclitaxel (300 nM) and ABT-737 (300 nM) for 12 h. Activation of BAX and BAK was analyzed by immunoprecipitation with active conformation-specific anti BAX (6A7) and anti-BAK (Ab-2) antibodies followed by immunoblot analysis of BAX and BAK. 5% of the input for immunoprecipitation was also subjected to immunoblot analysis. β-Actin was probed as a loading control. D, (left) MCF-7 TaxR50 cells were treated as in A, and activation of caspase-9 and caspase-8 was evaluated by fluorometric caspase activation assays. Data were expressed as relative fluorescence units (RFU). Columns, mean of three independent experiments; bars, SE. (middle) Total cell extracts were analyzed for the activation of caspase-9 and caspase-8 by immunoblotting. β-Actin was probed as a loading control. (right) MCF-7 TaxR50 cells were pretreated with 20 μM pancaspase inhibitor (Z-VAD-FMK), 20 μM caspase-9 inhibitor (Z-LEHD-FMK) and 20 μM caspase-8 inhibitor (Z-IETD-FMK) before treatment with paclitaxel plus ABT-737 for 48 h. The percent apoptotic response was evaluated by Annexin V staining. Columns, mean of three independent experiments; bars, SE.
To confirm the engagement of caspases downstream of mitochondria, we assayed the activation of caspase-9 and caspase-8 in MCF-7 TaxR50 cells using immunoblot analysis and fluorometric caspase activation assays, in response to treatment with paclitaxel, ABT-737 or combination of both. As shown in Fig. 4D (middle), elevated levels of active caspase 9 were induced by paclitaxel and ABT-737 when compared to treatment with paclitaxel or ABT-737 alone in MCF-7 TaxR50 cells. In contrast, caspase-8 was not activated in this cell line. Fluorometric caspase assays further confirmed the enhanced activation of caspase-9 by combination of paclitaxel and ABT-737 in MCF-7 TaxR50 cells (Fig. 4D, left). Additionally, pretreatment with pancaspase inhibitor z-VAD-FMK and caspase-9 inhibitor z-LEHD-FMK markedly abolished ABT-737-mediated sensitization of MCF-7 TaxR50 cells to paclitaxel-induced apoptosis, although caspase-8 inhibitor z-IETD-FMK did not elicit such a similar effect (Fig. 4D, right). Thus, ABT-737 is restoring taxane sensitivity to resistant MCF-7 cells via the mitochondrial apoptotic pathway controlled by BCL-2 family proteins, consistent with ABT-737 acting at the expected target.
Similarly, combination of paclitaxel and ABT-737 also led to a restored apoptotic response in MDA-MB-468 TaxR cells (Fig. 5A). On the contrary, no such effect was observed in parental MDA-MB-468 cells when ABT-737 was used in combination with paclitaxel (0-100 nM) (results not shown). Treatment of MDA-MB-468 parental cells with paclitaxel (100 nM) triggered apoptosis (Fig. 5A) and induced the release of cytochrome c into cytoplasm (Fig. 5B). In parallel to apoptotic response, paclitaxel plus ABT-737 treatment resulted in the release of cytochrome c into cytosol in MDA-MB-468 TaxR cells, even though treatment with paclitaxel or ABT-737 alone did not show such an effect (Fig. 5B). Activation of BAX and BAK were also drastically enhanced in MDA-MB-468 TaxR cells treated paclitaxel plus ABT-737 in comparison to treatment with paclitaxel or ABT-737 alone (Fig. 5C). In addition, activation of caspase-3, caspase-9 and caspase-8 was detected when MDA-MB-468 TaxR cells treated with paclitaxel plus ABT-737 as shown by immunoblot analysis and fluorometric caspase activation assays (Fig. 5D, left, middle). Correspondingly, pretreatment with z-VAD-FMK and z-LEHD-FMK attenuated ABT-737-mediated sensitization of MDA-MB-468 TaxR cells to paclitaxel-induced apoptosis; caspase-8 inhibitor z-IETD-FMK exerted only a partial protective effect (Fig. 5D, right).
Figure 5.



ABT-737 restores sensitivity to paclitaxel in paclitaxel-resistant MDA-MB-468 cells. A, MDA-MB-468 cells were treated with 100 nM paclitaxel for 48 h. MDA-MB-468 TaxR cells were treated with paclitaxel (100 nM), ABT-737 (200 nM) or combination of paclitaxel (100 nM) and ABT-737 (200 nM) for 48 h. The percent apoptotic response was evaluated by Annexin V staining. Columns, mean of three independent experiments; bars, SE. **, P < 0.01, paclitaxel plus ABT-737 treated with respect to paclitaxel-only treated. B, MDA-MB-468 cells were treated with paclitaxel (100 nM) for 0-48 h, MDA-MB-468 TaxR cells were treated as in A. Cytosolic and mitochondrial fractions were blotted for cytochrome c. CoxIV was probed as a loading control for mitochondrial fractions. C, MDA-MB-468 TaxR cells were treated with paclitaxel (100 nM), ABT-737 (200 nM) or combination of paclitaxel (100 nM) and ABT-737 (200 nM) for 12 h. Activation of BAX and BAK was analyzed by immunoprecipitation with active conformation-specific anti BAX (6A7) and anti-BAK (Ab-2) antibodies followed by immunoblot analysis of BAX and BAK. 5% of the input for immunoprecipitation was also subjected to immunoblot analysis. β-Actin was probed as a loading control. D, (left) MDA-MB-468 TaxR cells were treated as in A, and activation of caspase-3, caspase-9 and caspase-8 was evaluated by fluorometric caspase activation assays. Data were expressed as relative fluorescence units (RFU). Columns, mean of three independent experiments; bars, SE. (middle) Total cell extracts were analyzed for the activation of caspase-3, caspase-9 and caspase-8 by immunoblotting. β-Actin was probed as a loading control. (right) MDA-MB-468 TaxR cells were pretreated with 20 μM pancaspase inhibitor (Z-VAD-FMK), 20 μM caspase-9 inhibitor (Z-LEHD-FMK) and 20 μM caspase-8 inhibitor (Z-IETD-FMK) before treatment with paclitaxel plus ABT-737 for 48 h. The percent apoptotic response was evaluated by Annexin V staining. Columns, mean of three independent experiments; bars, SE.
To further demonstrate the cytotoxic effect of combining ABT-737 with paclitaxel in paclitaxel-resistant cell lines, MCF-7 TaxR50 and MDA-MB-468 TaxR cells were treated with paclitaxel, ABT-737 or combination of both for 16 h and survival capability of cells were evaluated by colony-formation assays. As shown in Fig. 6A, combination of paclitaxel with ABT-737 led to loss of colony formation capability of paclitaxel-resistant cells, although treatment with paclitaxel or ABT-737 did not exert a similar effect.
Figure 6. The effect of ABT-737 and paclitaxel on colony formation in paclitaxel-resistant MCF-7 and MDA-MB-468 cells.


A, MCF-7 TaxR50 cells were treated with paclitaxel (300 nM), ABT-737 (300 nM) or combination of paclitaxel (300 nM) and ABT-737 (300 nM) for 16 h. MDA-MB-468 TaxR cells were treated with paclitaxel (100 nM), ABT-737 (200 nM) or combination of paclitaxel (100 nM) and ABT-737 (200 nM) for 16 h. Clonogenic survival was assessed by colony-forming assay. Data presented are percentage of colony formation normalized to untreated control cells. B, Model of alterations in BCL-2 protein family members governing paclitaxel resistance. I, paclitaxel sensitive cells. II, Paclitaxel-resistant cells, similar to MCF-7 TaxR50. III, Paclitaxel resistant cells, similar to MDA-MB-468 TaxR.
Thus, alterations of BCL-2 family proteins may contribute to the acquired resistance to paclitaxel in breast cancer cells and ABT-737 could selectively restore paclitaxel-induced apoptosis via the mitochondrial pathway in these resistant cells. A summary of these alterations is presented in Fig. 6B. Briefly, in paclitaxel-sensitive cells prior to treatment, antiapoptotic BCL-2 proteins are present in sufficient quantity to sequester existing activator BH3-only proteins. Following paclitaxel treatment, additional BH3-only proteins are activated, by changes in subcellular localization, increase in protein abundance or posttranslational modifications. These new proteins compete for the BH3 binding site in antiapoptotic BCL-2 proteins, exceeding the total BH3 binding capacity. Unsequestered activator BH3-only proteins, including BIM, trigger apoptosis by activating BAX and BAK (Fig. 6A). We found that cancer cells could select to block this chain of events in two ways. First, in the MCF-7 cells, apoptosis could be blocked by increased expression of antiapoptotic BCL-2 proteins, preventing BAX and BAK activation through sequestering more activators and sensitizers effectively (Fig. 6B). Second, as in the MDA-MB-468 cells, decreased expression of BH3-only activators reduces the pro-apoptotic load so that death signaling induced by paclitaxel is insufficient to overwhelm the antiapoptotic BH3 domain binding capacity (Fig. 6C). ABT-737 can reverse these mechanisms of resistance by restoring increased competition for BH3 domain binding sites, thus yielding more unsequestered activator BH3-only proteins to activate BAX and BAK.
Discussion
Acquired resistance to treatment negatively affects the outcome of chemotherapy in solid tumors and hematologic malignancies (1). Many tumors show an initial sensitivity to chemotherapy, resulting in a good clinical response, even a complete remission with no remaining evidence of disease. All too often, however, the cancer recurs in a form that is newly resistant to the initially successful chemotherapy.
Many chemotherapeutics, including paclitaxel, have been shown to exert their effect through engagement and activation of the intrinsic apoptotic pathway (1, 32-34). However, most studies of paclitaxel resistance have focused on events upstream of engagement of the intrinsic apoptotic pathway, either on drug interacting with target or entry of drug into cell. Paclitaxel binds to the N-terminal domain of β-tubulin to initiate microtubule polymerization. Resistance to paclitaxel treatment was shown to be mediated by alterations in tubulin composition or mutations in the paclitaxel-binding region of β-tubulin (8, 10). Overexpression of P-glycoprotein (P-gp) was also reported in a variety of tumor cells and P-gp was proposed to play a role in resistance to paclitaxel treatment (7). Heretofore lacking is an examination of whether induced resistance to paclitaxel is often conferred by alterations in the control of apoptotic cell death. This question becomes more pressing as novel drugs that interact with BCL-2 proteins are now in clinical trials.
We provide evidence here that paclitaxel uses the mitochondrial apoptotic pathway to induce apoptosis in breast cancer cell lines consistent with previous reports (35, 36) When drugs kill using this pathway, they do so by increasing the balance of proapoptotic proteins compared to anti-apoptotic proteins at the mitochondria. BH3-only proteins are often the most dynamic participants in this process, and we and others have found that indeed, signaling by BH3-only proteins is required for paclitaxel-induced apoptosis (Kutuk and Letai, unpublished, (37-39). It seemed, therefore, that alterations in the intrinsic apoptotic pathway might well play an important and heretofore unexamined role in the acquisition of resistance to paclitaxel in cancer cells.
In fact, our findings indicate, in two independently derived paclitaxel resistant cell lines, a key mediator of acquired resistance is indeed alteration in control of the mitochondrial apoptotic pathway. As none of these changes was enforced, but rather selected by random mutation, it suggests that such alterations in apoptotic signaling may be quite important in cancers that acquire resistance to paclitaxel. The resistant MCF-7 lines exploit this strategy by increasing expression of BCL-2 and BCL-XL. In contrast, the MDA-MB-468 cells acquire resistance by decreasing BIM levels. They simultaneously lower BCL-XL levels as well, suggesting that the loss of BIM is more significant than the loss of BCL-XL, since the resulting phenotype is resistance to paclitaxel. Indeed, we have found that BIM plays a critical role in apoptosis due to paclitaxel in breast cancer cell lines (Kutuk and Letai, unpublished.)
ABT-737 opposes BCL-2 and BCL-XL function by acting as a mimetic of the BH3 domain of select BH3-only proteins. A test of whether the observed alterations in BCL-2, BCL-XL and BIM levels caused the observed resistance is whether treatment with ABT-737 would reverse the resistance. The restoration of sensitivity to paclitaxel by ABT-737 suggests that the observed changes are indeed responsible for the observed resistance.
Detailed understanding of pathways of paclitaxel resistance in cancer cells can provide important opportunities to design strategies for specifically targeting these pathways. Our results clearly rationalize the utilization of ABT-737 to augment apoptotic response induced by paclitaxel, whenever chemoresistance against paclitaxel was conferred by increased expression of BCL-2/BCL-XL or diminished levels of BIM. In practical terms, our results support clinical investigation of antagonism of BCL-2 and BCL-XL, for example with ABT-263, to reverse taxane resistance in breast cancer.
Acknowledgments
The authors would like to gratefully acknowledge financial support from NCI K08 CA102548 and R01 CA129974. We would like to further thank Hal Burstein for useful discussions. A. Letai is a founder of Eutropics Pharmaceuticals and a member of its scientific advisory board.
References
- 1.Pommier Y, Sordet O, Antony S, Hayward RL, Kohn KW. Apoptosis defects and chemotherapy resistance: molecular interaction maps and networks. Oncogene. 2004;23:2934–49. doi: 10.1038/sj.onc.1207515. [DOI] [PubMed] [Google Scholar]
- 2.Crotzer DR, Sun CC, Coleman RL, Wolf JK, Levenback CF, Gershenson DM. Lack of effective systemic therapy for recurrent clear cell carcinoma of the ovary. Gynecol Oncol. 2007;105:404–8. doi: 10.1016/j.ygyno.2006.12.024. [DOI] [PubMed] [Google Scholar]
- 3.Schrohl AS, Meijer-van Gelder ME, Holten-Andersen MN, et al. Primary tumor levels of tissue inhibitor of metalloproteinases-1 are predictive of resistance to chemotherapy in patients with metastatic breast cancer. Clin Cancer Res. 2006;12:7054–8. doi: 10.1158/1078-0432.CCR-06-0950. [DOI] [PubMed] [Google Scholar]
- 4.Seve P, Isaac S, Tredan O, et al. Expression of class III {beta}-tubulin is predictive of patient outcome in patients with non-small cell lung cancer receiving vinorelbine-based chemotherapy. Clin Cancer Res. 2005;11:5481–6. doi: 10.1158/1078-0432.CCR-05-0285. [DOI] [PubMed] [Google Scholar]
- 5.Wild PJ, Reichle A, Andreesen R, et al. Microsatellite instability predicts poor short-term survival in patients with advanced breast cancer after high-dose chemotherapy and autologous stem-cell transplantation. Clin Cancer Res. 2004;10:556–64. doi: 10.1158/1078-0432.ccr-0601-03. [DOI] [PubMed] [Google Scholar]
- 6.Wang TH, Wang HS, Soong YK. Paclitaxel-induced cell death: where the cell cycle and apoptosis come together. Cancer. 2000;88:2619–28. doi: 10.1002/1097-0142(20000601)88:11<2619::aid-cncr26>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 7.Penson RT, Oliva E, Skates SJ, et al. Expression of multidrug resistance-1 protein inversely correlates with paclitaxel response and survival in ovarian cancer patients: a study in serial samples. Gynecol Oncol. 2004;93:98–106. doi: 10.1016/j.ygyno.2003.11.053. [DOI] [PubMed] [Google Scholar]
- 8.Mozzetti S, Ferlini C, Concolino P, et al. Class III beta-tubulin overexpression is a prominent mechanism of paclitaxel resistance in ovarian cancer patients. Clin Cancer Res. 2005;11:298–305. [PubMed] [Google Scholar]
- 9.Sangrajrang S, Fellous A. Taxol resistance. Chemotherapy. 2000;46:327–34. doi: 10.1159/000007306. [DOI] [PubMed] [Google Scholar]
- 10.Hari M, Loganzo F, Annable T, et al. Paclitaxel-resistant cells have a mutation in the paclitaxel-binding region of beta-tubulin (Asp26Glu) and less stable microtubules. Mol Cancer Ther. 2006;5:270–8. doi: 10.1158/1535-7163.MCT-05-0190. [DOI] [PubMed] [Google Scholar]
- 11.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–19. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 12.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–92. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 13.Certo M, Del Gaizo Moore V, Nishino M, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–65. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 14.Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 15.Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest. 2007;117:112–21. doi: 10.1172/JCI28281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Del Gaizo Moore V, Schlis KD, Sallan SE, Armstrong SA, Letai A. BCL-2 dependence and ABT-737 sensitivity in acute lymphoblastic leukemia. Blood. 2008;111:2300–9. doi: 10.1182/blood-2007-06-098012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell. 2007;12:171–85. doi: 10.1016/j.ccr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 18.Kohl TM, Hellinger C, Ahmed F, et al. BH3 mimetic ABT-737 neutralizes resistance to FLT3 inhibitor treatment mediated by FLT3-independent expression of BCL2 in primary AML blasts. Leukemia. 2007;21:1763–72. doi: 10.1038/sj.leu.2404776. [DOI] [PubMed] [Google Scholar]
- 19.Witham J, Valenti MR, De-Haven-Brandon AK, et al. The Bcl-2/Bcl-XL family inhibitor ABT-737 sensitizes ovarian cancer cells to carboplatin. Clin Cancer Res. 2007;13:7191–8. doi: 10.1158/1078-0432.CCR-07-0362. [DOI] [PubMed] [Google Scholar]
- 20.Kang MH, Kang YH, Szymanska B, et al. Activity of vincristine, L-ASP, and dexamethasone against acute lymphoblastic leukemia is enhanced by the BH3-mimetic ABT-737 in vitro and in vivo. Blood. 2007;110:2057–66. doi: 10.1182/blood-2007-03-080325. [DOI] [PubMed] [Google Scholar]
- 21.van Delft MF, Wei AH, Mason KD, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–99. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Konopleva M, Contractor R, Tsao T, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–88. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 23.Trudel S, Stewart AK, Li Z, et al. The Bcl-2 family protein inhibitor, ABT-737, has substantial antimyeloma activity and shows synergistic effect with dexamethasone and melphalan. Clin Cancer Res. 2007;13:621–9. doi: 10.1158/1078-0432.CCR-06-1526. [DOI] [PubMed] [Google Scholar]
- 24.Kuroda J, Kimura S, Andreeff M, et al. ABT-737 is a useful component of combinatory chemotherapies for chronic myeloid leukaemias with diverse drug-resistance mechanisms. Br J Haematol. 2008;140:181–90. doi: 10.1111/j.1365-2141.2007.06899.x. [DOI] [PubMed] [Google Scholar]
- 25.Wesarg E, Hoffarth S, Wiewrodt R, et al. Targeting BCL-2 family proteins to overcome drug resistance in non-small cell lung cancer. Int J Cancer. 2007;121:2387–94. doi: 10.1002/ijc.22977. [DOI] [PubMed] [Google Scholar]
- 26.Kuroda J, Puthalakath H, Cragg MS, et al. Bim and Bad mediate imatinib-induced killing of Bcr/Abl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proc Natl Acad Sci U S A. 2006;103:14907–12. doi: 10.1073/pnas.0606176103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deng J, Shimamura T, Perera S, et al. Proapoptotic BH3-only BCL-2 family protein BIM connects death signaling from epidermal growth factor receptor inhibition to the mitochondrion. Cancer Res. 2007;67:11867–75. doi: 10.1158/0008-5472.CAN-07-1961. [DOI] [PubMed] [Google Scholar]
- 28.Brown I, Shalli K, McDonald SL, et al. Reduced expression of p27 is a novel mechanism of docetaxel resistance in breast cancer cells. Breast Cancer Res. 2004;6:R601–7. doi: 10.1186/bcr918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jackisch C, Hahm HA, Tombal B, et al. Delayed micromolar elevation in intracellular calcium precedes induction of apoptosis in thapsigargin-treated breast cancer cells. Clin Cancer Res. 2000;6:2844–50. [PubMed] [Google Scholar]
- 30.Leu JI, Dumont P, Hafey M, Murphy ME, George DL. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol. 2004;6:443–50. doi: 10.1038/ncb1123. [DOI] [PubMed] [Google Scholar]
- 31.Ruiz-Vela A, Opferman JT, Cheng EH, Korsmeyer SJ. Proapoptotic BAX and BAK control multiple initiator caspases. EMBO Rep. 2005;6:379–85. doi: 10.1038/sj.embor.7400375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Makin G, Dive C. Recent advances in understanding apoptosis: new therapeutic opportunities in cancer chemotherapy. Trends Mol Med. 2003;9:251–5. doi: 10.1016/s1471-4914(03)00084-4. [DOI] [PubMed] [Google Scholar]
- 33.Makin G, Dive C. Modulating sensitivity to drug-induced apoptosis: the future for chemotherapy? Breast Cancer Res. 2001;3:150–3. doi: 10.1186/bcr289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Green DR, Kroemer G. Pharmacological manipulation of cell death: clinical applications in sight? J Clin Invest. 2005;115:2610–7. doi: 10.1172/JCI26321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tudor G, Aguilera A, Halverson DO, Laing ND, Sausville EA. Susceptibility to drug-induced apoptosis correlates with differential modulation of Bad, Bcl-2 and Bcl-xL protein levels. Cell Death Differ. 2000;7:574–86. doi: 10.1038/sj.cdd.4400688. [DOI] [PubMed] [Google Scholar]
- 36.Sunters A, Madureira PA, Pomeranz KM, et al. Paclitaxel-induced nuclear translocation of FOXO3a in breast cancer cells is mediated by c-Jun NH2-terminal kinase and Akt. Cancer Res. 2006;66:212–20. doi: 10.1158/0008-5472.CAN-05-1997. [DOI] [PubMed] [Google Scholar]
- 37.Li R, Moudgil T, Ross HJ, Hu HM. Apoptosis of non-small-cell lung cancer cell lines after paclitaxel treatment involves the BH3-only proapoptotic protein Bim. Cell Death Differ. 2005;12:292–303. doi: 10.1038/sj.cdd.4401554. [DOI] [PubMed] [Google Scholar]
- 38.Sunters A, Fernandez de Mattos S, Stahl M, et al. FoxO3a transcriptional regulation of Bim controls apoptosis in paclitaxel-treated breast cancer cell lines. J Biol Chem. 2003;278:49795–805. doi: 10.1074/jbc.M309523200. [DOI] [PubMed] [Google Scholar]
- 39.Tan TT, Degenhardt K, Nelson DA, et al. Key roles of BIM-driven apoptosis in epithelial tumors and rational chemotherapy. Cancer Cell. 2005;7:227–38. doi: 10.1016/j.ccr.2005.02.008. [DOI] [PubMed] [Google Scholar]