

Abstract
The need to both understand and minimize the side effects of brain irradiation is heightened by the ever-increasing number of patients with brain metastases that require treatment with whole brain irradiation (WBI); some 200,000 cancer patients/year receive partial or WBI. At the present time, there are no successful treatments for radiation-induced brain injury, nor are there any known effective preventive strategies. Data support a role for chronic oxidative stress in radiation-induced late effects. However, the pathogenic mechanism(s) involved remains unknown. One candidate source of reactive oxygen species (ROS) is nicotinamide adenosine dinucleotide phosphate (NADPH) oxidase, which converts molecular oxygen (O2) to the superoxide anion (O2.−) upon activation. We hypothesize that brain irradiation leads to activation of NADPH oxidase. We report that irradiating rat brain microvascular endothelial cells in vitro leads to increased i] intracellular ROS generation, ii] activation of the transcription factor NFκB, iii] expression of ICAM-1 and PAI-1, and iv] expression of Nox4, p22phox, and p47phox. Pharmacologic and genetic inhibition of NADPH oxidase blocked the radiation-mediated upregulation of intracellular ROS, activation of NFκB, and upregulation of ICAM-1 and PAI-1. These results suggest that activation of NADPH oxidase may play a role in radiation-induced oxidative stress.
Keywords: ionizing radiation, radiation-induced brain injury, NADPH oxidase, oxidative stress
Introduction
In 2008, 1,437,180 new cancer cases will be diagnosed in the U.S. [1] with 10–30% of this population likely to develop brain metastases [2]. Of the treatment modalities available, whole brain irradiation (WBI) remains the standard treatment for most patients [3]. Currently, approximately 200,000 patients receive WBI each year [4], and over half of these patients will survive long enough to develop radiation-induced brain injury, including cognitive impairment that can markedly compromise their quality of life (QOL) [5].
Brain irradiation produces variable toxic effects, which are classified according to their time of expression, i.e., acute, early delayed and late delayed reactions [6–8]. Acute injury occurs days to weeks after irradiation, while early delayed injury occurs up to 6 months after irradiation. Late delayed injury can occur 6 months after radiation treatment but is known to occur several years after radiation exposure. Although acute and early delayed injuries are severe, they are normally reversible or managed effectively with steroid therapy. In contrast, late delayed injury, clinically expressed as chronic, progressive cognitive impairment in which patients may exhibit personality changes, fatigue, and impairment of short-term memory [8] that can lead to dementia and death [9], is currently untreatable. Presently, there are no effective therapies for radiation-induced brain injury. Therefore, understanding the molecular mechanisms of radiation-induced brain injury may help to develop strategies to ameliorate or prevent radiation-induced brain injury.
A growing body of evidence suggests that the development and progression of radiation-induced late effects involve, at least in part, an acute and chronic oxidative stress [10]. Irradiating late responding normal tissues leads to chronic increases in reactive oxygen species/reactive nitrogen oxide species (ROS/RNOS) that serve as intracellular signaling species to alter cell function/phenotype, leading to chronic inflammation and organ dysfunction. The brain is particularly sensitive to oxidative stress [11] due to its 1) high oxygen consumption [12], 2) content of oxidizable unsaturated lipids [12] and free iron [13], 3) limited capacity to perform anaerobic glycolysis [14], and 4) low levels of antioxidant defenses [15]. Indeed, recent data suggest a primary role for chronic oxidative stress/inflammation and ROS/RNOS in radiation-induced brain injury [16].
Cells can exhibit persistent and prolonged generation of ROS/RNOS that can last from minutes to days [17–18] with mitochondria as the primary site of increased ROS/RNOS generation [19]. However, in vascular cells, NADPH oxidases appear to be the primary physiological ROS producers [20]. Indeed, NADPH oxidase has been implicated in the radiation-induced bystander effect via persistent ROS production [21]. NADPH oxidase is a multi-subunit enzyme localized to cell membranes and consists of membrane-bound components (gp91phox and p22phox) and cytosolic components (p47phox, p67phox, and Rac1) that translocate to the membrane upon activation. Once the multi-subunit complex is formed, O2•− production is catalyzed by the transfer of a single electron from NADPH to molecular oxygen via the following reaction: NADPH + 2O2 → NADP+ + H+ + 2O2•− [22]. The O2•− anion is rapidly dismutated by superoxide dismutase (SOD) to produce H2O2, which itself is a signaling molecule [23]. H2O2 can also activate NADPH oxidase to cause oxidant injury via production of additional oxidant species, thus implicating the inappropriate activation of NADPH oxidase in chronic oxidative stress [22]. ROS derived from NADPH oxidase have been implicated in a variety of chronic CNS disorders, including Parkinson’s disease, Alzheimer’s disease, ischemic stroke, and HIV dementia [24].
The generation of ROS has been long thought of as a pathological process capable of initiating oxidative events that lead to cellular damage. However, physiological functions of ROS have now been identified, including host defense [25] and the modification of gene expression through intracellular redox signaling [26]. Redox-modulated genes involved in the radiation response include the transcription factor, nuclear factor kappaB (NFκB) [27], the endothelial Ig superfamily surface receptor, intercellular adhesion molecule-1 (ICAM-1) [28], and the serine protease inhibitor, plasminogen activator inhibitor-1 (PAI-1) [29]. We hypothesized that ionizing radiation can induce an oxidative stress via activation of NADPH oxidase. Since damage to the microvascular system is a primary event that may contribute to the late effects observed after radiation treatment [30–31], we chose to assess radiation-induced effects in rat brain microvascular endothelial cells (RBMEC). Results presented here indicate that pharmacologic and genetic inhibition of NADPH oxidase can abrogate radiation-induced oxidative stress as measured by increased intracellular ROS generation and activation/upregulation of redox-regulated genes. These data suggest a putative role for NADPH oxidase in the development and progression of radiation-induced oxidative stress and highlight the potential for development of novel antioxidant-based therapies that might preserve the cognitive function of brain cancer patients treated with WBI.
Experimental procedures
Reagents
Diphenyleneiodonium (DPI), apocynin, dichlorofluorescein diacetate (DCFH-DA), N-acetylcysteine (NAC), and Antimycin A (Anti A) were obtained from Sigma (St. Louis, MO). Dihydroethidium (DHE) and carboxy-DCFDA (C369) were obtained from Molecular Probes (Carlsbad, CA). p22phox and p47phox antibodies were obtained from Santa Cruz Biotechnology, Inc (Santa Cruz, CA). ICAM-1 antibodies were obtained from R&D Systems (Minneapolis, MN). PAI-1 antibodies were obtained from American Diagnostica (Stamford, CT). β-actin antibodies were obtained from Sigma. AdN17Rac1 was obtained from the Vector and Virus Vector Core of the University of Iowa.
Cell culture
Immortalized rat brain microvascular endothelial cells (RBMEC-GP8.3) were obtained from Bela Kis, PhD (Department of Physiology and Pharmacology, WFUSM), cultured in α-MEM/Ham’s F10 (Gibco; Carlsbad, CA), 10% FBS (Gibco), 50 IU/mL penicillin (Sigma), 50µg/mL streptomycin (Sigma), 200mM L-glutamine (Sigma), and 250µg/mL Geneticin (Gibco) and maintained in a humidified atmosphere containing 5% CO2 at 37°C.
Irradiation of brain cells
Prior to irradiation, cells were grown to 80–90% confluence and placed in 1% serum-containing media for 24 hours to achieve cell synchronization. Cells were then irradiated with a range of single doses (1–10 Gy) of γ rays with a dose rate of 3.91 Gy/min using a 137Cs irradiator. After irradiation, cells were maintained in 1% serum-containing media in a 5% CO2 atmosphere at 37°C for up to 24 h postirradiation.
Intracellular ROS measurement by dicholofluorescein fluorescence
Cells were preloaded with the fluorescence probe DCFH-DA (10 µM), irradiated with 1–10 Gy γ-rays, and intracellular ROS generation was measured 1 h postirradiation. The antioxidant, NAC (5mM), was used as a positive control.
O2•− measurement by dihydroethidium fluorescence
The effects of radiation on NADPH oxidase activity were determined using DHE to measure O2•− production. DHE is freely permeable to cells and undergoes a two-electron oxidation in the presence of O2•− to produce DNA-intercalating ethidium bromide (EtBr). Upon excitation at 488nm, EtBr fluoresces at 610nm. Cells were grown to 90% confluence, irradiated with 10 Gy γ-rays, and then labeled with 10 µM DHE for 40 min. Cells were washed and observed for O2•− generation 1 h postirradiation with fluorescence microscopy (Olympus IX70). Antimycin A was used as a positive control to block the mitochondrial electron transport chain.
Real-time PCR
Total RNA was isolated by the acid guanidinium thiocyanate-phenol-chloroform extraction method using TRIzol reagent (Life Technologies, Inc, Gaithersburg, MD). The RNA concentration was determined spectrophotometrically at 260 nm. The primer sequences for Nox4, p22phox, and β-actin were predesigned by Invitrogen (Carlsbad, CA). Real-time PCR was performed in a reaction volume of 25 µL, including 2 µL RT product as a template, 12.5 µL of Platinum® Quantitative PCR SuperMix-UDG w/ROX (Invitrogen), 10 µM of fluorophore-labeled primer, and 10 µM unlabeled primer. The PCR amplifications were performed with 40 cycles of 95°C for 15 s and 60°C for 30 sec using ABI 7000 (Applied Biosystems, Foster City, CA). Average Ct values of Nox4 and p22phox from triplicate analyses were normalized from average Ct values of β-actin. The ratios of expression levels of Nox4 and p22phox between irradiated and control groups at each time point were calculated as 2−ΔΔCt.
Electrophoretic mobility shift assay (EMSA)
Nuclear protein extraction was performed 1–8 h postirradiation. Cells were scraped and homogenized in 500 µL ice-cold buffer (10 mM HEPES (pH 7.9), 10 mM KCl, 1 mM MgCl, 1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 0.5 mM PMSF, 2 µg/mL aprotinin, 2 µg/mL leupeptin, and 1 mM Na3VO4), incubated on ice for 20 min, and centrifuged for 5 min at 14,000g at 4°C. The nuclear pellet was then resuspended in 15 µL of ice-cold extraction buffer (20 mM HEPES (pH 7.9), 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 0.5 mM PMSF, 2 µg/mL aprotinin, 2 µg /mL leupeptin, and 1 mM Na3VO4), incubated on ice for 30 min, and centrifuged at 5000g for 10 min at 4°C. The supernatant was collected and frozen at −80°C. Protein concentrations were determined using a Bio-Rad DC protein assay kit. NFκB DNA-binding activity was determined using Gel Shift Assay Systems (Promega, Madison, WI) according to the manufacturer's instructions. Briefly, 1.75 pmol of NFκB oligonucleotide was labeled with γ-32P-ATP (Perkin Elmer, Boston, MA) by incubation with 10 U of T4 polynucleotide kinase at 37°C for 10 min in a kinase buffer. The reaction was stopped by adding 1 µL of 0.5 M EDTA. Ten micrograms of nuclear protein were then preincubated in 1x DNA binding buffer for 10 min at room temperature followed by addition of the 32P-labeled oligonucleotide probe; incubation was continued for 20 min. For the specific competitor and supershift controls, 1.75 pmol of unlabelled NFκB oligonucleotide or 1 µL anti-p65 antibody (Cell Signaling Technology, Danvers, MA) was added to the reaction prior to the addition of radioactively labeled probe, respectively. The resulting DNA-protein complexes were electrophoresed on a 6% non-reducing polyacrylamide gel at 200 V in 0.5x Tris-borate-EDTA. The gel was dried and autoradiographed. Films were scanned, and densitometry was performed to quantify intensity of signal (Scion Image, Frederick, Maryland)
Northern blot analysis
Total RNA was isolated using TRIzol reagent. p22phox, PAI-1, and EF1α cDNA probes were labeled with α32P-dCTP (NEN) by the random primer extension method using a Random Primer DNA labeling kit (Boehringer Mannheim Biochemica, Indianapolis, IN). Autoradiographs were quantified by densitometry using Scion Image.
Western blot analysis
Cells were lysed in 50 mM Tris-HCl buffer, pH 7.0, containing 1% Triton X-100, 1 mM EDTA, 150 mM NaCl, 1 mM PMSF, 1 µg/mL aprotinin, 1 µg/mL leupeptin, 1 mM Na3VO4 and 1 mM NaF and stored in aliquots at −80°C. Protein concentrations were determined using a Bio-Rad DC protein assay kit. Cell lysates were incubated with an equal volume of sample buffer containing 62.5 mM Tris/HCl (pH 6.8), 10% glycerol, 2% SDS, 5% β-mercaptoethanol, and two or three drops of saturated bromophenol solution, denatured by boiling, and separated in a 10% polyacrylamide mini-gel at a constant voltage of 120 V for 2 h. The proteins were transferred by electrophoresis at 30 V for 16 h to polyvinylidene difluoride (PVDF) membranes (Perkin Elmer, Waltham, MA). The membranes were blocked for 1 h at room temperature in 5% (wt/vol) nonfat dry milk in TBS-T containing 20 mM Tris-HCl, pH 7.0, 137 mM NaCl, and 0.05% (vol/vol) Tween-20. The membrane was then incubated with commercially available primary antibodies (1:10,000 anti-ICAM-1 mouse monoclonal IgG, 1:1000 anti-PAI-1 rabbit polyclonal IgG, 1:500 anti-p22phox rabbit polyclonal IgG, and 1:1000 anti-p47phox mouse monoclonal IgG) overnight, washed, incubated with the secondary antibody conjugated to horseradish peroxidase (anti-rabbit/anti-mouse IgG, 1:10,000, Sigma, St. Louis, MO) for 1 h, and washed. Detection was accomplished using an enhanced chemiluminescence ECL plus kit (Amersham Pharmacia Biotech, Piscataway, NJ). Finally, bands were visualized as a function of optical density normalized to the housekeeping gene, β-actin.
Transfection of p22phox antisense oligonucleotides
Phosphorothioate-modified oligonucleotides were produced by the Wake Forest University Comprehensive Cancer Center DNA Sequencing Core Lab. The sequences of p22phox antisense and sense oligonucleotides were GATCTGCCCCATGGTGAGGACC and GGTCCTCACCATGGGGCAGATC, respectively. The cells were transfected with oligonucleotides using Effectene transfection reagent (Qiagen, Valencia, CA) and incubated for 24 h prior to collection.
Statistical analysis
All data are presented as mean ± SD. Significance was determined using Student’s t test or Repeated Measures ANOVA followed by Tukey’s or Dunnett’s Multiple Comparison Test as appropriate. A p-value <0.05 was considered significant.
Results
NADPH oxidase contributes to radiation-induced oxidative stress
RBMEC were preincubated with the nonfluorescent molecule, DCFH-DA, which is converted to the fluorescent DCF probe upon oxidation by ROS. As shown in Figure 1, irradiating RBMEC led to a significant dose-dependent increase in intracellular ROS generation (*p<0.05 and **p<0.01 vs. sham-irradiated control; Fig. 1A).
Fig. 1. NADPH oxidase inhibition ameliorates radiation-induced intracellular ROS generation in RBMEC.
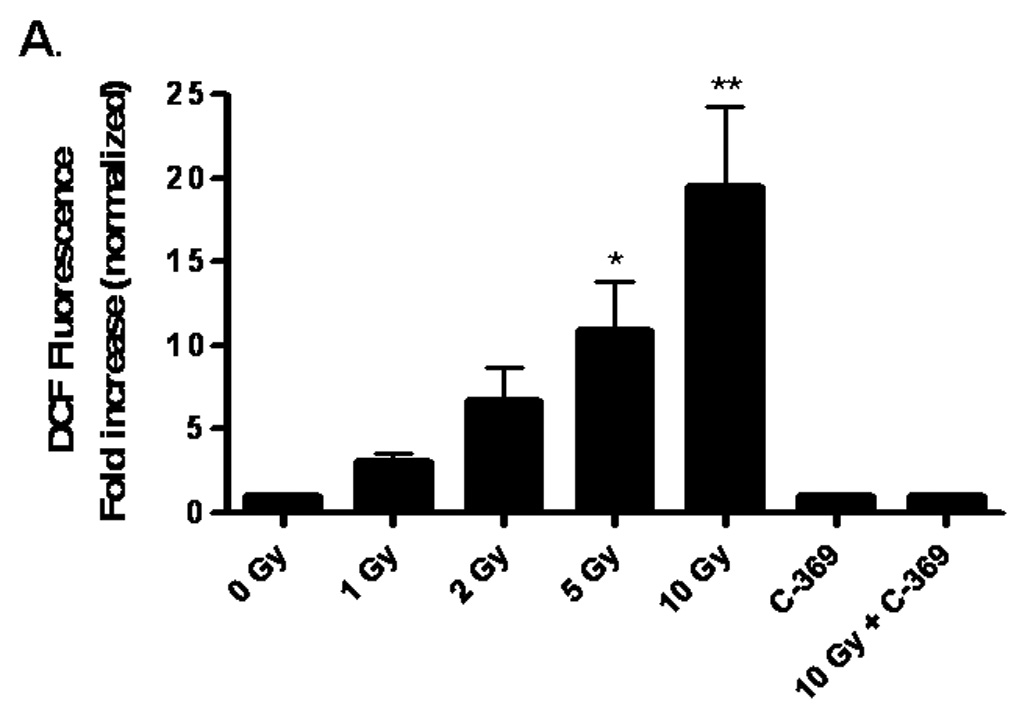
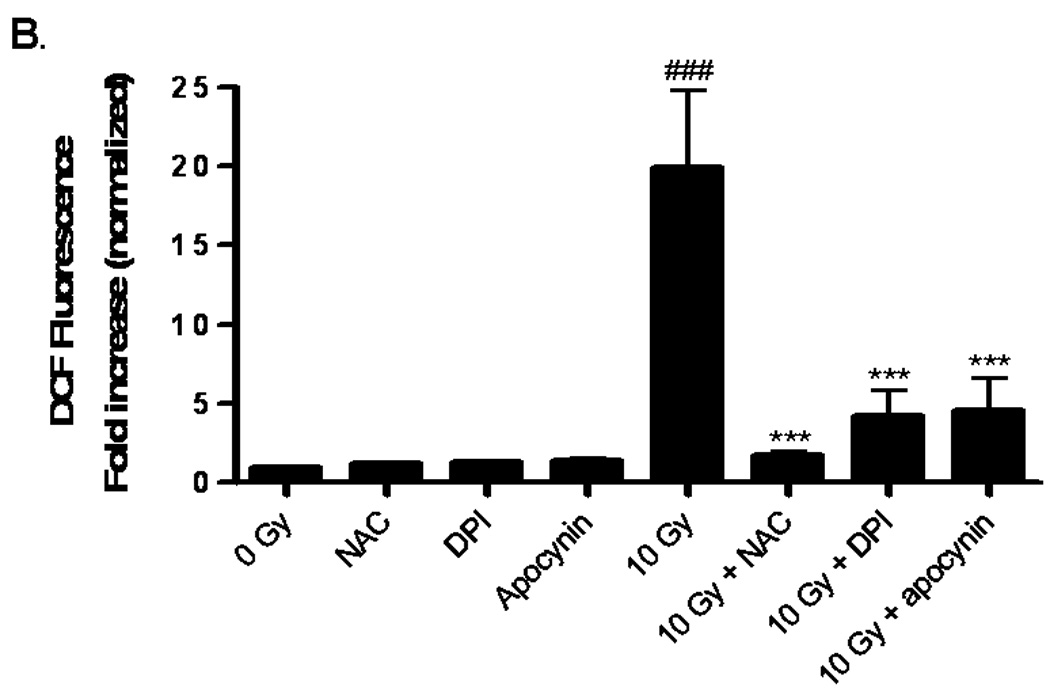
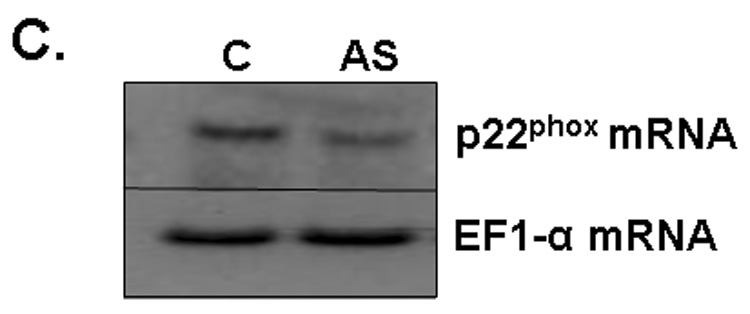
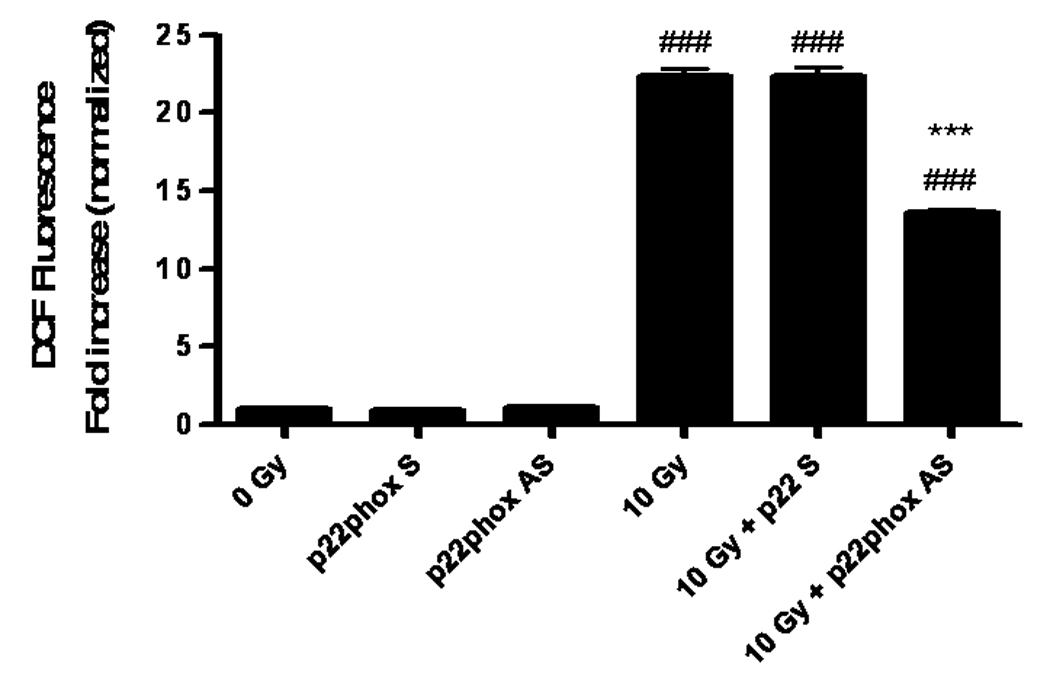
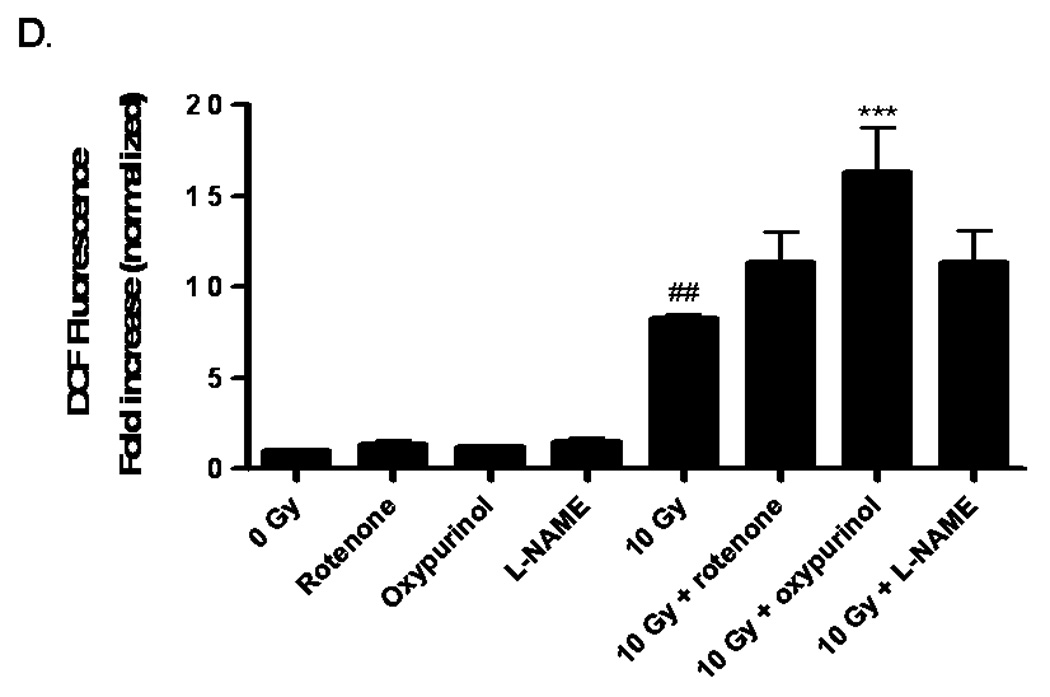
A) RBMEC were preloaded with DCFH-DA or C-369 (non-oxidizable control probe) for 30 min. Cells were then irradiated with 1–10 Gy γ-rays, and the relative fluorescence was measured 1 h postirradiation. Mean ± SD; n = 3; *p<0.05 and **p<0.01 vs. sham-irradiated control. B) RBMEC were pretreated with 5 mM NAC (30 min), 10 µM DPI, or 10 µM apocynin (1 h) and relative fluorescence was measured 1 h postirradiation. Mean ± SD; n=3; ### p< 0.001 vs. sham-irradiated control and ***p<0.001 vs. 10 Gy. C) RBMEC were transfected with 0.8 µg p22phox antisense oligonucleotides. Left panel: 24 h post-transfection, total RNA was collected and analyzed for p22phox gene expression by Northern blot. Right panel: relative fluorescence was measured 1 h postirradiation. Mean ± SD; n = 3; ### p< 0.001 vs. sham-irradiated control and ***p<0.001 vs. 10 Gy. D) RBMEC were pretreated with 2µM rotenone, 50 µM oxypurinol, or 1 mM L-NAME for 1 h prior to irradiation, and the relative fluorescence was measured 1 h postirradiation. Mean ± SD; n = 3; ## p< 0.01 vs. sham-irradiated control and ***p<0.001 vs. 10 Gy.
To explore the role of NADPH oxidase in this radiation-induced intracellular ROS generation, RBMEC were pretreated with two structurally and functionally distinct inhibitors of NADPH oxidase. DPI is a flavoprotein inhibitor that causes an irreversible phenylation of the flavin and heme moieties when reduced to its radical form, thus preventing the critical electron transport step required for NADPH oxidase activation [32]. Since DPI is capable of inhibiting any flavin-containing enzyme, apocynin was included in these studies as a specific inhibitor of NADPH oxidase. Apocynin is a methoxy-substituted catechol that interferes with the serine phosphorylation [33] and intracellular translocation of the cytosolic p47phox and p67phox subunits to the membrane [34]. Treatment with either pharmacologic inhibitor of NADPH oxidase prior to irradiation significantly abrogated the radiation-induced intracellular ROS generation (***p<0.001 vs. 10 Gy; Fig. 1B). The antioxidant, NAC, was used as a positive control. To address specificity concerns associated with the use of pharmacologic inhibitors, we also inhibited NADPH oxidase activity genetically using phosphorothioate-modified antisense oligonucleotides targeted against the p22phox subunit. After transfecting RBMEC with p22phox antisense oligonucleotides, we observed that knocking down p22phox gene expression (Fig. 1C, left panel) resulted in a significant decrease in the radiation-induced intracellular ROS generation (***p<0.001 vs. 10 Gy; Fig. 1C, right panel). To rule out other sources of O2•−, RBMEC were also pretreated with rotenone (mitochondrial e− transport inhibitor), oxypurinol (xanthine oxidase inhibitor), or L-NAME (nitric oxide synthase inhibitor) prior to irradiation. Each inhibitor failed to abrogate radiation-induced intracellular ROS generation (Fig. 1D). Together, these data suggest that NADPH oxidase is the primary source of intracellular ROS generation induced by ionizing radiation.
The DCF assay is a measure of generalized oxidative stress rather than a direct assay of any one particular ROS species. Therefore, an assay specific for detection of NADPH oxidase activity in terms of O2•− generation was utilized. DHE is a fluorescent probe specifically oxidized by O2•−, and its fluorescence was used to visualize the effect of radiation on intracellular O2•− generation. Ionizing radiation induced O2•− production 1 h postirradiation as demonstrated by an increase in ethidium bromide fluorescence; pretreating RBMEC with apocynin and transfecting with p22phox antisense oligonucleotides ameliorated the radiation-induced increase (Fig. 2A and 2B, respectively). Taken together, these results suggest that NADPH oxidase contributes to radiation-induced oxidative stress.
Fig. 2. NADPH oxidase inhibition abrogates radiation-induced intracellular O2.− generation in RBMEC.
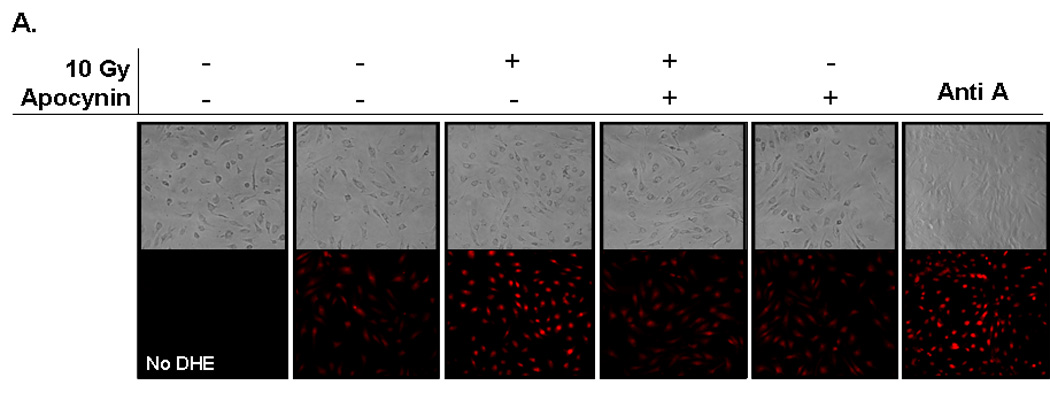
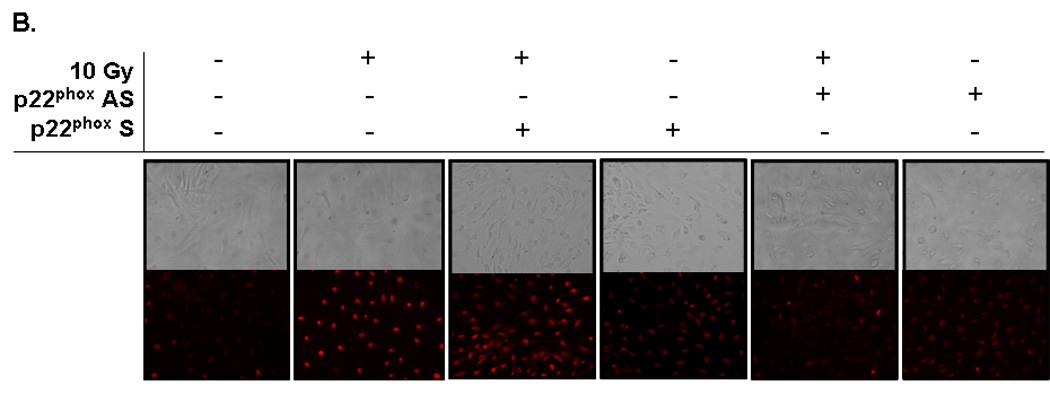
A) RBMEC were pretreated with 10 µM apocynin for 1 h and then irradiated with 10 Gy γ-rays. At 1 h postirradiation, cells were loaded with 10 µM DHE and observed for intracellular O2.− generation by confocal microscopy. Antimycin A was used as a positive control. N = 3. B) RBMEC were transfected with 2 µg p22phox sense or antisense oligonucleotides for 24 h, irradiated with 10 Gy γ-rays, and observed for intracellular O2.− generation 1 h postirradiation. N = 3.
To test the functional consequence of radiation-induced NADPH oxidase activation and its role in oxidative stress, we initially assessed activation of the redox-regulated transcription factor, NFκB. NFκB transcription complexes can be found as dimers sequestered in the cytoplasm by the inhibitor, IκB [35]. Upon stimulation, IκB is phosphorylated and degraded, thus exposing nuclear localization signals within the dimer and allowing NFκB translocation into the nucleus, where it binds consensus sequence sites within the promoter regions of target genes. Our results indicate that radiation significantly increased NFκB DNA-binding activity 4 h postirradiation (##p< 0.01 vs. sham-irradiated control; Fig. 3A and 3B); pretreatment with apocynin prevented the radiation-induced increase (**p<0.01 vs. 10 Gy; Fig. 3B). Genetic inhibition of NADPH oxidase also prevented the radiation-induced increase in NFκB DNA-binding activity (Fig. 3C).
Fig. 3. NADPH oxidase inhibition prevents radiation-induced NFκB DNA binding activity in RBMEC.
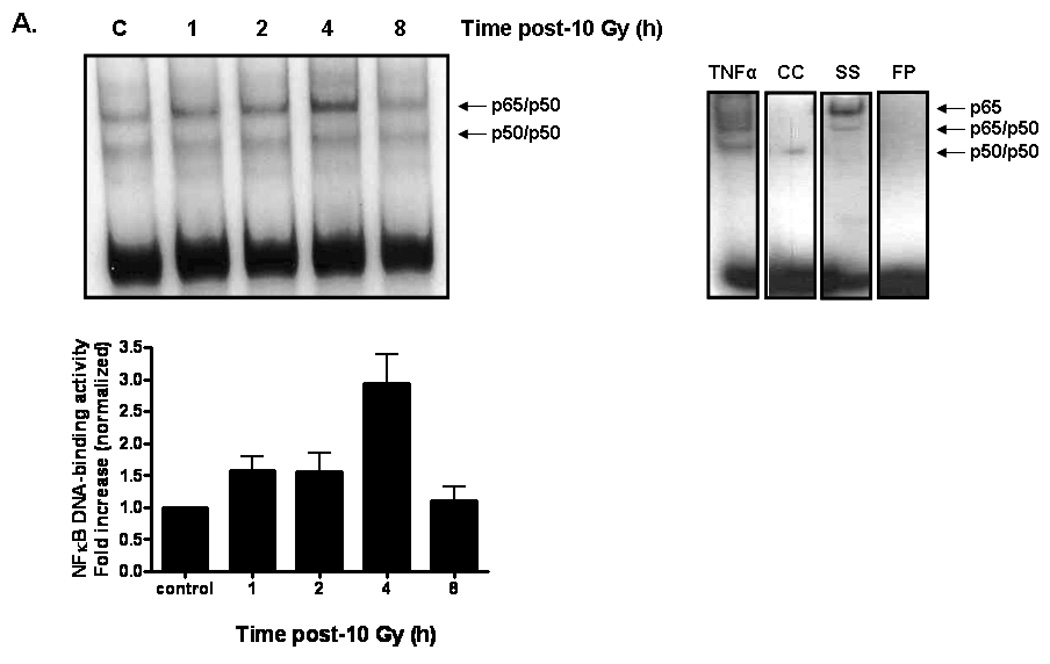
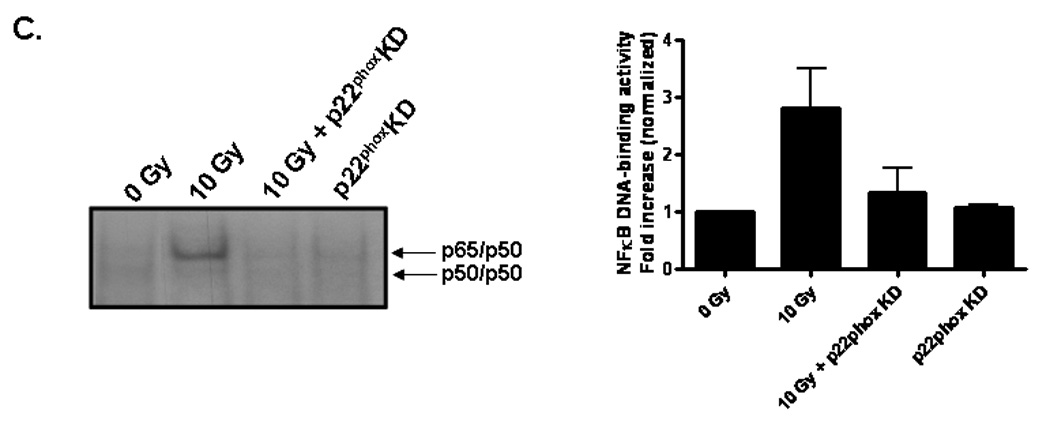
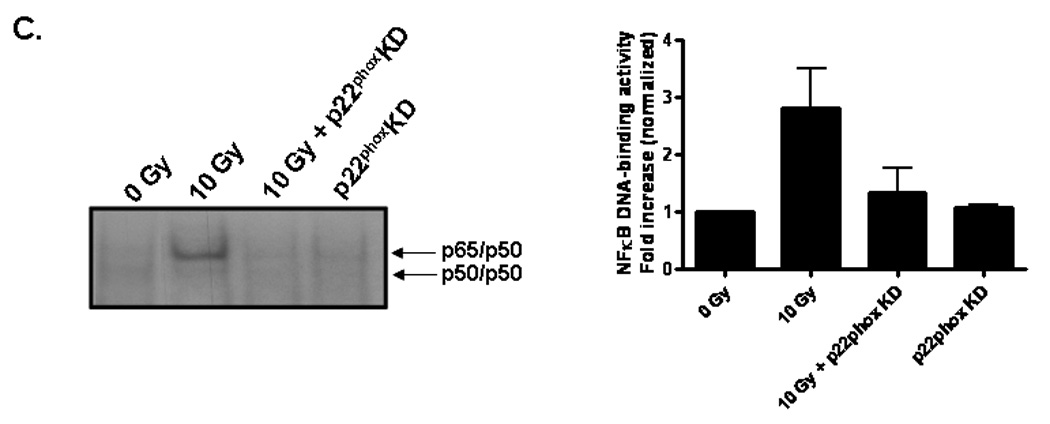
A) RBMEC were irradiated with 10 Gy γ-rays, and nuclear protein was extracted 1–8 h postirradiation and incubated with γ32P-labeled NFκB consensus oligonucleotides. NFκB DNA binding activity was measured using EMSA. Densitometric analysis is shown. N = 2. Controls are also shown. Tumor necrosis factor α (TNFα), a potent inducer of NFκB activation, was used as a positive control. The cold competitor (CC) served as a specificity control, in which excess unlabelled probe is added to the reaction and competes with the binding of the radioactively labeled probe. The supershift (SS) also served as a specificity control, in which anti-p65 antibody is added to the reaction, with its binding to the p65 subunit resulting in a band shift. The free probe (FP) contained no nuclear protein, serving as a negative control. B) RBMEC were pretreated with 10 µM apocynin for 1 h, irradiated with 10 Gy γ-rays, and nuclear protein was extracted 4 h postirradiation. Densitometric analysis is shown. Mean ± SD; n = 3; ##p< 0.01 vs. sham-irradiated control and **p<0.01 vs. 10 Gy. C) RBMEC were transfected with 2 µg p22phox antisense oligonucleotides, irradiated with 10 Gy γ-rays, and nuclear protein was extracted 4 h postirradiation. N = 2.
Brain irradiation has been shown to induce expression of redox-regulated genes, including ICAM-1 [28], an endothelial Ig superfamily surface receptor that plays a key role in leukocyte adhesion and migration. Irradiation led to a significant increase in ICAM-1 protein levels in RBMEC (*p <0.05 vs sham-irradiated control; Fig. 4A). ICAM-1 is upregulated by radiation-induced ROS in an NFκB-dependent manner [36]. To confirm that ICAM-1 is regulated in NFκB-dependent manner in our in vitro model, we pretreated RBMEC with BayII-7082, an inhibitor of NFκB, and assessed its effect on ICAM-1 protein levels. Indeed, NFκB inhibition abrogated the radiation-induced increase in ICAM-1 protein levels (Fig. 4B). As a control, we also assessed NFκB DNA-binding activity upon BayII-7082 treatment and confirmed its activity as an inhibitor of NFκB DNA-binding activity (Fig. 4C) via inhibition of IκB-α phosphorylation (Fig. 4D).
Fig. 4. Radiation-induced ICAM-1 upregulation is NFκB- and NADPH oxidase isoform-dependent.
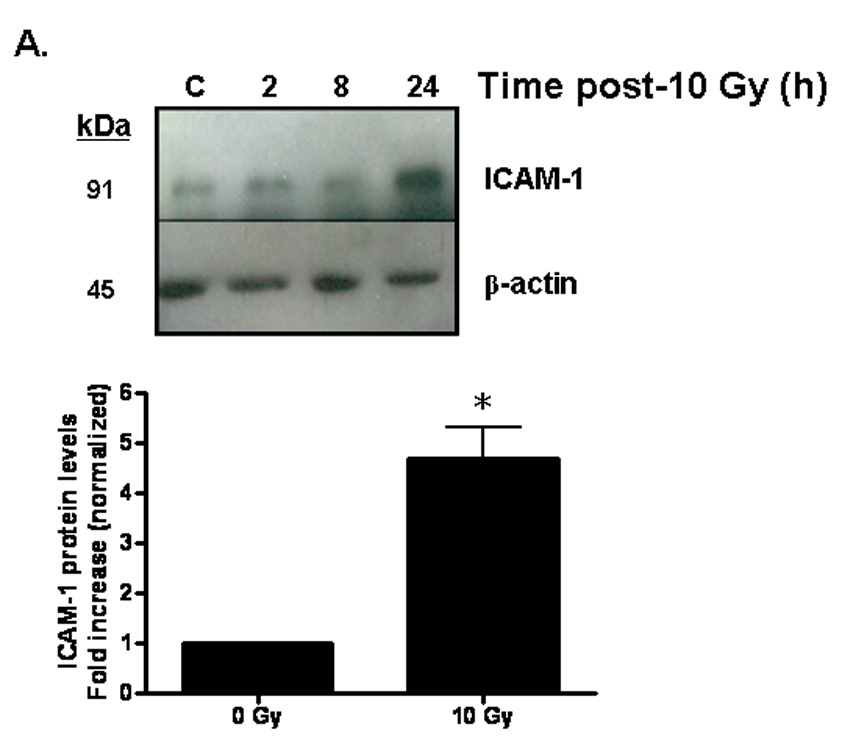
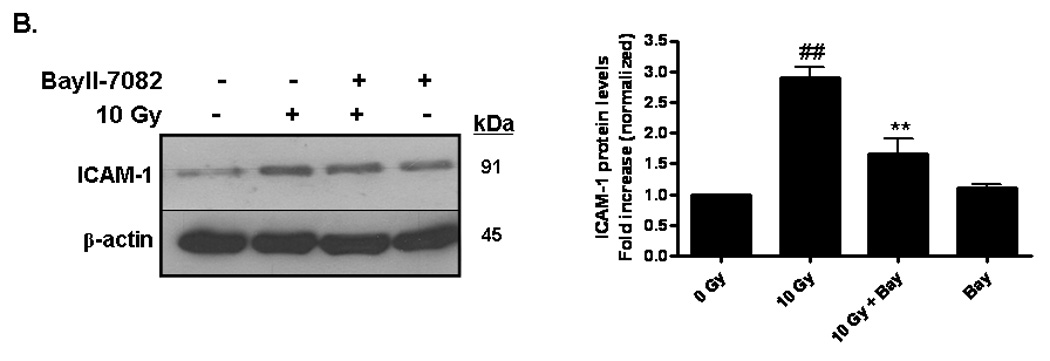

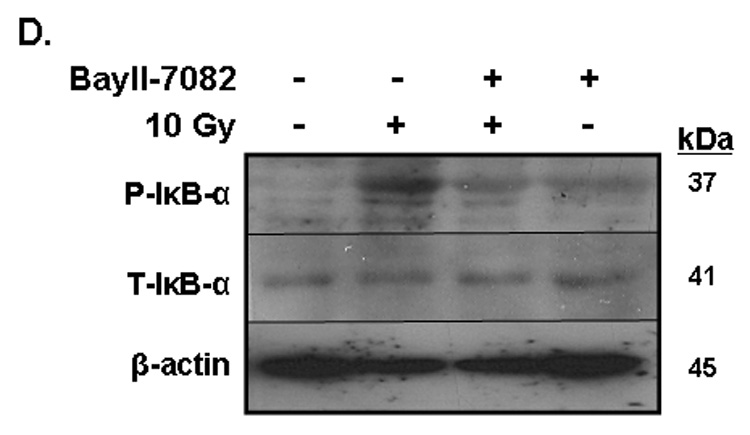

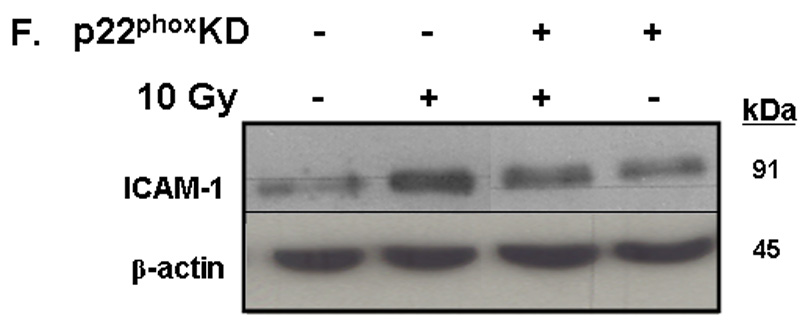
A) RBMEC were irradiated with 10 Gy γ-rays. Total cell lysates were collected 2–24 h postirradiation, separated by SDS-PAGE, and probed for ICAM-1; β-actin served as a loading control. Mean ± SD; n = 3; *p <0.05 vs. sham-irradiated control. B) RBMEC were pretreated with 5µM BayII-7082 for 1 h prior to irradiation. Total cell lysates were collected 24 h postirradiation and probed for ICAM-1. Mean ± SD; n = 3; ##p< 0.01 vs. sham-irradiated control and **p#0.01 vs. 10 Gy. C) RBMEC were pretreated with 5µM BayII-7082 for 1 h prior to irradiation. Nuclear protein was extracted 4 h postirradiation and assessed by EMSA. N = 2. D) RBMEC were pretreated with BayII-7082 for 1 h prior to irradiation. Total cell lysates were collected 1 h postirradiation and probed for phospho-IκB-α and total IκB-α. N = 1. E & F) RBMEC were pretreated with 10 µM apocynin for 1 h (E) or transfected with 2 µg p22phox antisense oligonucleotides (F) and then irradiated. After 24 h, total cell lysates were collected and probed for ICAM-1. N = 2.
To determine the role of NADPH oxidase-derived ROS in radiation-induced ICAM-1 expression, we transfected RBMEC with p22phox antisense oligonucleotides and treated the cells with apocynin prior to irradiation. As noted above, irradiating RBMECs led to upregulation of ICAM-1 gene expression (data not shown) and protein levels (Fig. 4A) 24 h postirradiation, and there was no observable abrogation upon apocynin pretreatment (Fig. 4E). However, knocking down p22phox gene expression appeared to abrogate the radiation-induced increase in ICAM-1 protein levels (Fig. 4F), which may suggest that NADPH oxidase-dependent regulation of ICAM-1 is isoform-specific.
PAI-1 served as an additional marker of radiation-induced oxidative stress in our studies. PAI-1 is secreted by endothelial cells in injury-related insults and is considered a marker of the on-going inflammatory process. PAI-1 has been shown to be highly concentrated adjacent to and within radiation-induced necrotic zones of the CNS [37]. Indeed, ionizing radiation led to upregulation of PAI-1 gene expression in RBMEC in a time-dependent manner, with highest expression occurring 24 h postirradiation (*p < 0.05 vs. sham-irradiated control; Fig. 5A). PAI-1 is also regulated in an NFκB-dependent manner, as radiation-induced upregulation of PAI-1 protein is inhibited upon pretreatment with BayII-7082 (*p < 0.05 vs. 10 Gy; Fig. 5B).
Fig. 5. NADPH oxidase inhibition blocks radiation-induced upregulation of PAI-1 expression in RBMEC.
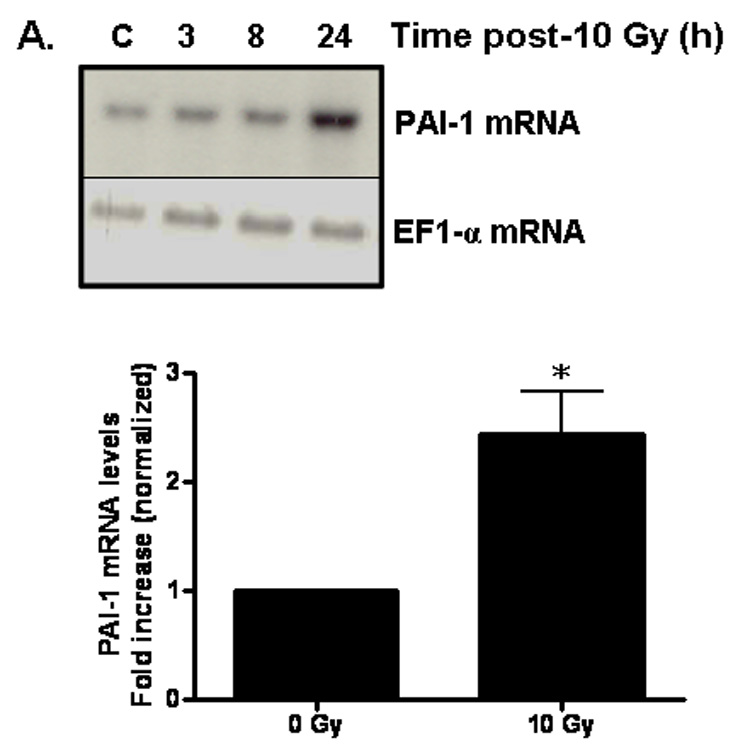
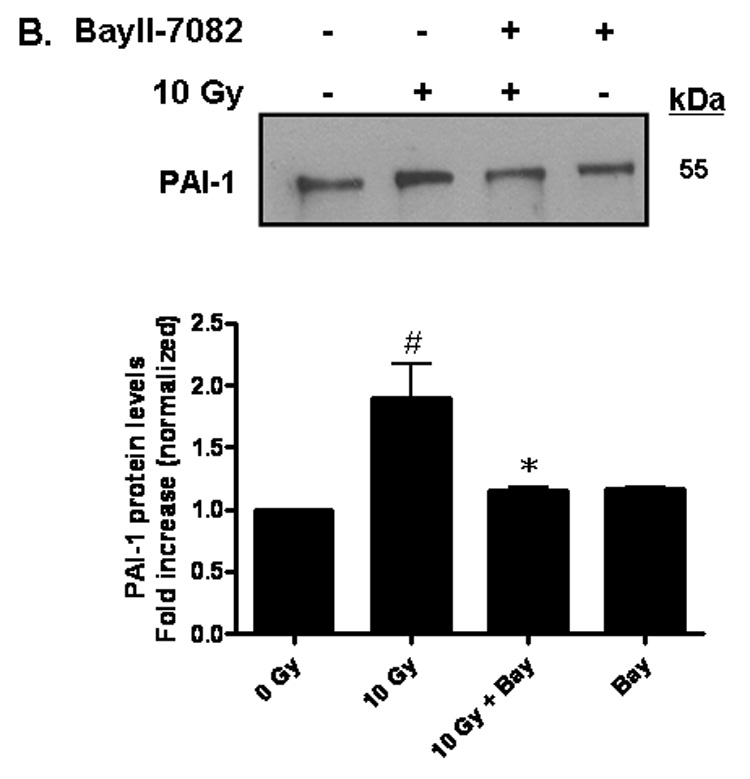
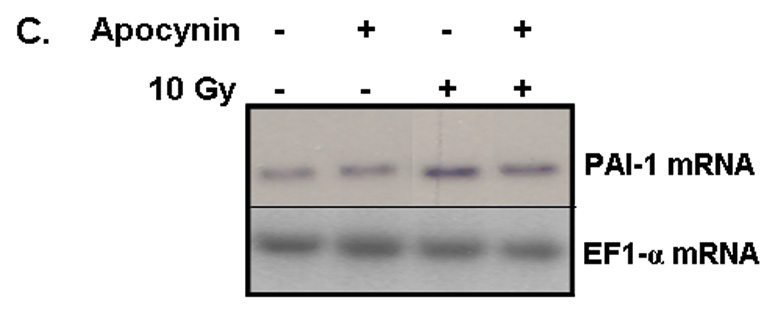

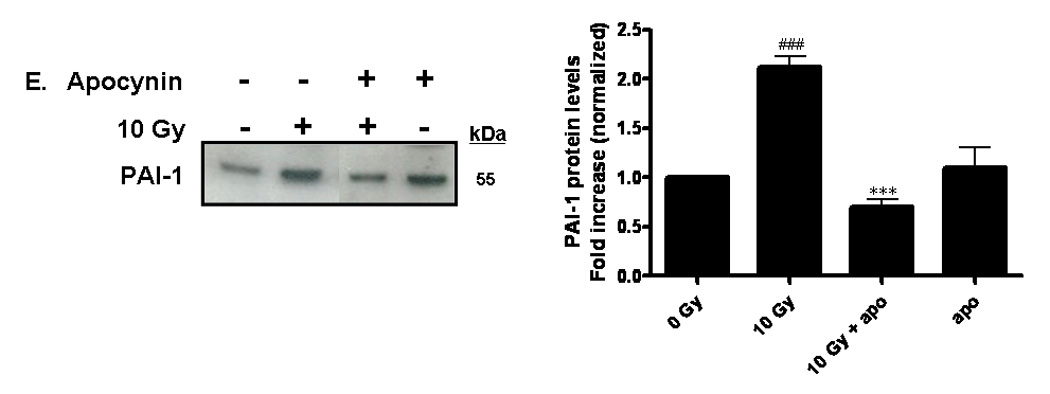
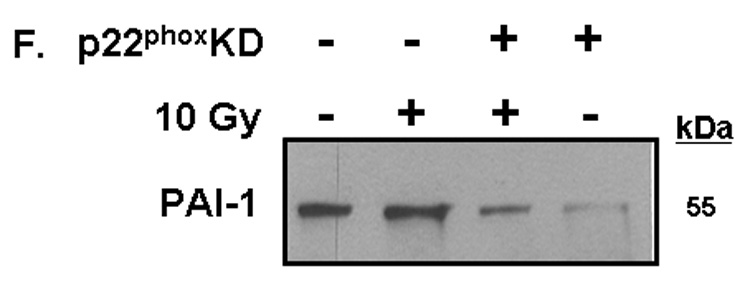
A) RBMEC were irradiated with 10 Gy γ-rays, and total RNA was collected 3–24 h postirradiation then analyzed by Northern blot. EF1-α is shown as a loading control. Mean ± SD; n=3; * p < 0.05 vs. sham-irradiated control. B) RBMEC were pretreated with 5µM BayII-7082 for 1 h prior to irradiation. Conditioned media was collected and concentrated 24 h postirradiation and probed for PAI-1. N = 3. C and D) RBMEC were pretreated with 10 µM apocynin for 1 h (C) or 30 MOI AdN17Rac1 for 24 h (D), irradiated with 10 Gy γ-rays, and total RNA was collected 24 h postirradiation then analyzed by Northern blot. N = 2. E) RBMEC were pretreated with 50 µM apocynin for 24 h and conditioned media was probed for PAI-1. Mean ± SD; n = 3; ###p<0.001 vs. sham-irradiated control and ***p#0.001 vs. 10 Gy. F) RBMEC were transfected with 2 µg p22phox antisense oligonucleotides and irradiated with 10 Gy γ-rays. Conditioned media was collected and concentrated 24 h postirradiation and probed for PAI-1.
To determine the role of NADPH oxidase-derived ROS in radiation-induced PAI-1 gene and protein expression, we treated RBMEC with apocynin prior to irradiation. This radiation-induced increase in PAI-1 mRNA levels was ameliorated upon apocynin pretreatment (Fig. 5C). As an alternative approach to inhibit NADPH oxidase molecularly, a dominant-negative form of Rac1 (AdN17Rac1) was used to infect RBMEC. Pretreatment with dominant-negative Rac1 ameliorated radiation-induced upregulation of PAI-1 gene expression (Fig. 5D). Ionizing radiation also significantly increased PAI-1 protein levels (###p<0.001 vs. sham-irradiated control), which was prevented upon pretreatment with apocynin (***p<0.001 vs. 10 Gy; Fig. 5E). Further, genetic inhibition of NADPH oxidase by p22phox antisense oligonucleotides prevented the radiation-induced increase in PAI-1expression (Figure 5F). Taken together, these results suggest that NADPH oxidase mediates radiation-induced upregulation of redox-regulated genes.
Ionizing radiation modulates individual NADPH oxidase components
The data outlined above demonstrate that ionizing radiation can acutely upregulate NADPH oxidase activity. Since chronic oxidative stress has been implicated in radiation-induced normal tissue injury [10], we hypothesized that NADPH oxidase might be chronically activated following irradiation. Indeed, evidence suggests that the complex can be repeatedly reactivated via continual replenishment of the complex components [38]. We therefore sought to determine whether ionizing radiation was capable of upregulating the individual components of the NADPH oxidase complex. Nox4, a homolog of gp91phox/Nox2, is highly expressed in brain arterial [39] and capillary [40] endothelium. Nox4 heterodimerizes with p22phox, forming the membrane-bound flavocytochrome b558. Using real-time PCR, we observed that ionizing radiation significantly induced Nox4 (*p<0.05 and **p<0.01 vs. sham-irradiated control) and p22phox (**p<0.01 vs. sham-irradiated control) mRNA expression up to 8 h postirradiation (Figures 6A and 6B, respectively). We further assessed p22phox protein levels and found that ionizing radiation appeared to modulate p22phox protein levels, with significant increases observed 12 h postirradiation (*p<0.05 vs. sham-irradiated control; Figure 6C). Ionizing radiation also induced protein levels (*p<0.05 vs. sham-irradiated control; Figure 6D) of the cytosolic component, p47phox, as observed by Western blot. These observations suggest that ionizing radiation is capable of regulating individual components of the NADPH oxidase complex, thus providing a putative mechanism for continual NADPH oxidase activation and chronic ROS production.
Fig. 6. Ionizing radiation can modulate the expression of NAPDH oxidase components in RBMEC.
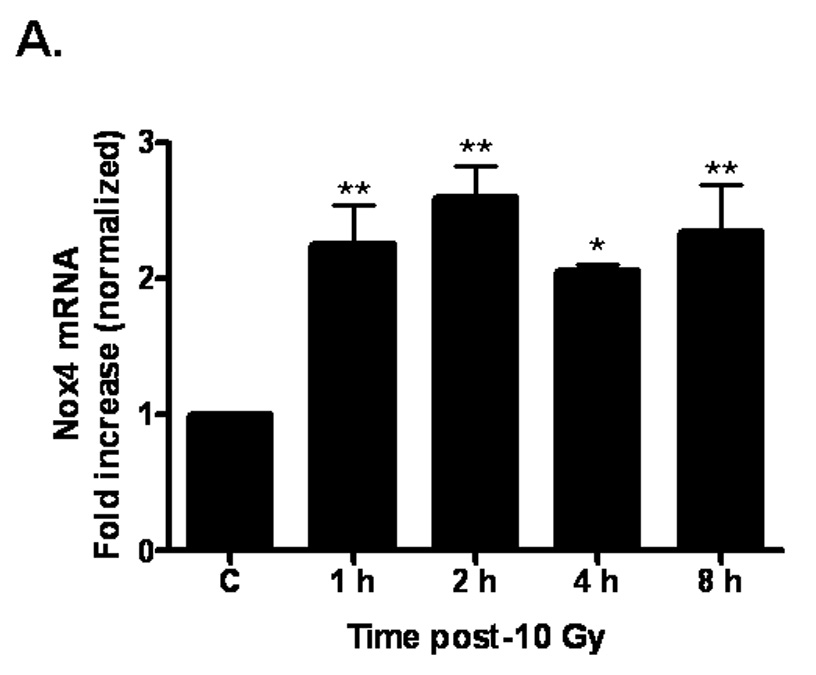
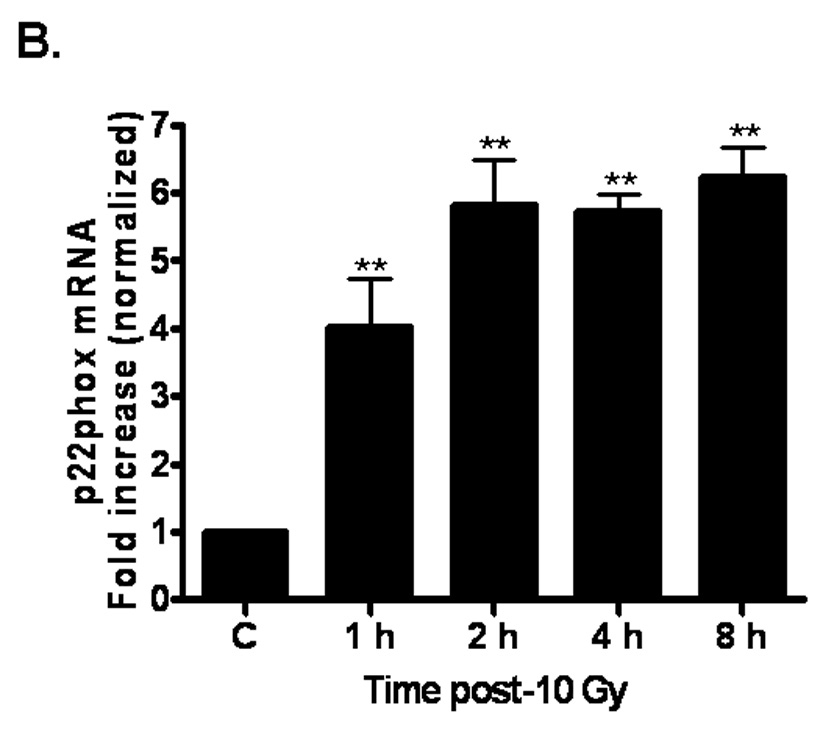
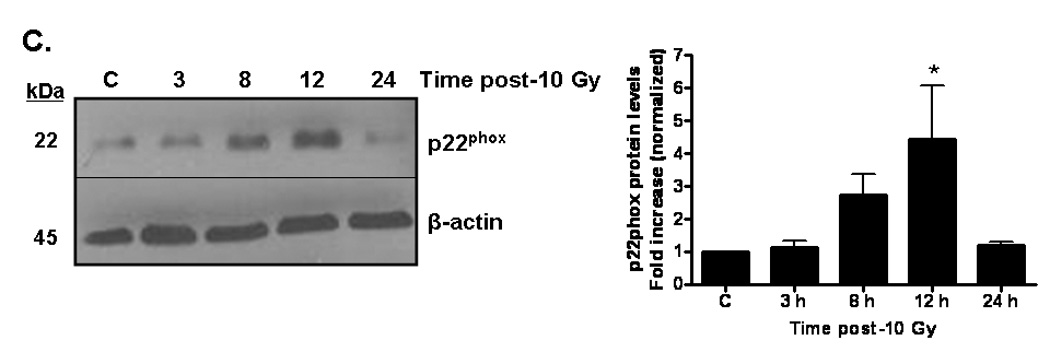

A and B) RBMEC were irradiated with 10 Gy γ-rays, and total RNA was collected 1–8 h postirradiation. mRNA expression of Nox4 (A) and p22phox (B) was assessed by real-time PCR. Values are expressed relative to β-actin. Mean ± SD; n = 3; *p<0.05 and **p<0.01 vs. sham-irradiated control. C) Total cell lysates were collected 3–24 h postirradiation, separated by SDS-PAGE, and probed for p22phox. β-actin served as a loading control. Densitometric analysis is shown. Mean ± SD; n = 3; *p<0.05 and vs. sham-irradiated control. D) Total cell lysates were collected 2–24 h postirradiation, separated by SDS-PAGE, and probed for p47phox. Densitometric analysis is shown. Mean ± SD; n = 3; *p<0.05 vs. sham-irradiated control.
Discussion
In the present study, we investigated whether NADPH oxidase plays a significant role in radiation-induced oxidative stress as measured by intracellular ROS generation, activation of NFκB, and upregulation of ICAM-1 and PAI-1. To our knowledge, this is the first time that endothelial NADPH oxidase-derived ROS have been implicated in radiation-induced oxidative stress.
Our findings indicate that NADPH oxidase contributes to radiation-induced oxidative stress; both pharmacologic (DPI and apocynin) and genetic (p22phox antisense oligonucleotides) inhibition of NADPH oxidase significantly abrogated the radiation-induced increase in intracellular ROS generation. We also demonstrated that NADPH oxidase is the primary source of ROS in RBMEC; inhibition of the mitochondrial electron transport chain, xanthine oxidase, and nitric oxide synthase failed to prevent the radiation-induced increase in intracellular ROS generation. It should be noted that these data only provide evidence indicating an indirect activation of NADPH oxidase by ionizing radiation. We made several attempts to test whether ionizing radiation was capable of directly activating NADPH oxidase. While our DHE assays demonstrated radiation-induced O2•− production that could be abrogated with apocynin and p22phox antisense oligonucleotides (thus, implicating NADPH oxidase activation by ionizing radiation), cytochrome c reduction, chemiluminescence, and electron spin resonance assays proved unsuccessful. This may be due to the fact that the detection limits of these assays may not be sensitive enough to detect changes in the relatively small amounts of ROS generated by non-phagocytic NADPH oxidases compared to those generated by phagocytic NADPH oxidases.
Our findings also indicate that NADPH oxidase contributes to the radiation-induced upregulation of redox-modulated genes. Both pharmacologic (apocynin) and genetic (p22phox antisense oligonucleotides) inhibition of NADPH oxidase abrogated the radiation-induced increase in i] NFκB DNA-binding activity, ii] ICAM-1 mRNA and protein expression, and iii] PAI-1 mRNA and protein expression. Interestingly, PAI-1 contains no classical inflammatory response element within its promoter; therefore it is unclear by what mechanism PAI-1 expression is upregulated during oxidative stress. Our studies demonstrate that PAI-1 is regulated in an NFκB-dependent manner since pretreatment with BayII-7082 abrogated radiation-induced upregulation of PAI-1 protein. This indicates that PAI-1 may contain an NFκB-like consensus sequence within its promoter, making it possible for PAI-1 to participate in oxidative stress induced by radiation.
Our data must be interpreted with some caution. Nox1, Nox2, and Nox4 isoforms are the catalytic components of the enzyme shown to be present in endothelial cells with Nox4 as the predominant isoform [41]. Of these isoforms, p47phox translocation is only required for activation of Nox1- and Nox2-containing NADPH oxidases [24]. The fact that apocynin can abrogate radiation-induced ROS generation, NFκB DNA-binding activity, and PAI-1 expression – presumably by inhibiting p47phox phosphorylation and translocation – suggests that a Nox1- or Nox2-containing NADPH oxidase mediates these radiation-induced responses. However, we were unable to detect Nox1 or Nox2 in our in vitro model using real-time PCR and western blotting techniques (data not shown). This may be explained by recent data suggesting that apocynin may be acting as an antioxidant in vascular cells rather than as a specific inhibitor of NADPH oxidase [42]. These data are based on the requirement of H2O2 and myeloperoxidase for apocynin activation and its subsequent inhibitory effect on NADPH oxidase. Vascular cells, at least in vitro, do not contain myeloperoxidase and therefore are not capable of activating apocynin appropriately. In that study, the authors clearly observed an abrogation of ROS generation in the absence of apocynin activation, suggesting that apocynin was acting as a radical scavenger rather than as a specific NADPH oxidase inhibitor. However, the possibility that other peroxidases present in vascular cells may be capable of activating apocynin should not be discounted.
Despite this debate regarding the specificity of apocynin, knocking down gene expression of p22phox, a subunit required for activation of most NADPH oxidase isoforms, clearly abrogated these radiation-mediated responses, further supporting a direct role of NADPH oxidase in radiation-induced oxidative stress. However, based on these data, we cannot rule out the possibility that various NADPH oxidase isoforms may simultaneously contribute to radiation-induced oxidative stress and inflammation. Future experiments to be performed with inhibition/knockout of the specific catalytic subunit are required to elucidate the details.
Despite not being able to show direct activation of NADPH oxidase by ionizing radiation as mentioned above, we were able to demonstrate for the first time that radiation is capable of regulating individual components of the endothelial NADPH oxidase complex, namely Nox4, p22phox, and p47phox. These data are supported by recent findings suggesting that ionizing radiation can induce Nox1 mRNA and Nox1-dependent ROS in salivary gland acinar cells [43]. The current finding that radiation can modulate individual components of NADPH oxidase at both the mRNA and protein level provides a mechanism for continual NADPH oxidase activation and chronic ROS production.
In summary, we have demonstrated that radiation can induce an oxidative stress in RBMEC with NADPH oxidase as a primary source of radiation-induced ROS. However, the ability to translate these finding to an in vivo setting is limited. While these results do not demonstrate a role for NADPH oxidase in radiation-induced brain injury, they do suggest a putative role for NADPH oxidase in the radiation response. Thus, NADPH oxidase may represent a novel target with which to potentially treat radiation-induced brain injury and improve QOL for long-term brain cancer survivors.
Acknowledgement
The project described was supported by NIH Grants CA117061 (JRCU) and CA122318 (MER). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jemal A, Siegal R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer Statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Khuntia D, Brown P, Li J, Mehta MP. Whole-brain radiotherapy in the management of brain metastasis. J Clin Oncol. 2006;24:1295–1304. doi: 10.1200/JCO.2005.04.6185. [DOI] [PubMed] [Google Scholar]
- 3.Brown PD, Asher AL, Farace E. Adjuvant whole brain radiotherapy: strong emotions decide but rational studies are needed. Int J Radiat Oncol Biol Phys. 2008;70:1305–1309. doi: 10.1016/j.ijrobp.2007.11.047. [DOI] [PubMed] [Google Scholar]
- 4.Shaw EG, Rosdhal R, D’Agostino RB, Jr, Lovato J, Naughton MJ, Robbins ME, Rapp SR. Phase II study of donepezil in irradiated brain tumor patients: effect on cognitive function, mood, and quality of life. J Clin Oncol. 2006;24:1415–1420. doi: 10.1200/JCO.2005.03.3001. [DOI] [PubMed] [Google Scholar]
- 5.Crossen J, Garwood D, Glatstein E, Neuwelt E. Neurobehavioral sequelae of cranial irradiation in adults: a review of radiation-induced encephalopathy. J Clin Oncol. 1994;12:627–642. doi: 10.1200/JCO.1994.12.3.627. [DOI] [PubMed] [Google Scholar]
- 6.Sheline G, Wara W, Smith V. Therapeutic irradiation and brain injury. Int J Radiat Oncol Biol Phys. 1980;6:1215–1228. doi: 10.1016/0360-3016(80)90175-3. [DOI] [PubMed] [Google Scholar]
- 7.Tofilon P, Fike J. The radioresponse of the central nervous system: a dynamic process. Radiat Res. 2000;153:357–370. doi: 10.1667/0033-7587(2000)153[0357:trotcn]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 8.Giglio P, Gilbert M. Cerebral Radiation Necrosis. Neurologist. 2003;9:180–188. doi: 10.1097/01.nrl.0000080951.78533.c4. [DOI] [PubMed] [Google Scholar]
- 9.DeAngelis L, Dellatre J, Posner J. Radiation-induced dementia in patients cured of brain metastases. Neurology. 1989;39:789–796. doi: 10.1212/wnl.39.6.789. [DOI] [PubMed] [Google Scholar]
- 10.Robbins MEC, Zhao W. Chronic oxidative stress and radiation-induced late normal tissue injury: a review. Int J Radiat Biol. 2004;80:251–259. doi: 10.1080/09553000410001692726. [DOI] [PubMed] [Google Scholar]
- 11.Schafer F, Qian S, Buettner G. Iron and free radical oxidations in cell membranes. Cell Mol Biol. 2000;46:657–662. [PMC free article] [PubMed] [Google Scholar]
- 12.Smith K, Kapoor R, Felts P. Demyelination: the role of reactive oxygen and nitrogen species. Brain Pathol. 1999;9:69–92. doi: 10.1111/j.1750-3639.1999.tb00212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Connor J, Menzies S. Cellular management of iron in the brain. J Neurol Sci. 1995;134:33–44. doi: 10.1016/0022-510x(95)00206-h. [DOI] [PubMed] [Google Scholar]
- 14.Bast A, Haenen G, Doelman C. Oxidants and antioxidants: state of the art. Am J Med. 1991;91:2S–13S. doi: 10.1016/0002-9343(91)90278-6. [DOI] [PubMed] [Google Scholar]
- 15.Peuchen S, Bolanos J, Heales S, Almeida A, Duchen M, Clark J. Interrelationships between astrocyte function, oxidative stress, and antioxidant status within the CNS. Prog Neurobiol. 1997;52:261–281. doi: 10.1016/s0301-0082(97)00010-5. [DOI] [PubMed] [Google Scholar]
- 16.Zhao W, Diz DI, Robbins ME. Oxidative damage pathways in relation to normal tissue injury. Br J Radiol. 2007;80:S23–S31. doi: 10.1259/bjr/18237646. [DOI] [PubMed] [Google Scholar]
- 17.Gallin E, Green S. Exposure to gamma-irradiation increases phorbol myristate acetate-induced H2O2 production in human macrophages. Blood. 1987;70:694–701. [PubMed] [Google Scholar]
- 18.Lyng F, Seymour C, Mothersill C. Oxidative stress in cell exposed to low level of ionizing radiation. Biochem Soc Trans. 2001;29:350–353. doi: 10.1042/0300-5127:0290350. [DOI] [PubMed] [Google Scholar]
- 19.Spitz DR, Azzam EI, Li JJ, Guis D. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: a unifying concept in stress response biology. Cancer Metastat Rev. 2004;23:311–322. doi: 10.1023/B:CANC.0000031769.14728.bc. [DOI] [PubMed] [Google Scholar]
- 20.Griendling K, Minieri C, Ollerenshaw J, Alexander R. Angiotensin II stimulated NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 21.Hamada N, Matsumoto H, Hara T, Kobayashi Y. Intercellular and intracellular signaling pathways mediating ionizing radiation-induced bystander effects. J Radiat Res. 2007;48:87–95. doi: 10.1269/jrr.06084. [DOI] [PubMed] [Google Scholar]
- 22.Li W-G, Miller F, Zhang H, Spitz D, Oberley L, Weintraub N. H2O2-induced O.2.− production by a non-phagocytic NADPH oxidase causes oxidant injury. J Biol Chem. 2001;276:29251–29256. doi: 10.1074/jbc.M102124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Finkel T. Signal transduction by reactive oxygen species in non-phagocytic cells. J Leukoc Biol. 1999;65:337–340. doi: 10.1002/jlb.65.3.337. [DOI] [PubMed] [Google Scholar]
- 24.Bedard K, Krause K-H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 25.Cross AR, Segal AW. The NADPH oxidase of professional phagocytes – prototype of the NOX electron transport chain systems. Biochem Biophys Acta. 2004;1657:1–22. doi: 10.1016/j.bbabio.2004.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Allen R, Tresini M. Oxidative stress and gene regulation. Free Radic Biol Med. 2000;28:463–499. doi: 10.1016/s0891-5849(99)00242-7. [DOI] [PubMed] [Google Scholar]
- 27.Schreck R, Albermann K, Beuerle P. Nuclear factor kappa B: an oxidative stress-responsive transcription factor of eukaryotic cells (a review) Free Radic Res Commun. 1992;17:221–237. doi: 10.3109/10715769209079515. [DOI] [PubMed] [Google Scholar]
- 28.Hong J-H, Chiang C-S, Campbell I, Sun J-R, Withers R, McBride W. Induction of acute phase gene expression by brain irradiation. Int J Radiat Oncol Biol Phys. 1995;33:619–626. doi: 10.1016/0360-3016(95)00279-8. [DOI] [PubMed] [Google Scholar]
- 29.Zhao W, Spitz D, Oberley L, Robbins M. Redox modulation of the pro-fibrogenic mediator plasminogen activator inhibitor-1 following ionizing radiation. Cancer Res. 2001;61:5537–5543. [PubMed] [Google Scholar]
- 30.Baker DG, Krochak RJ. The response of the microvascular system to radiation: a review. Cancer Invest. 1989;7:287–294. doi: 10.3109/07357908909039849. [DOI] [PubMed] [Google Scholar]
- 31.Brown WR, Thore CR, Moody DM, Robbins ME, Wheeler KT. Vascular damage after fractionated whole-brain irradiation in rats. Radiat Res. 2005;164:662–668. doi: 10.1667/rr3453.1. [DOI] [PubMed] [Google Scholar]
- 32.Pullar JM, Hampton MB. Diphenyleneiodonium triggers the efflux of glutathione from cultured cells. J Biol Chem. 2002;277:19402–19407. doi: 10.1074/jbc.M111053200. [DOI] [PubMed] [Google Scholar]
- 33.Ergul A, Johansen JS, Strømhaug C, Harris AK, Hutchinson J, Tawfik A, Rahimi A, Rhim E, Wells B, Caldwell RW, Anstadt MP. Vascular dysfunction of venous bypass conduits is mediated by reactive oxygen species in diabetes: role of endothelin-1. J Pharmacol Exp Ther. 2004;313:70–77. doi: 10.1124/jpet.104.078105. [DOI] [PubMed] [Google Scholar]
- 34.Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol. 1994;11:95–102. doi: 10.1165/ajrcmb.11.1.8018341. [DOI] [PubMed] [Google Scholar]
- 35.Pahl HL. Activators and target genes of Rel/NFκB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 36.Baeuml H, Behrends U, Peter RU, Mueller S, Kammerbauer C, Caughman SW, Degitz K. Ionizing radiation induces, via generation of reactive oxygen intermediates, intercellular adhesion molecule-1 (ICAM-1) gene transcription and NF kappa B-like binding activity in the ICAM-1 transcriptional regulatory element. Free Radic Res. 1997;27:127–142. doi: 10.3109/10715769709097846. [DOI] [PubMed] [Google Scholar]
- 37.Sawaya R, Rayford A, Kono S, Ang K, Feng Y, Stephens L, Rao J. Plasminogen activator inhibitor-1 in the pathogenesis of delayed radiation damage in rat spinal cord in vivo. J Neurosurg. 1994;81:381–387. doi: 10.3171/jns.1994.81.3.0381. [DOI] [PubMed] [Google Scholar]
- 38.DeCoursey TE, Ligeti E. Regulation and termination of NADPH oxidase activity. Cell Mol Life Sci. 2005;62:2173–2193. doi: 10.1007/s00018-005-5177-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ago T, Kitazono T, Kuroda J, Kumai Y, Kamouchi M, Ooboshi H, Wakisaka M, Kawahara T, Rokutan K, Ibayashi S, Iida M. NADPH oxidases in rat basilar arterial endothelial cells. Stroke. 2005;36:1040–1046. doi: 10.1161/01.STR.0000163111.05825.0b. [DOI] [PubMed] [Google Scholar]
- 40.Vallet P, Charnay Y, Steger K, Ogier-Denis E, Kovari E, Herrmann F, Michel JP, Szanto I. Neuronal expression of the NADPH oxidase NOX4, and its regulation in mouse experimental brain ischemia. Neuroscience. 2005;132:233–238. doi: 10.1016/j.neuroscience.2004.12.038. [DOI] [PubMed] [Google Scholar]
- 41.Ago T, Kitazono T, Ooboshi H, Iyama T, Han YH, Takada J, Wakisaka M, Ibayashi S, Utsumi H, Iida M. Nox4 as the major catalytic component of an endothelial NAD(P)H oxidase. Circulation. 2004;109:227–233. doi: 10.1161/01.CIR.0000105680.92873.70. [DOI] [PubMed] [Google Scholar]
- 42.Heumuller S, Wind S, Barbosa-Sicard E, Schmidt HHHW, Busse R, Schroder K, Brandes RP. Apocynin is not an inhibitor of vascular NADPH oxidases but as an antioxidant. Hypertension. 2008;51:211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- 43.Tateishi Y, Sasabe E, Ueta E, Yamamoto T. Ionizing radiation induces apoptotic damage of salivary gland acinar cells via NADPH oxidase-dependent superoxide production. Biochem Biophys Res Comm. 2008;366:301–308. doi: 10.1016/j.bbrc.2007.11.039. [DOI] [PubMed] [Google Scholar]