

One of the truisms in the medicinal chemistry of antibiotics is that there can never be enough scaffolds and structures, whether of natural or synthetic origin. No matter how successful a new antibiotic is upon clinical introduction, the inevitable selection for resistant microbes will limit its useful lifetime.[1] Resistance to all major classes of antibiotics has been well chronicled, and one of the bacterial pathogens best known for its resistance is Staphylococcus aureus. With high global mortality in human infections, S. aureus has been described as a professional pathogen that has overcome every antibiotic campaign. Methicillin-resistant S. aureus (MRSA) infections are particularly problematic in both community and clinical settings.[2]
The aureus designation (Latin for golden) comes from the pigment staphyloxanthin 1,[3] a glycosylated carotenoid whose biosynthetic pathway proceeds through the same early steps as sterol biosynthesis. A recent anti-MRSA drug discovery effort by Liu and coworkers[4] focuses on one step in particular: the head-to-head condensation of two molecules of farnesyl diphosphate (2) to the C30 hydrocarbon dehydrosqualene 3 by the S. aureus enzyme CrtM. This transformation parallels the eukaryotic pathway to cholesterol, including the intermediacy of the cyclopropane-containing molecule presqualene diphosphate 4.[5] However, a distinction occurs in the last step of catalysis: the eukaryotic squalene synthase[6] uses an NADPH-dependent reduction to quench an allylic cation and yield squalene 5 while the staphylococcal enzyme likely abstracts a proton to yield the central olefin in dehydrosqualene. Subsequent enzymatic dehydrogenations create the conjugated chromophore responsible for the golden color in the end product staphyloxanthin.[3]
Staphyloxanthin functions as a virulence factor for S. aureus. The conjugated polyene system is thought to quench reactive oxygen species such as superoxide, peroxide, and hypochlorite that are produced by the white cells of vertebrate hosts as they try to kill the staphylococci during the inflammatory response.[7] Thus, while staphyloxanthin biosynthesis does not appear to confer a growth advantage on S. aureus, it increases survival during vertebrate infections and therefore qualifies as a virulence factor.
In the search for the next generation of antibiotics, recent efforts have targeted virulence rather than essential gene functions.[8] A team of investigators that includes structural biologists, chemists, and microbiologists have recently discovered that inhibition of the S. aureus dehydrosqualene synthase reduces bacterial survival during infections, offering a proof-of-principle for such a virulence-targeted approach .[4]
The effort was based on determining of the x-ray structure of S. aureus CrtM, the dehydrosqualene synthase, after its heterologous expression in E. coli and subsequent purification and crystallization. Since prenyltransferases are involved in terpene and sterol biosynthesis and the posttranslational S-prenylation of proteins, many of these enzymes have been studied in mechanistic detail. The eukaryotic enzyme squalene synthase, a potential drug target for cholesterol-lowering therapy, has been the object of several medicinal chemistry campaigns to identify potent and specific small molecule inhibitors. Based on these efforts, a variety of known inhibitors could be resynthesized and tested as inhibitory ligands for S. aureus CrtM. One of these molecules, a sulfur-containing farnesyl analog 6, could be co-crystallized with CrtM. The x-ray structure of the complex shows two molecules of 6 in the active site, likely defining the orientation of the prenyl side chains of the natural CrtM intermediate presqualene diphosphate.[4]
Evaluation of several inhibitors, including other farnesyl diphosphate analogs and amine-containing hydrocarbons that had previously been prepared as mimics of cationic intermediates in the squalene/dehydrosqualene synthase reaction, led to the observation that phosphonosulfonate scaffolds are submicromolar inhibitors of CrtM and could also be co-crystallized. The biarylether phosphonosulfonate 7 was chosen for further evaluation for several reasons: it had a Ki value of 1.5 nM against CrtM, it inhibited staphyloxanthin production when administered to live S. aureus (IC50 = 110 nM), and it had already progressed through preclinical toxiciology and into human clinical studies as a cholesterol-lowering agent without significant adverse effects. Liu and coworkers found that 7 had no effect on the growth of three human cell lines in serum, a cholesterol-rich medium. While 7 caused S. aureus colonies to lose their golden color, it did not inhibit the growth of S. aureus per se, since staphyloxanthin is not essential for growth.[4]
The subsequent evaluation of CrtM as a virulence factor involved a model of systemic infection in mice. When 108 colony-forming units of wild-type S. aureus or a crtM knockout were inoculated intraperitoneally (i.p.), the crtM-deficient strain was successfully cleared 72 hours later while the wild-type strain, as expected, was far more resistant to the oxidant-based defenses of the mice. With this result as a backdrop, mice were then treated with 7 for four days and infected i.p. the second day, and bacteria remaining in the kidney were assessed at the end. The treatment worked successfully and the authors conclude that “on average this result corresponds to a 98% decrease in surviving bacteria in the treatment group”.[4]
What lessons should be taken from this report? First, the principle that inhibiting a virulence factor in a bacterial pathogen can have a dramatic effect on bacterial survival in a vertebrate host has been proven. (However, since S. aureus is famously difficult to defeat, it will be instructive to see whether repeated passaging of the staphyloxanthin-deficient S. aureus strain leads to compensatory mutations that restore evasion of oxidative host defenses.) A second lesson is that prior medicinal chemistry efforts on mammalian squalene synthases had great utility in this antibiotic drug development program. These efforts have produced a molecular inventory of inhibitors that served as valuable starting points for the evaluation of selectivity for the bacterial enzyme over the host enzyme, ability to penetrate into S. aureus cells, and lack of toxicity in mammalian cells. The definition of a new target is only the beginning of an antibacterial development program, but the existence of compounds that have already been tested in humans lends much confidence to the effort.
This story raises the broader question of the utility and advisability of narrow-spectrum vs. broad-spectrum antibiotics. Inhibitors of staphyloxanthin biosynthesis would likely be restricted to treating S. aureus; is this pathogen an important enough target to warrant the development of antibiotics specifically tailored to it? Probably - given the high mortality of S. aureus human infections, three of the antibiotics recently approved by the FDA (quinupristin/dalfopristin, linezolid, and daptomycin) share MRSA as a primary target.[9] In addition, combination therapies may become more prevalent in the face of infections by multidrug-resistant bacteria, so a staphyloxanthin biosynthesis inhibitor might become a useful agent in such an antibacterial cocktail.
The recommendations of a U.S. National Research Council committee in 2006 included the development of narrow-spectrum antibiotics to minimize the perturbation of normal microbial flora and to minimize resistance development.[10] While ecologically sound, such a discovery and development strategy will have its own challenges, including real-time diagnostic tests for rapid pathogen identification and a change in mindset about the acceptable market size for a new antibacterial. A breakthrough antibiotic targeted against virulence would advance such a debate.
Figure 1.
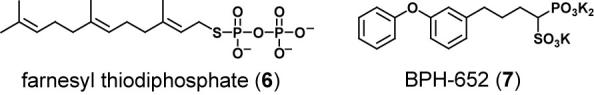
Chemical structures of CrtM inhibitors.
Scheme 1.

The role of CrtM in staphyloxanthin biosynthesis. The S. aureus squalene cyclase CrtM catalyzes the formation of dehydrosqualene from two molecules of farnesyl diphosphate. Dehydrosqualene is subsequently converted to staphyloxanthin.
Footnotes
This work was supported by NIH grants GM20011, GM49338, and AI 47238.
References
- [1].Walsh CT. Antibiotics: Actions, Origins, Resistance. ASM Press; Washington: 2003. [Google Scholar]
- [2].Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK. JAMA. 2007;298:1763. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- [3].Pelz A, Wieland KP, Putzbach K, Hentschel P, Albert K, Gotz F. J. Biol. Chem. 2005;280:32493. doi: 10.1074/jbc.M505070200. [DOI] [PubMed] [Google Scholar]
- [4].Liu CI, Liu GY, Song Y, Yin F, Hensler ME, Jeng WY, Nizet V, Wang AH, Oldfield E. Science. 2008;319:1391. doi: 10.1126/science.1153018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Edwards PA, Ericsson J. Annu. Rev. Biochem. 1999;68:157. doi: 10.1146/annurev.biochem.68.1.157. [DOI] [PubMed] [Google Scholar]
- [6].Pandit J, Danley DE, Schulte GK, Mazzalupo S, Pauly TA, Hayward CM, Hamanaka ES, Thompson JF, Harwood HJ., Jr. J. Biol. Chem. 2000;275:30610. doi: 10.1074/jbc.M004132200. [DOI] [PubMed] [Google Scholar]
- [7].Clauditz A, Resch A, Wieland KP, Peschel A, Gotz F. Infect. Immun. 2006;74:4950. doi: 10.1128/IAI.00204-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hung DT, Shakhnovich EA, Pierson E, Mekalanos JJ. Science. 2005;310:670. doi: 10.1126/science.1116739. [DOI] [PubMed] [Google Scholar]
- [9].Drew RH. Pharmacotherapy. 2007;27:227. doi: 10.1592/phco.27.2.227. [DOI] [PubMed] [Google Scholar]
- [10].National Research Council (U.S.) Treating infectious diseases in a microbial world: report of two workshops on novel antimicrobial therapeutics. National Academies Press; Washington, DC: 2006. Committee on New Directions in the Study of Antimicrobial Therapeutics: New Classes of Antimicrobials., National Research Council (U.S.). Committee on New Directions in the Study of Antimicrobial Therapeutics: Immunomodulation. [PubMed] [Google Scholar]