

Abstract
Early growth response-1 (Egr1) is a sequence-specific transcription factor (TF) which is induced under hypoxic conditions. We presently report that transient middle cerebral artery occlusion (MCAO) leads to increased expression of Egr1 in the brains of adult mice and rats between 2 h and 5 days of reperfusion with a peak increase of 8-12-fold at 1 day. When subjected to transient MCAO and 3 days of reperfusion, Egr1-/- mice showed significantly smaller infarcts (by 44.9 ± 8.4%, p < 0.05) and improved neurological function than Egr1+/+ littermates. Following transient MCAO, brains of Egr1-/- mice showed less water accumulation and decreased neutrophil infiltration (by 42 ± 8%, p < 0.05) compared to Egr1+/+ mice. The number of activated microglia/macrophages were also significantly lower (OX42+ cells by 53 ± 9%, p < 0.05 and ED1+ cells by 59 ± 11%) in the post-ischemic cortex of Egr1-/- mice compared to Egr1+/+ mice. In addition, post-ischemic inflammatory gene expression was less pronounced in the brains of Egr1-/- mice compared to Egr1+/+ mice. Preventing cerebral Egr1 protein induction with small interference RNAs that target Egr1 decreased inflammatory gene expression and led to smaller infarcts (by 40.2 ± 6.9%, p < 0.05) and reduced neurological deficits in rats subjected to transient MCAO. Conversely, transient MCAO following adenoviral-mediated Egr1 over-expression exacerbated the infarct volume (by 29 ± 5.3%, p < 0.05) and worsened the neurological deficits in rats. These studies indicate Egr1 as a significant contributor of inflammation and neuronal damage after stroke.
Keywords: edema, inflammation, ischemic brain damage, knockout, stroke, transcription factor
Massive inflammation is one of the putative promoters of stroke-induced secondary neuronal death. Transcription factors (TF) play an important role in inducing inflammatory genes, and several pro-inflammatory TF, such as interferon regulatory factor-1 (IRF1), nuclear factor kappa B (NF-κB), CCAAT enhancer binding protein-β (C/EBPβ), signal transduction and activator or transcription-3, and activating transcription factor-3 were shown to be induced rapidly following transient middle cerebral artery occlusion (MCAO) (Iadecola et al. 1999; Stephenson et al. 2000; Ohba et al. 2003; Kapadia et al. 2006; Satriotomo et al. 2006).
Egr1 (a.k.a. NGFI-A/zif268/krox24/Tis8) is an oxygen-sensing TF that binds to GC-rich regulatory elements in the promoter regions of several target genes (Yan et al. 2000). Following injury, Egr1 activation was shown to be vital for the induction of inflammatory genes in peripheral organs (Okada et al. 2001, 2002a; Ji et al. 2003; Liang et al. 2003). Furthermore, blocking Egr1 was thought to enhance organ endurance following injury (Yan et al. 2000; Okada et al. 2002b). A recent study showed that Egr1 protein is selectively required for behavioral responses to persistent inflammatory pain (Ko et al. 2005).
We presently observed that transient MCAO results in a rapid and persistent induction of Egr1 expression in rodent brain. To understand its functional significance, we evaluated infarct volume, neurological function, edema, macrophage/neutrophil infiltration and inflammatory gene expression in Egr1-/- and Egr1+/+ mice subjected to transient MCAO. As knockout mice often develop compensatory mechanisms, we also studied the effect of blocking post-ischemic cerebral Egr1 protein induction in rats using a pool of small interference RNAs (siRNAs) designed to target Egr1 mRNA. We further evaluated the effect of transient MCAO following adenoviral-mediated cerebral Egr1 over-expression in rats.
Materials and methods
The animals used in these studies were cared for in accordance with the Guide for the Care and Use of Laboratory Animals, US Department of Health and Human Services, Publication 85-23 (1985). The Research Animal Resources and Care Committee of the University of Wisconsin-Madison approved the surgical procedures.
Egr1 knockout mice
Homozygous Egr1-/- and Egr1+/+ mice were obtained by breeding heterozygous Egr1+/- mice on C57BL/6 background (kindly provided by Dr J. Milbrandt, Washington University School of Medicine, St Louis, MO, USA). To confirm the genotype, DNA extracted from a 2-3 mm piece of tail clipped on postnatal day 14 of each mouse was subjected to PCR (94°C 2 min, 35 cycles of 94°C 30 s, 60°C 30 s, 72°C 1 min) using primers (Neo929 F: CTCGTGCTTTACGGTATCGC; Egr1 F: AACCGGCCCAGCAAGACACC and Egr1 R: GGGCACAGGGGATGGGAATG). When the PCR products were resolved on an agarose gel, the wild-type allele gave a 400 bp band and the knockout allele gave a 420 bp band.
Egr1 siRNA studies
The siRNA experiments were designed and conducted as described earlier (Satriotomo et al. 2006). A pool of 4 siRNA sequences directed against Egr1 mRNA (NM_012551) contained symmetrical 3′-UU overhangs and 5′-phosphorylated antisense strand overhangs (siGENOME SMARTpool Reagents; Dharmacon, Lafayette, CO, USA). The sequences were designed and validated by using SMARTselection Design Algorithm (Dharmacon) and were submitted to a BLAST search to avoid possible targeting of other homologous genes. Sequences of the 4 Egr1 siRNA duplexes represented in the pool are as follows: (1) UAAAGGCUCUUAAUAACACUU-3′ (sense) and 5′-P.GUGUUAUUAAGAGCCUUUAUU (antisense), (2) CGACAGCAGUCCCAUUUACUU (sense) and 5′-P.GUAAAUGGGACUGCUGUCGUU (antisense), (3) UGACAUCGCUCUGAAUAACUU (sense) and 5-P.GUUAUUCAGAGCGAUGUCAUU (antisense) and (4) CGAAUCUGCAUGCGUAAUUUU (sense) and 5′-P.AAUUACGCAUGCAGAUUCGUU (antisense). To account for the non-sequence-specific effects, siControl Non-Targeting Pool (Dharmacon) comprised of four non-targeting siRNA duplexes with comparable GC content to that of the functional Egr1 siRNA but lack identity with known gene targets (confirmed by BLAST analysis to have at least four mismatches in each sequence with all known human, mouse and rat genes) was used. Dharmacon confirmed lack of effect of these control siRNAs on the expression of various mRNAs using microarray analysis. Egr1 or control siRNAs were suspended in siRNA universal buffer (Dharmacon) to yield a concentration of 50 μM. 17 μL siRNA was combined with 3 μL of oligofectamine and incubated at 20°C for 15 min. The siRNAs were slowly injected using a Hamilton syringe into cerebral cortex (0.2 mm from Bregma, 5 mm lateral-midline, 3 mm deep) of adult spontaneously hypertensive (SHR) rats. After 4 h, cohorts of Egr1 and control siRNA injected rats were subjected to either 1 h transient MCAO or sham operation.
Egr1 over-expression studies
Adenoviral recombinants were prepared essentially as described earlier by subcloning EGR1 I293F into pAC adenoviral transfer plasmid and inserted by homologous recombination into E1 region of adenovirus Ad5PacIGFP (Svaren et al. 2000). The viral stocks were multiplied in HEK293 cells, purified by Clontech Nucleospin Virus Purification System and quantified using a plaque forming assay. For in vivo gene transfer, purified Egr1 adenovirus suspended in saline (6 μL; 4.2 × 108 PFU) was injected into lateral ventricles (coordinates -1.5 mm, lateral; -0.8 mm, anterior-posterior; -4.8 mm, dorsal-ventral; based on Paxinos and Watson rat brain atlas) of adult SHR rats using a Hamilton microsyringe over 5 min. Cohorts of rat injected with same amount of LacZ adenovirus (AxCALacZ; 6 μL; 4.2 × 108 PFU) were used as controls. To study the effect of Egr1 over-expression on ischemic brain damage, cohorts of rats underwent either 1 h transient MCAO or sham surgery at 6 days after Egr1 or control adenovirus administration. A different cohort of 36 rats divided into four groups (naive/non-injected, aCSF injected, control adenovirus injected and Egr1 adenovirus injected; n = 9/group) were used to identify the expression of Egr1 as a function of time after adenovirus administration. These rats were killed at day 3, day 6 and day 10 (n = 3 of each group at each time point).
Transient MCAO
Focal ischemia was induced by intraluminal suture method of transient MCAO as described earlier (Vemuganti et al. 2004; Kapadia et al. 2006; Satriotomo et al. 2006; Tureyen et al. 2007; Yan et al. 2007; Zea Longo et al. 1989). In brief, a rat (275-325 g) or a mouse (23-27 g) was anesthetized with halothane (induction: 2%; maintenance: 1.2% in an oxygen and nitrous oxide 50 : 50 mixture), and placed in a stereotaxic frame fitted with a nose cone with halothane. The left femoral artery was cannulated for continuous monitoring of arterial blood pressure and to obtain measurements of pH, Pao2,Paco2, hemoglobin and blood-glucose concentration (i-STAT; Sensor Devices, Waukesha, WI, USA). The rectal temperature was controlled at 37.0 ± 0.5°C during surgery with a feedback-regulated heating pad. After a midline skin incision, the left external carotid artery (ECA) was exposed, and its branches were coagulated. A surgical monofilament nylon suture blunted at the end (3-0 for rats and 6-0 for mice) was introduced into ECA lumen and gently advanced to internal carotid artery until regional cerebral blood flow (rCBF) was reduced to 9-15% of the baseline (recorded by laser Doppler flowmeter; Vasamedics, LLC, St Paul, MN, USA) as described earlier (Vemuganti et al. 2004). The MCA was occluded for 1 h in rats and 2 h in mice. Restoration of the blood flow was confirmed by laser Doppler following withdrawal of the suture. After suturing the wound, animals were allowed to recover from anesthesia and returned to the cage with ad libitum access to food and water. During the surgery, animals were under spontaneous respiration. Animals were killed at various reperfusion periods (2 h to 5 days) as needed by the individual experiment.
Infarct volume estimation
Infarct volume was measured as described earlier using Cresyl violet staining (Vemuganti et al. 2004; Kapadia et al. 2006; Satriotomo et al. 2006). For estimating infarct volume, mice or rats subjected to transient MCAO were killed at 3 days of reperfusion. The animal was perfused transcardially with buffered paraformaldehyde, brain was post-fixed, cryoprotected and sectioned (coronal; 40 μm thick at an interval of 320 μm). The serial sections were stained with Cresyl violet and scanned using the NIH Image program. Volume of the ischemic lesion was computed by numeric integration of data from serial sections from each mouse or rat in respect to the sectional interval. To account for edema and differential shrinkage resulting from tissue processing, injury volumes were corrected by using the Swanson formula: corrected injury volume = contralateral hemisphere volume - (ipsilateral hemisphere volume - measured injury volume) (Swanson et al. 1990).
Neurological evaluation
All mice and rats used for infarct volume estimation were evaluated for post-ischemic neurological deficits on a five-point scale before transient MCAO, at 1 day and 3 days of reperfusion by an investigator blinded to the study groups. A score of 0 suggests no neurological deficit (normal), 1 suggests mild neurological deficit (failure to extend right forepaw fully), 2 suggests moderate neurological deficit (circling to the right), 3 suggests severe neurological deficit (falling to the right), 4 suggests very severe neurological deficit (animal could not walk spontaneously; depressed level of consciousness). This scoring system is similar to that we used before for rats and mice subjected to transient MCAO (Vemuganti et al. 2004; Kapadia et al. 2006; Satriotomo et al. 2006).
Brain edema
In the Egr1+/+ and Egr1-/- mice, cerebral water content was estimated in the contralateral and ipsilateral cortical tissue at 1 day of reperfusion following transient MCAO. Following a quick dissection and estimation of wet weight, tissue was placed at 105°C in an oven for 2 days. Tissue samples were reweighed and the brain water content was estimated by the formula: [(wet weight - dry weight)/wet weight] × 100 (Huang et al. 2007).
Myeloperoxidase activity
Activity of MPO (a lysosomal enzyme specific to leukocyte granules) was assayed as an index of neutrophil infiltration as described earlier (Candelario-Jalil et al. 2007). In brief, Egr1+/+ and Egr1-/- mice subjected to transient MCAO and 2 days of reperfusion were anesthetized with halothane and intracardially perfused with ice-cold isotonic saline. Sham-operated mice served as controls. The ipsilateral and the contralateral cortical samples (~60 mg each) were quickly homogenized in 1 mL of 5 mM potassium phosphate buffer (pH 6.0) and centrifuged (30 000 g; 30 min) at 4°C. Each pellet was resuspended in 0.5 mL of 50 mM phosphate buffer (pH 6.0) containing 0.5% hexadecyltrimethylammonium bromide (Sigma Chemical Co., St Louis, MO, USA). Samples were subjected to three freeze-thaw cycles on dry-ice with intermittent sonication to disrupt the granules. Samples were then incubated for 20 min at 4°C and centrifuged (12 500 g for 15 min) at 4°C. 50 μL of the supernatant was mixed with 1.45 mL of 50 mM potassium phosphate buffer containing o-dianisidine dihydrochloride (0.167 mg/mL; sigma) and 0.0005% hydrogen peroxide (H2O2). The change in absorbance was recorded in a spectrophotometer at 15-s intervals for 2 min at 467 nm. Each sample was assayed in triplicate. One unit of MPO activity was defined as the amount that degraded 1 μmol H2O2 X/min at 25°C. MPO activity was expressed as U/gm tissue.
OX42 and ED1 immunostaining
Parallel sets of 40 μm coronal brain sections from Egr1-/- and Egr1+/+ mice subjected to transient MCAO and 3 days of reperfusion were immunostained with antibodies against OX42 (rat anti-mouse Cd11b; 1 : 500; BD Biosciences, NJ, USA) and ED1 (rat-anti-mouse Cd68; 1 : 500; Serotec, Oxford, UK). In brief, sections were rinsed in 0.1 M Tris-buffered saline with 0.1% Triton-X100 (TBS-T; 3 × 5 min), incubated in 1% H2O2 for 30 min and washed in TBS-T (3 × 5 min). Sections were then incubated in the primary antibodies overnight at 4°C, washed in TBS-T (3 × 5 min), incubated for 1 h in anti-rat biotinylated secondary antibodies from Vector Labs (1 : 200; Burlingame, CA, USA). Conjugation with avidin-biotin complex (1 : 100; Vecstatin Elite ABC kit, Vector labs) was followed by visualization with 3,3′-diaminobenzidine-hydrogen peroxidase (Vector labs). Sections were dehydrated, cleared and mounted in Permount. Sections incubated without primary or secondary antibodies served as negative controls. ED1 and OX42 positive macrophages and activated microglia were counted in 3-4 × 300 fields in the ipsilateral cortex of each mouse. To ensure that homologous areas of injury were sampled between animals, parallel sets of sections from -3.0 to -5.0 mm from Bregma (covering the infarct area) were used.
Real-time PCR analysis
Gene expression analysis was conducted using quantitative real-time PCR as described earlier (Kapadia et al. 2006; Tureyen et al. 2007; Yan et al. 2007). The following transcripts were estimated: Egr1, interleukin (IL) 1β, IL6, monocyte chemoattractant protein1 (MCP1), macrophage inflammatory protein-1α (MIP1α), intracellular adhesion molecule1 (ICAM1) and complement component C3. In brief, total RNA was extracted from the ipsilateral cortex of each animal using Trizol reagent (Invitrogen, Carlsbad, CA, USA). 1 μg of RNA from each rat or mouse was reverse transcribed with oligo(dT)15 and random hexamer primers using M-MuLV reverse transcriptase (Life Technologies, Rockville, MD, USA). 10 ng of cDNA and gene-specific primers were added to SYBR Green PCR Master Mix (SYBR Green I Dye, AmpliTaq DNA polymerase, dNTPs with dUTP and optimal buffer components; Applied Biosystems) and subjected to PCR amplification in a Perkin-Elmer TaqMan 5700 Sequence Detection System (1 cycle at 50°C for 2 min, 1 cycle at 95°C for 10 min, and 40 cycles at 95°C for 15 s and 60°C for 1 min). PCR reactions were conducted in duplicate. The amplified transcripts were quantified with the comparative CT method using 18S rRNA and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as internal controls as described earlier (Kapadia et al. 2006; Vemuganti et al. 2006). The real-time PCR primers were designed using the Primer Express software (Applied Biosystems) based on rat and mouse GenBank accession numbers. The primer sequences are same as used previously (Vemuganti et al. 2004, 2006; Kapadia et al. 2006; Tureyen et al. 2007).
Western blotting
Post-ischemic Egr1 protein expression was evaluated using the ipsilateral cortical tissue of mice (Egr1+/+ and Egr1-/- cohorts) and SHR rats (treated with Egr1 siRNA, control siRNA, Egr1 adenovirus and control adenovirus) by western blotting. In brief, animals were killed at 1 day of reperfusion following transient MCAO and the tissue was homogenized in ice-cold 25 mM Tris-HCl buffer (pH 7.4) containing 2 mM EDTA and protease inhibitors (aprotinin, pepstatin-A, leupeptin, bestatin, 4-(2-aminoethyl)benzenesulfonyl fluoride and trans-epoxysuccinyl-L-leucylamido(4-guanidino) butane). 20 μg protein equivalent of each sample was electrophoresed on polyacrylamide gel, transferred onto nitrocellu-lose and probed with polyclonal anti-Egr1 antibodies (1 : 100; Cell Signaling Technology, Danvers, MA, USA) followed by horse radish peroxidase-conjugated goat anti-rabbit IgG (1 : 1000). The blots were stripped and reprobed with monoclonal anti-b-tubulin antibodies (Sigma Chemical Co. St Louis, MO, USA) and the protein bands recognized by antibodies were detected by enhanced chemiluminescence according to the manufacturer's instructions (Pierce, Rockford, IL, USA). For each sample, blots were prepared twice. Immunoblot intensities were quantified by densitometric scanning using the NIH Image program. Before immunodetection, blots were stained with Ponceau-S to confirm the protein loading and transfer efficiency.
Statistical analysis
Infarct volume estimations, neurological evaluations and the macrophage/microglia cell counts were conducted by an evaluator blinded to study groups. The data in each case was presented as mean ± SD and the statistical significance between groups was assessed using one-way ANOVA followed by Tukey-Kramer multiple comparisons post-test.
Results
Cerebral Egr1 mRNA expression following focal ischemia
Real-time PCR analysis showed a significant increase in the Egr1 mRNA levels in the ipsilateral cortex of rats and mice subjected to transient MCAO and 2 h to 5 days of reperfusion, compared to their respective sham controls (Fig. 1a). The peak increase in Egr1 expression was observed at 1 day of reperfusion in both rats (12.2 ± 2.6-fold over sham; p < 0.05; n = 5/group) and mice (8.3 ± 1.9-fold over sham; p < 0.05; n = 5/group) (Fig. 1a). Expression of the housekeeping transcript 18s rRNA showed no change after transient MCAO compared to sham in both rats and mice. Representative real-time PCR amplification plots for Egr1 and 18s rRNA in the sham, and MCAO/2 h reperfusion and MCAO/1 day reperfusion groups of mice were shown in Fig. 1b.
Fig. 1.
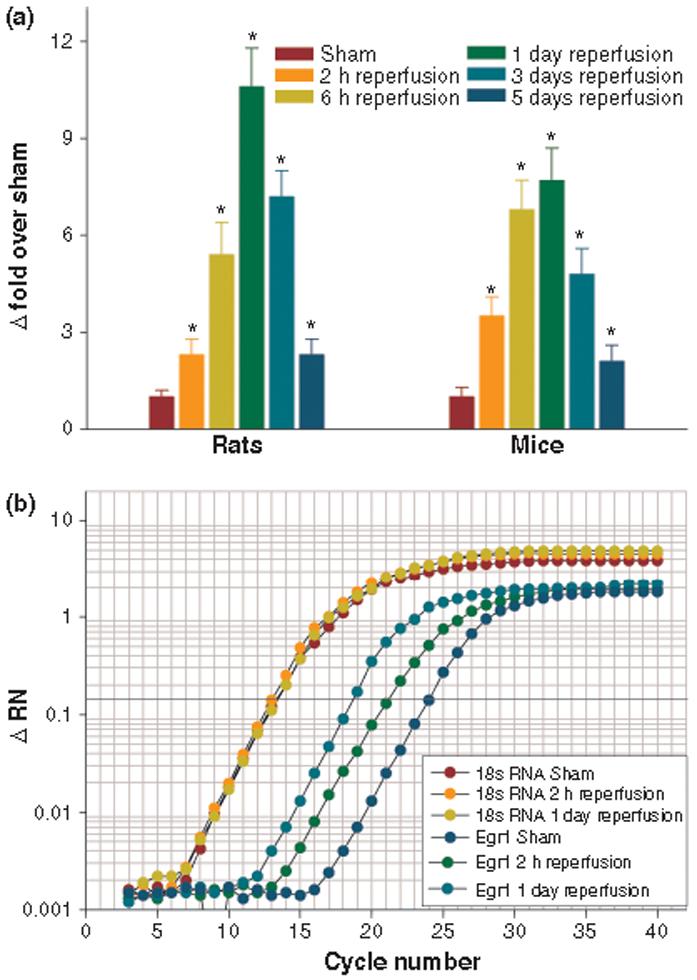
Following transient MCAO, Egr1 mRNA levels increased significantly in the ipsilateral cortex of adult rats and mice between 2 h to 5 days of reperfusion compared to respective sham controls. The bars in the histogram (A) represent mean ± SD (n = 5/group) of fold change over sham. For each sample, PCR was conducted in triplicate. *p < 0.05 versus sham by one-way ANOVA followed by Dunnett's multiple comparisons post-test. Real-time PCR amplification plots for 18s RNA and Egr1 of representative mice subjected to sham-operation and 2 h reperfusion and 1 day reperfusion following MCAO is shown in panel b.
Decreased post-ischemic infarction and neurological deficits in Egr1-/- mice
Egr1+/+ mice showed considerably bigger infarcts than the Egr1-/- mice (Fig. 2a). The total, cortical as well as striatal infarct volumes were significantly higher in the Egr1+/+ mice than the Egr1-/- mice (total by 44.9 ± 8.4%, p < 0.05, cortical by 44.7 ± 7.7%, p < 0.05, and striatal by 53.1 ± 8.4%, p < 0.05; n = 11 for Egr1+/+ and 13 for Egr1-/- genotypes) (Fig. 2b). Furthermore, the neurological deficits analyzed at 1 day as well at 3 days of reperfusion were severe in the Egr1+/+ mice as compared to mild in the Egr1-/- mice (n = 11-13 mice/group) (Fig. 2c). There was no worsening of neurological deficits between day 1 and day 3 of reperfusion in both genotypes, but in the Egr1+/+ cohort, 2 out of 13 mice died between day 1 and day 3 of reperfusion. The Egr1-/- and Egr1+/+ cohorts showed similar levels of rCBF during MCAO and reperfusion (n = 13/group) (Fig. 2d). There were no significant differences in the physiological parameters (brain and body temperature, hemoglobin levels, blood glucose, pH, PaCO2, PaO2) measured 20 min before occlusion, during occlusion and 20 min of reperfusion between the two groups (n = 13/ group) (Table 1).
Fig. 2.

Egr1-/- mice showed less brain damage following transient MCAO compared to Egr1+/+ littermates. Panel a shows 2,3,5-triphenyl tetrazolium chloride-stained serial brain sections of representative mice of Egr1+/+ and Egr1-/- genotype. Panel b shows the infarct volumes and panel c shows the neurological scores in the Egr1-/- and Egr1+/+ mice subjected to transient MCAO and 3 days of reperfusion. The values in B are mean ± SD (n = 11 and 13 for Egr1+/+ and Egr1-/- cohorts). A 5-point neurological scale was used in which a score of 0 suggests no neurological deficit (normal), 1 suggests mild neurological deficit (failure to extend right forepaw fully), 2 suggests moderate neurological deficit (circling to the right), 3 suggests severe neurological deficit (falling to the right), and 4 suggests very severe neurological deficit (the animal could not walk spontaneously; depressed level of consciousness). No mice entered grade 4. Neuroscores for the 2 Egr1+/+ mice that died between day 1 and 3 of reperfusion were not shown on day 3. In panels b and c, *p < 0.05 compared to Egr1+/+ mice by one-way ANOVA followed by Tukey-Kramer multiple comparisons post-test. The rCBF (mean ± SD; n = 13/group) measured by laser-Doppler during MCAO and reperfusion was not significantly different between the 2 genotypes (d). Panel e shows the tail DNA genotyping of representative mice from the Egr1+/+ and Egr1-/- cohorts. As expected, the PCR products were observed to be ~400 bp for Egr1+/+ and ~420 bp for Egr1-/- mice (e). Following transient MCAO and 1 day reperfusion, the cerebral cortex of the Egr1+/+ mice showed Egr1 immunoreactive protein band which was not observed in case of Egr1-/- mice (f).
Table 1.
Physiological parameters following transient MCAO in Egr1+/+ and Egr1-/- mice
| Pre-ischemia (30 min pre-MCAO) |
During ischemia (30 min of MCAO) |
Post-ischemia (30 min of reperfusion) |
||||
|---|---|---|---|---|---|---|
| Egr1+/+ | Egr1-/- | Egr1+/+ | Egr1-/- | Egr1+/+ | Egr1-/- | |
| Each value is mean ± SD (n = 13/group). MABP is mean arterial blood pressure. There were no statistically significant differences between the Egr1+/+ and Egr1-/- groups at different periods (pre-ischemia, during ischemia and post-ischemia) in any of the parameters. | ||||||
| MABP | 86.1 ± 7.2 | 84.7 ± 6.9 | 84.5 ± 6.5 | 82.5 ± 7.0 | 87.1 ± 6.5 | 86.3 ± 5.7 |
| Rectal Temp (°C) | 37.2 ± 0.4 | 36.8 ± 0.3 | 36.8 ± 0.2 | 37.1 ± 0.3 | 37.2 ± 0.4 | 36.9 ± 0.4 |
| pH | 7.40 ± 0.05 | 7.38 ± 0.06 | 7.41 ± 0.06 | 7.39 ± 0.06 | 7.38 ± 0.05 | 7.42 ± 0.06 |
| Glucose (mmol/L) | 6.9 ± 0.3 | 7.2 ± 0.3 | 7.1 ± 0.4 | 7.0 ± 0.3 | 6.8 ± 0.4 | 7.0 ± 0.3 |
| PaO2 (mm Hg) | 140 ± 35 | 135 ± 38 | 136 ± 37 | 139 ± 30 | 133 ± 37 | 140 ± 34 |
| PaCO2 (mm Hg) | 40.7 ± 4.1 | 39.3 ± 5.1 | 40.2 ± 5.6 | 38.5 ± 4.9 | 37.9 ± 5.4 | 40.3 ± 4.8 |
Egr1-/- mice showed reduced edema, curtailed activation of microglia, and decreased macrophage/neutrophil infiltration after focal ischemia
In the cerebral cortex of the sham-operated mice, the brain water content was observed to be 77.2 ± 1.1% and 77.7 ± 0.9% for the Egr1+/+ and Egr1-/- cohorts (Fig. 3a). At 1 day of reperfusion following transient MCAO, there was a significant increase in the cerebral water content in the ipsilateral cortex in both Egr1+/+ and Egr1-/- mice compared to their respective sham controls (Fig. 3a). However, Egr1-/- mice showed a significantly lower water accumulation (a 5.6 ± 0.3% increase in the Egr1-/- versus a 9.4 ± 0.8% increase in the Egr1+/+ mice; n = 6/group) (Fig. 3a). Following sham-operation, MPO activity was observed to be similar in the cortex of the Egr1+/+ nice (0.13 ± 0.05 U/gm tissue) and Egr1-/- mice (0.14 ± 0.05 U/gm tissue) (n = 6/group) (Fig. 3b). Whereas, following transient MCAO and 2 days of reperfusion, MPO activity observed in the ipsilateral cortex was significantly higher in the Egr1+/+ mice (by 23.7-fold, p < 0.05; 3.21 ± 0.48 U/gm tissue; n = 6) compared to the Egr1-/- mice (by 12.2-fold, p < 0.05; 1.71 ± 0.42 U/gm tissue; n = 6) (Fig. 3b).
Fig. 3.

Following transient MCAO, Egr1-/- mice showed significantly less edema (measured by water content) (a), decreased number of extravasated neutrophils (measured by MPO activity) (b), reduced numbers of activated microglia/macrophages (OX42 and ED1 positive; c and d) compared to Egr1+/+ mice. The values in the histograms are mean ± SD (n = 6/group). ap < 0.05 compared to respective sham and bp < 0.05 compared to respective Egr1+/+ group by ANOVA followed by Tukey-Kramer multiple comparisons post-test.
In the ipsilateral cortex of a separate cohort of Egr1+/+ mice subjected to transient MCAO and 3 days of reperfusion, the numbers of OX42+ activated microglial cells and the ED1+ macrophages increased significantly by 12.6-fold and 22.3-fold, respectively compared to the sham-operated mice (p < 0.05 for both cell types; n = 6/group). At the same reperfusion period, Egr1-/- mice also showed significantly increased OX42+ and the ED1+ cells compared to the sham control by 5.7-fold and 8.6-fold, respectively (p < 0.05 for both cell types; n = 6/group). Thus, the post-ischemic cerebral OX42+ and ED1+ cell number was significantly lower (by 53 ± 9% and 61 ± 12%, respectively; p < 0.05) in the Egr1-/- mice compared to Egr1+/+ mice (Fig. 3c and d).
Egr1-/- mice showed decreased post-ischemic inflammatory gene expression
Using real-time PCR, we estimated the expression of pro-inflammatory transcripts IL1β, IL6, MCP1, MIP1α, ICAM1, and complement component C3 (CC-C3) in the ipsilateral cortex of cohorts of Egr1+/+ and Egr1-/- mice subjected to transient MCAO and 6 h of reperfusion or sham surgery (n = 4/group). These transcripts are known to be up-regulated in rodent brain following transient MCAO (Vemuganti et al. 2002; Kapadia et al. 2006). At 6 h of reperfusion, the cortical infarct is still very small, but many inflammatory genes including the above are known to be induced significantly (Vemuganti et al. 2002; Tureyen et al. 2007). Hence, studying the effect of Egr1 knockdown at such an early time point will show if prevention of inflammatory gene expression is a cause, rather than an affect of post-ischemic neuroprotection. The ipsilateral cortex of both genotypes showed significantly increased expression of all the above transcripts compared to the respective sham controls (Table 2). However, the post-ischemic increases were significantly lower in the Egr1-/- mice compared to Egr1+/+ mice for IL1β (by 68 ± 11%, p < 0.05), IL6 (by 69 ± 13%, p < 0.05), MCP1 (by 74 ± 12%, p < 0.05) MIP1α (by 63 ± 9%, p < 0.05), ICAM1 (by 73 ± 14%, p < 0.05) and CC-C3 (by 81 ± 19%; Table 2). At 1 day of reperfusion following transient MCAO, these changes in gene expression persisted and all the transcripts showed significantly higher expression in Egr1+/+ mice compared to Egr1-/- mice (n = 4/group) (Table 2). On the other hand, three transcripts which are known to be induced after transient MCAO (HSP70, HSP27 and ornithine decarboxylase) showed similar levels of induction in both the genotypes at 6 h and 1 day reperfusion compared to respective shams (Table 2). The two house-keeping controls (18S rRNA and GAPDH) showed no significant change in either cohort at 6 h or 1 day reperfusion following transient MCAO compared to sham controls (Table 2).
Table 2.
Real-time PCR analysis of inflammatory mRNA expression in the Egr1+/+ and Egr1-/- mice subjected to transient MCAO
| 6 h reperfusion |
1 day reperfusion |
||||
|---|---|---|---|---|---|
| Transcript | GenBank# | Egr1+/+ | Egr1-/- | Egr1+/+ | Egr1-/- |
| The values are mean ± SD of fold differences of 16 cross-comparisons between four sham and four ischemic animals in each case. Total RNA was extracted from the ipsilateral cortex of each mouse. Real-time PCR was conducted in duplicate with each cDNA sample for each transcript. | |||||
| IL6 | NM_031168 | 17.1 ± 2.4 | 5.2 ± 1.4* | 56.7 ± 6.1 | 6.4 ± 1.1* |
| IL1β | NM_008361 | 5.7 ± 1.1 | 1.8 ± 0.7* | 8.9 ± 2.0 | 2.1 ± 0.6* |
| MIP1α | NM_011337 | 22.0 ± 3.3 | 8.2 ± 1.6* | 18.8 ± 2.5 | 6.3 ± 1.4* |
| MCP1 | AF065933 | 15.6 ± 2.2 | 4.1 ± 0.9* | 19.4 ± 2.9 | 5.3 ± 1.0* |
| ICAM1 | BC008626 | 7.7 ± 1.5 | 2.1 ± 0.5* | 9.6 ± 2.0 | 1.7 ±0.3* |
| CC-C3 | NM_009778 | 23.7 ± 3.1 | 4.5 ± 0.4* | 34.8 ± 5.4 | 5.7 ± 1.5* |
| HSP70 | AW763765 | 7.2 ± 1.3 | 6.4 ± 1.2 | 12.1 ± 2.0 | 10.6 ± 1.5 |
| HSP27 | U03561 | 4.5 ± 0.9 | 5.2 ± 1.3 | 5.1 ± 0.7 | 5.8 ± 1.1 |
| ODC | NM_013614 | 4.8 ± 1.4 | 4.1 ± 1.0 | 2.3 ± 0.4 | 1.8 ± 0.4 |
| 18S rRNA | M11188 | NC | NC | NC | NC |
| GAPDH | NM_008084 | NC | NC | NC | NC |
p < 0.05 compared to Egr1+/+ mice by one-way anova followed by Tukey-Kramer multiple comparisons post-test.
Egr1 siRNA decreased ischemic brain damage
Knockout mice are known to develop adaptations to compensate the lack of a specific protein from birth. For example, despite the paramount importance of Egr1 in growth and development, Egr1-/- mice survive to adulthood with no observable overt phenotypic changes. As their brains are incapable of inducing Egr1 mRNA and protein expression after focal ischemia, to understand the significance of post-ischemic induction of Egr1 against complete lack of Egr1, we employed the siRNA knockdown technique in adult rats. To assure the effectiveness of siRNA, we used a pool of four specific siRNA sequences that target rat Egr1. At 1 day of reperfusion following transient MCAO, the ipsilateral cortex of Egr1 siRNA treated rats showed a significantly decreased Egr1 mRNA (by 71.8 ± 9%, 0 < 0.05; Fig. 4a), and protein (by 83 ± 11%; p < 0.05; n = 4/group; Fig. 4b) levels compared to the ipsilateral cortex of the control siRNA treated rats. Neither Egr1 siRNA nor control siRNA had any effect on 18s rRNA levels or β-tubulin protein levels (housekeeping controls) (Fig. 4a and b).
Fig. 4.

The siRNA-mediated knockdown of post-ischemic Egr1 expression decreased infarction and inflammatory gene expression in adult rats subjected to transient MCAO. Egr1 siRNA depleted the Egr1 mRNA levels (a) as well as Egr1 protein levels (b) in the post-ischemic brain. Panel c shows Cresyl violet-stained serial sections from representative rats subjected to transient MCAO following treatment with Egr1 and control siRNA. Egr1 siRNA treatment also significantly curtailed the post-ischemic induction of the mRNA expression of cytokines, chemokines and ICAM1 (d). The values in the histograms are mean ± SD of n = 4/group. In panel a, ap < 0.05 compared to the respective sham and bp < 0.05 compared to the control siRNA group and in panel d, *p < 0.05 compared to the control siRNA group by ANOVA followed by Tukey-Kramer multiple comparisons post-test.
Cresyl violet stained serial coronal sections from representative rats from the control and Egr1 siRNA treated groups subjected to transient MCAO and 3 days of reperfusion were shown in Fig. 5c. Egr1 siRNA group showed significantly smaller cortical (by 42.4 ± 7.3%, p < 0.05), striatal (by 30.3 ± 6.1%, p < 0.05) and total (by 40.2 ± 6.9%, p < 0.05) infarct volume compared to control siRNA group (n = 9/group; Table 3). Furthermore, neurological deficits analyzed at 1 day of reperfusion were mild in the MCAO/Egr1 siRNA group compared to moderate-severe deficits observed in the MCAO/control siRNA group (median neuroscores were 1.31 ± 0.41 for Egr1 siRNA group and 2.79 ± 0.24 for control siRNA group; n = 9/group; Table 3). The rCBF as well as physiological parameters measured during MCAO and reperfusion were not significantly different between Egr1 siRNA and control siRNA groups (data not shown).
Fig. 5.

In adult rats, Egr1 adenovirus administration significantly induced Egr1 mRNA expression (a) and protein expression (b) in the cortex. Representative Cresyl violet-stained sections from a rat in the Egr1 adenovirus group show exacerbated ischemic infarct size compared to a rat in the control adenovirus group (c). The values in the histogram are mean ± SD of n = 4/group. *p < 0.05 compared to the control adenovirus group by ANOVA followed by Tukey-Kramer multiple comparisons post-test.
Table 3.
Effect of Egr1 knockdown and over-expression on transient MCAO-induced infarct volumes and neurological deficits in adult rats
| Infarct volume (mm3) |
|||||
|---|---|---|---|---|---|
| Treatment (n) | Cortex | Striatum | Total | Neuroscore | Neurological Deficit |
| Values are mean ± SD. MCAO was 1 h in all cases. Ischemic infarct volumes and neuroscores were measured at either 1 day or 3 days of reperfusion. The scale for neurological scoring is as follows: 0, no neurological deficit; 1, mild neurological deficit; 2, moderate neurological deficit; 3, severe neurological deficit and 4, very severe neurological deficit. None of the rats entered grad 4. | |||||
| None (12) | 178 ± 31 | 35 ± 5 | 217 ± 35 | 2.44 ± 0.31 | Moderate-severe |
| Control siRNA (9) | 172 ± 34 | 33 ± 7 | 209 ± 38 | 2.79 ± 0.24 | Moderate-severe |
| Egr1 siRNA (9) | 99 ± 19a,b | 23 ± 6a,b | 125± 25a,b | 1.31 ± 0.41a,b | Mild |
| Control adenovirus (10) | 169 ± 38 | 32 ± 8 | 204 ± 40 | 2.48 ± 0.53 | Moderate-severe |
| Egr1 adenovirus (10) | 225 ± 47a,b | 42 ± 8a,b | 264 ± 55a,b | 3.19 ± 0.58a,b | Severe |
p < 0.05 compared to none group.
p < 0.05 compared to the respective control group by one-way anova followed by Tukey-Kramer multiple comparisons post-test.
Egr1 siRNA prevented post-ischemic inflammatory gene expression
We estimated the expression of IL1β, IL6, MIP1α, MCP1, and ICAM1 in the rats subjected to transient MCAO and 6 h reperfusion following Egr1 siRNA or control siRNA administration. The ipsilateral cortical samples of Egr1 siRNA group showed significantly curtailed induction of all these transcripts compared to control siRNA group by 48-71%; p < 0.05 in all cases; n = 4/group) (Fig. 4d).
Over-expression of Egr1 protein increased post-ischemic infarction
As an opposing paradigm to knock-down, we evaluated the effect of focal ischemia following over-expression of Egr1 in rat brain by transfecting with Egr1 adenovirus. Intracerebral administration of Egr1 adenovirus significantly elevated Egr1 mRNA levels from 3 to 10 days (by 4.2 ± 0.8-fold at 3 days, by 7.7 ± 1.2-fold at 6 days and by 6.8 ± 0.9-fold at 10 days; n = 4/time point) compared to controls (adenovirus injected rats or naïve rats or aCSF injected rats; n = 4/group) (Fig. 5a). The Egr1 adenovirus group also showed a significantly higher cortical Egr1 protein levels (by 329 ± 71%; p < 0.05) at 6 days (n = 4/group) (Fig. 5b). No mortality or signs of distress were observed in the Egr1 or the control adenovirus administered rats. As adenovirus treatment can cause inflammation, we estimated the mRNA levels of the cytokine IL6 and the chemokine MCP1 in the adenovirus treated rats. At the low concentration of the adenovirus used presently, neither control adenovirus nor Egr1 adenovirus group showed any significant increased expression of IL6 or MCP1 over aCSF treated control at either 3 days or 6 days (data not shown).
In a separate cohort of rats, the total infarct volume was observed to be significantly greater in the Egr1 adenovirus group compared to the control adenovirus group at 3 days of reperfusion following transient MCAO (by 29 ± 5.3%; p < 0.05; n = 10/group) (Table 3). The cortical and the striatal infarct volumes were higher by 30.1 ± 5.8% (p < 0.05) and 31.2 ± 4.2% (p < 0.05), respectively in the Egr1 adenovirus group compared to the control adenovirus group (Table 3). At 3 days of reperfusion, the Egr1 adenovirus group also showed a significantly higher mean neuroscore (Egr1 adenovirus: 3.19 ± 0.58% and control adenovirus: 2.48 ± 0.53, n = 10/group; p < 0.05) reflecting worsened neurological deficits than the control adenovirus group (Table 3). Cresyl violet stained coronal sections from representative rats of control and Egr1 adenovirus groups are shown in Fig. 5c. There were no statistically significant differences in the rCBF and physiological parameters measured during MCAO and reperfusion between Egr1 adenovirus and control adenovirus treated groups (data not shown).
Discussion
In brief, the present study shows that transient focal ischemia leads to a rapid and sustained increase in the expression of the TF Egr1 in rodent brain. Focal ischemia in Egr1 knockout mice resulted in a significantly curtailed post-ischemic inflammatory gene expression, less infiltration of macrophages and neutrophils and smaller infarcts. In adult rats, Egr1 knockdown decreased and Egr1 overexpression exacerbated the inflammatory gene expression and infarction following focal ischemia. Thus, the present studies indicate that post-ischemic Egr1 induction is a promoter of strokeinduced inflammation and neuronal damage leading to neurological deficits.
By virtue of their ability to control gene expression, TFs play a significant role in modulating pathological events in the injured CNS. As stroke is a strong stimulator of several TFs in rodent brain, many studies attempted to understand their functional significance to post-ischemic outcome (Iadecola et al. 1999; Vemuganti et al. 2002; Kapadia et al. 2006; Rickhag et al. 2006; Satriotomo et al. 2006; Tureyen et al. 2007). On one hand, TFs like cyclic AMP response element binding protein, hypoxia inducible factor-1 (HIF1), nuclear factor E2-like factor (Nrf-2), c-fos, P53 and peroxisome proliferator-activated receptor (PPAR) isoforms PPARα and PPARγ, are known to prevent ischemic brain damage (Bergeron et al. 1999; Tanaka et al. 2000; Cho et al. 2001; Maeda et al. 2001; Sundararajan et al. 2005; Luo et al. 2006; Tureyen et al. 2007). On the other hand, TFs like nuclear factor-kappaB (NF-κB), activating transcription factor-3, CCAAT-enhancer binding protein-beta (C/EBPβ), interferon regulatory factor-1 (IRF1) and signal transduction and activator of transcription-3 were known to mediate the post-ischemic neuronal damage (Iadecola et al. 1999; Stephenson et al. 2000; Ohba et al. 2003; Kapadia et al. 2006; Satriotomo et al. 2006). Many of the above TFs including NF-κB, IRF1 and c/EBPβ are known to promote pro-inflammatory gene expression that precipitates secondary neuronal death (Yi et al. 2007). While not undermining the importance of any of the TFs in stroke pathophysiology, the present study is the first to show that Egr1 induction contributes significantly to post-ischemic inflammation and secondary brain damage.
The exact mechanisms by which Egr1 promotes ischemic brain damage are not known at present. However, rCBF or other physiological parameters which are major determinants of infarction after focal ischemia were not significantly different between Egr1 knockout mice and their wild-type littermates subjected to transient MCAO. Furthermore, neither Egr1 siRNA nor Egr1 adenovirus treated rats showed any significant changes in the post-ischemic rCBF compared to their respective controls. This excludes the possibility of vascular alterations as a cause for Egr1 induced ischemic neuronal damage. Our studies also exclude the possibility of genetic background as a causative factor of less brain damage in Egr1 knockout mice as the wild-type littermates that share the background showed no neuroprotection after focal ischemia. The effect of Egr1 on ischemic brain damage is also not species-specific as both mice that lack Egr1 as well as rats treated with Egr1 siRNA showed smaller infarcts compared to their respective controls.
Excessive inflammation during the acute phase after transient focal ischemia is known to be a major precipitator of the secondary neuronal death (Yi et al. 2007). Several inflammatory genes including cytokines, chemokines, adhesion molecules and pro-inflammatory enzymes are known to be up-regulated as early as 2 h and remain elevated for days following transient MCAO (Soriano et al. 2000; Vemuganti et al. 2002, 2004; Lu et al. 2004; Kapadia et al. 2006). Egr1 up-regulation might be one of the upstream promoters of this massive inflammatory gene expression in the post-ischemic brain. Previous studies that evaluated the mechanisms of inflammation following peripheral organ injury support this concept. In peripheral organs, several inflammation-related genes, such as tissue factor, tumor necrosis factor-α, ICAM1, vascular cell adhesion molecule-1, IL1β, IL6, MCP1, plasminogen activator inhibitor-1 and transforming growth factor-β are known to be induced down-stream to Egr1 (Maltzman et al. 1996; Svaren et al. 2000; Yan et al. 2000; Silverman et al. 2001; Pawlinski et al. 2003). Egr1 activation was shown to be vital for the onset of inflammation associated with pancreatitis, lung ischemia, organ transplantation and genitourinary diseases (Ji et al. 2003; Liang et al. 2003; Okada et al. 2001, 2002a,b). Lung ischemia/reperfusion in mice induces Egr1 and its target inflammatory mediators; and Egr1 gene deletion diminishes inflammatory gene expression and enhances animal survival (Yan et al. 2000). It was also shown that transplantation of heterotopic murine cardiac allografts induced the expression of Egr1 and its downstream inflammatory genes, and blocking Egr1 at the time of organ harvest is beneficial (Okada et al. 2002b). Lipopolysaccharide administration in mice rapidly increases the steady-state levels of Egr1 mRNA and promotes sustained expression of inflammatory genes in the kidneys and lungs (Pawlinski et al. 2003).
To determine whether promoting inflammation is one of the mechanisms by which Egr1 induction mediates brain damage, we analyzed inflammatory gene expression in the ipsilateral cortex of Egr1+/+ and Egr1-/- mice subjected to transient MCAO and 6 h of reperfusion. Egr1-/- mice showed significantly less induction of cytokines (IL1β and IL6), chemokines (MCP1 and MIP1α), the adhesion molecule ICAM1, and complement CC-C3 compared to Egr1+/+ mice. Furthermore, treating ischemic rats with Egr1 siRNA significantly curtailed the post-ischemic induction of IL1β, IL6, MIP1α, MCP1, and ICAM1. Post-ischemic expression of all these inflammatory transcripts were also significantly higher at 1 day of reperfusion in the Egr1+/+ mice compared to Egr1-/- mice. This indicates that prevention of inflammatory gene expression in the absence of Egr1 is not a transient phenomenon. On the other hand, expression of HSP70, HSP27, and ODC (three genes known to be induced significantly after focal ischemia) were observed to be expressed to the same extent in Egr1+/+ and Egr1-/- mice subjected to transient MCAO. This indicates that the effect of Egr1 on inflammatory gene expression is very specific. Furthermore, Egr1 siRNA treatment significantly prevented the post-ischemic induction of IL6, MIP1a, MCP1, and ICAM1, compared to control siRNA-treated rats. As adenoviral treatment can lead to inflammation, we also estimated IL6 and MCP1 3 days, 6 days, and 10 days following control and Egr1 adenovirus treatment in rats. The amount of adenovirus used in this study is not excessive (6 μL; 4.2 × 108 PFU) and neither adenovirus induced these two inflammatory transcripts to any significant level at any time point studied. This observation further supports that Egr1 induction is responsible for the increased expression of inflammatory genes in post-ischemic brain.
Within hours after an ischemic insult, increased levels of cytokines and chemokines enhance the expression of adhesion molecules on cerebral endothelial cells leading to transendothelial migration of the circulating neutrophils and monocytes into the injured brain (Okada et al. 1994). Of importance, ICAM1 is a major adhesion molecule that mediates leukocyte extravasation into brain parenchyma. The blood cells block capillaries in addition to releasing a host of neurotoxic compounds including free-radicals and lipid peroxidation products. We observed significantly less numbers of infiltrated macrophages and neutrophils in Egr1-/- mice show compared to Egr1+/+ mice subjected to transient MCAO. As cytokines enhance the expression of adhesion molecules, and chemokines attract macrophages and neutrophils, curtailed expression of IL6, IL1b, MCP1, and MIP1a might have contributed to the less infiltration of the blood-borne cells in the post-ischemic Egr1-/- mouse brain.
While Egr1 induction might be an important transcriptional event in starting inflammatory gene expression (Yan et al. 2000), other interacting TFs can control Egr1 function either positively or negatively. In particular, the TF C/EBPβ promotes and the TF PPARγ curtails Egr1 expression (Greenbaum et al. 1998; Okada et al. 2002a). Both these TFs are known to be induced after focal ischemia (Kapadia et al. 2006; Victor et al. 2006). In C/EBPβ knockout mice, Egr1 induction was reported to be six-fold less than wild-type mice following liver damage (Greenbaum et al. 1998). Recently, our laboratory reported that C/EBPβ knockout mice show significant neuroprotection and decreased inflammation following transient MCAO (Kapadia et al. 2006). We also observed that the post-ischemic Egr1 induction was of a lesser magnitude in the brains of C/EBPβ-/- mice (Kapadia et al. 2006). On the other hand, treating rodents after transient MCAO with PPARγ agonists induce significant neuroprotection and notably PPARγ agonists prevent post-ischemic Egr1 expression (Tureyen et al. 2007). The interaction of Egr1 with these TFs in promoting inflammation and the ensuing brain damage following focal ischemia needs to be evaluated further. In addition, a family of evolutionarily conserved proteins called NGFI-A binding (NAB) proteins acts as co-repressors of Egr1 transcriptional activation (Srinivasan et al. 2006). The role of NAB proteins in modulating Egr1 in the post-ischemic brain is yet to be evaluated. In conclusion, the present studies for the first time demonstrate that induction of Egr1 to an inappropriate level contributes to the inflammation and brain damage after stroke.
Acknowledgments
These studies were funded by grants to R. Vemuganti (NIH RO1 NS044173 and RO1 NS049448, American Heart Association Grant-in-Aid 0350164N) and to J. Svaren (NIH RO1 HD041590). The authors wish to thank J. Liang for the technical help.
Abbreviations used
- aCSF
artificial cerebrospinal fluid
- ANOVA
analysis of variance
- C/EBP
CCAAT/enhancer binding protein
- CC-C3
complement component-C3
- ECA
external carotid artery
- Egr1
early growth response-1
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- H2O2
hydrogen peroxide
- HEK293
human embryonic kidney-293
- HIF
hypoxia inducible factor
- HSP
heat shock protein
- ICAM1
intercellular adhesion molecule-1
- IL
interleukin
- IRF1
interferon regulatory factor-1
- MCA
middle cerebral artery
- MCAO
MCA occlusion
- MCP
monocyte chemoattractant protein
- MIP
macrophage inflammatory protein
- MPO
myeloperoxidase
- NF-κB
nuclear factor kappa B
- NGFI-A
nerve growth factor induced-A
- Nrf-2
nuclear factor E2-like factor
- ODC
ornithine decarboxylase
- PPAR
peroxisome proliferator-activated receptor
- rCBF
regional cerebral blood flow
- SD
Standard deviation
- SHR
spontaneously hypertensive
- siRNA
small interference RNA
- STAT3
signal transducer and activator of transcription-3
- TBS
Tris-buffered saline
- TBS-T
TBS with Triton X-100
- TF
transcription factor
References
- Bergeron M, Yu AY, Solway KE, Semenza GL, Sharp FR. Induction of hypoxia-inducible factor-1 (HIF-1) and its target genes following focal ischaemia in rat brain. Eur. J. Neurosci. 1999;11:4159–4170. doi: 10.1046/j.1460-9568.1999.00845.x. [DOI] [PubMed] [Google Scholar]
- Candelario-Jalil E, Gonzalez-Falcon A, Garcia-Cabrera M, Leon OS, Fiebich BL. Post-ischemic treatment with cyclooxygenase-2 inhibitor nimesulide reduces blood-brain barrier disruption and leukocyte infiltration following transient focal cerebral ischemia in rats. J. Neurochem. 2007;100:1108–1120. doi: 10.1111/j.1471-4159.2006.04280.x. [DOI] [PubMed] [Google Scholar]
- Cho S, Park EM, Kim Y, Liu N, Gal J, Volpe BT, Joh TH. Early c-Fos induction after cerebral ischemia: a possible neuroprotective role. J. Cereb. Blood Flow Metab. 2001;21:550–556. doi: 10.1097/00004647-200105000-00009. [DOI] [PubMed] [Google Scholar]
- Greenbaum LE, Li W, Cressman DE, Peng Y, Ciliberto G, Poli V, Taub R. CCAAT enhancer-binding protein beta is required for normal hepatocyte proliferation in mice after partial hepatectomy. J. Clin. Invest. 1998;102:996–1007. doi: 10.1172/JCI3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZX, Kang ZM, Gu GJ, Peng GN, Yun L, Tao HY, Xu WG, Sun XJ, Jhang ZH. Therapeutic effects of hyperbaric oxygen in a rat model of endothelin-1-induced focal cerebral ischemia. Brain Res. 2007;1153:204–213. doi: 10.1016/j.brainres.2007.03.061. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Salkowski CA, Zhang F, Aber T, Nagayama M, Vogel SN, Ross ME. The transcription factor interferon regulatory gactor-1 is expressed after cerebral ischemia and contributes to ischemic brain injury. J. Exp. Med. 1999;189:719–727. doi: 10.1084/jem.189.4.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji B, Chen XQ, Misek DE, Kuick R, Hanash S, Ernst S, Najarian R, Logsdon CD. Pancreatic gene expression during the initiation of acute pancreatitis: identification of EGR-1 as a key regulator. Physiol. Genomics. 2003;14:59–72. doi: 10.1152/physiolgenomics.00174.2002. [DOI] [PubMed] [Google Scholar]
- Kapadia R, Tureyen K, Bowen KK, Kalluri H, Johnson PF, Vemuganti R. Decreased brain damage and curtailed inflammation in transcription factor CCAAT/enhancer binding protein beta knockout mice following transient focal cerebral ischemia. J. Neurochem. 2006;98:1718–1731. doi: 10.1111/j.1471-4159.2006.04056.x. [DOI] [PubMed] [Google Scholar]
- Ko SW, Vadakkan KI, Ao H, Gallitano-Mendel A, Wei F, Milbrandt J, Zhuo M. Selective contribution of Egr1 (zif/268) to persistent inflammatory pain. J. Pain. 2005;6:12–20. doi: 10.1016/j.jpain.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Liang TB, Man K, Kin-Wah Lee T, Hong-Teng Tsui S, Lo CM, Xu X, Zheng SS, Fan ST, Wong J. Distinct intragraft response pattern in relation to graft size in liver transplantation. Transplantation. 2003;75:673–678. doi: 10.1097/01.TP.0000048490.24429.89. [DOI] [PubMed] [Google Scholar]
- Lu XC, Williams AJ, Yao C, et al. Microarray analysis of acute and delayed gene expression profile in rats after focal ischemic brain injury and reperfusion. J. Neurosci. Res. 2004;77:843–857. doi: 10.1002/jnr.20218. [DOI] [PubMed] [Google Scholar]
- Luo Y, Yin W, Signore AP, Zhang F, Hong Z, Wang S, Graham SH, Chen J. Neuroprotection against focal ischemic brain injury by the peroxisomal proliferator-activated receptor-γ agonist rosiglitazone. J. Neurochem. 2006;97:435–448. doi: 10.1111/j.1471-4159.2006.03758.x. [DOI] [PubMed] [Google Scholar]
- Maeda K, Hata R, Gillardon F, Hossman KA. Aggravation of brain injury after focal cerebral ischemia in p53-deficient mice. Mol. Brain Res. 2001;88:54–61. doi: 10.1016/s0169-328x(01)00017-1. [DOI] [PubMed] [Google Scholar]
- Maltzman JS, Carman JA, Monroe JG. Transcriptional regulation of the ICAM-1 gene in antigen receptor- and phorbol ester-stimulated B lymphocytes: role for transcription factor EGR1. J. Exp. Med. 1996;183:1747–1759. doi: 10.1084/jem.183.4.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohba N, Maeda M, Nakagomi S, Muraoka M, Kiyama H. Biphasic expression of activating transcription factor-3 in neurons after cerebral ischemia. Mol. Brain Res. 2003;115:147–156. doi: 10.1016/s0169-328x(03)00181-5. [DOI] [PubMed] [Google Scholar]
- Okada Y, Copeland BR, Mori E, Tung MM, Thomas WS, Del Zoppo GJ. P-selectin and intercellular adhesion molecule-1 expression after focal brain ischemia and reperfusion. Stroke. 1994;25:202–211. doi: 10.1161/01.str.25.1.202. [DOI] [PubMed] [Google Scholar]
- Okada M, Fujita T, Sakaguchi T, Olson KE, Collins T, Stern DM, Yan SF, Pinsky DJ. Extinguishing Egr-1-dependent inflammatory and thrombotic cascades after lung transplantation. FASEB J. 2001;15:2757–2759. doi: 10.1096/fj.01-0490fje. [DOI] [PubMed] [Google Scholar]
- Okada M, Yan SF, Pinsky DJ. Peroxisome proliferator-activated receptor-gamma (PPAR-gamma) activation suppresses ischemic induction of Egr-1 and its inflammatory gene targets. FASEB J. 2002a;16:1861–1868. doi: 10.1096/fj.02-0503com. [DOI] [PubMed] [Google Scholar]
- Okada M, Wang CY, Hwang DW, Sakaguchi T, Olson KE, Yoshikawa Y, Minamoto K, Mazer SP, Yan SF, Pinsky DJ. Transcriptional control of cardiac allograft vasculopathy by early growth response gene-1 (Egr-1) Circ. Res. 2002b;91:135–142. doi: 10.1161/01.res.0000027815.75000.33. [DOI] [PubMed] [Google Scholar]
- Pawlinski R, Pedersen B, Kehrle B, Aird WC, Frank RD, Guha M, Mackman N. Regulation of tissue factor and inflammatory mediators by Egr-1 in a mouse endotoxemia model. Blood. 2003;101:3940–3947. doi: 10.1182/blood-2002-07-2303. [DOI] [PubMed] [Google Scholar]
- Rickhag M, Wieloch T, Gido G, et al. Comprehensive regional and temporal gene expression profiling of the rat brain during the first 24 h after experimental stroke identifies dynamic ischemia-induced gene expression patterns, and reveals a biphasic activation of genes in surviving tissue. J. Neurochem. 2006;96:14–29. doi: 10.1111/j.1471-4159.2005.03508.x. [DOI] [PubMed] [Google Scholar]
- Satriotomo I, Bowen K, Vemuganti R. JAK2 and STAT3 activation contributes to neuronal damage following transient focal cerebral ischemia. J. Neurochem. 2006;98:1353–1368. doi: 10.1111/j.1471-4159.2006.04051.x. [DOI] [PubMed] [Google Scholar]
- Silverman ES, De Sanctis GT, Boyce J, et al. The transcription factor early growth-response factor 1 modulates tumor necrosis factor-α, immunoglobulin E, and airway responsiveness in mice. Am. J. Resp. Crit. Care Med. 2001;163:778–785. doi: 10.1164/ajrccm.163.3.2003123. [DOI] [PubMed] [Google Scholar]
- Soriano MA, Tessier M, Certa U, Gil R. Parallel gene expression monitoring using oligonucleotide probe arrays of multiple transcripts with an animal model of focal ischemia. J. Cereb. Blood Flow Metab. 2000;20:1045–1055. doi: 10.1097/00004647-200007000-00004. [DOI] [PubMed] [Google Scholar]
- Srinivasan R, Mager GM, Ward RM, Mayer J, Svaren J. NAB2 represses transcription by interacting with the CHD4 subunit of the nucleosome remodeling and deacetylase (NuRD) complex. J. Biol. Chem. 2006;22:15129–15137. doi: 10.1074/jbc.M600775200. [DOI] [PubMed] [Google Scholar]
- Stephenson D, Yin T, Smalstig EB, Hsu MA, Panetta J, Little S, Clemens J. Transcription factor nuclear factor-kappa B is activated in neurons after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2000;20:592–603. doi: 10.1097/00004647-200003000-00017. [DOI] [PubMed] [Google Scholar]
- Sundararajan S, Gamboa JL, Victor AN, Wanderi EW, Lust D, Landreth GE. Peroxisome proliferator-activated receptor-γligands reduce inflammation and infarction size in transient focal ischemia. Neuroscience. 2005;130:685–696. doi: 10.1016/j.neuroscience.2004.10.021. [DOI] [PubMed] [Google Scholar]
- Svaren J, Ehrig T, Abdulkadir SA, Ehrengruber MU, Watson MA, Milbrandt J. EGR1 target genes in prostate carcinoma cells identified by microarray analysis. J. Biol. Chem. 2000;275:38524–38531. doi: 10.1074/jbc.M005220200. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semi-automated method for measuring brain infarct volume. J. Cereb. Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Nogawa S, Ito D, Suzuki S, Dembo T, Kosakai A, Fukuchi Y. Activated phosphorylation of cyclic AMP response element binding protein is associated with preservation of striatal neurons after focal cerebral ischemia in the rat. Neuroscience. 2000;100:345–354. doi: 10.1016/s0306-4522(00)00289-x. [DOI] [PubMed] [Google Scholar]
- Tureyen K, Kapadia R, Bowen KK, Satriotomo R, Liang J, Feinstein DL, Vemuganti R. Peroxisome proliferator-activated receptor-gamma agonists induce neuroprotection following transient focal ischemia in normotensive, normoglycemic as well as hypertensive and type-2 diabetic rodents. J. Neurochem. 2007;101:41–56. doi: 10.1111/j.1471-4159.2006.04376.x. [DOI] [PubMed] [Google Scholar]
- Vemuganti R, Bowen KK, Dhodda VK, Song G, Franklin JL, Gavva NR, Dempsey RJ. Gene expression analysis of spontaneously hypertensive rat cerebral cortex following transient focal ischemia. J. Neurochem. 2002;83:1072–1086. doi: 10.1046/j.1471-4159.2002.01208.x. [DOI] [PubMed] [Google Scholar]
- Vemuganti R, Dempsey RJ, Bowen KK. Inhibition of intercellular adhesion molecule-1 protein expression by antisense oligonucleotides is neuroprotective after transient middle cerebral artery occlusion in rat. Stroke. 2004;35:179–184. doi: 10.1161/01.STR.0000106479.53235.3E. [DOI] [PubMed] [Google Scholar]
- Vemuganti R, Kalluri H, Yi JH, Bowen KK, Hazell AS. Gene expression changes in thalamus and inferior colliculus associated with inflammation, cellular stress, metabolism and structural damage in thiamine deficiency. Eur. J. Neurosci. 2006;23:1172–1188. doi: 10.1111/j.1460-9568.2006.04651.x. [DOI] [PubMed] [Google Scholar]
- Victor NA, Wanderi EW, Gamboa J, Zhao X, Aronowski J, Deininger K, Lust WD, Landreth GE, Sundararajan S. Altered PPAR-gamma expression and activation after transient focal ischemia in rats. Eur. J. Neurosci. 2006;24:1653–1663. doi: 10.1111/j.1460-9568.2006.05037.x. [DOI] [PubMed] [Google Scholar]
- Yan SF, Fujita T, Lu J, Okada K, Shan Zou Y, Mackman N, Pinsky DJ, Stern DM. Egr-1, a master switch coordinating upregulation of divergent gene families underlying ischemic stress. Nature Med. 2000;6:1355–1361. doi: 10.1038/82168. [DOI] [PubMed] [Google Scholar]
- Yan YP, Sailor KA, Lang BT, Park SW, Vemuganti R, Dempsey RJ. Monocyte chemoattractant protein-1 plays a critical role in neuroblast migration after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2007;27:1213–1224. doi: 10.1038/sj.jcbfm.9600432. [DOI] [PubMed] [Google Scholar]
- Yi JH, Park SW, Kapadia R, Vemuganti R. Role of transcription factors in mediating post-ischemic cerebral inflammation and brain damage. Neurochem. Int. 2007;50:1014–1027. doi: 10.1016/j.neuint.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zea Longa E, Weinstein PR, Carlson S, Cummnins R. Reversible middle cerebral artery occlusion without craniectomy in rat. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]