

Abstract
Human reticulocyte 15-lipoxygenase (15-hLO-1) and epithelial 15-lipoxygenase (15-hLO-2) have been implicated in a number of human diseases, with differences in their substrate specificity potentially playing a central role. In the current paper, we present a novel method to accurately measure the substrate specificity of the two 15-hLO isozymes and demonstrate that both cholate and specific LO products affect substrate specificity. The linoleic acid (LA) product, 13-hydroperoxyoctadienoic acid (13-HPODE), changes the [kcat/Km]AA/[kcat/Km]LA ratio over 5-fold for 15-hLO-1 and 3-fold for 15-hLO-2, while the arachidonic acid (AA) product, 12-(S)-hydroperoxyeicosatetraenoic acid (12-HPETE), only affects the ratio of 15-hLO-1 (over 5-fold). In addition, the reduced products, 13-(S)-hydroxyoctadecadienoic acid (13-HODE) and 12-(S)-hydroxyeicosatetraenoic acid (12-HETE), also affect substrate specificity, indicating that iron oxidation is not responsible for the change in the [kcat/Km]AA/[kcat/Km]LA ratio. These results, coupled with the dependence of the 15-hLO-1, kcat/Km kinetic isotope effect (Dkcat/Km) on the presence of 12-HPETE and 12-HETE, indicate that the allosteric site, previously identified in 15-hLO-1 [Mogul, R., Johansen, E., Holman, T. R. (1999) Biochemistry 39, 4801-4807], is responsible for the change in substrate specificity. The ability of LO products to regulate substrate specificity may have relevancy to cancer progression and warrants further investigation into the role of this product-feedback loop in the cell.
Human lipoxygenases (hLO) are a family of structurally related enzymes that catalyze the hydroperoxidation of polyunsaturated fatty acids using molecular oxygen (Scheme 1) (1). There are three main isozymes of pharmacological interest: 5-hLO, 12-hLO and 15-hLO, which are named according to their positional specificity on arachidonic acid (AA), producing their respective hydroperoxyeicosatetraenoic acid (HPETE) products. Each of these lipoxygenase isozymes plays a distinct biological role in human disease; 5-hLO is implicated in asthma (2) and cancer (3, 4), 12-hLO is implicated in psoriasis (5) and cancer (4, 6, 7) and 15-hLO is implicated in atherosclerosis (8) and cancer (4, 9).
Scheme 1.

Defining the exact role of LO in human disease is complicated by the incomplete understanding of three fundamental biochemical properties of hLO: substrate specificity, activation specificity and allosteric regulation. With regards to substrate specificity, enzymes are typically highly specific and react with only one particular substrate, such as 5-hLO and platelet 12-hLO, which only react with AA. However, reticulocyte 15-hLO-1 and epithelial 15-hLO-2 react with both LA and AA, albeit with different efficiencies. 15-hLO-1 reacts preferentially towards LA (10), while 15-hLO-2 reacts preferentially towards AA (11), although there is a discrepancy in the literature (12) (vida infra). Interestingly, soybean lipoxygenase-1 (sLO-1), a plant homologue and model enzyme of 15-hLO-1, also reacts preferentially towards AA over LA, even though AA is not a native substrate in soybeans (13). The promiscuity in substrate preference for 15-hLO-1 and 15-hLO-2 has relevancy to cellular biology since the products of LA, 13-hydroperoxyoctadienoic acid (13-HPODE), and AA, 15-HPETE, both affect cellular function. For example, in prostate cancer 13-HPODE up-regulates the MAP kinase signaling pathway, which causes cell proliferation and differentiation (14, 15), while 15-HPETE, down-regulates MAP kinase activity and lowers cell proliferation (16), implying a possible role for 15-hLO in cancer progression.
The fact that 15-hLO-1 and 15-hLO-2 react with both AA and LA is also interesting in a structural sense because it is unclear exactly how either substrate binds in the active site or how substrate recognition occurs. It has previously been shown for 15-hLO-1 that there are three factors that participate in substrate recognition (17, 18). First, the depth of the active site (F352, I417 and M418) adjusts how far the methyl end of the substrate can enter, dictating which hydrogen atom is abstracted (regio-selectivity). Second, R402 interacts with the carboxylic acid end of the substrate and affects the reactivity with AA and LA equally. This is an intriguing result since it indicates that R402 interacts with both AA and LA, even though the substrates are different in length by two carbons (∼3 Å). This suggests that in order to accommodate the different lengths of AA and LA, either a global/local structural change of the enzyme, or a conformational adjustment of the substrate, or both, is required. Finally, F414 appears to facilitate pi-pi stacking with the ω-9 unsaturation, present in both AA and LA. In contrast, little is known about 15-hLO-2 substrate recognition, however, positional specificity has been studied, with D602, V603 and A416 of 15-hLO-2 affecting product distribution of AA products (19, 20). Notably, a comparison of 15-hLO-1 and 15-hLO-2 by sequence alignments indicates that only F352, discussed previously for 15-hLO-1, is conserved in 15-hLO-2, suggesting a possible difference in their molecular mechanism for substrate recognition.
The activation specificity of LO is another property which may potentially have relevancy toward the role of LO in cellular function. LO isozymes are inactive in the as-isolated, ferrous state, however, one equivalent of the hydroperoxide product converts the enzyme to the active, ferric state (Scheme 1) (1). Interestingly, the specific LO products which activate particular LO's is quite broad. 5-HPETE, 12-HPETE, 15-HPETE, and 13-HPODE, all have been shown to activate 5-hLO (21, 22), while both 15-HPETE and 13-HPODE have been shown to activate 15-hLO-1 (17) and 15-hLO-2 (12). These data suggest that enzymatic products from one isozyme of LO can be used to activate another LO isozyme, potentially regulating each other's activity in the cell.
Allosteric regulation of protein activity is a common phenomenon in biology, and is observed in various LO's. For example, ATP and CaII have been shown to bind to non-catalytic sites in 5-hLO, resulting in the activation of enzyme catalysis (23). For sLO-1, our laboratory has shown that an allosteric site exists, which can be specifically targeted by the allosteric inhibitor, oleyl sulfate (OS), to lower the enzymatic activity (24). In addition, we have observed an increase in the kinetic isotope effect of kcat/Km (Dkcat/Km) for 15-hLO-1 with the addition of OS, further indicating the presence of an allosteric site (24). These experimental results for 15-hLO-1, in conjunction with the other allosteric results for sLO-1 and 5-hLO, led us to surmise that the allosteric site in 15-hLO-1 may have biological relevance and warranted further study.
The current investigation utilizes a novel competitive substrate capture method to determine the substrate specificity of both 15-hLO-1 and 15-hLO-2. The use of this method resulted in the discovery that various LO products elicit changes in the substrate specificity for both 15-hLO isozymes and also change the Dkcat/Km for 15-hLO-1, indicating the presence of a product-activated, allosteric regulatory site for both 15-hLO isozymes.
Material and Methods
Materials
All commercial fatty acids (Sigma-Aldrich Chemical Company) and LO products were repurified using a Higgins HAIsil Semi-Preparative (5μm, 250 × 10 mm) C-18 column. Solution A was 99.9% MeOH and 0.1% acetic acid; solution B was 99.9% H2O and 0.1% acetic acid. An isocratic elution of 85% A:15% B was used to purify all fatty acids and then they were stored at −80 °C for a maximum of 6 months. Perdeuterated LA (D31-LA), 98% deuterated, Cambridge Isotope Labs) was purified as previously described (25). All other chemicals were reagent grade or better and were used without further purification.
Overexpression, and Purification of sLO-1 and 15-Human Lipoxygenase
Overexpression and purification of sLO-1 followed a protocol outlined previously (26, 27). Human reticulocyte 15-lipoxygenase-1 (15-hLO-1) is an N-terminus, His6-tagged protein which was expressed and purified as previously published (28). Human prostate epithelial 15-lipoxygenase-2 (15-hLO-2), without a His6-tag, was expressed and purified as previously published for high-throughput screening (29). All enzymes were purified to greater than 90% purity, as evaluated by SDS-PAGE analysis. Iron content of sLO-1 and the 15-hLO isozymes were determined with a Finnigan inductively coupled plasma mass spectrometer (ICP-MS), using cobalt-EDTA as an internal standard. Iron concentrations were compared to standardized iron solutions.
Determination of the Substrate Specificity of sLO-1 using the Competitive Substrate Capture Method with AA and LA as Substrates
The competitive substrate capture method experiments were performed using reaction mixtures of AA:LA of known molar ratio (1:1) and were initiated with sLO-1 (∼1.0 nM, normalized to iron content) using the following buffer conditions, 100 mM Borate, pH 9.2, 22 °C (no cholate added). The ratio of the simultaneous product formation (15-HPETE and 13-HPODE) by sLO-1 was determined at 1 μM total substrate concentration (substrate-limiting conditions). The reaction was monitored at 234 nm with a Perkin-Elmer Lambda 40 using an over-sized reaction cuvette (path length = 5.4 cm) with 60 mls total volume, and stopped with an acetic acid quench at ∼5% total substrate consumption (∼0.05 μM), similar to our previous experiments (25). The acidified reaction mixture was extracted with dichloromethane, evaporated to dryness under vacuum, reconstituted in 50 μl of MeOH and injected onto a Phenomenex Luna (5 μm, 250 × 4.6 mm) C-18 column. The elution protocol consisted of 1 ml/min, isocratic mobile phase of 54.9% ACN:45% H2O:0.1% acetic acid. The molar amount of 15-HPETE and 13-HPODE formation was equated to the corresponding peak areas determined by HPLC. The ratio of the peak areas was then used to determine the [kcat/Km]AA/[kcat/Km]LA ratio, modeled after our previous report (25).
Determination of the Substrate Specificity of 15-hLO Isozymes using the Competitive Substrate Capture Method with an AA and LA Reaction Mixture
Competitive substrate capture experiments with the 15-hLO isozymes were performed as described above. Sodium cholate was not present since it hampers product extraction. Briefly, reactions were initiated by addition of 15-hLO-1 and 15-hLO-2 (∼4 nM and ∼20 nM, respectively, normalized to iron content) to a mixture of AA:LA of known molar ratio (1:1) (25 mM Hepes, pH 7.5, 22 °C). The ratio of the simultaneous formation of 15-HPETE and 13-HPODE by the 15-hLO enzymes was determined as described above for sLO-1. However, it is important to note that 15-hLO-1 also produces 12-HPETE (13 ± 4% of total AA turnover), which was observable by the HPLC analysis and was considered a component of the total AA product concentration when calculating the competitive substrate capture [kcat/Km]AA/[kcat/Km]LA ratios.
Determination of the Substrate Specificity of 15-hLO-1 and 15-hLO-2 using Steady-State Kinetics with and without Sodium Cholate
Steady-state kinetics was performed with 15-hLO-1 and 15-hLO-2 with and without the presence of sodium cholate (0.2%). Lipoxygenase rates were determined by following the formation of the conjugated diene product at 234 nm (ε = 25,000 M-1 cm-1) with a Perkin-Elmer Lambda 40 UV/Vis spectrophotometer. All reactions were 2 ml in volume and constantly stirred using a magnetic stir bar at room temperature (22 °C). Assays were carried out in 25 mM Hepes buffer (pH 7.5, 22 °C) with substrate concentrations ranging from 1 μM – 50 μM and 1 μM – 30 μM for cholate and no cholate, respectively. Assays performed in the presence of sodium cholate were initiated by addition of 15-hLO-1 (∼90 nM for AA, and ∼20 nM for LA, enzyme concentration normalized to iron content) and 15-hLO-2 (∼350 nM for both substrates, normalized to iron content). Assays performed without cholate were initiated by 15-hLO-1 (∼25 nM for AA and ∼15 nM for LA, normalized to iron content) and 15-hLO-2 (∼300 nM for AA and ∼750 nM for LA, normalized to iron content). Assays for each substrate were done concurrently to allow direct comparison of AA and LA kinetic data with and without detergent. Substrate concentrations were quantitatively determined by allowing the enzymatic reaction to go to completion. Kinetic data were obtained by recording initial enzymatic rates at each substrate concentration, which were then fitted to the Michaelis-Menton equation using the KaleidaGraph (Synergy) program to determine kcat and kcat/Km kinetic parameters.
Determination of the Steady-State Substrate Specificity Using Activated 15-hLO-1 and 15-hLO-2 with Lipoxygenase Products
To establish the concentration of product needed for the steady-state kinetic analysis, the purified products (13-HPODE, 15-HPETE and 12-HPETE) were titrated to determine their effects on the 15-hLO isozymes rate of catalysis under substrate-limiting conditions (2.5 μM AA and LA). It was determined that the greatest effect on the rates, with the least amount of product, to avoid product inhibition, was with 1 μM for 15-hLO-1 and 10 μM for 15-hLO-2. The difference in the amount of product required is due to the difference in protein concentration between the two isozymes. There was little change in the rates for both enzymes beyond these concentrations (data not shown). Steady-state kinetic data were obtained for both 15-hLO-1 and 15-hLO-2 in the presence of the three different LO products (13-HPODE, 15-HPETE and 12-HPETE) to determine if activating the enzymes with products (i.e. oxidizing the active site ferrous ion and removing the lag phase) had an affect on substrate specificity. Steady-state kinetics for non-activated enzyme was done concurrently for direct comparison and to minimize error. Enzymatic assays were conducted using the same conditions as described in the steady-state kinetics without cholate present (25 mM Hepes, pH 7.5, 22 °C, no cholate); however, enzymes were incubated with LO products to facilitate enzyme-product interaction, followed by initiation of enzymatic assays with the addition of substrate. There was no observable difference in the initial rates by either adding enzyme first, then initiating the reaction with substrate, or visa versa, as described above. The effects of product-activated enzyme on the kinetic parameters ([kcat/Km]Act/[kcat/Km] and [kcat]Act/[kcat]), as well as the changes in the activated ratios ([kcat/Km]AA/[kcat/Km]LA and [kcat]AA/[kcat]LA) were subsequently determined.
Determining the Effect of 12-HPETE and 12-HETE on the Substrate Specificity using the Competitive Substrate Capture Method with 15-hLO-1
The competitive substrate capture reactions were carried out using the conditions described above (25 mM Hepes, pH 7.5, 22 °C) with the addition of 12-HPETE or 12-HETE (0.1, 0.5, 1.0, 2.0, and 5.0 μM). The enzymatic reactions were initiated by the addition of 1 μM substrate, after pre-incubation of product with 15-hLO-1 (∼4 nM, normalized to iron content). The [kcat/Km]AA/[kcat/Km]LA ratio was determined from the 15-HPETE/13-HPODE HPLC peak area ratio, as previously described. Since 12-HPETE and 12-HETE are the last products to elute from the C-18 column using the HPLC method, it was possible to add them without affecting the 13-HPODE and 15-HPETE peak areas, which is the key property that made this experiment possible. The amount of 12-HPETE generated by the isozyme was assumed to be constant (13% of the total AA turnover) because it could not be determined directly (the added 12-HPETE or 12-HETE saturated the signal) and because the 12-HPETE/15-HPETE peak ratio showed no significant change under all experimental conditions including temperature or pH (data not shown). The reaction mixture was reduced with trimethylphosphite for HPLC analysis to ensure co-elution of the 12-product added with the 12-product generated during the reaction. The determined amount of total product present in the final reaction included 3.5 nM of 12-HPETE, an approximation from the 5% enzymatic turnover.
Determining the Effect of Perdeuterated 13-HPODE and 13-HODE on the Substrate Specificity using the Competitive Substrate Capture Method with 15-hLO-1 and 15-hLO-2
The competitive substrate capture reactions were carried out using the conditions described above (25 mM Hepes, pH 7.5, 22 °C), with the addition of perdeuterated 13-HPODE or 13-HODE (0.1, 0.5, 1.0, 2.0, and 5.0 μM). The enzymatic reactions were initiated by the addition of 1 μM substrate concentrations, after pre-incubation of product and enzyme, 15-hLO-1 and 15-hLO-2 (∼4 nM and ∼20 nM, respectively, normalized to iron content). The [kcat/Km]AA/[kcat/Km]LA ratio was determined using the competitive substrate capture method as described above, however, the reduced products were quantitated with a Finnigan LTQ liquid chromatography - tandem mass spectrometer (LC-MS/MS). The titration of 13-HPODE or 13-HODE could not be performed directly since the added products would affect the [kcat/Km]AA/[kcat/Km]LA ratio calculation by modifying their HPLC peaks. However, perdeuterated 13-HPODE or 13-HODE can be distinguished from the other LO products using LC-MS ion peaks, which is the fundamental property that made this experiment possible. A Thermo Electron Corporation Aquasil (3 μm, 100 × 2.1 mm) C-18 column was used to detect the reduced LO products with an elution protocol consisting of 0.2 ml/min, isocratic mobile phase of 59.9% ACN:40% H2O:0.1% THF. The corresponding reduced product ion peak ratio was determined using negative ion MS/MS (collision energy = 35 eV), with the following masses; 15-HETE, parent m/z = 319, fragments m/z = 175 and 219, 12-HETE, parent m/z = 319, fragments m/z = 179 and 257, 13-HODE, parent m/z = 295, fragments m/z = 183 and 251, and perdeuterated 13-HODE, parent m/z = 325, fragments m/z = 213 and 281 (30). Using controls, we determined there was a difference in the MS detector response between the HODE and HETE products. A one-to-one 13-HODE:15-HETE control mixture, with similar concentration as produced in our competitive experiment, yielded a ratio of 0.7 ± 0.05, instead of a ratio of 1.0. There was a linear correlation between product concentration and ionization peak areas throughout the range of concentrations used in the competitive substrate capture experiments for both AA and LA products, therefore, all LC-MS data were systematically adjusted by 0.7. The amount of total product present in the final reaction includes 20 nM of 13-HPODE, an approximation from the 5% enzymatic turnover.
Determining the Effects of 12-HPETE and 12-HETE on the Competitive Kinetic Isotope Effect of 15-hLO-1 with LA as Substrate
The competitive KIE was performed as described above for the competitive substrate capture method (25), however, reactions were initiated by addition of a mixture of LA:D31-LA, of known molar ratio (1:2), to a pre-incubated mixture of 15-hLO-1 (∼10 nM, normalized to iron content) and 12-HPETE or 12-HETE (5 μM). Enzymatic assays were performed under substrate-limiting conditions (5 μM total substrate concentration), using buffer conditions described above (25 mM Hepes, pH 7.5). The addition of 12-HPETE or 12-HETE was performed at 22 °C, for comparison to previously published results (25). All extracted reaction mixtures were reduced with trimethylphosphite for HPLC analysis.
Construction of 15-hLO-1 and 15-hLO-2 Homology Models, Substrate Docking Studies, and Cavity Visualization
Open (PDB ID: 1P0M chain A: no inhibitor bound) and closed (PDB ID: 1P0M chain B: inhibitor bound) 15-hLO-1 and 15-hLO-2 homology models were built using the revised 2.4 Å resolution 15-rLO structure as a template (31). Coordinate from the Chain A (open) were translated to match Chain B (closed) of 15-rLO prior to creating the homology models, using Combinatorial Extension, which calculates structural alignment of two models in 3D using the combinatorial extension method (32). 15-hLO-1 and 15-hLO-2 share 80% and 37% sequence identity to 15-rLO, respectively. As previously described (33), the homology models were created using PLOP (distributed commercially as PRIME) which uses loop prediction (34), side chain prediction (35, 36), and energy minimization to construct the models. The homology model construction is based on alignments obtained from NCBI-BLAST (blastp) (37). Briefly, primary structural modifications that must be made during the initial construction are the closing of chain breaks associated with gaps in the sequence alignment and side chain optimization for all residues not identical between the target and template sequences. Chain break closure is accomplished using an iterative application of a loop prediction algorithm as previously described in detail (34). After closing chain breaks, side chain optimization (35, 36) and complete energy minimization are performed on all portions of the protein whose coordinates were either not taken from the template at all, or were modified during the building procedure. The homology modeling procedure uses the OPLS all-atom force field (35, 38, 39) and the Generalized Born solvent model (40, 41) for choosing low energy structures. Similarly to previous studies (33), flexible ligand docking of the substrates was performed using the Glide (Schrödinger, Inc.) program (42, 43), which uses a modified version of the Chemscore energy function to score the protein-ligand interactions (44). The optimized force field reduces the net ionic charge on formally charged groups by ∼50% to make the gas-phase Coulombic interaction energy a better predictor of binding. The charge-charge interaction is not a qualification, but a contributor to docked poses. Molecules were docked using either the standard precision mode (SP) or the extra precision mode (XP), which uses a more optimized scoring function as well as a more extensive search of ligand conformations than the SP mode. Clefts and interior surfaces for 15-hLO-1 and 15-hLO-2 homology models were calculated using Hollow (45). Hollow generates volume-filling atoms which can be visualized as surfaces. A surface probe of 0.8 Å was used for the calculations and the limits of the grid were chosen as [-57, 170, 43] and [-48, 161, 31], which allow visualization (PYMOL) of only the active site cavity (46).
Results
Overexpression, and Purification of sLO-1 and 15-Human Lipoxygenase
Protein expression yields were similar to previously published results (26, 27, 47). Metal content was determined to be 90 ± 5 %, 33 ± 2 % and 43 ± 2 %, for sLO-1, 15-hLO-1 and 15-hLO-2, respectively. All kinetic data was normalized to metal content.
Determination of the Substrate Specificity of sLO-1 using the Competitive Substrate Capture Method with AA and LA as Substrates
The [kcat/Km]AA/[kcat/Km]LA ratio for sLO-1 using the competitive substrate capture method (100 mM Borate, pH 9.2, 22 °C) was determined to be 1.8 ± 0.2. This is in agreement with previous steady-state kinetic data (1.8 ± 0.5, 100 mM Borate, pH 9.2, 22 °C) (13) and confirms the validity of the competitive substrate capture method.
Determination of the Substrate Specificity of 15-hLO-1 and 15-hLO-2 using the Competitive Substrate Capture Method with an AA and LA Reaction Mixture
The competitive substrate capture experiment (25 mM Hepes, pH 7.5, 22 °C) was utilized as an alternative method for the determination of the substrate specificity for both 15-hLO isozymes. There was no change in the [kcat/Km]AA/[kcat/Km]LA ratio from 5 – 10% total substrate turnover and between 1 – 5 μM total substrate mixture concentrations. Therefore, experiments were conducted with 1 μM initial substrate concentration (∼5% total substrate turnover and a 1:1, AA-to-LA mixture), with no inactivation of either 15-hLO isozyme being observed. The [kcat/Km]AA/[kcat/Km]LA ratio was thus determined to be 1.3 ± 0.1 and 2.2 ± 0.2 for 15-hLO-1 and 15-hLO-2, respectively.
Determination of the Substrate Specificity of 15-hLO-1 and 15-hLO-2 using Steady-State Kinetics with and without Sodium Cholate
The kinetic data with and without sodium cholate (25 mM Hepes, pH 7.5, 22 °C) for 15-hLO-1 and 15-hLO-2 are presented in Table 1 (standardized to iron content) and compared to the competitive substrate capture method (Figure 1).
Table 1.
Comparison of Steady-State Kinetic Parameters for 15-hLO Isozymesa
| 15-hLO-1 | ||||||
|---|---|---|---|---|---|---|
| AA | LA | AA/LA | ||||
|
kcat/Km (μM-1s-1) |
kcat (s-1) |
kcat/Km (μM-1s-1) |
kcat (s-1) |
kcat/Km | kcat | |
| Cholate | 0.45
± 0.04 |
1.7
± 0.08 |
1.8
± 0.2 |
8.5
± 0.6 |
0.25
± 0.05 |
0.20
± .02 |
| No Detergent | 2.0
± 0.2 |
5.3
± 0.4 |
2.5
± 0.2 |
7.8
± 0.4 |
0.80
± 0.1 |
0.68
± 0.09 |
| 15-hLO-2 | ||||||
| Cholate | 0.026
± 0.002 |
0.33
± 0.02 |
0.029
± 0.002 |
0.31
± 0.02 |
0.90
± 0.1 |
1.1
± 0.1 |
| No Detergent | 0.10
± 0.01 |
0.74
± 0.03 |
0.013
± 0.001 |
0.14
± 0.01 |
8.0
± 1.0 |
5.3
± 0.5 |
Enzymatic assays were performed in 25 mM Hepes (pH 7.5) at 22 °C.
Figure 1.

Comparison of [kcat/Km]AA/[kcat/Km]LA ratio from steady-state kinetics and competitive substrate capture method for 15-hLO-1 and 15-hLO-2 (grey and white, respectively). Dotted line represents no substrate specificity (ratio = 1). Ratio > 1 represents AA specificity; ratio < 1 represents LA specificity. Enzymatic assays were performed in 25 mM Hepes at pH 7.5 at 22 °C.
Unlike the sLO-1 data, the ratios for the 15-hLO isozymes are markedly different from that of the competitive method (Figure 1). Briefly, cholate reduced both the kcat/Km and kcat of 15-hLO-1 and 15-hLO-2 with AA as substrate; however, cholate increased both the kcat/Km and kcat of 15-hLO-2 with LA as substrate. Consequently, the AA/LA ratios were reduced for both the kinetic parameters and both the isozymes with cholate present. The 15-hLO-1 substrate specificity ratio is in excellent agreement with the previous steady-state result (0.25 and 0.20. respectively) (10), however, our [kcat/Km]AA/[kcat/Km]LA ratio for 15-hLO-2, with cholate present, is two-fold greater than that of Kilty et al. (0.9 ± 0.1 and 0.5, respectively) (12). Considering that our 15-hLO-1 data is consistent with the literature, we propose that the difference between our 15-hLO-2 data and that of Kilty et al. is due to experimental differences. Kilty et al. added 13-HPODE (2 μM) to product-activate the enzyme (12), which we currently show affects the LO kinetics (vida infra), and utilized exceedingly high substrate concentrations (>150 μM), which may form micelles at pH 7.4.
Determination of the Steady-State Substrate Specificity Using Activated 15-hLO-1 and 15-hLO-2 with Lipoxygenase Products
The discrepancy between the steady-state kinetic data and the competitive substrate capture method data was investigated using three LO products (13-HPODE, 12-HPETE, and 15-HPETE) to activate the 15-hLO isozymes. The data is presented in two forms; the ratio of kinetic parameters from Product-Activated to Unactivated ([kcat/Km]Act/[kcat/Km] and [kcat]Act/[kcat], Tables 2 and 3, respectively) and the ratio of kinetic parameters from the Product-Activated Enzyme with AA and LA as substrates (Product-Activated [kcat/Km]AA/[kcat/Km]LA and Product-Activated [kcat]AA/[kcat]LA, Tables 2 and 3, respectively).
Table 2.
Comparison of the Steady-State Product-Activated Kinetic Parameter [kcat/Km]Act/[kcat/Km] and Activated [kcat/Km]AA/[kcat/Km]LAa
| 15-hLO-1 | |||
|---|---|---|---|
| [kcat/Km]Act/[kcat/Km] | [kcat/Km]/[kcat/Km] | ||
| Product | AA | LA | AA/LA |
| None | --- | --- | 0.8 ± 0.1 |
| 13-HPODE | 1.8 ± 0.4 | 1.2 ± 0.4 | 1.6 ± 0.5 |
| 15-HPETE | 1.2 ± 0.4 | 1.1 ± 0.4 | 1.0 ± 0.4 |
| 12-HPETE | 2.7 ± 0.5 | 1.0 ± 0.1 | 2.0 ± 0.5 |
| 15-hLO-2 | |||
| Product | AA | LA | AA/LA |
| None | --- | --- | 8.0 ± 1.0 |
| 13-HPODE | 0.8 ± 0.2 | 1.7 ± 0.6 | 3.9 ± 0.9 |
| 15-HPETE | 1.4 ± 0.4 | 1.3 ± 0.3 | 8.0 ± 3.0 |
| 12-HPETE | 1.2 ± 0.4 | 1.4 ± 0.4 | 7.0 ± 2.0 |
Enzymatic assays were performed in 25 mM Hepes (pH 7.5) at 22 °C, with [1 μM] and [10 μM] product added to 15-hLO-1 and 15-hLO-2, respectively.
Table 3.
Comparison of Steady-State Product-Activated Kinetic Parameter [kcat]Act/[kcat] and Activated [kcat]AA/[kcat]LAa
| 15-hLO-1 | |||
|---|---|---|---|
| [kcat]Act/[kcat] | [kcat]/[kcat] | ||
| Product | AA | LA | AA/LA |
| None | --- | --- | 0.68 ± 0.09 |
| 13-HPODE | 0.9 ± 0.1 | 0.9 ± 0.1 | 0.8 ± 0.2 |
| 15-HPETE | 1.0 ± 0.1 | 0.9 ± 0.1 | 0.7 ± 0.2 |
| 12-HPETE | 1.1 ± 0.2 | 0.7 ± 0.1 | 0.9 ± 0.2 |
| 15-hLO-2 | |||
| Product | AA | LA | AA/LA |
| None | --- | --- | 5.3 ± 0.5 |
| 13-HPODE | 0.8 ± 0.1 | 1.0 ± 0.1 | 5.3 ± 0.4 |
| 15-HPETE | 0.9 ± 0.1 | 0.9 ± 0.1 | 4.6 ± 0.9 |
| 12-HPETE | 0.9 ± 0.1 | 0.8 ± 0.2 | 4.6 ± 0.9 |
Enzymatic assays were performed in 25 mM Hepes (pH 7.5) at 22 °C, with [1 μM] and [10 μM] product added to 15-hLO-1 and 15-hLO-2, respectively.
The kcat/Km values increased with the addition of most of the three products, however, only certain LO products elicited a change in the [kcat/Km]AA/[kcat/Km]LA ratio (Table 2). For 15-hLO-1, 13-HPODE increased the steady-state kcat/Km for both LA and AA, but the increase for AA was greater, resulting in a net increase in the [kcat/Km]AA/[kcat/Km]LA ratio towards AA. The LO product, 12-HPETE, however, did not affect LA kinetics, but did increase the kcat/Km for AA, resulting in an increased [kcat/Km]AA/[kcat/Km]LA ratio towards AA. For 15-hLO-2, 13-HPODE increased the kcat/Km for LA, but decreased the kcat/Km for AA, which resulted in a net decrease in the [kcat/Km]AA/[kcat/Km]LA ratio towards AA. However, 15-hLO-2 displayed little change in the [kcat/Km]AA/[kcat/Km]LA ratio with the addition of either HPETE product. The kcat ratios were only slightly changed with the addition of LO products for both 15-hLO-1 and 15-hLO-2 (Table 3), indicating that the effect of product addition is principally targeting substrate capture, a common feature in allosteric regulation (48).
Determining the Effect of 12-HPETE and 12-HETE on the Substrate Specificity using the Competitive Substrate Capture Method with 15-hLO-1
The addition of 12-HPETE or 12-HETE displayed a hyperbolic saturating effect on the substrate specificity for 15-hLO-1, demonstrating [kcat/Km]AA/[kcat/Km]LA ratios ranging from 1.3 ± 0.2 to 4.1 ± 0.3 and 1.3 ± 0.2 to 3.0 ± 0.3, respectively (Figure 2). The data were fit to a saturation curve by plotting Δ[kcat/Km]AA/[kcat/Km]LA versus [12-HPETE] or [12-HETE] μM (data not shown) and the half saturation points were determined to be 1.0 ± 0.2 μM and 1.1 ± 0.1 μM, with the maximum effect being 4.6 ± 0.2 and 3.5 ± 0.2, at saturating 12-HPETE and 12-HETE, respectively.
Figure 2.

Competitive substrate capture method saturation curves of [kcat/Km]AA/[kcat/Km]LA versus [Product] (μM) for 15-hLO-1. Data demonstrates similar effects on the substrate specificity for the AA products, 12-HPETE (closed squares) and 12-HETE (open squares), and for the perdeuterated LA products, 13-HPODE (closed circles) and 13-HODE (open circles). Enzymatic assays were performed in 25 mM Hepes at pH 7.5 at 22 °C.
Determining the Effect of Perdeuterated 13-HPODE and 13-HODE on the Substrate Specificity using the Competitive Substrate Capture Method with 15-hLO-1 and 15-hLO-2
The addition of perdeuterated 13-HPODE or 13-HODE products displayed a hyperbolic saturating effect on the substrate specificity for 15-hLO-1, demonstrating [kcat/Km]AA/[kcat/Km]LA ratios ranging from 1.4 ± 0.2 to 3.6 ± 0.3 and 1.4 ± 0.2 to 2.9 ± 0.3, respectively (Figure 2). The data were fit to a saturation curve by plotting Δ[kcat/Km]AA/[kcat/Km]LA versus [13-HPODE] or [13-HODE] μM (data not shown) and the half saturation points were determined to be 1.2 ± 0.2 μM and 0.8 ± 0.1 μM, with the maximum effect being 4.1 ± 0.3 and 3.2 ± 0.2, respectively, at saturating conditions. For 15-hLO-2 however, the perdeuterated 13-HPODE had no effect on the [kcat/Km]AA/[kcat/Km]LA ratio, with an average ratio of 2.3 ± 0.3, at all concentrations of perdeuterated 13-HPODE added (data not shown). It should be noted that the 15-hLO-2 competitive ratio, with 5 μM 13-HPODE added, does not match the steady-state ratio, with 10 μM 13-HPODE added (2.2 ± 0.2 and 3.9 ± 0.9, respectively). This discrepancy is most likely because the concentration of the 15-hLO-2 enzyme that is necessary to detect the steady-state initial rates is comparable to the substrate concentration. Consequently, the enzyme is no longer under steady-state approximation conditions, which leads to high error in the [kcat/Km]AA/[kcat/Km]LA ratio.
Determining the Effects of 12-HPETE and 12-HETE on the Competitive Kinetic Isotope Effect of 15-hLO-1 with LA as Substrate
Previous mechanistic characterization, using the competitive KIE experiment, demonstrated that 15-hLO-1 displayed a temperature dependent Dkcat/Km, with LA as substrate. The addition of 12-HPETE or 12-HETE (5 μM) saturated the Dkcat/Km by increasing it from 24 ± 4 to 48 ± 10 and 44 ± 8 at 22 °C, respectively. These results are comparable to the previously published Dkcat/Km results at high temperature (25) and similar to the effect of the allosteric inhibitor, oleyl sulfate (OS), on the Dkcat/Km of 15-hLO-1 (24), demonstrating allosteric binding of LO products to 15-hLO-1.
Construction of 15-hLO-1 and 15-hLO-2 Homology Models, Substrate Docking Studies, and Cavity Visualization
Homology models, docking simulations and molecular visualization were carried out as described in the Materials and Methods section. AA and LA were successfully docked in the closed 15-hLO-1 and closed 15-hLO-2 homology models using the SP mode; however, the substrates did not dock to the open models of either isozyme. The modes of binding for AA and LA with the closed 15-hLO-1 model were consistent with mutagenesis studies (17, 18). The carboxylate end of the fatty acid formed a salt bridge with R402, which has no interaction with other residues prior to substrate binding in either the 15-rLO structure or our 15-hLO-1 model. The methyl end has hydrophobic interactions with I417 and M418, and ω-6/ω-9 are in close proximity to F414 (Figure 3). The LA and AA poses are similar to previous docking studies with the charge interaction at R402 (R707 in sLO) and the hydrophobic interaction at the methyl terminus (17, 49, 50). Furthermore, it has been previously demonstrated through mutagenesis that F352 is a primary determinant for the regio-specificity of mammalian 15-LOs (18), which is also found at the methyl end of the homology model active site of 15-hLO-1 (residue not shown in Figure 3 for clarity). These observations, along with our previous use of a 15-hLO-1 model for LO inhibitor discovery (33), leads to the credibility of this docking model.
Figure 3.
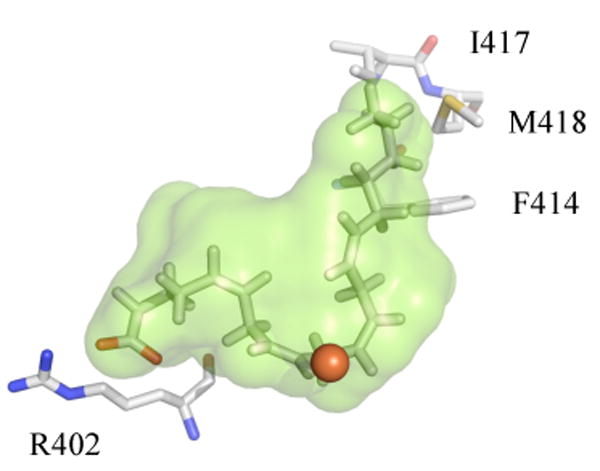
Depiction of the 15-hLO-1 homology model, active site cavity (green), with a docked AA molecule. Selected residues, observed to be catalytically important, are displayed (R402, F414, I417, M418, and the red sphere as the iron).
The fatty acid docking to the closed 15-hLO-2 model, however, is less straightforward. The majority of substrate docking poses of both AA and LA were in the reverse orientation as that seen for 15-hLO-1, with S430 forming a hydrogen bond with the carboxylate of the substrate. This is most likely due to the fact that there is no positively charged residue in the 15-hLO-2 active site, such as R402 in 15-hLO-1, which is the primary determinate for the substrate orientation (17). In fact, none of the residues found by mutagenesis to be important for substrate recognition in 15-hLO-1 are conserved in 15-hLO-2, except for F365 (F352 in 15-hLO-1). The substrates were subsequently docked in the more accurate XP mode, due to this unusual orientation of the substrate in the closed 15-hLO-2 model, and a salt bridge between the carboxylate of AA and R429 was observed (data not shown). However, the long distance of R429 from the active site (> 17 Å) does not allow proper positioning of the substrate for hydrogen atom abstraction, with the methyl end of the substrate being near the iron, indicating this is an unlikely substrate orientation for catalysis.
Representations of the active site cavity for both 15-hLO isozymes were created using Hollow, with the active site dimensions being constrained to depict only the substrate-binding cavity. A similar approach was used previously with various plant LO isozymes, to demonstrate differences in their active sites (51). As observed in Figure 4, the superposition of the open (red) and closed (green) active site cavities of 15-hLO-1, clearly show a marked change in the 15-hLO-1 structure, similar to that seen for the 15-rLO structure (31).
Figure 4.
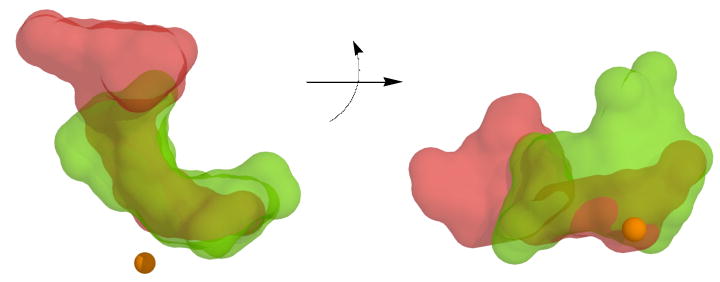
The 15-hLO-1, homology model cavities from the open form (red) and closed form (green), are superimposed and viewed from two perspectives, with an approximate 90° rotation about the horizontal axis. Note that the closed 15-hLO-1 cavity perspective, on the right side, is the same as that seen in Figure 3. The ferrous ion is represented as a sphere and the cavities were created using Hollow.
The closed active site demonstrates the classic “U” or “boot” shape of the cavity, allowing the substrate to bend around the active site iron. The open form is markedly different, with an expansion of the cavity near the substrate entrance by the α-2 helix and a contraction of the cavity in the interior, which appears to hamper our ability to dock substrates to the open form. In order to understand this movement better, a six Å sphere was created around the docked AA to 15-hLO-1 and the active site amino acids with RMSD values greater than 2.0 Å were recorded (Table 4), with the majority being near the carboxylate end of the bound substrate. The flexibility of this region of the active site may have relevance to the substrate specificity/recognition, since the carboxylate end of the substrate is where AA and LA have the greatest difference.
Table 4.
Change in distance of α-carbon and moiety for residues within 6 Å of docked AA to 15-hLO-1a
| Residue | α -Carbon ΔDistance
(Å) |
Residue Moiety | Moiety ΔDistance
(Å) |
|---|---|---|---|
| K170 | 5.6 | C(NH2) | 3.4 |
| R171 | 6.2 | C(NH2)2 | 16 |
| V172 | 9.4 | C(CH3) | 12 |
| D173 | 9.4 | C(CO2) | 10 |
| F174 | 10 | Ring C1 | 3.4 |
| E175 | 10 | C(CO2) | 11 |
| S177 | 12 | COH | 13 |
| L178 | 12 | C(CH3)2 | 12 |
| R402 | 0.6 | C(NH2)2 | 0.8 |
| W594 | 2.3 | Ring C1 | 3.2 |
| Q595 | 4.0 | C(C=O)NH2 | 4.8 |
| L596 | 5.6 | C(CH3)2 | 5.6 |
| G597 | 5.8 | CH2 | 5.8 |
| R598 | 5.0 | C(NH2)2 | 10 |
| Q600 | 2.2 | C(C=O)NH2 | 3.5 |
Changes in distances were determined from the open vs. closed forms of 15-hLO-1 homology models.
In comparing the closed cavities of 15-hLO-1 and 15-hLO-2, they display similar shapes and volumes, with the cavity of 15-hLO-2 being slightly larger (Figure 5). However, there are no positively charged residues, within a reasonable vicinity of the 15-hLO-2 active site, to facilitate binding of the substrate's carboxylate moiety, as seen in 15-hLO-1. This lack of similarity between the electrostatic surfaces of the two cavities, as well as a lack of literature regarding known residues involved in substrate recognition for 15-hLO-2, makes it difficult to assess the catalytically productive substrate orientation and the corresponding structural constraints for 15-hLO-2.
Figure 5.
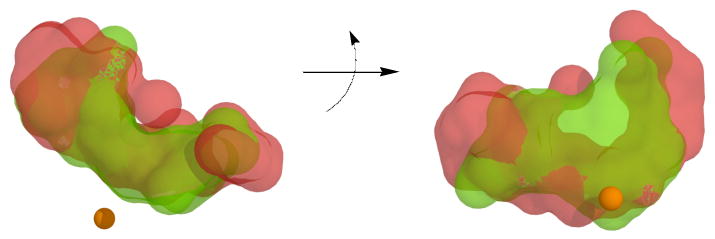
The homology model cavities for the closed forms of 15-hLO-1 (green) and 15-hLO-2 (red) are superimposed and viewed from two perspectives, with an approximate 90° rotation about the horizontal axis. Note that the closed 15-hLO-1 cavity perspective, on the right side, is the same as that seen in Figure 3 and 4. The ferrous ion is represented as a sphere and the cavities were created using Hollow.
Discussion
Our laboratory has had a continued interest in the molecular mechanism of substrate recognition and allostery for lipoxygenase, and whether there are similarities between all lipoxygenases, be they from the plant or mammalian kingdom. One of the more thoroughly investigated LO isozymes, with respect to substrate recognition, is 15-hLO-1. For this reason, it was especially interesting that the substrate specificity of 15-hLO-2 was found to be different from that of 15-hLO-1 (11, 12, 17), suggesting a possible difference in their substrate recognition mechanism. We therefore decided to investigate the substrate specificity of both 15-hLO-1 and 15-hLO-2 in further detail, however, the facts that the catalytic activity of 15-hLO-2 is exceedingly low against LA and that the addition of product is required to remove its long lag phase, prompted us to develop a novel method to measure the substrate specificity ([kcat/Km]AA/[kcat/Km]LA). This method is based on competitive substrate capture and measures the ratio of product turnover in the presence of both substrates, which is directly related to the [kcat/Km]AA/[kcat/Km]LA ratio. In addition, the competitive experiment is performed at low substrate concentrations (1 μM total substrate concentration) and is therefore a better mimic of the cellular conditions as compared to steady-state methods.
The reliability of the novel competitive substrate capture method was confirmed using the model lipoxygenase, sLO-1, which demonstrated a [kcat/Km]AA/[kcat/Km]LA ratio similar to that of previously published steady-state results (1.8 ± 0.2 and 1.8 ± 0.5, respectively) (13). The competitive substrate capture data for the two 15-hLO isozymes, however, did not match their published steady-state kinetic data. The competitive substrate capture method demonstrated that both 15-hLO-1 and 15-hLO-2 display specificity toward AA ([kcat/Km]AA/[kcat/Km]LA = 1.3 ± 0.1 and 2.2 ± 0.2, respectively), which was significantly different from the LA preference previously determined using steady-state kinetics ([kcat/Km]AA/[kcat/Km]LA = 0.2 and 0.5, respectively) (10, 12). Considering that the sLO-1 competitive results were consistent with the steady-state results, the discrepancy of the 15-hLO data was suspected to be due to experimental differences between the two methods; the presence of cholate and the number of substrate/product pairs (e.g. LA/13-HPODE or AA/15-HPETE) in the reaction mixture.
With cholate present (0.2% or 4.6 mM), it was observed that the steady-state [kcat/Km]AA/[kcat/Km]LA ratio for both 15-hLO-1 and 15-hLO-2 decreased (Table 1, Figure 1). These data indicate that cholate is not simply a substrate solublizer, but that it also affects enzymatic activity. The consequence of detergents has already been investigated with respect to plant lipoxygenases (52), but this is the first example of a direct comparison of the cholate effect on both the 15-hLO-1 and 15-hLO-2 kinetic parameters. Nevertheless, it is not straightforward to assign the role of cholate in LO catalysis. Cholate acts as an inhibitor towards 15-hLO-1 and 15-hLO-2 with AA as the substrate, but as an activator towards 15-hLO-2 with LA as the substrate (Table 1), effectively changing the [kcat/Km]AA/[kcat/Km]LA ratio for both 15-hLO-1 (from 0.8 ± 0.1 without cholate to 0.25 ± 0.05 with cholate) and 15-hLO-2 (from 8.0 ± 1.0 without cholate to 0.9 ± 0.1 with cholate) (Table 1, Figure 1). It should be noted that even though the concentration of cholate is below its critical micelle concentration, suggesting a individual molecular interaction, it is still high enough (4.6 mM) to indicate a weak, non-specific interaction. These data offer an explanation for the discrepancy in the literature for the substrate specificity of 15-hLO-2 (11, 12), since experimental conditions, such as the presences of cholate or LO products (vida infra), have large effects on substrate specificity.
The dual nature of cholate (activator and inhibitor), along with the lack of correlation between the competitive and the steady-state results, led us to suspect that an allosteric site, as already observed for 15-hLO-1 (24), was affecting the substrate specificity of the 15-hLO isozymes. These facts compelled us to consider the second difference between the competitive method and the steady-state method; the presence of two substrate/product pairs. To determine if LO products affect substrate specificity, the steady-state kinetic method was performed with the addition of purified hydroperoxide products (13-HPODE, 12-HPETE and 15-HPETE), without cholate present. The first observation was that all three products removed the kinetic lag phase for both 15-hLO-1 and 15-hLO-2, indicating that all three products oxidize the active site iron and are pseudoperoxidase substrates. These results are consistent with previous activation data for 5-hLO, 15-hLO-1 and 15-hLO-2 (12, 17, 21, 22), in which a variety of LO products are pseudoperoxidase substrates for these hLO isozymes.
The more significant result from this experiment was that certain LO products changed the substrate specificity of the 15-hLO isozymes (Table 2). The LA product, 13-HPODE, increased the [kcat/Km]AA/[kcat/Km]LA ratio towards AA for 15-hLO-1, but decreased the [kcat/Km]AA/[kcat/Km]LA ratio towards AA for 15-hLO-2. The AA product, 12-HPETE, also increased the [kcat/Km]AA/[kcat/Km]LA ratio towards AA for 15-hLO-1, but did not affect the ratio for 15-hLO-2. Furthermore, little effect was seen with the addition of 15-HPETE to either 15-hLO enzymes. These data indicate that alteration of the substrate specificity is a highly selective event, with discrimination between structurally similar LO products, such as 12-HPETE and 15-HPETE. In addition, the LO products have opposite effects depending on the 15-hLO isozyme, with the [kcat/Km]AA/[kcat/Km]LA ratio increasing for 15-hLO-1, yet decreasing for 15-hLO-2 with the addition of 13-HPODE. These combined results suggest that these specific LO products are binding to an allosteric site, which directly affects substrate specificity.
We subsequently utilized our novel competitive method for confirmation of these unusual results. The competitive substrate capture method, in which 12-HPETE or perdeuterated 13-HPODE was titrated to 15-hLO-1, demonstrated saturating effects on the [kcat/Km]AA/[kcat/Km]LA ratio, and revealed a half saturation point of 1.0 ± 0.1 μM and 1.2 ± 0.2 μM and a maximum effect on the [kcat/Km]AA/[kcat/Km]LA ratio of 4.6 ± 0.2 and 4.1 ± 0.2, respectively (Figure 2). This is a greater than 5-fold change in the substrate specificity ratio with the addition of either 12-HPETE or 13-HPODE, and confirms that addition of LO product directly affects substrate specificity for 15-hLO-1. However, when perdeuterated 13-HPODE was added to 15-hLO-2 using the competitive substrate capture method, no effect was seen. This is an intriguing result since the substrate specificity for the competitive method is clearly different than that of the steady-state method (2.2 ± 0.2 and 8.0 ± 1.0, respectively) (Figure 1). We speculate that the small amount of 13-HPODE generated from LA being present in the competitive method may be sufficient to saturate the effect on the 15-hLO-2 substrate specificity. This would imply that the 13-HPODE has a tighter affinity for 15-hLO-2 than that seen for 15-hLO-1. We are currently investigating the affinity of the LO products to the allosteric site to both 15-hLO-1 and 15-hLO-2 directly, but due to the low percentage of iron loading and multiple possible binding sites for the LO products, catalytic, activation and allosteric, this has proved to be more difficult than first perceived. Finally, the LO products do not affect either the regio-selectivity (12-HPETE versus 15-HPETE), nor the oxygenation selectivity (11-HPETE versus 15-HPETE) (53), indicating that the allosteric effect is very specific to substrate specificity.
These results are strong evidence that both 15-hLO-1 and 15-hLO-2 have allosteric sites which bind LO products, however, there is a possibility that the substrate specificity change is linked to the ability of the LO products to oxidize the active site iron. It has been previously shown that under certain conditions, the radical intermediate can be lost during catalysis, leaving the inactive, ferrous species (54). If the rate of loss of the intermediate is different for LA versus AA, and the rate of re-activation by the various products is different as well, then it is conceivable that the change in substrate specificity could be coupled to iron oxidation, not to allosteric effector properties of the LO products. In order to rule out this possibility, the aforementioned competitive method titration experiments were repeated using the reduced products, perdeuterated 13-HODE and 12-HETE. As shown in Figure 2, the reduced products exhibit similar effects on the substrate specificity as the hydroperoxide products, both in terms of half-saturation and maximal effect, which clearly indicate that the substrate specificity effect is unrelated to oxidation of the active site iron. Another possible explanation for the substrate specificity effect is that the products induce substrate, micelle formation, which subsequently affects the substrate specificity. This is unlikely since LO products have been shown to disrupt lipid bilayers (55), and the substrate specificity effect is only seen with a limited number of LO products and not the entire family of hydroperoxides. In total, these data strongly supports the hypothesis that the change in substrate specificity by the addition of LO products, is due to an allosteric interaction and not due to active site iron oxidation or product aggregation.
Allosteric regulation is a common feature in biology that can either activate or inhibit enzymatic activity. In the case of 5-hLO, ATP and CaII bind to allosteric effector sites and activate the enzyme catalysis (23). Allostery has also been observed with sLO-1, in which oleyl sulfate (OS) partially inhibits the enzyme activity, but does not interfere with enzyme activation (24, 25, 27). In addition, allosteric binding of OS increases the observed Dkcat/Km for both sLO-1 and 15-hLO-1. This change in Dkcat/Km can be explained by examining the proposed mechanism of LO, minimally described using Scheme 2 (24, 25, 27).
Scheme 2.

| (1) |
According to Scheme 2, Dkcat/Km would be described by eq 1, where substrate release (k-1) and C-H bond cleavage (k2) are the primary determinants for Dkcat/Km (this assumes k2 = kcat and the multiple steps observed at low temperature are included in k2). The Dkcat/Km increases to a maximum of k2H/k2D when commitment (k2/k-1) is small, and decreases to approaching 1, when commitment is large, assuming that the intrinsic k2H/k2D remains unchanged. With respect to 12-HPETE and 12-HETE, both products have a similar effect on the Dkcat/Km of 15-hLO-1 as OS does (24), increasing the Dkcat/Km at 22 °C from 24 ± 4 to 48 ± 10 and 44 ± 8, respectively. These Dkcat/Km data indicate that the LO products lower commitment (k2/k-1), which is supported by the steady-state data where the addition of 12-HPETE lowers the kcat (i.e. k2) of 15-hLO-1 (Table 2). These Dkcat/Km data also support the substrate specificity data, which indicate that specific LO products bind as allosteric effectors to the 15-hLO isozymes and change the enzymatic mechanism, however, it is unclear what conformational change occurs to elicit such a change. The allosteric regulation of substrate specificity by an enzymes own product is a relatively rare occurrence in biology, with ribonucleotide reductase (RNR) being one of the few enzymes in the literature to regulate its substrate specificity via binding its own products as allosteric effectors (56). The mechanism for this effect in RNR involves the product binding close to the active site and modulating the conformation of a protein loop, which in turn affects the active site and substrate specificity (57).
In an attempt to hypothesize possible structural changes in the active site which could change substrate specificity, we have utilized molecular modeling to compare the active sites of both 15-hLO-1 and 15-hLO-2. As mentioned in the introduction, there are specific residues in 15-hLO-1 that play a role in substrate recognition which are not present in the 15-hLO-2 enzyme. Therefore, we have generated computer models for both isozymes based on the 15-rLO crystal structures (both, closed/inhibitor bound and open/no inhibitor) (31), and docked both AA and LA into their active sites to better understand the binding modes of these substrates. The first observation is that both LA and AA were successfully docked into the closed active site of 15-hLO-1, with clear interactions to R402, F352, F414, I417 and M418, corroborating the results from previous mutagenesis studies (Figure 3). Considering that the methyl end of both AA and LA are identical up to ω-9, it is reasonable to assume that any discrimination between the two substrates would occur on the carboxylate end of the fatty acids, which is longer and more unsaturated for AA than LA. Therefore, one could hypothesize that subtle changes in this region of the protein structure, such as changing the volume of the active site or changing the charge interaction with the carboxylate on the fatty acid, such as R402 in 15-hLO-1, could affect the substrate specificity. With this hypothesis in mind, we compared the active site of the closed structure (inhibitor bound) to the open structure (no inhibitor) for 15-hLO-1, and observed 16 residues within 6 Å of the carboxylate end of the fatty acid that are different between the two structures (open and closed) (Figure 4, Table 4). These findings indicate structural mobility in 15-hLO-1 and identify residues that could potentially affect substrate specificity. Interestingly, R402 does not move significantly between the open and closed structures, suggesting it is not involved in substrate specificity. This conclusion is supported by the fact that the 15-hLO-1(R402L) mutant does not elicit a change in the [kcat/Km]AA/[kcat/Km]LA ratio compared to 15-hLO-1(WT) (17).
The analysis of the 15-hLO-2 is more complex. The active site cavities of the two 15-hLO isozymes are similar in shape, despite the fact that the two isozymes have only 55% sequence similarity (37% identity) and that most of the conserved residues discussed above for 15-hLO-1, are not present in the 15-hLO-2 cavity (Figure 5). Of the key residues suspected of substrate interaction for 15-hLO-1, only F365 is present in 15-hLO-2 (F352 in 15-hLO-1) and there is no charged residue in the active site of 15-hLO-2 that could substitute for R402 in 15-hLO-1, nor is there a positively charged amino acid within 10 Å of the docked substrates (data not shown). These results suggest that substrate binding/recognition for 15-hLO-2 is fundamentally different from that of 15-hLO-1. This could explain the difference in their substrate specificity and their different responses to the addition of 13-HPODE; with 15-hLO-1 becoming more reactive against AA, while 15-hLO-2 becomes less reactive. We are currently investigating the mechanism of substrate binding for both 15-hLO-1 and 15-hLO-2 using site-directed mutagenesis of the residues listed in Table 4, in the hopes of understanding substrate recognition more thoroughly and how it can be modified by allosteric regulation.
With respect to the cancer biology relevance of this discovery, it is possible that the product regulation of 15-hLO substrate specificity may have a direct effect on prostate cancer progression. As mentioned in the introduction, 13-HPODE and 15-HPETE have opposing effects on cell proliferation, so a change in the substrate specificity of a particular 15-hLO isozyme could initiate a change in the 13-HPODE/15-HPETE ratio in the cell and affect certain cellular responses. This hypothesis is consistent with the half saturation points of 12-HPETE, 12-HETE, 13-HPODE and 13-HODE (approximately 1 μM for each LO product) on 15-hLO-1 substrate specificity, since they are within the LO product concentration range seen in cells (from 0.6 to 2.0 μM) (58). The magnitude of the change in substrate specificity for the 15-hLO isozymes (3- to 5-fold) is also in the range seen in biology, 2-fold to 10-fold for RNR (59), suggesting that the allosteric effect in both 15-hLO-1 and 15-hLO-2 is feasible in the cell. We are currently investigating if LO products regulate substrate specificity in the cell and if there is any effect on cancer progression.
In conclusion, the key finding of this report is that LO products, and their reduced counterparts, bind to an allosteric site in the 15-hLO isozymes, affecting their substrate specificity. Considering that the allosteric effector molecule is an LO product itself, these in vitro results suggest that the 15-hLO isozymes manifest a product-feedback loop which could affect cellular biology and possibly be targeted for allosteric inhibition against cancer progression.
Acknowledgments
The authors acknowledge Dr. J. Klinman, Dr. S. Rubin and Dr. E. Skrzypczak-Jankun for helpful discussions.
Abbreviations
- LO
lipoxygenase
- sLO-1
soybean lipoxygenase-1
- 15-hLO-1
human reticulocyte 15-lipoxygenase-1
- 15-hLO-2
human epithelial 15-lipoxygenase-2
- 15-rLO
rabbit 15-lipoxygenase
- AA
arachidonic acid
- 15-HPETE
15-(S)-hydroperoxyeicosatetraenoic acid (the major oxidation product of AA with 15-hLO-1)
- 15-HETE
15-(S)-hydroxyeicosatetraenoic acid
- 12-HPETE
12-(S)-hydroperoxyeicosatetraenoic acid (the minor oxidation product of AA with 15-hLO-1)
- 12-HETE
12-(S)-hydroxyeicosatetraenoic acid
- LA
linoleic acid
- 13-HPODE
13-(S)-hydroperoxyoctadecadienoic acid (the oxidation product of LA)
- 13-HODE
13-(S)-hydroxyoctadecadienoic acid
- D31-LA
fully deuterated LA
- perdeuterated 13-HPODE
fully deuterated 13-(S)-HPODE
- perdeuterated 13-HODE
fully deuterated 13-(S)-HODE
- kcat
the rate constant for product release
- kcat/Km
the rate constant for substrate capture
- [kcat/Km]AA/[kcat/Km]LA
substrate capture ratio between AA and LA
- [kcat]AA/[kcat]LA
product release ratio between AA and LA
- [kcat/Km]Act/[kcat/Km]
substrate capture ratio between product-activated enzyme and unactivated enzyme
- [kcat]Act/[kcat]
product release ratio between product-activated enzyme and unactivated enzyme
- Dkcat/Km
kinetic isotope effect for kcat/Km
Footnotes
This work was supported by the National Institutes of Health (GM56062 and S10-RR20939 (MS equipment grant))
References
- 1.Solomon EI, Zhou J, Neese F, Pavel EG. New insights from spectroscopy into the structure/function relationships of lipoxygenases. Chem Biol. 1997;4:795–808. doi: 10.1016/s1074-5521(97)90113-7. [DOI] [PubMed] [Google Scholar]
- 2.Nakano H, Inoue T, Kawasaki N, Miyataka H, Matsumoto H, Taguchi T, Inagaki N, Nagai H, Satoh T. Synthesis and biological activities of novel antiallergic agents with 5-lipoxygenase inhibiting action. Bioorg Med Chem. 2000;8:373–380. doi: 10.1016/s0968-0896(99)00291-6. [DOI] [PubMed] [Google Scholar]
- 3.Ghosh J, Myers CE. Inhibition of arachidonate 5-lipoxygenase triggers massive apoptosis in human prostate cancer cells. Proc Natl Acad Sci U S A. 1998;95:13182–13187. doi: 10.1073/pnas.95.22.13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steele VE, Holmes CA, Hawk ET, Kopelovich L, Lubet RA, Crowell JA, Sigman CC, Kellof GJ. Lipoxygenase Inhibitors as Potential Cancer Chemopreventives. Cancer Epidemiology, Biomarkers & Prevention. 1999;8:467–483. [PubMed] [Google Scholar]
- 5.Hussain H, Shornick LP, Shannon VR, Wilson JD, Funk CD, Pentland AP, Holtzman MJ. Epidermis contains platelet-type 12-lipoxygenase that is overexpressed in germinal layer keratinocytes in psoriasis. Am J Physiol. 1994;266:C243–253. doi: 10.1152/ajpcell.1994.266.1.C243. [DOI] [PubMed] [Google Scholar]
- 6.Connolly JM, Rose DP. Enhanced angiogenesis and growth of 12-lipoxygenase gene-transfected MCF-7 human breast cancer cells in athymic nude mice. Cancer Lett. 1998;132:107–112. doi: 10.1016/s0304-3835(98)00171-2. [DOI] [PubMed] [Google Scholar]
- 7.Natarajan R, Nadler J. Role of lipoxygenases in breast cancer. Front Biosci. 1998;3:E81–88. doi: 10.2741/a369. [DOI] [PubMed] [Google Scholar]
- 8.Harats D, Shaish A, George J, Mulkins M, Kurihara H, Levkovitz H, Sigal E. Overexpression of 15-lipoxygenase in vascular endothelium accelerates early atherosclerosis in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2000;20:2100–2105. doi: 10.1161/01.atv.20.9.2100. [DOI] [PubMed] [Google Scholar]
- 9.Kamitani H, Geller M, Eling T. Expression of 15-lipoxygenase by human colorectal carcinoma Caco-2 cells during apoptosis and cell differentiation. J Biol Chem. 1998;273:21569–21577. doi: 10.1074/jbc.273.34.21569. [DOI] [PubMed] [Google Scholar]
- 10.Sloane DL, Sigal E. On the positional specificity of 15-lipoxygenase. Ann N Y Acad Sci. 1994;744:99–106. doi: 10.1111/j.1749-6632.1994.tb52727.x. [DOI] [PubMed] [Google Scholar]
- 11.Brash AR, Boeglin WE, Chang MS. Discovery of a second 15S-lipoxygenase in humans. Proc Natl Acad Sci U S A. 1997;94:6148–6152. doi: 10.1073/pnas.94.12.6148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kilty I, Logan A, Vickers PJ. Differential characteristics of human 15-lipoxygenase isozymes and a novel splice variant of 15S-lipoxygenase. Eur J Biochem. 1999;266:83–93. doi: 10.1046/j.1432-1327.1999.00818.x. [DOI] [PubMed] [Google Scholar]
- 13.Ruddat VC, Mogul R, Chorny I, Chen C, Perrin N, Whitman S, Kenyon V, Jacobson MP, Bernasconi CF, Holman TR. Tryptophan 500 and arginine 707 define product and substrate active site binding in soybean lipoxygenase-1. Biochemistry. 2004;43:13063–13071. doi: 10.1021/bi0489098. [DOI] [PubMed] [Google Scholar]
- 14.Shappell SB, Manning S, Boeglin WE, Guan YF, Roberts RL, Davis L, Olson SJ, Jack GS, Coffey CS, Wheeler TM, Breyer MD, Brash AR. Alterations in lipoxygenase and cyclooxygenase-2 catalytic activity and mRNA expression in prostate carcinoma. Neoplasia. 2001;3:287–303. doi: 10.1038/sj.neo.7900166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Butler RM, S H, Tindall DJ, Young CY. Nonapoptotic cell death associated with S-phase arrest of prostate cancer cells via the peroxisome proliferator-activated receptor gamma ligand, 15-deoxy-delta12,14-prostaglandin J2. Cell Growth Diff. 2000;11:49–61. [PubMed] [Google Scholar]
- 16.Hsi LC, Wilson L, Nixon J, Eling TE. 15-lipoxygenase-1 metabolites down-regulate peroxisome proliferator-activated receptor gamma via the MAPK signaling pathway. J Biol Chem. 2001;276:34545–34552. doi: 10.1074/jbc.M100280200. [DOI] [PubMed] [Google Scholar]
- 17.Gan QF, Browner MF, Sloane DL, Sigal E. Defining the arachidonic acid binding site of human 15-lipoxygenase. Molecular modeling and mutagenesis. J Biol Chem. 1996;271:25412–25418. doi: 10.1074/jbc.271.41.25412. [DOI] [PubMed] [Google Scholar]
- 18.Borngraber S, Kuban RJ, Anton M, Kuhn H. Phenylalanine 353 is a primary determinant for the positional specificity of mammalian 15-lipoxygenases. J Mol Biol. 1996;264:1145–1153. doi: 10.1006/jmbi.1996.0702. [DOI] [PubMed] [Google Scholar]
- 19.Coffa G, Brash AR. A single active site residue directs oxygenation stereospecificity in lipoxygenases: stereocontrol is linked to the position of oxygenation. Proc Natl Acad Sci U S A. 2004;101:15579–15584. doi: 10.1073/pnas.0406727101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coffa G, Schneider C, Brash AR. A comprehensive model of positional and stereo control in lipoxygenases. Biochem Biophys Res Commun. 2005;338:87–92. doi: 10.1016/j.bbrc.2005.07.185. [DOI] [PubMed] [Google Scholar]
- 21.Rouzer CA, Samuelsson B. The importance of hydroperoxide activation for the detection and assay of mammalian 5-lipoxygenase. FEBS Lett. 1986;204:293–296. doi: 10.1016/0014-5793(86)80831-6. [DOI] [PubMed] [Google Scholar]
- 22.Riendeau D, Falgueyret JP, Nathaniel DJ, Rokach J, Ueda N, Yamamoto S. Sensitivity of immunoaffinity-purified porcine 5-lipoxygenase to inhibitors and activating lipid hydroperoxides. Biochem Pharmacol. 1989;38:2313–2321. doi: 10.1016/0006-2952(89)90471-1. [DOI] [PubMed] [Google Scholar]
- 23.Falgueyret JP, Denis D, Macdonald D, Hutchinson JH, Riendeau D. Characterization of the arachidonate and ATP binding sites of human 5-lipoxygenase using photoaffinity labeling and enzyme immobilization. Biochemistry. 1995;34:13603–13611. doi: 10.1021/bi00041a040. [DOI] [PubMed] [Google Scholar]
- 24.Mogul R, Johansen E, Holman TR. Oleyl sulfate reveals allosteric inhibition of soybean lipoxygenase-1 and human 15-lipoxygenase. Biochemistry. 2000;39:4801–4807. doi: 10.1021/bi992805t. [DOI] [PubMed] [Google Scholar]
- 25.Lewis E, Johnson E, Holman T. Large Competitive Kinetic Isotope Effects in Human 15-Lipoxygenase Catalysis Measured by a Novel HPLC Method. J Am Chem Soc. 1999;121:1395–1396. [Google Scholar]
- 26.Holman TR, Zhou J, Solomon EI. Spectroscopic and functional characterization of a ligand coordination mutant of soybean lipoxygenase: First coordination sphere analogue of human 15-lipoxygenase. J Am Chem Soc. 1998;120:12564–12572. [Google Scholar]
- 27.Ruddat VC, Whitman S, Holman TR, Bernasconi CF. Stopped-flow kinetic investigations of the activation of soybean lipoxygenase-1 and the influence of inhibitors on the allosteric site. Biochemistry. 2003;42:4172–4178. doi: 10.1021/bi020698o. [DOI] [PubMed] [Google Scholar]
- 28.Amagata T, Whitman S, Johnson TA, Stessman CC, Loo CP, Lobkovsky E, Clardy J, Crews P, Holman TR. Exploring sponge-derived terpenoids for their potency and selectivity against 12-human, 15-human, and 15-soybean lipoxygenases. J Nat Prod. 2003;66:230–235. doi: 10.1021/np020462l. [DOI] [PubMed] [Google Scholar]
- 29.Deschamps JD, Gautschi JT, Whitman S, Johnson TA, Gassner NC, Crews P, Holman TR. Discovery of platelet-type 12-human lipoxygenase selective inhibitors by high-throughput screening of structurally diverse libraries. Bioorg Med Chem. 2007;15:6900–6908. doi: 10.1016/j.bmc.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deems R, Buczynski MW, Bowers-Gentry R, Harkewicz R, Dennis EA. Detection and Quantitation of Eicosanoids via High Performance Liquid Chromatography-Electrospray Ionization-Mass Spectrometry. Methods Enzymol. 2007;432:59–82. doi: 10.1016/S0076-6879(07)32003-X. [DOI] [PubMed] [Google Scholar]
- 31.Choi J, Chon JK, Kim S, Shin W. Conformational flexibility in mammalian 15S-lipoxygenase: Reinterpretation of the crystallographic data. Proteins. 2008;70:1023–1032. doi: 10.1002/prot.21590. [DOI] [PubMed] [Google Scholar]
- 32.Shindyalov IN, Bourne PE. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998;11:739–747. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- 33.Kenyon V, Chorny I, Carvajal WJ, Holman TR, Jacobson MP. Novel human lipoxygenase inhibitors discovered using virtual screening with homology models. J Med Chem. 2006;49:1356–1363. doi: 10.1021/jm050639j. [DOI] [PubMed] [Google Scholar]
- 34.Jacobson MP, Pincus DL, Rapp CS, Day TJ, Honig B, Shaw DE, Friesner RA. A hierarchical approach to all-atom protein loop prediction. Proteins. 2004;55:351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 35.Jacobson MP, Kaminski GA, Friesner RA, Rapp CS. Force Field Validation Using Protein Side Chain Prediction. J Phys Chem B. 2002;106:11673–11680. [Google Scholar]
- 36.Jacobson MP, Friesner RA, Xiang Z, Honig B. On the role of the crystal environment in determining protein side-chain conformations. J Mol Biol. 2002;320:597–608. doi: 10.1016/s0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- 37.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 39.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 40.Gallicchio E, Zhang LY, Levy RM. The SGB/NP hydration free energy model based on the surface generalized born solvent reaction field and novel nonpolar hydration free energy estimators. J Comput Chem. 2002;23:517–529. doi: 10.1002/jcc.10045. [DOI] [PubMed] [Google Scholar]
- 41.Ghosh A, Rapp CS, Friesner RA. Generalized Born Model Based on a Surface Integral Formulation. J Phys Chem B. 1998;102:10983–10990. [Google Scholar]
- 42.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 43.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 44.Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J Comput Aided Mol Des. 1997;11:425–445. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- 45.Gruswitz BKHaF. HOLLOW: A Tool for Generating Interior Surface Representations of Channels and Pockets in Molecular Structures. http://hollow.sourceforge.net/
- 46.DeLano WL. The PyMOL Molecular Graphics System. http://www.pymol.org.
- 47.Deschamps JD, Kenyon VA, Holman TR. Baicalein is a potent in vitro inhibitor against both reticulocyte 15-human and platelet 12-human lipoxygenases. Bioorg Med Chem. 2006;14:4295–4301. doi: 10.1016/j.bmc.2006.01.057. [DOI] [PubMed] [Google Scholar]
- 48.Perutz MF. Mechanisms of cooperativity and allosteric regulation in proteins. Q Rev Biophys. 1989;22:139–237. doi: 10.1017/s0033583500003826. [DOI] [PubMed] [Google Scholar]
- 49.Prigge ST, Boyington JC, Gaffney BJ, Amzel LM. Structure conservation in lipoxygenases: structural analysis of soybean lipoxygenase-1 and modeling of human lipoxygenases. Proteins. 1996;24:275–291. doi: 10.1002/(SICI)1097-0134(199603)24:3<275::AID-PROT1>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 50.Borngraber S, Browner M, Gillmor S, Gerth C, Anton M, Fletterick R, Kuhn H. Shape and specificity in mammalian 15-lipoxygenase active site. The functional interplay of sequence determinants for the reaction specificity. J Biol Chem. 1999;274:37345–37350. doi: 10.1074/jbc.274.52.37345. [DOI] [PubMed] [Google Scholar]
- 51.Youn B, Sellhorn GE, Mirchel RJ, Gaffney BJ, Grimes HD, Kang C. Crystal structures of vegetative soybean lipoxygenase VLX-B and VLX-D, and comparisons with seed isoforms LOX-1 and LOX-3. Proteins. 2006;65:1008–1020. doi: 10.1002/prot.21182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schilstra MJ, Veldink GA, Vliegenthart JF. Effect of nonionic detergents on lipoxygenase catalysis. Lipids. 1994;29:225–231. doi: 10.1007/BF02536325. [DOI] [PubMed] [Google Scholar]
- 53.Jacqout C, Wecksler AT, McGinley CM, Segraves EN, Holman TR, van der Donk WA. Isotope Sensitive Branching in the Reaction of Deuterated Arachidonic Acids with Human 12- and 15-Lipoxygenases. Biochemistry. 2008 doi: 10.1021/bi800308q. Accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Berry H, Debat H, Garde VL. Oxygen concentration determines regiospecificity in soybean lipoxygenase-1 reaction via a branched kinetic scheme. J Biol Chem. 1998;273:2769–2776. doi: 10.1074/jbc.273.5.2769. [DOI] [PubMed] [Google Scholar]
- 55.van Leyen K, Duvoisin RM, Engelhardt H, Wiedmann M. A function for lipoxygenase in programmed organelle degradation. Nature. 1998;395:392–395. doi: 10.1038/26500. [DOI] [PubMed] [Google Scholar]
- 56.Kolberg M, Strand KR, Graff P, Andersson KK. Structure, function, and mechanism of ribonucleotide reductases. Biochim Biophys Acta. 2004;1699:1–34. doi: 10.1016/j.bbapap.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 57.Nordlund P, Reichard P. Ribonucleotide reductases. Annu Rev Biochem. 2006;75:681–706. doi: 10.1146/annurev.biochem.75.103004.142443. [DOI] [PubMed] [Google Scholar]
- 58.Guichardant M, Thevenon C, Pageaux JF, Lagarde M. Basal concentrations of free and esterified monohydroxylated fatty acids in human blood platelets. Clin Chem. 1997;43:2403–2407. [PubMed] [Google Scholar]
- 59.Reichard P. Ribonucleotide reductases: the evolution of allosteric regulation. Arch Biochem Biophys. 2002;397:149–155. doi: 10.1006/abbi.2001.2637. [DOI] [PubMed] [Google Scholar]