

Summary
Type A (I) lantibiotics are cationic antimicrobial peptides that have a potential usefulness in treating infectious diseases. They are known to have a potent and broad spectrum of activity, an insignificant cytotoxicity, and demonstrated efficacy in animal infection models, suggesting therapeutic potential. In this review, topics pertaining to their basic structure, mode of bactericidal activity, pharmacology, and methods of manufacture are described.
Introduction
A group of cationic peptide antibiotics, called Type A (I) lantibiotics, has received considerable attention because of the broad spectrum of activity, high potency, low frequency of antimicrobial resistance, and a cytotoxicity of two to three orders of magnitude higher than those required for antimicrobial activity. This class of antibiotics has been known for decades but has not been extensively tested for their potential usefulness in treating infectious diseases. The principal reason for this is the general difficulty of obtaining these molecules in sufficient, cost effective amounts to enable their testing and commercialization. Lantibiotic synthesis is a complex process involving multiple enzymes that leads to the formation of several unique amino acid residues. The post-translational modifications are believed to be, in part, responsible for low production by fermentation and make synthetic synthesis of these antibiotics difficult. In this review, before discussing topics pertaining to mode of bactericidal action, pharmacology, and manufacture of type A (I) lantibiotics, a general discussion of lantibiotic structure and synthesis is provided to give the reader the basic knowledge and understanding of these compounds.
Type A (I) Lantibiotic Structure and Synthesis
There are 5 subclasses of lantibiotics based on differences in their chemistry and biosynthesis: Type A(I), Type A(II), Type B, Two-Component and those of unknown structures. Type A (I) lantibiotics (Class I bacteriocins) fall into two subgroups: those that are structurally similar to nisin A [1], which is produced by L. lactis, and those that share structural similarities to mutacin 1140 [2], which is produced by Streptococcus mutans (Figure 1). Gram positive bacteria are responsible for biosynthesis of the known lantibiotics. Lantibiotics are rich in the sulfur-containing amino acids, lanthionine (Lan, ala-S-ala) and, frequently, 3-methyl-lanthionine (MeLan, abu-S-ala). The occurrence of the unusual amino acids Lan and MeLan define lantibiotics and give them their name. In addition to the Lan and MeLan residues, there may be other post-translationally modified amino acids. Some of the other modified amino acids found in mutacin 1140 include 2,3-didehydroalanine (Dha), 2,3 didehydrobutyrine (Dhb), and the unsaturated lanthionine derivatives such as S-amino vinyl-D-cysteine (AviCys) (Figure 2).
Figure 1.
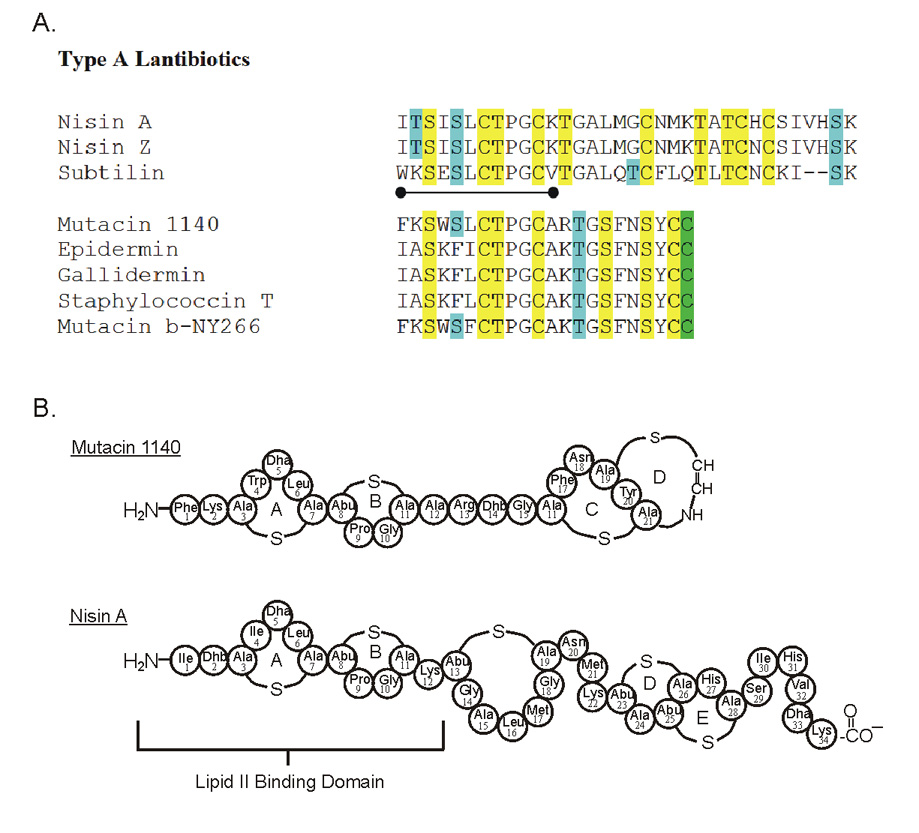
A. Sequence alignment of Type A (I) lantibiotics belonging to the Nisin A and mutacin 1140 structural group. There is a considerable amount of similarity between the first eleven amino acids in the nisin A and mutacin 1140 group, represented by the dumbbell. Residues highlighted in yellow represent amino acids involved in thioether linkages. Residues highlighted in blue designate the location of the dehydrated residues 2, 3-didehydroalanine (Dha) and 2, 3-didehydrobutyrine (Dhb). The residues that are decarboxylated are highlighted in green. B. Representation of the covalent structures of nisin A and mutacin 1140.
Figure 2.

The structure of the modified residues in type A (I) lantibiotics of nisin and mutacin 1140 subgroup.
The mature lantibiotic molecule is made using a series of sequential enzymatic steps that act on a ribosomally synthesized prepropeptide (Figure 3, step 1) [3]. The genes responsible for encoding the modifying enzymes are typically clustered on an 8–10 Kb DNA fragment that may reside on the chromosome, a plasmid, or as part of a transposon. In Type A lantibiotics, the serine and threonine residues in the ribosomally synthesized prepeptide encoded by the lanA gene are dehydrated by an enzyme encoded by the lanB gene and these dehydrated amino acids are involved in the formation of thioether linkages to a nearby cysteine residue that is situated more toward the carboxyl end of the molecule. This reaction is catalyzed by the protein expressed by the lanC gene (Figure 3, step 2). In the case of certain mutacin 1140-like type A (I) lantibiotics the C-terminal cysteine is decarboxylated by the enzyme expressed by the lanD gene and converted into an S-amino vinyl-D-cysteine (Figure 3, step 2). Following transport out of the cell by the product of the lanT gene, the leader sequence of the modified prepropeptide is then cleaved by an extracellular protease encoded by lanP to produce the mature antibiotic (Figure 3, step 3). Lastly, the production is regulated through the binding of the lantibiotic to a membrane bound kinase (Figure 3, step 4). The mechanism of regulating lantibiotic production is still not well understood.
Figure 3.
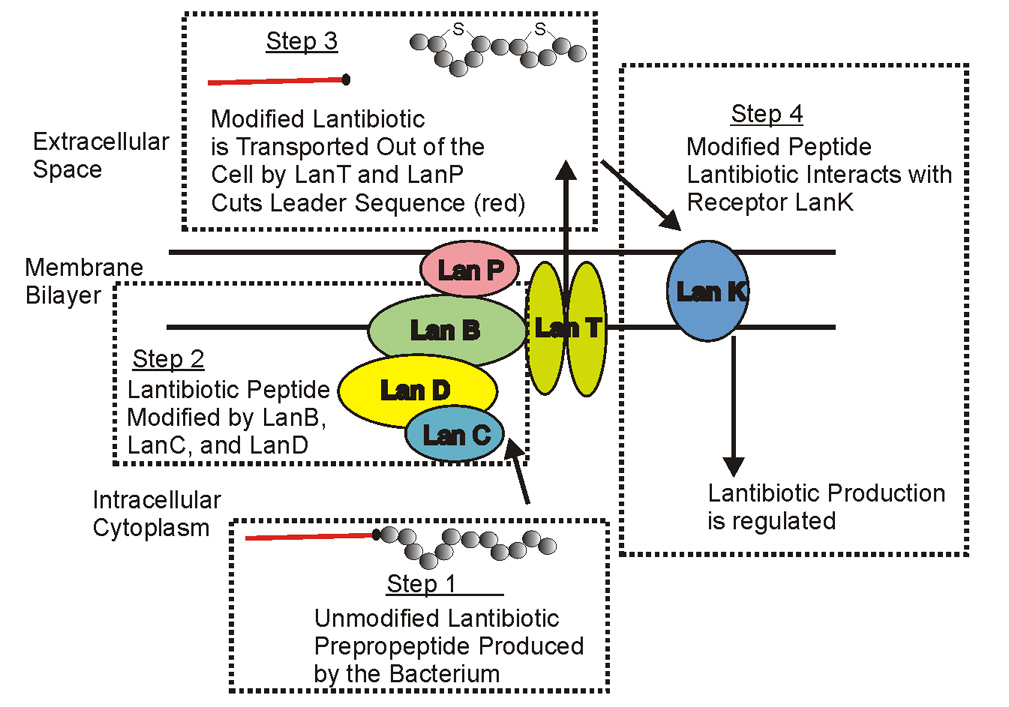
Schematic of the synthetase complex of Type A (I) lantibiotics.
Modes of Activity for Type A (I) Lantibiotics
Lantibiotics have multiple modes of bactericidal activity. Transmembrane pore formation, lipid II-mediated pore formation, and lipid II abduction from physiological domains have been reported [4–8]. Given that a single lantibiotic monomer is too small to form a pore across a lipid bilayer, they must have a mechanism for lateral assembly in which they aggregate into complexes that can span the bilayer, an event that is followed by the disruption of the cytoplasmic membrane causing efflux of ions, ATP, and other essential cellular components. Two possible mechanisms for pore formation for nisin A have been reported, a barrel-stave model [9,10] and a wedge-like model [6,11]. Other cationic peptide antibiotics, including magainins, melittin and protegrins, all appear to induce transmembrane pores that conform to the toroidal model [12,13]. Several good reviews have been written on these mechanisms of pore formation [14,15].
Electrostatic interactions between the positive charge of type A (I) lantibiotics and the negative charge of the bacterial membrane are believed to be required for initial binding. Experiments with nisin have demonstrated that the sensitivity of the host bacterium is dependent on the charged state of its cell wall and membrane [9,16–18]. However, the bacterium Listeria monocytogenes was shown to be relatively insensitive to nisin (MIC between 200–1000 µg/L) even though the negatively charged lipid composition of the membrane is relatively high (50–88%) [16,19]. Therefore, other factors besides anionic content are likely to affect the sensitivity of a bacterium to this class of lantibiotics. The thickness and composition of the cell wall and the accessibility of the peptidoglycan precursor lipid II are other potentially important determinants for bacterial susceptibility [4,5,20].
Nisin’s interactions with bacterial cells are enhanced by the presence of lipid II [5,20,21]. Early experiments with nisin showed an interference with cell wall biosynthesis of in vitro systems [22], and that it forms complexes with the lipid bound peptidoglycan precursors lipid I and lipid II [23,24]. These effects of inhibiting cell wall biosynthesis were once believed to be secondary to the primary mode of action of membrane disruption and pore formation. However, membrane pore-formation by nisin and epidermin was shown to be inhibited by preincubation of M. luteus or S. simulans with ramoplanin, a lipopeptide that binds lipid II [25,26]. In addition, model membranes containing lipid II were shown to increase nisin activity three fold [5]. More recently, the mutacin 1140-like lantibiotics gallidermin and epidermin have also been shown to interfere with cell wall biosynthesis via lipid II binding as well as binding to lipid II precursor, lipid I [4].
The formation of a highly stable complex of nisin with lipid II has been reported [27]. Recently, a novel lipid II binding motif for nisin has been characterized by NMR [28], in which the N-terminal portion of nisin, lanthionine rings A and B, interact with the pyrophosphate, peptidoglycan MurNAc, and first isoprene of lipid II. The N-terminal portion of nisin A and mutacin 1140 share a high degree of similarity (Figure 1B), and it is believed that this portion of mutacin 1140 is also the lipid II binding motif [7,8].
A novel mode of action for type A lantibiotics that interact with lipid II was recently published [7]. Using confocal fluorescence microscopy, fluorescein-labeled nisin was found to induce the clustering of lipid II into large patches in model membrane vesicles and in in vivo membrane studies. From these observations, it was clear that nisin A and presumably other type A (I) lantibiotics, have a novel mechanism of bactericidal activity that not only involves lipid II binding, but also abduction of lipid II from its physiological domain in the bacterial membrane where active cell wall formation is occurring. This mechanism ensures that the peptidoglycan subunits carried by lipid II will not be available for cell wall synthesis, thereby inhibiting bacterial cell growth.
Moreover, the abduction of lipid II from the physiological domain by mutacin 1140 is sufficient to inhibit target cell growth, even in the absence of pore formation. Lipid II-containing model membrane vesicles revealed no detectable pore formation by mutacin 1140, indicating that lipid II abduction, and not pore formation is responsible for its observed bactericidal activity [7,8]. Vancomycin also targets lipid II, although the interaction occurs at a different site on lipid II. This glycopeptide also does not abduct lipid II from its normal physiological locations in the bacterial cell membrane [29].
Differences in the lipid II-lantibiotic complex were observed in model membrane experiments between nisin A and mutacin 1140. Nisin has a distinctive pyrene excimer signal when added to model membrane vesicles containing pyrene labeled lipid II. Conversely, there was no induced excimer signal following the addition of mutacin 1140 to the model membrane vesicles containing pyrene labeled lipid II [8]. The lack of an excimer signal is most probably attributed to differences in the way lipid II is oriented during formation of the mutacin-lipid II and nisin-lipid II complexes. Lipid II monomers must be located farther apart in the mutacin complexes than in the nisin complexes, and the observed nisin induced excimer signal observed may actually be attributed to an actual pore complex. The peptide sequence variability outside of the lipid II binding domain (Figure 1B), in all probability, confers the functionality of the lantibiotic including whether or not it is a pore former and how it positions lipid II during complex formation. In addition to the lipid II binding domain, there must be a binding motif for lateral association of these lantibiotics to account for the large mutacin or nisin complexes observed in the florescence studies and transmission electron microscopy studies [7,8]. This dual activity of binding to lipid II followed by lateral assembly of the lantibiotic may explain the high potency of these antimicrobial compounds. The binding affinity for nisin to lipid II has been calculated to be in the order of 107 [5]. This binding affinity does not take into consideration the lateral assembly of the lantibiotic which would further trap lipid II into large lantibiotic islands in the bilayer. This bactericidal mechanism of activity ensures that lipid II will not become available for cell wall synthesis.
Pharmacology of Type A (I) lantibiotics
This class of antibiotics has been known for decades but has not been extensively tested for their potential usefulness in treating infectious diseases even though many are known to be both potent and have a broad spectrum of activity. The principal reason for this is the general difficulty of obtaining these molecules in sufficient, cost effective amounts to enable their testing and commercialization. Numerous studies have shown that they have low MICs against many clinically relevant Gram positive bacterial spp., such as Enterococcus spp., Listeria spp., Staphylococcus spp., Streptococcus spp., Bacillus spp., and others [30–32]. As early as 1952, nisin A was shown to be as effective as penicillin in treating mice infected with Mycobacterium tuberculosis, Streptococcus pyogenes, and Staphylococcus aureus [33]. However, the authors commented that nisin most likely would not find a place in therapeutics given the cost of production and the rapid clearance of the antibiotic from the blood. The rise of antibiotic resistant pathogens like MRSA and the 8 billion dollar cost attributed to the treatment of this pathogen [34] counters the authors remark, but this comment does illustrate the differences in the mindset of 1952 and today.
Cytotoxicity of gallidermin and nisin A on intestinal epithelial cells following 24 hour incubation was determined by MTT assay and neutral red dye uptake assay [35]. The IC50 was greater than 210 µM in both assays for gallidermin and greater than 89 µM in both assays for nisin. Hemolytic potential of gallidermin and nisin A on live sheep erythrocytes, following a one hour exposure to their respective IC50 concentration, showed <2% and <5% hemolysis for gallidermin and nisin, respectively, which are comparable to the activity seen with vancomycin. In a separate study, no haemolytic activity for nisin A or mutacin B-Ny266 was observed when incubated with sheep erythrocytes at concentrations 30 to 60 times greater than is required for in vitro bactericidal activity [32]. Nisin was also shown to have no significant cytoxicity on promyelocytic leukemia HL-60 cells after 24 hour exposure [36] and no significant hemolytic activity against human lung fibroblast cells [37] and human erythrocytes [36].
Nisin was shown to be more effective than vancomycin in an S. pneumoniae mouse infection model [31]. Nisin was 100% effective with two intravenous doses of 0.16 mg/kg body weight, while vancomycin was only effective 80% of the time with two intravenous doses 1.25 mg/kg body weight. These authors also noted the rapid clearance of nisin from the blood, and provided a predicted half life of 0.9 hours. However, they noted that nisin is very effective despite the short half life. The assumption was made that nisin may have a more rapid bactericidal action compared to vancomycin, since both antibiotics have comparable MICs against S. pneumoniae. Intraperotineal injection of 1 mg/kg body weight of Mutacin B-Ny266 in mice infected intraperitoneally with 6.2 × 107 CFU of S. aureus demonstrated efficacy, but the experimental design in which the drug was injected into the same compartment as the bacterial challenge makes interpretation of the results difficult [38].
Oni Biopharma Inc. is currently conducting preclinical trials on the lantibiotic mutacin 1140 (MU1140). A particularly interesting feature of mutacin 1140 is the relative difficulty of inactivating mutacin 1140 by trypsin or pronase treatment [39,40]. The potentially susceptible arginine at residue 13, was later found by 3-dimensional NMR structural analyses to be buried in a horseshoe-like confirmation [41], thus protecting it from protease cleavage. Presumably, bacterial resistance to mutacin 1140 by acquiring a protease activity would be difficult. Another interesting feature was noted when none of four sensitive species of bacteria tested were able to acquire genetically stable, spontaneous resistance. Incubation of 1011 cells of multidrug resistant (MDR) strains of S. aureus and E. faecalis, as well as L. casei and an S. rattus indicator strain, with a threshold killing concentration of mutacin 1140 failed to lead to the recovery of resistant mutants (unpublished data). By contrast, spontaneous mutants of L. monocytogenes that were resistant to nisin were isolated at frequencies that ranged from 10−6 to 10−8 [42,43]. Furthermore, following a 21 daily, sequential passages in subinhibitory concentrations of mutacin 1140, there was only a slight (≤4-fold) increase in the MIC against S. pneumoniae or S. aureus. The producer strain, Streptococcus mutans JH1140, has no significant immunity to mutacin 1140 as evidenced by the fact that its MIC is comparable to MICs of other viridans streptococci tested.
Time-kill studies against S. aureus, S. pneumoniae, and E. faecalis show that mutacin 1140 is bactericidal against S. aureus and S. pneumoniae, and bacteriostatic against E. faecalis (unpublished data). Vancomycin also exhibits this species-dependent difference in activity [44,45]. In vitro, the addition of human or rat serum causes a reduction in mutacin 1140 activity against S. pneumoniae, presumably due to significant (92%) binding to serum protein(s), but serum was found to cause an unexplained increase in activity against S. aureus. The half-life of this lantibiotic was approximately 1.5 hr in a rat model. Other aspects of safety and efficacy that have been tested, including maximum tolerated dose, immunogenicity, cytotoxicity, efficacy in S. aureus sepsis models, all suggest the potential usefulness of mutacin 1140 for the treatment of Gram positive infectious diseases. Presumably, other lantibiotics will demonstrate potential for clinical application as well. The current challenge is to develop an approach for their production in cost effective amounts to enable their testing and commercialization.
Manufacture of Lantibiotics
Several interesting approaches to manufacturing lantibiotics have been tried over the last few decades. These include the development of fermentation methods, semi-synthetic methods, and organochemistry synthesis approaches. Fermentation methods have been reported for gallidermin, in which the authors report improvements in its production [46,47] with a yield of 249 mg/L in a 200 L bioreactor [46]. Fermentation methods for mutacin B-Ny266 have also demonstrated higher yields of production [48]. Common components in their production appear to be 10% inoculums in late log growth, and the media component, yeast extract. These reports demonstrate the possibility of fermentation-based methods for the manufacture of lantibiotics and also demonstrate the efficiency of the lantibiotic modification machinery for a high yield production. Proof of principal for a semi-synthetic approach was demonstrated for a Type A lantibiotic lacticin 481 [49], which belongs to the A (II) subgroup of lantibiotics. An enzyme, LctM, characteristic of this subgroup contains both LanB and LanC enzymatic activities. Using purified E. coli recombinant expression products of the LctA protein and LctM, the authors demonstrated in vitro modification of the prepropeptide. The authors also demonstrated permissive substrate specificity for the LctM enzyme on several LctA mutant peptides, suggesting that the enzyme may also be useful in other antibiotic engineering experiments. In contrast, cell extracts and recombinant epidermin LanB and LanC enzymes have no in vitro activity [50–52]. The researchers postulate that the post-translational modifications brought about by LanB and LanC happen only if the lantibiotic synthetase complex is formed in the bacterial membrane.
Several organic synthesis schemes have been developed to produce the lanthionine rings found in the lantibiotics [53]. None of these methods have lead to the synthesis of a completely functional lantibiotic. One reason for this is the complexity of the overlapping rings (e.g., rings D and E in nisin or C and D in mutacin 1140; Figure 1B). Using a ring closing metathesis approach, one group produced rings A, B, and C (ABC) mimics of nisin that contained alkene bridges instead of thioether linkages [54]. The substitution of the thioether linkage for an alkene bridge was shown not to interfere with the lipid II binding activity. The ring ABC mimics did not induce CF leakage in model membrane studies, but did compete for lipid II in a competition study between nisin A and the ring ABC mimics. Presumably the synthesis of the complete nisin molecule will be necessary for CF leakage and biological activity. To accomplish a complete synthesis of nisin A or mutacin 1140, an approach that can synthesize the overlapping rings of these antibiotics is needed. One approach, currently under development at Oni Biophama utilizes a novel technology called differentially protected orthogonal lanthionine technology (DPOLT). This technology uses two differentially protected orthogonal lanthionine residues in standard peptide synthesis chemistry (United States Patent Application), which will enable the synthesis of the overlapping rings found in type A (I) lantibiotics.
Another technology called functional enhancement of antimicrobials (FEAM) holds promise for the manufacture of novel lantibiotic analogs (United States Patent Application). The premise for this technology involves understanding that defined and undefined constraints on lantibiotics prevent Nature from making the most effective bactericidal compound. For instance, in the case of nisin, conformational constraints required for its interaction with the lantibiotic synthetase enzymes, immunity gene product [55–58], and membrane receptor for its autoinducing activity [56,59], may prevent it from evolving into the most effective antibiotic. A single addition of a functional group, such as a charge group, polar group, or a hydrophobic group can have significant effects on the bioactivities, pharmacokinetics, and/or pharmacodynamics of an antimicrobial compound. For instance, ring A of nisin showed mutational freedom, and the incorporation of a positive charge or a hydrophobic group by site directed mutagenesis had a profoundly positive effect on the spectrum of activity and level of antimicrobial activity against some target bacterial species when compared to wild-type Nisin [60]. As described above, 2,3-didehydroalanine (Dha) and 2,3 didehydrobutyrine (Dhb) residues are commonly found in lantibiotics, as well as other ribosomally and non-ribosomally synthesized antimicrobials. FEAM makes use of the alpha, beta unsaturated carbonyl group found in these residues, which lend themselves to the addition of thiol compounds containing novel functional groups in a highly selective fashion. The single step additions are easily optimized and can be made in aqueous solvents with greater than a 90% yield. Furthermore, Dha and Dhb residues are easily engineered in lantibiotics by site directed mutagenesis or by an organosynthesis method, further facilitating the production of unique analogs with enhanced function.
Conclusion
The paucity of new antibiotics in the drug development pipeline has prompted serious concern from the scientific community that was nicely articulated by the Infectious Disease Society of America (IDSA) in a report entitled “Bad Bugs, No Drugs”. This report as well as letters to congressional members from IDSA and from the President of the American Society of Microbiology (ASM) has prompted the US Congress to pass the Antibiotic Access and Innovation Amendment in hopes of increasing the amount of antibiotics being developed for therapeutic use. In the cases tested, Type A (I) lantibiotics have a well characterized mechanism of bactericidal activity that is not easily amenable to the development of bacterial resistance. Furthermore, they exhibit the pharmacological characteristics for therapeutic use. Given the current innovations in their manufacture, they are well suited to help fill the antibiotic pipeline.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gross E, Morell JL. Structure of Nisin. J Amer Chem Soc. 1971;93:4634–4639. doi: 10.1021/ja00747a073. [DOI] [PubMed] [Google Scholar]
- 2.Smith L, Novak J, Rocca J, McClung S, Hillman JD, Edison AS. Covalent structure of mutacin 1140 and a novel method for the rapid identification of lantibiotics. Eur J Biochem. 2000;267:6810–6816. doi: 10.1046/j.1432-1033.2000.01777.x. [DOI] [PubMed] [Google Scholar]
- 3.Chatterjee C, Paul M, Xie LL, van der Donk WA. Biosynthesis and mode of action of lantibiotics. Chem Rev. 2005;105:633–683. doi: 10.1021/cr030105v. [DOI] [PubMed] [Google Scholar]
- *4. Bonelli RR, Schneider T, Sahl HG, Wiedemann I. Insights into in vivo activities of lantibiotics from gallidermin and epidermin mode-of-action studies. Antimicrob Agents Chemother. 2006;50:1449–1457. doi: 10.1128/AAC.50.4.1449-1457.2006. The work presented shows that the bactericidal activity of the lantibiotic gallidermin is primarily attributed to blocking cell wall synthesis and that a pore forming mechanism of bactericidal activity depended on the relative thickness of the membrane. This article suggests that the alternative modes of activity presumably contribute in different degrees to their ability to kill various bacteria.
- 5.Breukink E, Wiedemann I, van Kraaij C, Kuipers OP, Sahl HG, de Kruijff B. Use of the cell wall precursor lipid II by a pore-forming peptide antibiotic. Science. 1999;286:2361–2364. doi: 10.1126/science.286.5448.2361. [DOI] [PubMed] [Google Scholar]
- 6.Driessen AJM, van den Hooven HW, Kuiper W, van de Kamp M, Sahl HG, Konings RNH, Konings WN. Mechanistic Studies of Lantibiotic-Induced Permeabilization of Phospholipid-Vesicles. Biochemistry. 1995;34:1606–1614. doi: 10.1021/bi00005a017. [DOI] [PubMed] [Google Scholar]
- *7. Hasper HE, Kramer NE, Smith JL, Hillman JD, Zachariah C, Kuipers OP, de Kruijff B, Breukink E. An alternative bactericidal mechanism of action for lantibiotic peptides that target lipid II. Science. 2006;313:1636–1637. doi: 10.1126/science.1129818. The authors describe a novel mechanism of bactericidal activity. It has previously been shown that antibiotics can block cell wall synthesis by binding to the cell wall precursor molecule lipid II. In this study the authors show, using fluorescent microscopy experiments, that the lantibiotic nisin not only binds to lipid II but actually abducts it from cell septum, where the molecule is needed for the synthesis of new cell wall. In essence, nisin hijacks lipid II.
- *8. Smith L, Hasper H, Breukink E, Novak J, Cerkasov J, Hillman JD, Wilson-Stanford S, Orugunty RS. Elucidation of the Antimicrobial Mechanism of Mutacin 1140. Biochemistry. 2008;17:3308–3314. doi: 10.1021/bi701262z. These authors describe the bioactivity of the lantibiotic mutacin 1140. These experiments show that a lateral assembly mechanism for the antibiotic traps lipid II into a cage-like complex, thus increasing the affinity of the lantibiotic to lipid II.
- 9.Abee T. Pore-Forming Bacteriocins of Gram-Positive Bacteria and Self- Protection Mechanisms of Producer Organisms. FEMS Microbiol Lett. 1995;129:1–9. doi: 10.1016/0378-1097(95)00137-T. [DOI] [PubMed] [Google Scholar]
- 10.Breukink E, de Kruijff B. The lantibiotic nisin, a special case or not? Biochim Biophys Acta. 1999;1462:223–234. doi: 10.1016/s0005-2736(99)00208-4. [DOI] [PubMed] [Google Scholar]
- 11.Moll GN, Clark J, Chan WC, Bycroft BW, Roberts GCK, Konings WN, Driessen AJM. Role of transmembrane pH gradient and membrane binding in nisin pore formation. J Bacteriol. 1997;179:135–140. doi: 10.1128/jb.179.1.135-140.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin Y, Harroun TA, Weiss TM, Ding L, Huang HW. Barrel-Stave Model or Toroidal Model? A Case Study on Melittin Pores. Biophys J. 2001;81:1475–1485. doi: 10.1016/S0006-3495(01)75802-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ludtke SJ, He K, Heller WT, Harroun TA, Yang L, Huang HW. Membrane pores induced by magainin. Biochemistry. 1996;35:13723–13728. doi: 10.1021/bi9620621. [DOI] [PubMed] [Google Scholar]
- 14.Moll GN, Konings WN, Driessen AJM. Bacteriocins: mechanism of membrane insertion and pore formation. Antonie Leeuwenhoek. 1999;76:185–198. [PubMed] [Google Scholar]
- 15.Yeaman MR, Yount NY. Mechanisms of Antimicrobial Peptide Action and Resistance. Pharmacol Rev. 2003;55:27–55. doi: 10.1124/pr.55.1.2. [DOI] [PubMed] [Google Scholar]
- 16.Breukink E, van Kraaij C, Demel RA, Siezen RJ, Kuipers OP, de Kruijff B. The C-terminal region of nisin is responsible for the initial interaction of nisin with the target membrane. Biochemistry. 1997;36:6968–6976. doi: 10.1021/bi970008u. [DOI] [PubMed] [Google Scholar]
- 17.Breukink E, van Kraaij C, van da Len A, de Mel RA, Siezen RJ, de Kruijff B, Kuipers OP. The orientation of nisin in membranes. Biochemistry. 1998;37:8153–8162. doi: 10.1021/bi972797l. [DOI] [PubMed] [Google Scholar]
- 18.Montville TJ, Chen Y. Mechanistic action of pediocin and nisin: recent progress and unresolved questions. Appl Microbiol Biotechnol. 1998;50:511–519. doi: 10.1007/s002530051328. [DOI] [PubMed] [Google Scholar]
- 19.Winkowski K, Ludescher RD, Montville TJ. Physicochemical characterization of the nisin-membrane interaction with liposomes derived from Listeria monocytogenes. Appl Environ Microbiol. 1996;62:323–327. doi: 10.1128/aem.62.2.323-327.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Breukink E, Bonev BB, Wiedemann I, Sahl HG, Watts A, de Kruijff B. Specific interaction of the lantibiotic nisin with lipid II leads to highly efficient pore formation. Biophys J. 2001;80:7. [Google Scholar]
- 21.Breukink E, de Kruijff B. Lipid II as a target for antibiotics. Nature Rev Drug Disc. 2006;5:321–332. doi: 10.1038/nrd2004. [DOI] [PubMed] [Google Scholar]
- 22.Linnett PE, Stroming Jl. Additional Antibiotic Inhibitors of Peptidoglycan Synthesis. Antimicrob Agents Chemother. 1973;4:231–236. doi: 10.1128/aac.4.3.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reisinger P, Seidel H, Tschesche H, Hammes WP. The Effect of Nisin On Murein Synthesis. Arch Microbiol. 1980;127:187–193. doi: 10.1007/BF00427192. [DOI] [PubMed] [Google Scholar]
- 24.Sahl HG, Bierbaum G. Lantibiotics: Biosynthesis and biological activities of uniquely modified peptides from gram-positive bacteria. Annu Rev Microbiol. 1998;52:41–79. doi: 10.1146/annurev.micro.52.1.41. [DOI] [PubMed] [Google Scholar]
- 25.Brotz H, Josten M, Wiedemann I, Schneider U, Gotz F, Bierbaum G, Sahl HG. Role of lipid-bound peptidoglycan precursors in the formation of pores by nisin, epidermin and other lantibiotics. Mol Microbiol. 1998;30:317–327. doi: 10.1046/j.1365-2958.1998.01065.x. [DOI] [PubMed] [Google Scholar]
- 26.Somner EA, Reynolds PE. Inhibition of Peptidoglycan Biosynthesis by Ramoplanin. Antimicrob Agents Chemother. 1990;34:413–419. doi: 10.1128/aac.34.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Breukink E, van Heusden HE, Vollmerhaus PJ, Swiezewska E, Brunner L, Walker S, Heck AJR, de Kruijff B. Lipid II is an intrinsic component of the pore induced by nisin in bacterial membranes. J Biol Chem. 2003;278:19898–19903. doi: 10.1074/jbc.M301463200. [DOI] [PubMed] [Google Scholar]
- **28. Hsu STD, Breukink E, Tischenko E, Lutters MAG, de Kruijff B, Kaptein R, Bonvin A, van Nuland NAJ. The nisin-lipid II complex reveals a pyrophosphate cage that provides a blueprint for novel antibiotics. Nat Struct Mol Biol. 2004;11:963–967. doi: 10.1038/nsmb830. The authors present structural data that provides the first understanding of the molecular interactions involved in the lantibiotic nisin - lipid II complex. The data shows a novel lipid II binding motif in which the pyrophosphate moiety of lipid II interacts with the N-terminal backbone amides of rings A and B via intermolecular hydrogen bonds.
- 29.Daniel RA, Errington J. Control of cell morphogenesis in bacteria: Two distinct ways to make a rod-shaped cell. Cell. 2003;113:767–776. doi: 10.1016/s0092-8674(03)00421-5. [DOI] [PubMed] [Google Scholar]
- 30.Brumfitt W, Salton MRJ, Hamilton-Miller JMT. Nisin, alone and combined with peptidoglycan modulating antibiotics: activity against methicillin resistant Staphylococcus aureus and vancomycin-resistant enterococci. J Antimicrob Chemother. 2002;50:731–734. doi: 10.1093/jac/dkf190. [DOI] [PubMed] [Google Scholar]
- 31.Goldstein B, Wei J, Greenberg K, Novick R. Activity of nisin against S. pneumoniae, in vitro, and in mouse infection model. J Antimicrob Chemother. 1998;42:277–278. [PubMed] [Google Scholar]
- 32.Mota-Meira M, LaPointe G, Lacroix C, Lovoie M. MICs of Mutacin B-Ny266, Nisin A, Vancomycin, and Oxacillin against Bacterial Pathogens. Antimicrob Agent Chemother. 2000;44:24–29. doi: 10.1128/aac.44.1.24-29.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bavin EM, Beach AS, Falconer R, Friedman R. Nisin in experimental tuberculosis. The Lancet. 1952;259:127–129. doi: 10.1016/s0140-6736(52)92429-x. [DOI] [PubMed] [Google Scholar]
- 34.Yoneyama H, Katsumata R. Antibiotic resistance in bacteria and its future for novel antibiotic development. Biosci Biotechnol Biochem. 2006;70:1060–1075. doi: 10.1271/bbb.70.1060. [DOI] [PubMed] [Google Scholar]
- 35.Maher S, McClean S. Investigation of the cytotoxicity of eukaryotic and prokaryotic antimicrobial peptides in intestinal epithelial cells in vitro. Biochem Pharmacol. 2006;71:1289–1298. doi: 10.1016/j.bcp.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 36.Cruz-Chamorro L, Puertollano MA, Puertollano E, de Cienfuegos GAI, de Pablo MA. In vitro biological activities of magainin alone or in combination with nisin. Peptides. 2006;27:1201–1209. doi: 10.1016/j.peptides.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 37.Kordel M, Sahl HG. Susceptibility of Bacterial, Eukaryotic and Artificial Membranes to the Disruptive Action of the Cationic Peptides Pep-5 and Nisin. FEMS Microbiol Lett. 1986;34:139–144. [Google Scholar]
- 38.Mota-Meira M, Morency H, Lavoie MC. In vivo activity of mutacin B-Ny266. J Antimicrob Chemother. 2005;56:869–871. doi: 10.1093/jac/dki295. [DOI] [PubMed] [Google Scholar]
- 39.Hillman JD, Johnson KP, Yaphe BI. Characterization of a Streptococcus mutans Bacteriocin with Novel Properties. J Dent Res. 1983;62:241–241. [Google Scholar]
- 40.Hillman JD, Johnson KP, Yaphe BI. Isolation of a Streptococcus mutans Strain Producing a Novel Bacteriocin. Infect Immun. 1984;44:141–144. doi: 10.1128/iai.44.1.141-144.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith L, Zachariah C, Thirumoorthy R, Rocca J, Novak J, Hillman JD, Edison AS. Structure and dynamics of the lantibiotic mutacin 1140. Biochemistry. 2003;42:10372–10384. doi: 10.1021/bi034490u. [DOI] [PubMed] [Google Scholar]
- 42.Schillinger U, Chung HS, Keppler K, Holzapfel WH. Use of bacteriocinogenic lactic acid bacteria to inhibit spontaneous nisin-resistant mutants of Listeria monocytogenes. J Appl Microbiol. 1998;85:657–663. doi: 10.1111/j.1365-2672.1998.00573.x. [DOI] [PubMed] [Google Scholar]
- 43.Kramer NE, Smid EJ, Kok J, de Kruijff B, Kuipers OP, Breukink E. Resistance of Gram-positive bacteria to nisin is not determined by Lipid II levels. FEMS Microbiol Lett. 2004;239:157–161. doi: 10.1016/j.femsle.2004.08.033. [DOI] [PubMed] [Google Scholar]
- 44.Bailey EM, Rybak MJ, Kaatz GW. Comparative effect of protein binding on the killing activities of teicoplanin and vancomycin. Antimicrob Agents Chemother. 1991;35:1089–1092. doi: 10.1128/aac.35.6.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fekety R. Vancomycin and teicoplan. New York: Churchill Livingstone; 1990. [Google Scholar]
- 46.Kempf M, Theobald U, Fiedler HP. Economic improvement of the fermentative production of gallidermin by Staphylococcus gallinarum. Biotechnol Lett. 1999;21:663–667. [Google Scholar]
- 47.Kempf M, Theobald U, Fiedler HP. Production of the antibiotic gallidermin by Staphylococcus gallinarum - development of a scale-up procedure. Biotechnol Lett. 2000;22:123–128. [Google Scholar]
- 48.Nicolas G, Auger I, Beaudoin M, Halle F, Morency H, LaPointe G, Lavoie MC. Improved methods for mutacin detection and production. J Microbiol Methods. 2004;59:351–361. doi: 10.1016/j.mimet.2004.07.013. [DOI] [PubMed] [Google Scholar]
- *49. Xie LL, Miller LM, Chatterjee C, Averin O, Kelleher NL, van der Donk WA. Lacticin 481: In vitro reconstitution of lantibiotic synthetase activity. Science. 2004;303:679–681. doi: 10.1126/science.1092600. The authors demonstrate for the first time that synthetic lantibiotics may be synthesized by a recombinant expression system. The enzyme LctM was shown to have permissive substrate specificity, demonstrating that it may be useful in the production of other lantibiotics.
- 50.Kupke T, Gotz F. Expression, purification, and characterization of EpiC, an enzyme involved in the biosynthesis of the lantibiotic epidermin, and sequence analysis of Staphylococcus epidermidis epiC mutants. J Bacteriol. 1996;178:1335–1340. doi: 10.1128/jb.178.5.1335-1340.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peschel A, Ottenwalder B, Gotz F. Inducible production and cellular location of the epidermin biosynthetic enzyme EpiB using an improved Staphylococcal expression system. FEMS Microbiol Lett. 1996;137:279–284. doi: 10.1111/j.1574-6968.1996.tb08119.x. [DOI] [PubMed] [Google Scholar]
- 52.Schnell N, Engelke G, Augustin J, Rosenstein R, Ungermann V, Gotz F, Entian KD. Analysis of Genes Involved in the Biosynthesis of Lantibiotic Epidermin. Eur J Biochem. 1992;204:57–68. doi: 10.1111/j.1432-1033.1992.tb16605.x. [DOI] [PubMed] [Google Scholar]
- 53.Paul M, van der Donk WA. Chemical and enzymatic synthesis of lanthionines. Mini-Reviews in Organic Chemistry. 2005;2:23–37. [Google Scholar]
- **54. Ghalit N, Reichwein JF, Hilbers HW, Breukink E, Rijkers DTS, Liskamp RMJ. Synthesis of Bicyclic Alkene-/Alkane-Bridged Nisin Mimics by Ring-Closing Metathesis and their Biochemical Evaluation as Lipid II Binders: toward the Design of Potential Novel Antibiotics. ChemBioChem. 2007;8:1540–1554. doi: 10.1002/cbic.200700244. The authors demonstrate an alternative approach for lantibiotic synthesis. They synthesized rings AB and C of nisin which contain alkene bridges instead of the naturally occurring lanthionine bridges. In a fluorescent competition assay, these experiments show that the alkene rings AB and C can still bind to lipid II.
- 55.Engelke G, Gutowskieckel Z, Kiesau P, Siegers K, Hammelmann M, Entian KD. Regulation of Nisin Biosynthesis and Immunity in Lactococcus Lactis. Appl Environ Microbiol. 1994;60:814–825. doi: 10.1128/aem.60.3.814-825.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ra SR, Qiao MQ, Immonen T, Pujana I, Saris PEJ. Genes responsible for nisin synthesis, regulation and immunity form a regulon of two operons and are induced by nisin in Lactococcus lactis. Microbiology. 1996;142:1281–1288. doi: 10.1099/13500872-142-5-1281. [DOI] [PubMed] [Google Scholar]
- 57.Takala TM, Koponen O, Qiao MQ, Saris PEJ. Lipid-free NisI: interaction with nisin and contribution to nisin immunity via secretion. FEMS Microbiol Lett. 2004;237:171–177. [Google Scholar]
- 58.Takala TM, Saris PEJ. C terminus of Nisl provides specificity to nisin. Microbiology. 2006;152:3543–3549. doi: 10.1099/mic.0.29083-0. [DOI] [PubMed] [Google Scholar]
- 59.van Kraaij C, Breukink E, Rollema HS, Siezen RJ, Demel RA, de Kruijff B, Kuipers OP. Influence of charge differences in the C-terminal part of nisin on antimicrobial activity and signaling capacity. Eur J Biochem. 1997;247:114–120. doi: 10.1111/j.1432-1033.1997.00114.x. [DOI] [PubMed] [Google Scholar]
- **60. Rink R, Wierenga J, Kuipers A, Kluskens LD, Driessen AJM, Kuipers OP, Moll GN. Dissection and Modulation of the Four Distinct Activities of Nisin by Mutagenesis of Rings A and B and by C-Terminal Truncation. Appl Environ Microbiol. 2007;73:5809–5816. doi: 10.1128/AEM.01104-07. This article presents a series of mutations in rings A and B of nisin. The authors show that ring A is amenable to mutagenesis and that some of these variants have enhanced bactericidal activity, but at the cost of the producing bacterium strain losing immunity to the antibiotic.