

Summary
Cerebral malaria is a major complication of Plasmodium falciparum infection in children. The pathogenesis of cerebral malaria involves vascular inflammation, immune stimulation and obstruction of cerebral capillaries. Platelets have a prominent role in both immune responses and vascular obstruction. We now demonstrate that the platelet derived chemokine, platelet factor 4 (PF4)/CXCL4, promotes the development of experimental cerebral malaria. Plasmodium infected red blood cells (RBC) activated platelets independent of vascular effects, resulting in increased plasma PF4. PF4 or CXCR3 null mice had less ECM, decreased brain T-cell recruitment, and platelet depletion or aspirin treatment reduced the development of ECM. We conclude that Plasmodium infected RBC can activate platelets and platelet derived PF4 then contributes to immune activation and T-cell trafficking as part of the pathogenesis of ECM.
Introduction
Cerebral malaria is a major complication of Plasmodium falciparum infection, particularly in children. Thirty-three percent of children admitted to a hospital in Kenya were reported to have had malaria, and of these, 47% had neurologic symptoms (Idro et al., 2007). There were an estimated 515 million clinical episodes of acute P. falciparum infection in the world in 2002, mainly affecting children less than 5 years of age, making the impact of cerebral malaria (CM) staggering (Idro et al., 2007).
Cerebral malaria results from a combination of vascular and immune system dysfunction. Brain tissue from patients that die of CM reveals multifocal capillary obstruction with parasitized red blood cells (pRBCs), platelets, and leukocytes (van der Heyde et al., 2006). Several hypotheses have attempted to explain the noted pathology. Some view CM as mechanical obstruction of microvessels, the result of an increase in endothelial cell adhesion molecule expression and cell attachment (Chakravorty and Craig, 2005; Tripathi et al., 2006; Tripathi et al., 2007). Others emphasize an immunological basis caused by excessive immune stimulation and pro-inflammatory cytokine release (van der Heyde et al., 2006). A unification of these two ideas includes cell adhesion to the endothelium promoting pro-thrombotic immune responses, resulting in further vascular inflammation, immune stimulation and obstruction of cerebral capillaries (van der Heyde et al., 2006). Platelets have a prominent role in both immune responses and vascular obstruction.
In addition to their vital hemostatic function, platelets are also active in inflammation. Platelet granules contain many inflammatory and adhesion molecules that are either released or expressed upon activation. Platelets can interact with an intact inflamed endothelial cell layer and upon activation initiate interactions with quiescent endothelial and immune cells (Gawaz et al., 2005). These interactions promote further platelet localization and release of more inflammatory molecules setting up a cycle of continued vascular inflammation (Gawaz et al., 2005). Platelets are known to contribute to the progression of diverse vascular and inflammatory diseases and may be involved in the pathogenesis of CM (Grau et al., 2003; Wassmer et al., 2006; Wassmer et al., 2003).
Platelet and RBC aggregates are found in cerebral blood vessels of individuals with fatal malaria (Grau et al., 2003; van der Heyde et al., 2005). Platelets in co-culture with P. falciparum infected RBCs (PfRBC) facilitate the binding of PfRBC to endothelial cells (Wassmer et al., 2004), suggesting a role for platelets in cooperating with pRBCs to promote cerebral vascular obstruction. TNFα increased in CM and TNFα has recently been demonstrated to increase platelet binding to the brain microvasculature in experimental cerebral malaria (ECM) (von Zur Muhlen et al., 2008), further demonstrating this important interplay between platelets and immune responses in cerebral malaria. However, a direct early effect of infected RBC on platelets and platelet derived mediators in CM immune activation has not been addressed.
PF4 was the first described CXC class chemokine and is a platelet granule constituent of great abundance (about 25% of total alpha granule protein content) (Lambert et al., 2007; Sachais et al., 2004). Basal PF4 concentration in human plasma is 2–20 ng/mL, but in acute thrombosis has been reported elevated 1000 times to 5–10 µg/mL (Eslin et al., 2004; Sachais et al., 2004). During acute P. falciparum infection plasma concentrations of PF4 are significantly elevated from a mean control concentration of 18 ng/mL to greater than 75 ng/mL (Essien and Ebhota, 1983), demonstrating a potentially significant role of PF4 in mediating vascular effects in malaria infection. PF4 has many known cell targets including neutrophils, monocytes/macrophages, NK cells, and T-cells (Sachais et al., 2004). PF4 is a ligand for the chemokine receptor CXCR3B, a G Protein Coupled Receptor (GPCR) expressed on T cells and some endothelial cells (Lasagni et al., 2003; Mueller et al., 2008; Sachais et al., 2004) and stimulates neutrophils and macrophages through surface chondroitin (Boehlen and Clemetson, 2001; von Hundelshausen et al., 2005).
We now demonstrate that the Plasmodium infected RBC directly activate platelets and the release of the chemokine PF4. PF4 is a mediator of the pathogenesis of ECM by promoting a pro-inflammatory environment that includes increasing T-cell CXCR3 expression and trafficking to the brain.
Results
Platelets are activated by PfRBC
Platelets contribute to CM by promoting RBC adhesion and vessel obstruction subsequent to PfRBC induced vascular inflammation (Wassmer et al., 2006; Wassmer et al., 2004). To demonstrate that platelets are also directly activated by PfRBC, control human platelet rich plasma (PRP) was incubated with purified washed control RBC, percoll purified intact trophozoite stage infected erythrocytes (PfRBC), or control buffer. As a positive control comparison, PRP was also treated with a moderate concentration of a thrombin receptor activating peptide (TRAP, 5 µM). Platelet activation was determined by measuring activated GPIIb/IIIa receptor expression (PAC-1 antibody) and P-selectin surface expression using flow cytometry. Platelets incubated with control RBC had no increase in GPIIb/IIIa activation (Figure 1A, white vs. light grey bars). However, platelets incubated with PfRBC had significantly increased PAC-1 expression (Figure 1A, white vs. dark grey bars) and increased P-selectin expression (Figure 1B). Platelet activation was also determined by measuring PF4 release from PRP incubated with buffer, control RBC, or PfRBC. Plasma PF4 concentration was similar in platelets incubated with buffer and control RBC (Figure 1C, grey vs. white bars). PfRBC however stimulated platelet activation and PF4 release (Figure 1C, black vs grey bars). Similarly, washed RBC from C57Bl6/J mice infected with P. berghei ANKA had increased platelet activation as compared to platelets incubated with control mouse RBC (Supplemental Figure S1). These data demonstrate that parasitized RBC can directly activate platelets.
Figure 1. Platelets are activated by Plasmodium infected RBC.

(A) P. falciparum infected RBC increase platelet activation. GPIIb/IIIa. Human platelets were incubated with buffer, control RBC, or PfRBC for 30 mins and platelet activation measured by GPIIb/IIIa receptor activation (PAC-1 antibody) using flow cytometry. TRAP (5 µM) treated platelets were used as positive controls (n=4, mean ± S.D. *P<0.01). (B) P-selectin. Human platelets were incubated with control RBC, or PfRBC and surface P-selectin determined using flow cytometry (n=4, mean ± S.D. *P<0.03). (C) PF4 Release. Human platelets were incubated with buffer solution, control RBC, or PfRBC for 30 mins and supernatant PF4 measured by ELISA (n=4, mean ± S.D. *P<0.01). (D) P. falciparum infected RBC membranes activate platelets. Control or PfRBC red blood cell ghosts were prepared and incubated with platelets. Platelet activation was determined by FACS for PAC-1 binding (n=4, mean ± S.D. *P<0.01). (E) PfRBC activates platelets via CD36. Human platelets were incubated with control RBC or PfRBC in the presence of control IgG or CD36 blocking antibody Fab fragments for 30 mins and platelet activation determined by FACS (n=4, mean ± S.D. *P<0.05).
To begin to determine how Plasmodium may stimulate platelet activation we incubated PRP with saponin purified P. falciparum trophozoites or purified hemozoin. Neither pure trophozoite nor hemozoin stimulated platelet activation (data not shown). Purified membranes (RBC ghosts) from control RBC or PfRBC were also incubated with platelets and platelet activation determined. Platelets incubated with PfRBC ghosts have significantly greater platelet activation as compared to platelets incubated with control RBC ghosts (Figure 1D), demonstrating that Plasmodium infected RBC express membrane proteins that are pro-thrombotic.
Plasmodium infected RBC can engage CD36 on endothelial cells (Wassmer et al., 2003). Recently, ligand binding of platelet CD36 has been shown to stimulate platelet activation (Podrez et al., 2007). To determine whether PfRBC stimulates platelet activation via CD36 mediated signaling, we incubated platelets with control RBC or PfRBC pre-treated with Fab fragements of non-specific IgG or a CD36 blocking antibody. As anticipated, PfRBC significantly increased platelet activation (Figure 1E) in the presence of IgG. However, PfRBC do not activate platelets when platelets are pretreated with CD36 blocking antibody (Figure 1E). These data demonstrate that PfRBC can stimulate platelet activation through platelet CD36 mediated signaling events.
Taken together, these data demonstrate that Plasmodium infected RBC directly induced platelet activation, in part by parasitized RBC interactions with platelets through a CD36 mediated mechanism.
Platelets are activated early in ECM
In the mouse model of cerebral malaria P. berghei ANKA infected C57Bl6/J mice typically develop neurological symptoms on day 5 and begin to die day 6 post infection (p.i.), with some investigators finding death rates approaching 90%. To identify cerebral vascular thrombi as an early component of ECM, control and P. berghei infected mice were sacrificed on day 5 p.i., brains isolated and histochemistry performed. Using standard hematoxylin and eosin (H&E) staining, cerebral vessels of control mice had no inflammatory cells (Figure 2A, top left). Immunohistochemistry for von Willebrand factor (vWf), a constituent of platelet and endothelial granules was also performed and in control mice vWf is confined to the endothelial cells lining blood vessels (Figure 2A, bottom left). In contrast, cerebral vessels of P. berghei infected mice had multifocal, peri-vascular hemorrhage, inflammatory cell aggregates (Figure 2A, top right) and numerous vWf positive thrombi (arrows) lining vessel walls (Figure 2A, bottom right). CD61 (β3 integrin) immunohistochemistry confirms the presence of non-occlusive adherent immune cells lining cerebral blood vessels of P. berghei infected mice (Supplementary S2)
Figure 2. Platelets are activated in ECM.

(A) Cerebral thrombi in ECM. P. berghei infected mice have cerebral vascular inflammation and hemorrhage (arrowheads top) and vWf positive microvascular thrombi (arrows bottom). (B) P. berghei infected mice have increased brain PF4. Brains were harvested from control and malaria infected mice on day 5 p.i. PF4 concentration was determined in brain lysates by ELISA (n=5 mean ± S.D. *P<0.01 vs control). (C) Malaria infected mice have increased circulating activated platelets. Platelets from P. berghei infected mice were isolated on day 2 and 4 post infection, surface P-selectin and fibrinogen binding determined by flow cytometry and expressed as change in fluorescence vs control platelets (n=5, mean ± S.D. *P<0.01 vs Control). (D) P. berghei infected mice have increased plasma PF4. Plasma from infected and control mice was isolated and PF4 concentration determined by ELISA (n=5 pre-infection and Day 4, n=3 day 6 mean ± S.D. *P<0.01 vs pre-infection).
Platelet factor 4 (PF4/CXCL4) is a major platelet granule constituent. To further demonstrate that during the development of ECM there is an increase in platelet cerebral vascular localization we measured day 5 p.i. whole brain PF4 concentrations using lysates from homogenized control and P. berghei infected mice. PF4 was elevated greater than 2 times above control levels in brain lysates from P. berghei infected mice (Figure 2B). These data clearly demonstrate that platelets are cerebral vascular localized early in the pathogenesis of ECM.
Platelet activation can exert both local and systemic vascular immune effects. We therefore next determined the relative increase in circulating activated platelets by comparing infected mouse platelet P-selectin expression and fibrinogen binding to control mice using flow cytometry (FACS) on days 2 and 4 p.i. On day 4 p.i. circulating platelets had an increase in both surface P-selectin expression and FITC-fibrinogen binding (Figure 2C). Activated platelets tend to marginate or get cleared from the circulation so a measure of circulating activated platelets under-represents total platelet activation. To further demonstrate the potential systemic influence of platelet activation in ECM, plasma was collected from mice before P. berghei ANKA infection and again on days 4 and 6 p.i. and PF4 measured by ELISA. PF4 is elevated more than 2 times in P. berghei infected mice (Figure 2D).
PF4 helps drive the development of ECM
To demonstrate that PF4 is not only a marker of ECM associated platelet activation, but also a mediator of the pathogenesis of ECM, we next infected wild-type (WT) and PF4−/− mice with P. berghei. PF4−/− mice have been well characterized (Eslin et al., 2004; Sachais et al., 2007) and have normal complete blood counts and platelet activation (Supplemental Table 1 and Figure S3). Additionally, CXCR3−/− mice were infected; CXCR3 is a T-cell chemokine receptor recently found necessary for the development of ECM (Campanella et al., 2008; Miu et al., 2008; Van den Steen et al., 2008). Infected WT mice began to die on day 6 p.i., and by day 10 only 30% of mice survived (Figure 3A). In contrast, PF4−/− mice have equal parasitemia (Figure 3B) but decreased ECM mortality with greater than 60% survival by day 10 p.i. CXCR3−/− mice are also protected (Figure 3A). To demonstrate PF4 specific effects in the PF4 null mice, P. berghei infected PF4−/− mice were given recombinant PF4 (5 µg/mouse) daily. Exogenous PF4 restores the PF4−/− mouse phenotype to WT status (Figure 3A).
Figure 3. PF4 drives the development of ECM.
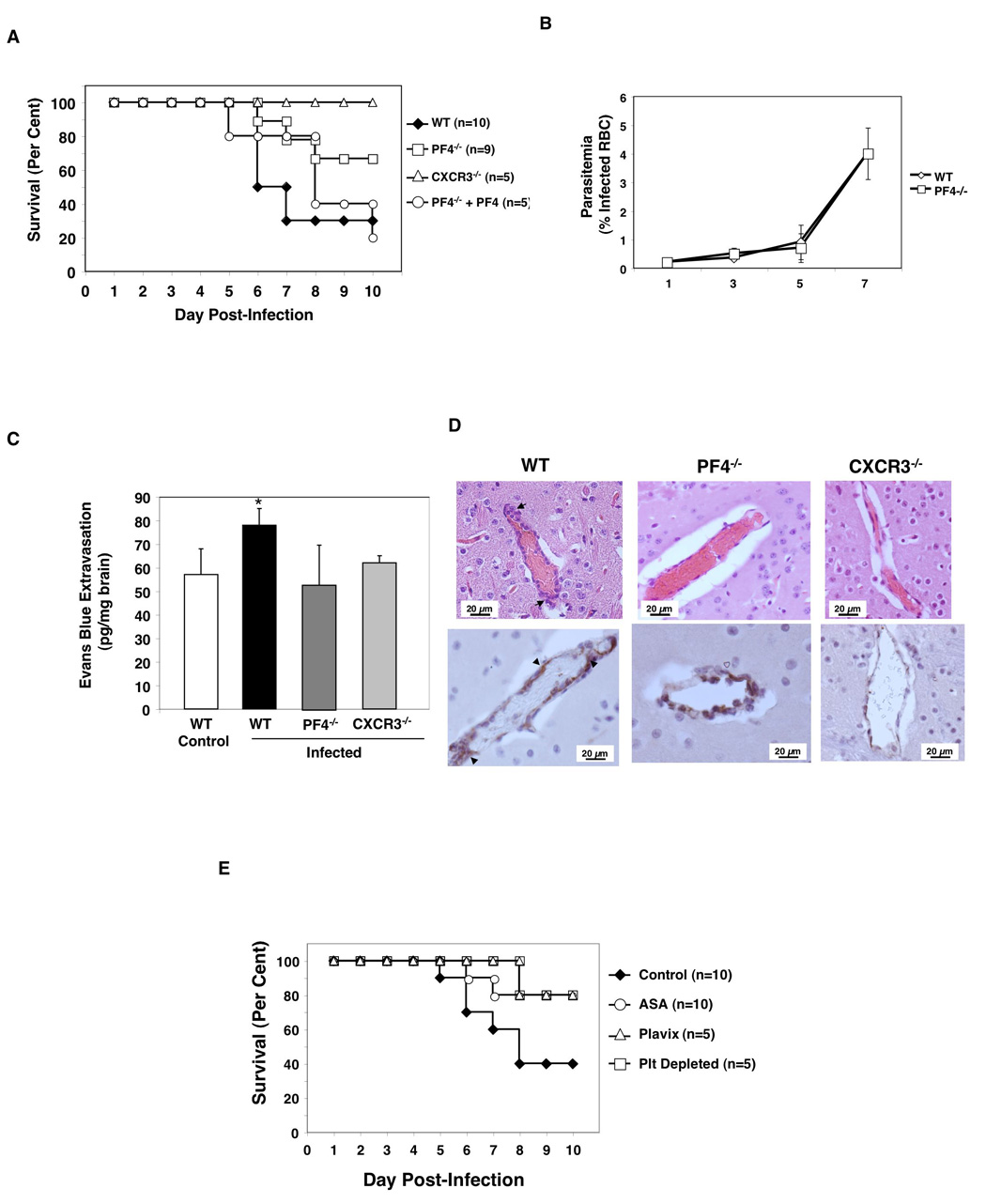
(A) PF4 increases ECM. WT, PF4−/−, CXCR3−/−, and PF4 reconstituted PF4−/− mice were infected with P. berghei and monitored for death (*P<0.01, log-rank test). (B) WT and PF4−/− P. berghei infected mice have equal parasite burden (n=5 days 1–5, n=3 day 7, mean ± S.D.). (C) BBB integrity is protected in PF4−/− and CXCR3−/− mice. On day 5 p.i., P. berghei infected and control mice were injected with 200 µL of 2% Evans Blue Dye IV and dye extravasation determined (n=5 mean ± S.D., *P<0.05 vs WT). (D) P. berghei infected PF4−/− and CXCR3−/− mice have decreased cerebral vascular damage compared to WT infected mice. H&E top, vWf immunohistochemistry bottom. (E) Platelet inhibition decreases ECM. P. berghei infected mice were treated with ASA, Plavix/clopidogrel, or platelet depleted (*P<0.01, log-rank test).
A major step in the pathogenesis of ECM associated death is loss of blood brain barrier (BBB) integrity. BBB integrity was determined in WT control, and infected WT, PF4−/−, and CXCR3−/− mice by Evans Blue dye extravasation on day 5 p.i. At this early time point before mice begin to die WT infected mice already have an increase in dye extravasation (Figure 3C, black bar). In contrast, the BBB in PF4−/− and CXCR3−/− mice is intact and dye extravasation equal to control uninfected mice (Figure 3C). Histological examination confirms that PF4−/− and CXCR3−/− mice have less cerebral vascular inflammation, hemorrhage, and fewer microvascular thrombi (Figure 3D). These data demonstrate that PF4 contributes to cerebral vascular damage in ECM.
If platelets have a role in ECM, then platelet inhibition may ameliorate cerebral vascular injury associated with Plasmodium infection. To explore this, beginning one day after infection mice were either platelet depleted or treated with the platelet inhibitors acetylsalicylic acid (ASA, the active component of aspirin) or Plavix (clopidogrel). Control mice began to die on day 5 with 60% mortality by day 10 (Figure 3E). Mice treated with platelet inhibitors or platelet depleted had delayed onset of death and only 20% mortality (Figure 3E). Initiation of anti-platelet therapy on day 3 p.i. did not improve survival (data not shown) indicating that platelet mediated effects begin early in the pathogenesis of ECM.
PF4 drives ECM associated immune stimulation
TNF family members TNFα and lymphotoxin α (LT, also called TNFβ) are strongly associated with the development of CM (Hunt and Grau, 2003). TNFα has recently been demonstrated to be important in the development of ECM by exerting cerebral vascular effects that lead to further platelet localization and cerebral inflammation (Grau and Lou, 1993; Grau et al., 1993; von Zur Muhlen et al., 2008). To determine whether PF4 exerts its effects in part by increasing plasma TNF, TNFα was measured in control, or infected WT, PF4−/− and CXCR3−/− mice on day 5 p.i. TNFα is elevated in the plasma of WT infected mice on day 5 p.i. (Figure 4A). However, TNFα was not elevated in P. berghei infected PF4−/− or CXCR3−/− mice (Figure 4A) demonstrating that PF4 signaling promotes an inflammatory response to P. berghei including increased TNFα. IFNγ has also been associated with ECM (Chakravorty and Craig, 2005; Tripathi et al., 2006) and its plasma concentrations mirrored that of TNFα (Figure 4B).
Figure 4. PF4 Promotes Immune Stimulation and T-Cell Trafficking in ECM.

(A and B) Plasma TNFα and IFNγ are not increased in infected PF4−/− mice. Plasma was isolated from control or P. berghei infected mice on day 5 p.i. and (A) TNFα and (B) IFNγ concentrations determined by ELISA (n=5, *P<0.01 vs WT control). (C) PF4 increases macrophage TNFα production. Peritoneal macrophages were incubated with 1 µg of PF4 for 48 hrs and TNFα measured in the culture supernatant by ELISA (n=4, mean ± S.D., *P<0.01 vs Control). (D) PF4 increases T-cell TNFα. TNFα was measured in splenocyte culture supernatants by ELISA (n=3–6, mean ± S.D., *P<0.01 vs Control). (E) PF4 increases the number of CXCR3 positive T-cells in vivo. T-cells were isolated from the spleens of P. berghei infected mice on day 5 p.i. CXCR3 expression was determined by FACS (n=5 mean ± S.D., mean ± S.D., *P<0.01 vs WT control). (F) PF4 directly increases T-cell CXCR3 expression. Splenocytes were incubated in anti-CD3 coated wells with anti-CD28 antibody in the presence of control or 1 µg/mL of recombinant PF4 for 4 days. CXCR3 surface expression was determined by FACS (n=4, mean ± S.D., *P<0.05 vs Control). (G) PF4 signaling increases T-cell brain infiltration. Brains were isolated and CD8+ and CD4+ T-cells determined by FACS (n=4–5, mean ± S.D., *P<0.05 vs WT Control). (H) Aspirin treated mice have fewer T-cell infiltrates when P. berghei infected as compared to control infected mice (n=5, mean ± S.D., *P<0.02 vs WT Control).
To determine if increased plasma TNFα was driven in part by a direct PF4 effect on immune cells, we first incubated peritoneal derived macrophages with control or 1 µg/mL of recombinant PF4 for 48 hrs in culture and TNFα in the supernatant determined by ELISA. PF4 greatly increased macrophage TNFα production in vitro (Figure 4C). We next explored whether PF4 can also directly stimulate T-cell TNFα production. Splenocytes were isolated and incubated with control or 1 µg/mL of recombinant PF4 in anti-CD3 coated wells with anti-CD28 antibody for 4 days. TNFα was increased in the cell culture media of PF4 treated splenocytes (Figure 4D). IFNγ was unchanged (data not shown).
CXCR3 has been strongly implicated as a key mediator of T-cell migration to the brain in cerebral malaria (Campanella et al., 2008; Hansen et al., 2007; Miu et al., 2008; Van den Steen et al., 2008). To determine whether PF4 also contributes to the important increase in CXCR3 T-cell response, spleens from control and infected WT and PF4−/− mice were isolated on day 5 p.i. and T-cell CXCR3 expression determined by flow cytometry. The number of CXCR3 positive CD4+ and CD8+ T-cells is more than doubled in P. berghei infected WT mice (Figure 4E); however, T-cells from infected PF4−/− mice do not have an increase in CXCR3 expression (Figure 4E). This is in part a direct PF4 effect. Splenocytes incubated with control or PF4 in anti-CD3 coated wells with anti-CD28 antibody have an increase CD4+ and CD8+ T-cell CXCR3 surface expression (Figure 4F).
T-cell trafficking to the brain is central to the pathogenesis of cerebral malaria. On day 5 p.i. brain mononuclear cells were isolated and T-cells quantified to determine whether PF4 mediated an increase in T-cell trafficking to the brain in ECM. CD4+ and CD8+ T-cells are approximately 2 times elevated in brains of infected WT mice as compared to uninfected control mice (Figure 4G, white vs black bars). The number of T-cells in the brains of PF4−/− or CXCR3−/− mice however is not significantly increased (Figure 4G, white vs dark and light grey bars). Additionally, mice treated with ASA had a decreased number of brain T-cell infiltrates compared to control mice (Figure 4H). These data demonstrate that platelet activation and PF4 secretion early in infection is an important step that drives the ECM T-cell response.
Discussion
We have discovered that malaria infected RBC directly activated platelets and stimulated the release of the chemokine PF4. Platelets were activated early in the pathogenesis of ECM, platelet inhibition reduced the development of ECM, and PF4 is an important mediator of this disease. PF4 helps drive ECM in part by increasing TNF production in vitro and in vivo and T-cell trafficking. Our data is novel in many aspects. This is the first demonstration that Plasmodium infected RBC can directly activate platelets and that this platelet activating effect is dependent on platelet CD36 signaling. Furthermore, we present the first demonstration that a platelet derived chemokine, PF4, is a key mediator of the development of ECM.
PF4 stimulated macrophage and T-cell TNFα production. TNFα and LTα are key pro-inflammatory molecules that have been closely linked to the development of CM (Gimenez et al., 2003; Lou et al., 1997; Wassmer et al., 2006). TNFα contributes to the increased expression of cell adhesion molecules that can localize immune cells to the cerebral vasculature. Furthermore, TNFα has been importantly demonstrated to increase platelet adhesion to cerebral vasculature (von Zur Muhlen et al., 2008). This suggests a self-perpetuating cycle involving TNF and other inflammatory mediators of platelet activation, immune stimulation, cerebral vascular inflammation, and further platelet cerebral localization and vascular damage.
Our study helps to extend the role of platelets in cerebral malaria beyond vascular contact dependent mediated effects, to a systemic pro-inflammatory role. There has recently been a rapidly expanding appreciation for the wide range of platelet immune functions and their role in directing immune responses. Platelets are now known to contribute significantly to the pathogenesis of diverse diseases such as atherosclerosis (Huo and Ley, 2004), transplant vasculopathy (Morrell et al., 2008; Morrell et al., 2007), and T-cell recruitment in viral hepatitis (Lang et al., 2008). Platelets can also alter their inflammatory molecule content by rapidly synthesizing protein products, such as IL-1β, in response to stimulation (Denis et al., 2005; Zimmerman and Weyrich, 2008). This indicates that platelets may alter their inflammatory phenotype under different environmental conditions. More work is needed to determine if platelets are not only activated in malaria infection, but also alter their inflammatory phenotype in response to Plasmodium infected RBC.
Our studies indicate that platelet derived PF4 may be a key player in ECM T-cell recruitment. Although PF4 is known to be associated with thrombosis and the pathogenesis of heparin induced thrombocytopenia (Eslin et al., 2004) other functions of PF4 have remained somewhat elusive. PF4 is only now beginning to be associated with other vascular disease processes that have an inflammatory basis, such as atherosclerosis (Sachais et al., 2007). Our findings demonstrate an important role for PF4 in ECM that may extend to other vascular diseases with an immune basis.
CXCR3, has received recent much recent attention for its role in T-cell trafficking in ECM (Campanella et al., 2008; Miu et al., 2008; Van den Steen et al., 2008). These important studies highlighted the critical role of CXCR3 chemokine ligands and CXCR3 signaling in the pathogenesis of CM. Our work demonstrates an important connection in ECM between innate platelet driven immune responses and T-cell responses mediated by PF4 increased CXCR3 signaling and cerebral localization.
The timing of anti-platelet therapeutic intervention in ECM is important; we have found that platelet depletion or inhibition on day 3 p.i. offers no help in prolonging the survival of infected mice. This emphasizes the importance of reducing early platelet activation to limit a deleterious exaggerated immune response. Our data is also the first to demonstrate that Plasmodium infected RBC can directly activate platelets, potentially accounting for the need for early intervention as RBC first begin to express Plasmodium derived molecules. Using the ECM mouse model we have also demonstrated that platelet derived inflammatory molecules such as PF4 contribute greatly to the development of cerebral malaria. Although the P. berghei ANKA mouse model recapitulates most aspects of human CM (van der Heyde et al., 2006), mouse and human immune responses can differ somewhat and further study must be undertaken to determine the role of PF4 in the development of cerebral malaria.
An appreciation for platelets as mediators of host immune responses is a relatively new concept, particularly in the context of microbial blood borne pathogens. Our work demonstrates a novel role for platelets in directing an immune response to Plasmodium that leads to the development of cerebral malaria. Future study is necessary to discern the potential for simple platelet directed therapeutic strategies in the prevention of cerebral malaria.
Experimental Procedures
Reagents
All ELISA kits, recombinant mouse PF4, and CXCR3 antibody were purchased from R and D systems. Anti-P-selectin, PAC-1, CD4 and CD8 antibodies were purchased from BD Pharmingen. Fibrinogen (Sigma) was FITC (Sigma) labeled using described methods (Xia et al., 1996). Ant-CD3 and CD28 antibodies were purchased from Ebioscience. Fab CD36 antibody fragments were prepared using a Pierce ImmunoPure Fab Preparation kit according to manufacturer instructions and CD36 blocking antibody (Santa Cruz).
Ex Vivo platelet activation
Percoll purified washed P. falciparum (clone 3D7) trophozoite stage infected RBC (approximately 94% parasitemia) or control RBC from the same O+ donor were incubated for 30 mins with platelet rich plasma (PRP) in an approximately physiologic cell ratio of 10:1 (RBC:platelet) and final volume ratio of 1:1. FITC-PAC-1 antibody (BD Pharmingen) was then added for 20 mins prior to fixation with 1% formalin. Washed mouse platelets were isolated as described prior (Morrell et al., 2005) and mouse plasma isolated from blood obtained by retro-orbital bleed into EDTA coated tubes (BD Pharmingen) and centrifugation. Plasma was incubated with washed platelets for 30 mins prior to incubation with FITC anti-P-selectin antibody. RBC ghosts were prepared as described by others (Heinz and Hoffman, 1965).
Mouse studies
Mice were on a C57Bl6 background greater than 10 generations. Mice were infected with P. berghei ANKA by injection of approximately 0.5×106 parasites intraperitoneal (IP). Mice were platelet depleted by injection of 100 µg of anti-GPIb antibody (Emfret) IP as described prior(Kisucka et al., 2006) on day 1 post infection. Clopidogrel (Plavix) was injected IP daily (10 mg/kg) and acetylsalicylic acid (ASA) was provided in the drinking water at 200 mg/L such that mice receive approximately 500 µg/day. Each treatment was initiated 1 day p.i. Recombinant mouse PF4 was purchased from R&D Systems and injected intravenously via the retro-orbital plexus.
In Vitro T-cell Culture
For in vitro T-cell studies, wells were coated with 0.5 µg/mL of anti-CD3 antibody overnight and washed prior to plating splenocytes. Anti-CD28 antibody (0.5 µg/mL) and recombinant PF4 were added and the media changed replacing PF4 on day 2. We isolated peritoneal macrophages from C57Bl6/J mice by peritoneal lavage with saline and plated 1×106 cells/well in a 24 well plate for 2 hrs. Non-adherent cells were washed away to eliminate non-macrophage cells in the culture. Cells were then incubated with 0 or 1 µg/mL of PF4 for 48 hrs and the culture supernatant collected and TNFα production measured by an ELISA (BioLegend).
Immunohistochemistry was performed using prior published methods, reagents and protocols (Yamakuchi et al., 2007).
Brain Mononuclear Cell Isolation
Brain mononuclear cells were isolated by removing brains on day 5 p.i. and a single cell suspension obtained by grinding the tissue and mincing it with a razor blade in Dulbecco modified Eagle medium with 10% fetal bovine serum while on ice. Cell suspensions were placed in 15-ml conical tubes and Percoll (Sigma) added to a final concentration of 30%. One mL of Percoll was underlaid and cells spun at 1,300 × g for 30 min at 4°C. Cells at the interface were isolated, washed twice, resuspended in Tyrode’s buffer, and labeled with anti-CD4 and CD8 antibodies for flow cytometry.
Blood Brain Barrier Studies
Blood brain barrier permeability was determined by injecting 200 µL of 2% Evans Blue dye IV into mice on day 5 p.i. One hour later brains were harvested, incubated in 10% buffered formalin for 48 hrs and dye extravasation determined by optical density (Gramaglia et al., 2006).
Data analysis
Data are expressed as means ± standard deviation. Statistical comparisons between two groups were performed using Student's t-test. Statistics for survival curves were performed using a log-rank test.
Supplementary Material
Acknowledgements
This work was supported by a National Institutes of Health (NIH) grant to C.N.M (5K08HL074945). NCCR grant GCRC RR0052 supported the culturing of P. falciparum for the production of malaria parasites. Studies were also supported by the Johns Hopkins Malaria Research Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Boehlen F, Clemetson KJ. Platelet chemokines and their receptors: what is their relevance to platelet storage and transfusion practice? Transfus Med. 2001;11:403–417. doi: 10.1046/j.1365-3148.2001.00340.x. [DOI] [PubMed] [Google Scholar]
- Campanella GS, Tager AM, El Khoury JK, Thomas SY, Abrazinski TA, Manice LA, Colvin RA, Luster AD. Chemokine receptor CXCR3 and its ligands CXCL9 and CXCL10 are required for the development of murine cerebral malaria. Proc Natl Acad Sci U S A. 2008;105:4814–4819. doi: 10.1073/pnas.0801544105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravorty SJ, Craig A. The role of ICAM-1 in Plasmodium falciparum cytoadherence. Eur J Cell Biol. 2005;84:15–27. doi: 10.1016/j.ejcb.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Denis MM, Tolley ND, Bunting M, Schwertz H, Jiang H, Lindemann S, Yost CC, Rubner FJ, Albertine KH, Swoboda KJ, et al. Escaping the nuclear confines: signal-dependent pre-mRNA splicing in anucleate platelets. Cell. 2005;122:379–391. doi: 10.1016/j.cell.2005.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eslin DE, Zhang C, Samuels KJ, Rauova L, Zhai L, Niewiarowski S, Cines DB, Poncz M, Kowalska MA. Transgenic mice studies demonstrate a role for platelet factor 4 in thrombosis: dissociation between anticoagulant and antithrombotic effect of heparin. Blood. 2004;104:3173–3180. doi: 10.1182/blood-2003-11-3994. [DOI] [PubMed] [Google Scholar]
- Essien EM, Ebhota MI. Platelet secretory activities in acute malaria (Plasmodium falciparum) infection. Acta Haematol. 1983;70:183–188. doi: 10.1159/000206720. [DOI] [PubMed] [Google Scholar]
- Gawaz M, Langer H, May AE. Platelets in inflammation and atherogenesis. J Clin Invest. 2005;115:3378–3384. doi: 10.1172/JCI27196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez F, Barraud de Lagerie S, Fernandez C, Pino P, Mazier D. Tumor necrosis factor alpha in the pathogenesis of cerebral malaria. Cell Mol Life Sci. 2003;60:1623–1635. doi: 10.1007/s00018-003-2347-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramaglia I, Sobolewski P, Meays D, Contreras R, Nolan JP, Frangos JA, Intaglietta M, van der Heyde HC. Low nitric oxide bioavailability contributes to the genesis of experimental cerebral malaria. Nat Med. 2006;12:1417–1422. doi: 10.1038/nm1499. [DOI] [PubMed] [Google Scholar]
- Grau GE, Lou J. TNF in vascular pathology: the importance of platelet-endothelium interactions. Res Immunol. 1993;144:355–363. doi: 10.1016/s0923-2494(93)80080-i. [DOI] [PubMed] [Google Scholar]
- Grau GE, Mackenzie CD, Carr RA, Redard M, Pizzolato G, Allasia C, Cataldo C, Taylor TE, Molyneux ME. Platelet accumulation in brain microvessels in fatal pediatric cerebral malaria. J Infect Dis. 2003;187:461–466. doi: 10.1086/367960. [DOI] [PubMed] [Google Scholar]
- Grau GE, Tacchini-Cottier F, Vesin C, Milon G, Lou JN, Piguet PF, Juillard P. TNF-induced microvascular pathology: active role for platelets and importance of the LFA-1/ICAM-1 interaction. Eur Cytokine Netw. 1993;4:415–419. [PubMed] [Google Scholar]
- Hansen DS, Bernard NJ, Nie CQ, Schofield L. NK cells stimulate recruitment of CXCR3+ T cells to the brain during Plasmodium berghei-mediated cerebral malaria. J Immunol. 2007;178:5779–5788. doi: 10.4049/jimmunol.178.9.5779. [DOI] [PubMed] [Google Scholar]
- Heinz E, Hoffman JF. Phosphate Incorporation And Na, K-Atpase Activity In Human Red Blood Cell Ghosts. J Cell Physiol. 1965;65:31–43. doi: 10.1002/jcp.1030650106. [DOI] [PubMed] [Google Scholar]
- Hunt NH, Grau GE. Cytokines: accelerators and brakes in the pathogenesis of cerebral malaria. Trends Immunol. 2003;24:491–499. doi: 10.1016/s1471-4906(03)00229-1. [DOI] [PubMed] [Google Scholar]
- Huo Y, Ley KF. Role of platelets in the development of atherosclerosis. Trends Cardiovasc Med. 2004;14:18–22. doi: 10.1016/j.tcm.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Idro R, Ndiritu M, Ogutu B, Mithwani S, Maitland K, Berkley J, Crawley J, Fegan G, Bauni E, Peshu N, et al. Burden, features, and outcome of neurological involvement in acute falciparum malaria in Kenyan children. Jama. 2007;297:2232–2240. doi: 10.1001/jama.297.20.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisucka J, Butterfield CE, Duda DG, Eichenberger SC, Saffaripour S, Ware J, Ruggeri ZM, Jain RK, Folkman J, Wagner DD. Platelets and platelet adhesion support angiogenesis while preventing excessive hemorrhage. Proc Natl Acad Sci U S A. 2006;103:855–860. doi: 10.1073/pnas.0510412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Rauova L, Bailey M, Sola-Visner MC, Kowalska MA, Poncz M. Platelet factor 4 is a negative autocrine in vivo regulator of megakaryopoiesis: clinical and therapeutic implications. Blood. 2007;110:1153–1160. doi: 10.1182/blood-2007-01-067116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang PA, Contaldo C, Georgiev P, El-Badry AM, Recher M, Kurrer M, Cervantes-Barragan L, Ludewig B, Calzascia T, Bolinger B, et al. Aggravation of viral hepatitis by platelet-derived serotonin. Nat Med. 2008 doi: 10.1038/nm1780. [DOI] [PubMed] [Google Scholar]
- Lasagni L, Francalanci M, Annunziato F, Lazzeri E, Giannini S, Cosmi L, Sagrinati C, Mazzinghi B, Orlando C, Maggi E, et al. An alternatively spliced variant of CXCR3 mediates the inhibition of endothelial cell growth induced by IP-10, Mig, and I-TAC, and acts as functional receptor for platelet factor 4. J Exp Med. 2003;197:1537–1549. doi: 10.1084/jem.20021897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou J, Donati YR, Juillard P, Giroud C, Vesin C, Mili N, Grau GE. Platelets play an important role in TNF-induced microvascular endothelial cell pathology. Am J Pathol. 1997;151:1397–1405. [PMC free article] [PubMed] [Google Scholar]
- Miu J, Mitchell AJ, Muller M, Carter SL, Manders PM, McQuillan JA, Saunders BM, Ball HJ, Lu B, Campbell IL, Hunt NH. Chemokine gene expression during fatal murine cerebral malaria and protection due to CXCR3 deficiency. J Immunol. 2008;180:1217–1230. doi: 10.4049/jimmunol.180.2.1217. [DOI] [PubMed] [Google Scholar]
- Morrell CN, Matsushita K, Chiles K, Scharpf RB, Yamakuchi M, Mason RJ, Bergmeier W, Mankowski JL, Baldwin WM, 3rd, Faraday N, Lowenstein CJ. Regulation of platelet granule exocytosis by S-nitrosylation. Proc Natl Acad Sci U S A. 2005;102:3782–3787. doi: 10.1073/pnas.0408310102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrell CN, Murata K, Swaim AM, Mason E, Martin TV, Thompson LE, Ballard M, Fox-Talbot K, Wasowska B, Baldwin WM., 3rd In vivo platelet-endothelial cell interactions in response to major histocompatibility complex alloantibody. Circ Res. 2008;102:777–785. doi: 10.1161/CIRCRESAHA.107.170332. [DOI] [PubMed] [Google Scholar]
- Morrell CN, Sun H, Swaim AM, Baldwin WM., 3rd Platelets an inflammatory force in transplantation. Am J Transplant. 2007;7:2447–2454. doi: 10.1111/j.1600-6143.2007.01958.x. [DOI] [PubMed] [Google Scholar]
- Mueller A, Meiser A, McDonagh EM, Fox JM, Petit SJ, Xanthou G, Williams TJ, Pease JE. CXCL4-induced migration of activated T lymphocytes is mediated by the chemokine receptor CXCR3. J Leukoc Biol. 2008;83:875–882. doi: 10.1189/jlb.1006645. [DOI] [PubMed] [Google Scholar]
- Podrez EA, Byzova TV, Febbraio M, Salomon RG, Ma Y, Valiyaveettil M, Poliakov E, Sun M, Finton PJ, Curtis BR, et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med. 2007;13:1086–1095. doi: 10.1038/nm1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachais BS, Higazi AA, Cines DB, Poncz M, Kowalska MA. Interactions of platelet factor 4 with the vessel wall. Semin Thromb Hemost. 2004;30:351–358. doi: 10.1055/s-2004-831048. [DOI] [PubMed] [Google Scholar]
- Sachais BS, Turrentine T, Dawicki McKenna JM, Rux AH, Rader D, Kowalska MA. Elimination of platelet factor 4 (PF4) from platelets reduces atherosclerosis in C57Bl/6 and apoE−/− mice. Thromb Haemost. 2007;98:1108–1113. [PubMed] [Google Scholar]
- Tripathi AK, Sullivan DJ, Stins MF. Plasmodium falciparum-infected erythrocytes increase intercellular adhesion molecule 1 expression on brain endothelium through NF-kappaB. Infect Immun. 2006;74:3262–3270. doi: 10.1128/IAI.01625-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi AK, Sullivan DJ, Stins MF. Plasmodium falciparum-infected erythrocytes decrease the integrity of human blood-brain barrier endothelial cell monolayers. J Infect Dis. 2007;195:942–950. doi: 10.1086/512083. [DOI] [PubMed] [Google Scholar]
- Van den Steen PE, Deroost K, Aelst IV, Geurts N, Martens E, Struyf S, Nie CQ, Hansen DS, Matthys P, Damme JV, Opdenakker G. CXCR3 determines strain susceptibility to murine cerebral malaria by mediating T lymphocyte migration toward IFN-gamma-induced chemokines. Eur J Immunol. 2008;38:1082–1095. doi: 10.1002/eji.200737906. [DOI] [PubMed] [Google Scholar]
- van der Heyde HC, Gramaglia I, Sun G, Woods C. Platelet depletion by anti-CD41 (alphaIIb) mAb injection early but not late in the course of disease protects against Plasmodium berghei pathogenesis by altering the levels of pathogenic cytokines. Blood. 2005;105:1956–1963. doi: 10.1182/blood-2004-06-2206. [DOI] [PubMed] [Google Scholar]
- van der Heyde HC, Nolan J, Combes V, Gramaglia I, Grau GE. A unified hypothesis for the genesis of cerebral malaria: sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends Parasitol. 2006;22:503–508. doi: 10.1016/j.pt.2006.09.002. [DOI] [PubMed] [Google Scholar]
- von Hundelshausen P, Koenen RR, Sack M, Mause SF, Adriaens W, Proudfoot AE, Hackeng TM, Weber C. Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood. 2005;105:924–930. doi: 10.1182/blood-2004-06-2475. [DOI] [PubMed] [Google Scholar]
- von Zur Muhlen C, Sibson NR, Peter K, Campbell SJ, Wilainam P, Grau GE, Bode C, Choudhury RP, Anthony DC. A contrast agent recognizing activated platelets reveals murine cerebral malaria pathology undetectable by conventional MRI. J Clin Invest. 2008;118:1198–1207. doi: 10.1172/JCI33314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassmer SC, Combes V, Candal FJ, Juhan-Vague I, Grau GE. Platelets potentiate brain endothelial alterations induced by Plasmodium falciparum. Infect Immun. 2006;74:645–653. doi: 10.1128/IAI.74.1.645-653.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassmer SC, Combes V, Grau GE. Pathophysiology of cerebral malaria: role of host cells in the modulation of cytoadhesion. Ann N Y Acad Sci. 2003;992:30–38. doi: 10.1111/j.1749-6632.2003.tb03135.x. [DOI] [PubMed] [Google Scholar]
- Wassmer SC, Lepolard C, Traore B, Pouvelle B, Gysin J, Grau GE. Platelets reorient Plasmodium falciparum-infected erythrocyte cytoadhesion to activated endothelial cells. J Infect Dis. 2004;189:180–189. doi: 10.1086/380761. [DOI] [PubMed] [Google Scholar]
- Xia Z, Wong T, Liu Q, Kasirer-Friede A, Brown E, Frojmovic MM. Optimally functional fluorescein isothiocyanate-labelled fibrinogen for quantitative studies of binding to activated platelets and platelet aggregation. Br J Haematol. 1996;93:204–214. doi: 10.1046/j.1365-2141.1996.445980.x. [DOI] [PubMed] [Google Scholar]
- Yamakuchi M, Kirkiles-Smith NC, Ferlito M, Cameron SJ, Bao C, Fox-Talbot K, Wasowska BA, Baldwin WM, 3rd, Pober JS, Lowenstein CJ. Antibody to human leukocyte antigen triggers endothelial exocytosis. Proc Natl Acad Sci U S A. 2007;104:1301–1306. doi: 10.1073/pnas.0602035104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman GA, Weyrich AS. Signal-dependent protein synthesis by activated platelets: new pathways to altered phenotype and function. Arterioscler Thromb Vasc Biol. 2008;28:s17–s24. doi: 10.1161/ATVBAHA.107.160218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.