

Abstract
Each year in the United States approximately 700,000 individuals are afflicted with a stroke and currently there are almost 2 million survivors of stroke living in the US with prolonged disability. In China 1.5 million people die from stroke each year and in developed nations stroke is the third leading cause of death, only surpassed by heart disease and cancer. Brain injury following stroke results from the complex interplay of multiple pathways including excitotoxicity, acidotoxicity, ionic imbalance, oxidative/nitrative stress, inflammation, apoptosis and peri-infarct depolarization. Here, we discuss the underlying pathophysiology of this devastating disease and reveal the intertwined pathways that are the target of therapeutic intervention.
Introduction
Each year in the United States approximately 700,000 individuals are afflicted with a stroke. Currently there are approximately 2 million survivors of stroke living in the US with prolonged disability, many unable to work or resume personal relationships. In China 1.5 million people die from stroke each year and in developed nations stroke is the third leading cause of death, only surpassed by heart disease and cancer. In the US health care costs reach 62 billion dollars annually. The economic, social and psychological costs of stroke are substantial (Fisher and Bogousslavsky 1998; Pancioli, Broderick et al. 1998; Stephenson 1998; Caplan 2000; Rosamond, Flegal et al. 2007; Flynn et al, this issue). There are very few treatments for stroke and the development of new therapeutics is imperative. Here, we discuss the underlying pathophysiology of this devastating disease and reveal the intertwined pathways that are the target of therapeutic intervention.
Stroke Pathophysiology
Stroke can be subdivided into two categories, ischemic and hemorrhagic. Ischemic strokes are more prevalent than hemorrhagic making up approximately 87% of all cases and have been the target of most drug trials (Rosamond, Flegal et al. 2007). A thrombosis, an embolism or systemic hypo-perfusion, all of which result in a restriction of blood flow to the brain, can cause an ischemic stroke, which results in insufficient oxygen and glucose delivery to support cellular homoestasis. This elicits multiple processes that lead to cell death: excitotoxicity, acidotoxicity and ionic imbalance, oxidative/nitrative stress, inflammation, apoptosis and peri-infarct depolarization (Gonzalez, Hirsch et al. 2006).
Each of the above pathophysiological processes has a distinct time frame, some occurring over minutes, others over hours and days. These processes share overlapping and redundant features and cause injury to neurons, glia and endothelial cells. Within the core of the ischemic territory, where blood flow is most severely restricted, excitotoxic and necrotic cell death occurs within minutes. In the periphery of the ischemic area, where collateral blood flow can buffer the full effects of the stroke, the degree of ischemia and the timing of reperfusion determine the fate of individual cells. In this ischemic penumbra cell death occurs less rapidly via active cell death mechanisms such as apoptosis. Targeting these mechanisms provides therapeutic opportunity (Gonzalez, Hirsch et al. 2006).
Excitotoxcity, Acidotoxicity and Ionic Imbalance
The human brain comprises 2% of body weight but requires 20% of total oxygen consumption (Edvinsson and Krause 2002). The brain requires this large amount of oxygen to generate sufficient ATP by oxidative phosphorylation to maintain and restore ionic gradients. One estimate suggests that the Na+/K+ATPase found on the plasma membrane of neurons, consumes 70% of the energy supplied to the brain (Edvinsson and Krause 2002). This ion pump maintains the high intracellular K+ concentration and the low intracellular Na+ concentration necessary for the propagation of action potentials. After global ischemia, mitochondrial inhibition of ATP synthesis leads to ATP being consumed within two minutes, this causes neuronal plasma membrane depolarization, release of potassium into the extracellular space and entry of sodium into cells (Caplan 2000). Energy failure also prevents the plasma membrane Ca2+ ATPase from maintaining the very low concentrations of calcium that are normally present within each cell.
The extracellular calcium concentration is approximately 1.2mM and most cellular processes regulated by calcium have a Km value in the range of 0.1 to 1μM. During ischemia intracellular calcium levels rise to 50-100μM, activating many, if not all calcium dependent proteases, lipases and DNAses (Edvinsson and Krause 2002). Activation of these enzymes causes many cells in the ischemic core to die from simple catabolism. Because no ATP is available for the re-synthesis of cellular constituents these catabolic enzymes cause the necrosis of essential cellular structures.
Membrane depolarization also leads to neurotransmitter release, with the release of the excitatory neurotransmitter glutamate playing a critical role in ischemic pathology. A large concentration gradient of glutamate is maintained across the plasma membrane by sodium dependent glutamate transporters located on presynaptic and postsynaptic membranes. The synaptic glutamate concentration is in the micromolar range, whereas the cytosolic concentration of glutamate is approximately 10mM (Hsu 1998). Membrane depolarization and accumulation of sodium inside cells during ischemia causes reversal of glutamate transporters and allows glutamate to exit cells along its concentration gradient.
The effect of an increase in synaptic glutamate concentration is the activation of N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. NMDA receptors are calcium permeable and the opening of these channels leads to further membrane depolarization and greater calcium influx, exacerbating intracellular calcium overload (excitotoxicity) (Olney 1969). AMPA receptors are not normally calcium permeable by virtue of their GluR2 subunit, however, this subunit is reduced after ischemia increasing the calcium permeability of these receptors by up to 18 fold, allowing AMPA receptors to contribute to delayed calcium dependent cell death (Liu, Liao et al. 2006; Peng, Zhong et al. 2006). Blocking glutamate binding sites on NMDA and AMPA receptors has repeatedly been shown to provide robust neuroprotection in models of focal ischemia, where receptor blockade is thought to prevent calcium entry from reaching a toxic threshold (Hsu 1998).
Metabotropic glutamate (mGlu) receptors also contribute to excitotoxicity. MGlu receptors are G-protein coupled receptors that modulate excitatory synaptic transmission. There are 8 subtypes divided into 3 groups. The group I mGlu receptors (mGlu1 and mGlu5) are predominantly found at the postsynaptic membrane of glutamatergic synapses where they increase neuronal excitability by modulating NMDA and AMPA receptors. Evidence that these receptors enhance the induction and progression of excitotoxic neuronal death is provided by the finding that pharmacologic blockade of group I mGlu receptors provides neuroprotection in in vitro and in vivo models of ischemia (Bruno, Battaglia et al. 2001).
MGlu1 receptors are expressed in GABAergic neurons where their activation suppresses GABA release. Therefore mGlu1 antagonists may be neuroprotective by enhancing the release of the inhibitory neurotransmitter GABA. MGlu5 receptors are physically and functionally connected to NMDA receptors. Therefore, mGlu5 antagonists may limit excitotoxicity by reducing NMDA receptor activation (Pellegrini-Giampietro 2003).
Group II (mGlu2 and mGlu3) and group III (mGlu4, mGlu6, mGlu7, mGlu8) mGlu receptors are predominantly found at presynaptic terminals where they inhibit the release of glutamate. Group II receptors are also widely expressed on astrocytes. Both group II and group III receptor agonists have been found to be neuroprotective by limiting the induction of excitotoxicty, however, group II receptor agonists also confer neuroprotection by increasing production of neurotrophic factors such as nerve growth factor and TGF-β in astrocytes (Bruno, Battaglia et al. 2001).
Concurrent to the induction of excitotoxicity, calcium overload is further exacerbated by acidosis, one of the hallmark neurochemical elements of the anaerobic metabolism of ischemia. Hyperglycemia increases lactate in the ischemic environment further depressing pH. Dissociated protons activate sodium selective acid sensing ion channels (ASICs) that are permeable to calcium. There are 4 ASIC genes encoding 6 polypeptides (ASIC1a, ASIC1b, ASIC2a, ASIC2b, ASIC3 and ASIC4), each of which has a distinct pH sensitivity. The pH0.5 of the calcium permeable homomeric ASIC1a channel is 6.2 and because the pH in ischemic brain falls to 6.0 to 6.5 it is likely that in ischemia this channel opens and allows further calcium entry into the cell (acidotoxicity) (Simon 2006). This phenomenon is glutamate independent, thus it is not prevented by administration of NMDA antagonists. However, administration of the selective ASIC1a blocker PcTx1 can prevent ASIC1a activation and has been shown to reduce lesion volume in experimental stroke (Pignataro, Simon et al. 2007).
Peri-infarct Depolarizations
Cortical spreading depression (CSD) is a self-propagating wave of electrochemical activity that progresses through cortical tissue in intact brain. CSD causes sustained (1-5 minutes) cellular depolarization, depressed neuro-electrical activity, increased glutamate release and loss of membrane ionic gradients (Gonzalez, Leroux et al. 1992). Peri-infarct depolarizations (PIDs) are spontaneous waves of depolarization with all of the characteristic features of CSD that propagate through the penumbra following focal stroke. PIDs may be caused by the release of potassium and excitatory amino acids from the ischemic core. Although CSD in the normally perfused brain does not lead to cell death, recurrent PIDs in the ischemic brain are associated with increased ischemic injury.
Repeated depolarization in the penumbra may mediate tissue damage by allowing calcium to accumulate within neurons. A critical threshold of calcium could be reached in the case of PID due to the compromised energy supply of the tissue, thus causing damage in the case of PID but without evidence of lasting damage in the case of CSD.
PIDs are known to occur in mouse, rat and cat stroke models, where the incidence and duration of spreading depression correlates with infarct maturation (Gill, Andine et al. 1992; Strong, Smith et al. 2000). Recently Fabricius and colleagues demonstrated the existence of PIDs in the acutely injured human brain, which suggests that inhibition of spreading depression using a therapeutic approach such as hypothermia or glutamate receptor antagonism could be an important strategy to limit development of ischemic injury within the penumbra (Chen, Chopp et al. 1993; Fabricius, Fuhr et al. 2006).
Oxidative and Nitrative Stress
High levels of intracellular Ca2+, Na+ and ADP cause mitochondria to produce deleterious levels of reactive oxygen species. Unlike other organs the brain is especially vulnerable to reactive oxygen species due to neurons having relatively low levels of endogenous antioxidants (Coyle and Puttfarcken 1993). Overly abundant oxygen radicals cause the destruction of cellular macromolecules and participate in signaling mechanisms that result in apoptotic cell death (Halliwell 1994; Sugawara and Chan 2003). Ischemia activates nitric oxide synthase (NOS) and increases the generation of nitric oxide (NO), which combines with superoxide to produce peroxynitrite, a potent oxidant. The production of NO and oxidative stress is also linked to over-activation of poly(ADP-ribose)polymerase-1 (PARP-1), a DNA repair enzyme. In response to DNA strand breaks PARP-1 catalyzes the transformation of β-nicotinamide adenine dinucleotide (NAD+) into nicotinamide (NA) and long polymers of poly(ADP-ribose). When PARP-1 is over-activated it depletes cells of NAD+, impairing NAD+ dependent processes such as anaerobic glycolysis and mitochondrial respiration, which leads to ATP starvation, energy failure and neuronal death (Gonzalez, Hirsch et al. 2006).
Following reperfusion there is a surge in production of superoxide, NO and peroxynitrate. Formation of these radicals in the vicinity of blood vessels plays an important role in reperfusion-induced injury. These radicals activate matrix metalloproteases (MMPs), which degrade collagen and laminins in the basal lamina, which disrupts the integrity of the vascular wall and increases blood brain barrier (BBB) permeability. Oxidative and nitrative stress also triggers recruitment and migration of neutrophils and other leukocytes to the cerebral vasculature, which release enzymes that further increase basal lamina degradation and vascular permeability. These events can lead to parenchymal hemorrhage, vasogenic brain edema and neutrophil infiltration into the brain (Crack and Taylor 2005).
Thrombolytic therapy has a 3 hour time window of efficacy. Part of the reason for this limited time window is that the surge in production of free radicals associated with delayed reperfusion brings a second wave of oxidative and nitrative stress that increases the risk of brain hemorrhage and edema. Combined administration of thrombolytic agents with free radical or peroxynitrite scavengers, or NO synthase inhibitors is a potential strategy for reducing reperfusion-induced injury and extending the time window available for thrombolysis (Crack and Taylor 2005).
Inflammation
Inflammation contributes to stroke related brain injury. However, the effect of individual components of the inflammatory cascade can be beneficial depending on the stage of tissue injury, the magnitude of the response and whether the inflammatory component also activates neuroprotective pathways (Bruce, Boling et al. 1996; Nawashiro, Brenner et al. 2000; Zhang, Zhang et al. 2000). The inflammatory response is a composite process that involves many different cell types, inflammatory mediators and extracellular receptors. Below, the inflammatory response to stroke is sub-divided into the cellular response, the cytokine response and the response to toll-like receptor (TLR) activation.
i.) The Cellular Inflammatory Response
Stroke causes neutrophilia, lymphocytopenia and an increase in the number of circulating monocytes (Ross, Hurn et al. 2007). Although the increase in neutrophil and monocyte number is likely to contribute to ischemic damage, this change is also evident in patients that have experienced a transient ischemic attack (TIA), which indicates that these cellular elements alone are insufficient by themselves to mediate damage (Ross, Hurn et al. 2007). Instead, access to the brain may be the key determinant of whether this change contributes to ischemic injury.
Neutrophils accumulate in the brain as early as 30 minutes after permanent middle cerebral artery occlusion (MCAO). Transmigration is mediated by three classes of cell adhesion molecules: selectins, integrins, and immunoglobulins, the expression of which is regulated both intracellularly and by cytokine signaling (Huang, Upadhyay et al. 2006). The recruitment of neutrophils to ischemic brain begins with neutrophil rolling on activated endothelial blood vessel walls, mediated by selectins, followed by neutrophil activation and adherence, mediated by integrins and immunoglobins. When adhered to cerebral blood vessel walls, neutrophils transmigrate into the cerebral parenchyma, a process facilitated by blood brain barrier (BBB) disruption. The recruitment of neutrophils can obstruct the microcirculation and prevent complete restoration of cerebral blood flow after reperfusion. This blockage may cause further tissue damage after ischemia and is described as the ischemic no-reflow phenomenon (Huang, Upadhyay et al. 2006).
Once neutrophils penetrate into ischemic brain they cause tissue damage by releasing oxygen-free radicals and proteolytic enzymes. Neutrophil depletion, inhibition of neutrophil adhesion, and inhibition of neutrophil function are all strategies that have been shown to reduce infarct volume and improve outcome. For example, protein kinase C has been shown to play a significant role in neutrophil adhesion, degranulation, and superoxide generation. Mice that are deficient for protein kinase C have diminished infarct volumes when subjected to transient cerebral ischemia (Chou, Choi et al. 2004; Huang, Upadhyay et al. 2006).
Lymphocytes also appear to be responsible for mediating damage in response to brain ischemia. Although lymphocytes are ordinarily excluded from the central nervous system (CNS), they appear within 24 hours in post ischemic brain (Schroeter, Jander et al. 1994). While the mechanism producing their infiltration into the brain remains unclear, it is likely that BBB disruption plays a major role in the influx, either by directly allowing free lymphocyte movement, or by leakage of brain antigens resulting in the transmigration of activated lymphocytes. It has been shown recently, using severe combined immunodeficiency (SCID) mice deficient for T and B lymphocytes, that these cells contribute to development of the lesion in the cortex. In these mice Hurn et al find that following focal cerebral ischemia, striatal infarction is not altered, suggesting that the core of an evolving infarct is not protected by a lack of T and B lymphocytes, while cortical infarct volume is reduced by as much as 40% (Hurn, Subramanian et al. 2007).
The infiltration of bone marrow derived cells into the ischemic brain persists for weeks following stroke, and while the initial infiltration leads to worsening of tissue damage and exacerbation of neurological deficits, subsequent aspects of the infiltration are beneficial. For example, the phagocytosis of debris and the release of cytokines that promote glial scar formation are critical for effective wound healing.
ii.) The Cytokine Inflammatory Response
Cytokines and chemokines contribute to stroke related brain injury (Gong, Qin et al. 1998). During ischemia, cytokines, such as IL-1, IL-6, TNF-α, TGF-β and chemokines such as CINC and MCP-1 are produced by a variety of activated cell types, including endothelial cells, microglia, neurons, platelets, leukocytes, and fibroblasts (Huang, Upadhyay et al. 2006).
Production of IL-1 is increased after permanent or transient cerebral ischemia in microglia, astrocytes, and neurons. The exact role of IL-1 in propagating tissue damage is unclear, although possible deleterious effects of IL-1 include fever, arachidonic acid release, enhancement of NMDA mediated excitotoxicity, and stimulation of nitric oxide synthesis (Huang, Upadhyay et al. 2006). An additional role of IL-1 may be recruitment and adhesion of neutrophils. IL-1 has been shown to cause up-regulation of E-selectin, ICAM-1, ICAM-2, and VCAM-1 on cerebral endothelial cells and the induction of such adhesion molecules may explain why elevated IL-1 levels after ischemia increases neutrophil infiltration (Yamasaki, Matsuo et al. 1997; Huang, Upadhyay et al. 2006). That the effects of IL-1 are deleterious was demonstrated by Garcia and Relton who showed that administration of recombinant IL-1 receptor antagonist reduces the severity of neurologic deficits and tissue necrosis in rats subjected to permanent MCAO (Garcia, Liu et al. 1995; Relton JK, Martin D et al. 1996; Huang, Wang et al. 2003). Insight into the deleterious effect of IL-1 also comes from data in rats administered recombinant human IL-1β via intracerebroventricular (icv) injection after MCAO. These rats show increased brain edema and lesion size, as well as an increased influx of neutrophils (Yamasaki Y, Shozuhara H et al. 1994; Yamasaki, Matsuura et al. 1995; Huang, Upadhyay et al. 2006).
In rats subjected to permanent MCAO, IL-6 mRNA expression is up-regulated as early as 3 hours after occlusion, peaks at 12 hours, and continues for at least 24 hours (Wang, Yue et al. 1995). The biological activity of IL-6 overlaps with those of IL-1, and data from human studies suggest a proinflammatory role for IL-6 in stroke. Peripheral blood levels of IL-6 are higher in stroke patients and detectable within a few hours of stroke onset and higher CSF and serum levels of IL-6 correlate with larger infarct size and poorer clinical outcome (Tarkowski, Rosengren et al. 1995; Huang, Upadhyay et al. 2006). However, IL-6 also has anti-inflammatory properties due to its ability to induce IL-1 receptor antagonist synthesis (Schindler, Mancilla et al. 1990; Relton JK, Martin D et al. 1996). Thus, it is unclear whether the overall effect of IL-6 is beneficial or detrimental in the context of stroke.
Up-regulation of TNF-α mRNA parallels that of IL-1 and IL-6 mRNA within the first hours after ischemia (Huang, Upadhyay et al. 2006). Both experimental and human data indicate a positive correlation between TNF-α and the extent of ischemic injury. For example, a study of 24 patients with ischemic stroke found that CSF levels of TNF-α were markedly increased within 24 hours of ischemic stroke and that the levels of CSF and serum TNF-α were positively correlated with infarct volume (Zaremba, Skrobanksi et al. 2001). Like IL-1, TNF-α induces adhesion molecule expression in cerebral endothelial cells and promotes neutrophil accumulation and transmigration. In addition TNF-α stimulates acute-phase protein production, disrupts the blood-brain barrier and stimulates the induction of other inflammatory mediators. Administration of TNF-α neutralizing antibody reduces brain injury after focal ischemia in rats and TNF-α targeted therapies hold great promise as a stroke treatment (Nawashiro, Martin et al. 1997; Nawashiro, Martin et al. 1997).
Growing evidence suggests that TGF-β plays a neuroprotective role in the pathogenesis of stroke. In rodent models of cerebral ischemia, increased expression of TGF-β mRNA is demonstrated in ischemic tissues as early as 1 to 6 hours after the ischemic event and remains elevated for up to 15 to 21 days (Wiessner, Gehrmann et al. 1993). The effects of TGF-β upon stroke volume have been studied with intracarotid and icv administration and TGF-β has been found to be neuroprotective if administered before or after the ischemic insult (McNeill, Williams et al. 1994; Huang, Upadhyay et al. 2006).
It is likely that the neuroprotective effect of TGF-β is the concerted result of the activation of several neuroprotective pathways. TGF-β1 has a concentration-dependent protective effect against neuronal injury caused by glutamate excitotoxicity in vitro, and recent evidence indicates that administration of a TGF-β1 blocking agent increases the extent of excitotoxic lesions after focal cerebral ischemia (Ruocco, Nicole et al. 1999; Huang, Upadhyay et al. 2006). Additionally, intracarotid administration of TGF-β has been shown to reduce the number of circulating neutrophils, which may ameliorate the post ischemic no-reflow state (Mori, Del Zoppo et al. 1992). Data also exists to support a role for TGF-β in diminishing ischemia-induced endothelial dysfunction (Lefer, Ma et al. 1993).
Increased expression of CINC and MCP-1 mRNA is detected in the brain of rats as early as 6 hours after permanent MCAO, reaches a maximal level at 12 hours and is decreased by 24 hours (Minami and Satoh 2003). CINC and MCP-1 expression attracts neutrophils to ischemic tissue, evidence for which comes from rodent experiments using transient ischemia, where CINC and MCP-1 are detectable in cerebral tissue before neutrophil infiltration (Huang, Upadhyay et al. 2006). In rats, administration of anti-CINC antibody decreases cerebral edema and infarction, which further supports a role for CINC in mediating neutrophil infiltration and demonstrates another therapeutic opportunity (Yamasaki, Matsuo et al. 1997).
iii.) The Role of TLRs in the Inflammatory Response
TLRs function as a first-line defense against pathogen invasion. TLRs recognize pathogen-associated molecules such as the bacterial cell wall components peptidoglycan (TLR2) and lipopolysaccharide (TLR4), as well as dsRNA (TLR3), ssRNA (TLR7), and nonmethylated Cytosine-Guanosine (CpG) DNA (TLR9). Upon activation TLRs, induce downstream signals that lead to cytokine and chemokine production, which initiates a localized inflammatory response. In the periphery, TLRs are expressed on B cells, dendritic cells, and macrophages. Within the CNS they are expressed on endothelial cells, microglia, astrocytes, oligodendrocytes, and neurons (Marsh and Stenzel-Poore 2007).
Endogenous molecules associated with tissue damage can also activate TLRs. Therefore, in addition to playing a role in pathogen detection and defense, TLRs function as sensors of tissue damage. Fibrinogen, heat shock proteins and components of the extracellular matrix activate TLR4, while host DNA and mRNA are ligands for TLR9 and TLR3, respectively. Several recent studies implicate TLR activation by endogenous ligands as a detrimental response to cerebral ischemia. Endogenous TLR ligands such as Hsp70 are upregulated in the brain following ischemia and mice lacking either TLR2 or TLR4 have significantly smaller infarcts than wild-type mice (Kinouchi, Sharp et al. 1993; Cao, Yang et al. 2007; Ziegler, Harhausen et al. 2007).
TLRs present a novel therapeutic target for stroke pretreatment. Tasaki et al show that administration of a low dose of the TLR4 agonist LPS, can induce tolerance to cerebral ischemia. LPS induced tolerance to brain ischemia has since been demonstrated in a mouse model of stroke and in a porcine model of deep hypothermic circulatory arrest (Rosenzweig, Lessov et al. 2004; Hickey, You et al. 2007). Tolerance induction appears to require a small inflammatory response to LPS, as it can be blocked by simultaneous administration of cyclohexamide, dexamethasone, and TNF-α inhibitors (Tasaki, Ruetzler et al. 1997; Bordet, Deplanque et al. 2000; Rosenzweig, Minami et al. 2007). Antecedent treatment with LPS also appears to confer protection by protecting against the cytotoxic effects of TNF-α following cerebral ischemia. Mice that have been pretreated with LPS show reduced TNF-α in the serum, decreased levels of TNFR1 and enhanced levels of neutralizing soluble TNFR1 following stroke (Rosenzweig, Minami et al. 2007). LPS induced tolerance is comparable to ischemic preconditioning, the paradigm of a brief non-injurious period of ischemia conferring neuroprotection against a subsequent bout of injurious ischemia. Both these phenomena appear to protect in part by reprogramming the genomic response to ischemic injury (Stenzel-Poore, Stevens et al. 2003).
Apoptosis
Mild ischemic injury preferentially induces cell death via an apoptotic-like mechanism rather than necrosis. Because the ischemic penumbra sustains milder injury and preserves ATP, apoptosis predominates in this region (Kerr 1965; Kerr, Wyllie et al. 1972; Gonzalez, Hirsch et al. 2006). Triggers of apoptosis include oxygen free radicals, death receptor ligation, DNA damage, protease activation and ionic imbalance.
The release of cytochrome c from the outer mitochondrial membrane plays a central role in mediating apoptosis in response to ischemia. Release of cytochrome c is caused by ionic imbalance and mitochondrial swelling or by formation of a pore in the outer mitochondrial membrane. The complex interplay of the Bcl-2 family of proteins either promotes (Bax, Bak, Bad, Bim, Bid) or prevents (Bcl-2, Bcl-XL, Bcl-w) pore formation. The pore is formed by oligomerization of Bax and/or Bak in the outer membrane, with Bax transcriptionally induced by p53, which in turn is activated by DNA damage. The anti-apoptotic Bcl-2 proteins, Bcl-2 and Bcl-XL, can form heterodimers with Bax, thereby preventing pore formation (Edwards and Dean 1977; Miyashita and Reed 1995; Hengartner 2000; Kroemer and Reed 2000; Adams and Cory 2001; Antonsson, Montessuit et al. 2001).
Bad, Bim and Bid also influence pore formation, although the exact manner by which these molecules do so is unclear. Bim is bound to dynein and actin and is released by dissociation of the cytoskeleton. Upon mobilization, Bim translocates to the mitochondrial membrane and promotes the release of cytochrome c. Bid is present in a proform in the cytosol and is cleaved by caspase 8 after TNF/Fas receptor activation. Once cleaved, Bid also translocates to the mitochondrial membrane and promotes the release of cytochrome c. In its phosphorylated form Bad is bound to protein 14-3-3. Bad is dephosphorylated by calcineurin, a calcium dependent serine/threonine phosphatase. When Bad is dephosphorylated it is released from 14-3-3 and translocates to the mitochondria where it also promotes cytochrome c release. Unlike Bax and Bak, Bid, Bad and Bim do not have the ability to directly mediate cytochrome c release by forming a pore. Instead they appear to function as sensors of cell stress that may promote apoptosis by heterodimerizing and antagonizing the function of anti-apoptotic Bcl-2/Bcl-XL and/or activating the function of Bax and Bak (Edwards and Dean 1977; Hengartner 2000; Kroemer and Reed 2000; Adams and Cory 2001).
Cytochrome c release activates downstream caspases of the intrinsic pathway through formation of the apoptosome, a complex of dATP, cytochrome c, procaspase 9 and Apaf1. Effector caspases 3 and 7 then target substrates that dismantle the cell by cleaving homeostatic, cytoskeletal, repair, metabolic, and cell signaling proteins. These caspases also cause further DNA fragmentation by activating capase-activated deoxyribonuclease (CAD) by cleaving the inhibitor protein ICAD. Caspase activation can be modulated by protein inhibitors of apoptosis (IAP) and indirectly by secondary mitochondria-derived activator of caspase (Smac/Diablo).
Activation of the extrinsic pathway of death receptors can induce caspase activation independent of the release of cytochrome c. Death receptor ligation results in activation of caspase-8 and caspase-10, which in turn can activate effector caspase 3 (Namura, Zhu et al. 1998). Activation of death receptors such as Fas/CD95, TNFR1, and the TRAIL receptor is promoted by the TNF family of ligands, including FASL, TNF, LT-alpha, LT-beta, CD40L, LIGHT, RANKL, and TRAIL, which are released as part of the inflammatory response to ischemia (Zoppo 1997; Zoppo, Ginis et al. 2000).
Susceptibility to ischemia often correlates with expression of Bcl-2. For example in the hippocampus, basal Bcl-2 immunoreactivity is high in ischemia resistant pyramidal neurons of the CA3 region, but very low in the ischemia sensitive CA1 region (Chen, Graham et al. 1995). Bcl-2 expression is also high in the brainstem where autonomic function is often preserved following ischemia and low in the selectively vulnerable neurons of the cortex (Chen, Graham et al. 1995). This correlation of Bcl-2 expression and resistance to apoptosis may be explained by recent findings that suggest in addition to physically trapping pro-apototic proteins, Bcl-2 has other properties that enable it to attenuate cell death. For example, Ellerby et al find that Bcl-2 is sensitive to redox changes and has antioxidant properties during calcium stress (Ellerby, Ellerby et al. 1996). As Bcl-2 is found in the ER, the plasma membrane, and the nuclear membrane, additional Bcl-2 functions may be revealed.
Several experimental studies, have shown that inhibition of apoptosis reduces ischemic injury (Graham and Chen 2001). For example, activation of the extracellular signaling protein kinase (ERK) pathway and the phosphatidylinositol-3 (PI3) kinase pathway has been found to be neuroprotective. These pathways activate transcription factors such as CREB and NFκB related to cell survival and phosphorylate Bax and Bad, thereby preventing the release of cytochrome c. Additionally, caspase 3 inhibitors, gene deletions of Bid, the use of peptide inhibitors and viral vector mediated gene transfer of Bcl-2 and Bcl-XL are strategies that are known to be neuroprotective (Shinoura, Satou et al. 2000; Zhao, Yenari et al. 2003; Gonzalez, Hirsch et al. 2006; Guan, Pei et al. 2006; Guan QH, Pei et al. 2006).
A summary of the most salient features of mitochondria-dependent and mitochondria-independent activation of cell death is provided in figure 1.
Figure 1. Apoptosis in Stroke.
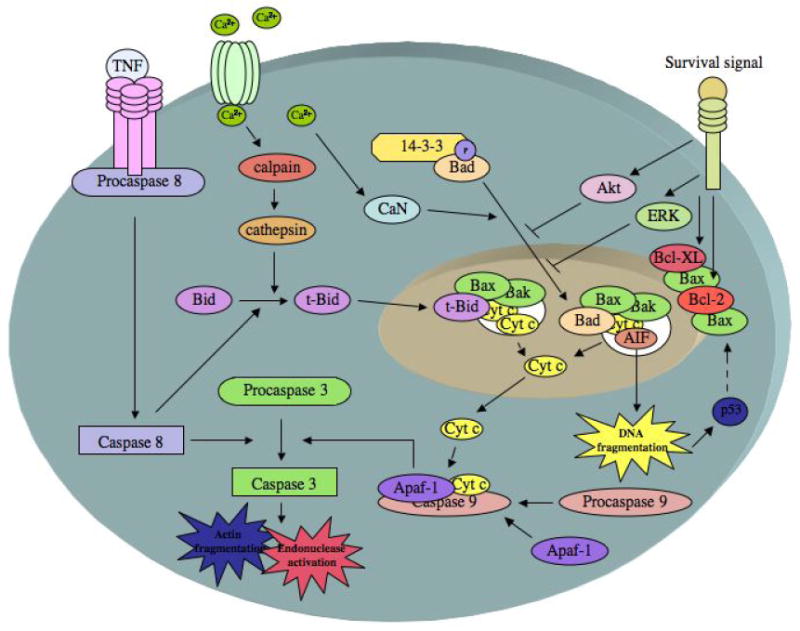
Cell death pathways relevant to apoptosis in cerebral ischemia. The release of cytochrome c (cyt c) from the mitochondria is mediated by the proapoptotic proteins Bax and/or Bak forming a pore in the mitochondrial membrane. Pore formation is facilitated by Bad and Bid. Calcium influx causes the dephosphorylation of Bad by calcineurin (CaN), which releases BAD from 14-3-3 and allows its translocation to the mitochondria. Calcium can also activate calpains, which can activate cathepsins that mediate the limited proteolysis of Bid, allowing truncated Bid (t-Bid) to translocate to the mitochondria. Caspase 8, which is activated by TNF receptor ligation, also mediates the limited proteolysis of Bid, allowing truncated Bid (t-Bid) to translocate to the mitochondria. Once present in the cytosol, cyt c forms the apoptosome complex by binding to Apaf-1 and procaspase 9. The apoptosome complex cleaves and activates caspase 3, which causes actin fragmentation, and endonuclease activation. Caspase 3 can also be activated by caspase 8. Apoptosis-inducing factor (AIF) can also be released from the pore created in the mitochondria, causing DNA degradation. DNA damage activates p53, which further increases Bax expression. The antiapoptotic proteins Bcl-XL and Bcl-2 prevent Bax mediated pore formation and cyt c release. Various survival factors also prevent pore formation and cytochrome C release by activation of Akt and ERK pathways (adapted from (Edvinsson and Krause 2002)).
Conclusion and Perspectives
Cell death following stroke results from the complex interplay of excitotoxicity, acidosis, inflammation, oxidative stress, peri-infarct depolarization and apoptosis. On the basis of the complexity of events in cerebral ischemia and the disappointing results from single agent trials, it may be unrealistic to expect that a single neuroprotective drug will demonstrate benefit in human stroke. In light of this complexity, it is likely that effective stroke therapy will require a combinatorial approach. This point is particularly salient when one considers that stroke is also a heterogeneous disorder that can be ischemic or hemorrhagic, involve small or large blood vessels and be exacerbated by hypotension, fever and hyperglycemia. Age, gender, racial background, comorbidity and concurrent medications also influence stroke; thus individual differences among patients undoubtedly influence the impact and temporal profile of ischemic injury.
Several experimental studies of combination drug therapy for stroke have shown that drugs that target different pathways can act together to have a synergistic or additive effect. For example tirilazad mesylate, a scavenger of free radicals, has been successfully combined with MK-801, a glutamate receptor antagonist. Tirilazad mesylate has also been successfully combined with insulin, which reduces hyperglycemia following acute stroke and diazepam, a GABA-ergic drug that inhibits nitric oxide formation in the brain. Nimodipine, a calcium channel blocker, has been successfully combined with MK-801. FGF, which strengthens antiapoptotic pathways in neurons has been successfully combined with citicoline, an essential intermediate in the biosynthetic pathway of neuronal membranes that inhibits membrane phosphlipases and can restore Na+/K+ ATPase function (Uematsu, Araki et al. 1991; Matsumoto, Scheller et al. 1993; Meden, Overgaard et al. 1993; Auer 1995; Lyden, Lonzo et al. 1995; Meden, Overgaard et al. 1996; Schabitz, Fuhai et al. 1999). Additional studies have shown that the effect of a single drug can be enhanced or the time window available for administration extended if applied in combination with hypothermia, a powerful therapy that targets multiple mechanisms of damage. For example, Matsumoto et al found synergistic effects of s-emopamil, which reduces brain edema, and nimopidine with hypothermia and Zhao et al used hypothermia to extend the time window available for Bcl-2 gene therapy (Zausinger, Scholler et al. 2003; Kollmar, Henninger et al. 2004; Zhao, Yenari et al. 2004; Zhao, Shimohata et al. 2005). Therefore, due to the extensive overlap that exists with the mechanisms of ischemic damage activated by stroke, combinatorial therapy may represent the most promising direction for future stroke research.
Acknowledgments
Support: This work was supported by NIH grant NS046827 (MS-P)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams J, Cory S. Life or death decisions by the Bcl-2 protein family. Trends Biochem Sci. 2001;26:61–66. doi: 10.1016/s0968-0004(00)01740-0. [DOI] [PubMed] [Google Scholar]
- Antonsson B, Montessuit S, et al. Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. Journal of Biological Chemistry. 2001;276:11615–11623. doi: 10.1074/jbc.M010810200. [DOI] [PubMed] [Google Scholar]
- Auer R. Combination therapy with U74006F (tirilazad mesylate), MK-801, insulin and diazepam in transient forebrain ischemia. Neurology Research. 1995;17:132–136. doi: 10.1080/01616412.1995.11740301. [DOI] [PubMed] [Google Scholar]
- Bordet R, Deplanque D, et al. Increase in endogenous brain superoxide dismutase as a potential mechanism of lipopolysaccharide-induced brain ischemic tolerance. J Cereb Blood Flow Metab. 2000;20:1190–6. doi: 10.1097/00004647-200008000-00004. [DOI] [PubMed] [Google Scholar]
- Bruce AJ, Boling W, et al. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nature Medicine. 1996;2:788–794. doi: 10.1038/nm0796-788. [DOI] [PubMed] [Google Scholar]
- Bruno V, Battaglia G, et al. Metabotropic glutamate receptor subtypes as targets for neuroprotective drugs. Journal of Cerebral Blood Flow & Metabolism. 2001;21:1013–1033. doi: 10.1097/00004647-200109000-00001. [DOI] [PubMed] [Google Scholar]
- Cao C, Yang Q, et al. Reduced cerebral ischemia- reperfusion injury in Toll-like receptor 4 deficient mice. Bioch Biophys Res Comm. 2007;353:509–514. doi: 10.1016/j.bbrc.2006.12.057. [DOI] [PubMed] [Google Scholar]
- Caplan L. A Clinical Approach. Butterworth Heinemann; 2000. Caplan's Stroke. [Google Scholar]
- Chen J, Graham S, et al. Bcl-2 is expressed in neurons that survive focal ischemia in the rat. Neuroreport. 1995;6:394–398. doi: 10.1097/00001756-199501000-00040. [DOI] [PubMed] [Google Scholar]
- Chen Q, Chopp M, et al. Temperature modulation of cerebral depolarization during focal cerebral ischemia in rats: correlation with ischemic injury. Journal of Cerebral Blood Flow and Metabolism. 1993;13:389–394. doi: 10.1038/jcbfm.1993.52. [DOI] [PubMed] [Google Scholar]
- Chou W, Choi D, et al. Neutrophil protein kinase C delta as a mediator of stroke reperfusion injury. Journal of Clinical Investigation. 2004;114:49–56. doi: 10.1172/JCI21655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle J, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- Crack P, Taylor J. Reactive oxygen species and the modulation of stroke. Free Radical Biology and Medicine. 2005;38:1433–1444. doi: 10.1016/j.freeradbiomed.2005.01.019. [DOI] [PubMed] [Google Scholar]
- Edvinsson L, Krause DN. Cerebral Blood Flow and Metabolism. Lippincott Williams and Wilkins; 2002. [Google Scholar]
- Edwards EA, Dean LM. Effects of crowding of mice on humoral antibody formation and protection to lethal antigenic challenge. Psychosomatic Medicine. 1977;39:19–24. doi: 10.1097/00006842-197701000-00003. [DOI] [PubMed] [Google Scholar]
- Ellerby L, Ellerby H, et al. Shift of the cellular oxidation reduction potential in neural cells expressing Bcl-2. Journal of Neurochemistry. 1996;67:1259–1267. doi: 10.1046/j.1471-4159.1996.67031259.x. [DOI] [PubMed] [Google Scholar]
- Fabricius M, Fuhr S, et al. Cortical spreading depression and peri-infarct depolarization in acutely injured human cerebral cortex. Brain. 2006;129:778–790. doi: 10.1093/brain/awh716. [DOI] [PubMed] [Google Scholar]
- Fisher M, Bogousslavsky J. Further evolution toward effective therapy for acute ischemic stroke. JAMA. 1998;279:1298–1303. doi: 10.1001/jama.279.16.1298. [DOI] [PubMed] [Google Scholar]
- Garcia J, Liu K, et al. Interleukin-1 receptor antagonsist decreases the number of necrotic neurons in rats with middle cerebral artery occlusion. American Journal of Pathology. 1995;147:1477–86. [PMC free article] [PubMed] [Google Scholar]
- Gill R, Andine P, et al. The effect of MK-801 on cortical spreading depression in the penumbral zone following focal ischemia in the rat. Journal of Cerebral Blood Flow and Metabolism. 1992;12:371–379. doi: 10.1038/jcbfm.1992.54. [DOI] [PubMed] [Google Scholar]
- Gong C, Qin Z, et al. Cellular localization of tumor necrosis factor alpha following focal cerebral ischemia in mice. Brain Research. 1998;801(12):1–8. doi: 10.1016/s0006-8993(98)00489-2. [DOI] [PubMed] [Google Scholar]
- Gonzalez B, Leroux P, et al. Somatostatin receptors are expressed by immature cerebellar granule cells. PNAS. 1992;89:9627–9631. doi: 10.1073/pnas.89.20.9627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez R, Hirsch J, et al. Imaging and intervention. Springer; 2006. Acute ischemic stroke. [Google Scholar]
- Graham S, Chen J. Programmed cell death in cerebral ischemia. Journal of Cerebral Blood Flow and Metabolism. 2001;21:99–109. doi: 10.1097/00004647-200102000-00001. [DOI] [PubMed] [Google Scholar]
- Guan Q, Pei D, et al. Neuroprotection against ischemic brain injury by SP600125 via suppressing the extrinsic and intrinsic pathways of apoptosis. Brain Research. 2006;1092(1):36–46. doi: 10.1016/j.brainres.2006.03.086. [DOI] [PubMed] [Google Scholar]
- Guan QH, Pei D, et al. Neuroprotection against ischemic brain injury by a small peptide inhibitor of c-Jun N terminal kinase (JNK) via nuclear and non-nuclear pathways. Neuroscience. 2006;139(2):609–27. doi: 10.1016/j.neuroscience.2005.11.067. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Free radicals, antioxidants, and human disease: curiousity, cause, or consequence? Lancet. 1994;344:721–4. doi: 10.1016/s0140-6736(94)92211-x. [DOI] [PubMed] [Google Scholar]
- Hengartner M. The biochemisty of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- Hickey E, You X, et al. Lipopolysaccharide preconditioning induces robust protection against brain injury resulting from deep hypothermic circulatory arrest. Stroke. 2007;133:1588–1596. doi: 10.1016/j.jtcvs.2006.12.056. [DOI] [PubMed] [Google Scholar]
- Hsu CY. Ischemic Stroke: From basic mechanisms to new drug development, Karger 1998 [Google Scholar]
- Huang FP, Wang ZQ, et al. Early NFkappaB activation is inhibited during focal cerebral ischemia in interleukin-1beta-converting enzyme deficient mice. J Neurosci Res. 2003;73(5):698–707. doi: 10.1002/jnr.10654. [DOI] [PubMed] [Google Scholar]
- Huang J, Upadhyay U, et al. Inflammation in stroke and focal cerebral ischemia. Surgical Neurology. 2006;66:232–245. doi: 10.1016/j.surneu.2005.12.028. [DOI] [PubMed] [Google Scholar]
- Hurn P, Subramanian S, et al. T and B cell deficient mice with experimental stroke have reduced lesion size and inflammation. Journal of Cerebral Blood Flow & Metabolism. 2007:1–8. doi: 10.1038/sj.jcbfm.9600482. advance online publication 28 March 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr J. A histochemical study of hypertrophy and iscaemic injury of rat liver with special reference to changes in lysosomes. Journal of Pathology and Bacteriology. 1965;90:419–435. doi: 10.1002/path.1700900210. [DOI] [PubMed] [Google Scholar]
- Kerr J, Wyllie A, et al. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. British Journal of Cancer. 1972;26(4):239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinouchi H, Sharp FR, et al. Induction of heat shock hsp 70 mRNA and HSP70 kDa protein in neurons in the ‘penumbra’ following focal cerebral ischemia in the rat. Brain Research. 1993;619(12):334–338. doi: 10.1016/0006-8993(93)91630-b. [DOI] [PubMed] [Google Scholar]
- Kollmar R, Henninger N, et al. Combination therapy of moderate hypothermia and thrombolysis in experimental thromboembolic stroke- an MRI study. Experimental Neurology. 2004;190(1):204–12. doi: 10.1016/j.expneurol.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Reed J. Mitochondrial control of cell death. Nature Medicine. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- Lefer A, Ma X, et al. Mechanism of the cardioprotective effect of transforming growth factor beta 1 in feline myocardial ischemia and reperfusion. Proceeding of the National Academy of Science. 1993;90(3):1018–1022. doi: 10.1073/pnas.90.3.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Liao M, et al. Ischemic insults direct glutamate receptor subunit 2-lacking AMPA receptors to synaptic sites. Journal of Neuroscience. 2006;26(20):5309–19. doi: 10.1523/JNEUROSCI.0567-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyden P, Lonzo L, et al. Combination chemotherapy extends the therapeutic window to 60 minutes after stroke. Journal of Neurotrauma. 1995;12:223–230. doi: 10.1089/neu.1995.12.223. [DOI] [PubMed] [Google Scholar]
- Marsh B, Stenzel-Poore M. Toll-like receptors: Novel pharmacological targets for the treatment of neurological diseases. Current Opinion in Pharmacology. 2007 doi: 10.1016/j.coph.2007.09.009. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Scheller M, et al. Effects of s-emopamil, nimopidine, and mild hypothermia on hippocampal glutamate concentrations after repeated cerebral ischemia in rabbtits. stroke. 1993;24:1228–1234. doi: 10.1161/01.str.24.8.1228. [DOI] [PubMed] [Google Scholar]
- McNeill H, Williams C, et al. Neuronal rescue with TGFB1 after hypoxic-ischemic brain injury. Neuroreport. 1994;5:901–4. doi: 10.1097/00001756-199404000-00012. [DOI] [PubMed] [Google Scholar]
- Meden P, Overgaard K, et al. Effect of early treatment with Tirilazad (U74006F) combined with delayed thrombolytic therapy in rat embolic stroke. Cerebrovascular Disease. 1996;6:141–148. [Google Scholar]
- Meden P, Overgaard K, et al. Enhancing the efficacy of thrombolysis by AMPA receptor blockade NBQX in a rat embolic stroke model. Journal of Neurological Sciences. 1993;119:209–216. doi: 10.1016/0022-510x(93)90136-m. [DOI] [PubMed] [Google Scholar]
- Minami M, Satoh M. Chemokines and their receptors in the brain: pathophysiological roles in ischemic brain injury. Life Science. 2003;74:321–7. doi: 10.1016/j.lfs.2003.09.019. [DOI] [PubMed] [Google Scholar]
- Miyashita T, Reed J. Tumor suppressor gene p53 is a direct transcriptional activator of the human Bax gene. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- Mori E, Del Zoppo G, et al. Inhibition of polymorphonuclear leukocyte adherence suppresses no-reflow after focal cerebral ischemia in baboons. Stroke. 1992;23:712–8. doi: 10.1161/01.str.23.5.712. [DOI] [PubMed] [Google Scholar]
- Namura S, Zhu J, et al. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. Journal of Neuroscience. 1998;18(10):3659–68. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawashiro H, Brenner M, et al. High susceptibility to cerebral ischemia in GFAP-null mice. Journal of Cerebral Blood Flow and Metabolism. 2000;20:1040–1044. doi: 10.1097/00004647-200007000-00003. [DOI] [PubMed] [Google Scholar]
- Nawashiro H, Martin D, et al. Inhibition of tumor necrosis factor and amelioration of brain infarction in mice. Journal of Cerebral Blood Flow & Metabolism. 1997;17(2):229–32. doi: 10.1097/00004647-199702000-00013. [DOI] [PubMed] [Google Scholar]
- Nawashiro H, Martin D, et al. Neuroprotective effects of TNF binding protein in focal cerebral ischemia. Brain Research. 1997;778(2):265–71. doi: 10.1016/s0006-8993(97)00981-5. [DOI] [PubMed] [Google Scholar]
- Olney J. Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science. 1969;164(880):719–721. doi: 10.1126/science.164.3880.719. [DOI] [PubMed] [Google Scholar]
- Pancioli A, Broderick J, et al. Public perception of stroke warning signs and knowledge of potential risk factors. JAMA. 1998;279:1288–1292. doi: 10.1001/jama.279.16.1288. [DOI] [PubMed] [Google Scholar]
- Pellegrini-Giampietro D. The distinct role of mGlu1 receptors in post-ischemic neuronal death. Trends in Pharmacological Sciences. 2003;24(9):461–470. doi: 10.1016/S0165-6147(03)00231-1. [DOI] [PubMed] [Google Scholar]
- Peng P, Zhong X, et al. ADAR2-dependent RNA editing of AMPA receptor subunit GluR2 determines vulnerability of neurons in forebrain ischemia. Neuron. 2006;49(5):719–33. doi: 10.1016/j.neuron.2006.01.025. [DOI] [PubMed] [Google Scholar]
- Pignataro G, Simon R, et al. Prolonged activation of ASIC1a and the time window for neuroprotection in cerebral ischaemia. Brain. 2007;130:151–8. doi: 10.1093/brain/awl325. [DOI] [PubMed] [Google Scholar]
- Relton JK, Martin D, et al. Peripheral administration of Interleukin-1 Receptor antagonist inhibits brain damage after focal cerebral ischemia in the rat. Experimental Neurology. 1996;138(2):206–13. doi: 10.1006/exnr.1996.0059. [DOI] [PubMed] [Google Scholar]
- Rosamond W, Flegal K, et al. Heart disease and stroke statistics-2007 Update. A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2007;115:69–171. doi: 10.1161/CIRCULATIONAHA.106.179918. [DOI] [PubMed] [Google Scholar]
- Rosenzweig H, Minami M, et al. Endotoxin preconditioning protects against the cytotoxic effect of TNF-alpha after stroke: a novel role for TNF-alpha in LPS-ischemic tolerance. Journal of Cerebral Blood Flow & Metabolism. 2007;27(10):1663–74. doi: 10.1038/sj.jcbfm.9600464. [DOI] [PubMed] [Google Scholar]
- Rosenzweig HL, Lessov NS, et al. Endotoxin preconditioning prevents the cellular inflammatory response during ischemic neuroprotection in mice. Stroke. 2004;35(11):2576–2581. doi: 10.1161/01.STR.0000143450.04438.ae. [DOI] [PubMed] [Google Scholar]
- Ross A, Hurn P, et al. Evidence of the peripheral inflammatory response in patients with transient ischeimc attack. Journal of Stroke and Cerebrovascular Diseases. 2007;16(5):203–7. doi: 10.1016/j.jstrokecerebrovasdis.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruocco A, Nicole O, et al. A TGF-Beta antagonist unmasks the neuroprotective role of this endogenous cytokine in excitotoxic and ischemic brain injury. Journal of Cerebral Blood Flow & Metabolism. 1999;19:1345–53. doi: 10.1097/00004647-199912000-00008. [DOI] [PubMed] [Google Scholar]
- Schabitz W, Fuhai L, et al. Synergistic effects of a combination of low-dose basic fibroblast growth factor and citicoline after temporary experimental focal ischemia. Stroke. 1999;30:427–432. doi: 10.1161/01.str.30.2.427. [DOI] [PubMed] [Google Scholar]
- Schindler R, Mancilla J, et al. Correlations and interactions in the production of IL-6, IL-1, and TNF in human blood mononuclear cells: IL-6 suppresses IL-1 and TNF. Blood. 1990;75:40–47. [PubMed] [Google Scholar]
- Schroeter M, Jander S, et al. Local immune responses in the rat cerebral cortex after middle cerebral artery occlusion. Journal of Neuroimmunology. 1994;55(2):195–203. doi: 10.1016/0165-5728(94)90010-8. [DOI] [PubMed] [Google Scholar]
- Shinoura N, Satou R, et al. Adenovirus mediated transfer of Bcl-X(L) protects neuronal cells from Bax-induced apoptosis. Experimental Cell Research. 2000;254(2):221–31. doi: 10.1006/excr.1999.4751. [DOI] [PubMed] [Google Scholar]
- Simon R. Acidotoxicity trumps excitotoxicity in ischemic brain. Archives of Neurology. 2006;63:1368–1370. doi: 10.1001/archneur.63.10.1368. [DOI] [PubMed] [Google Scholar]
- Stenzel-Poore MP, Stevens SL, et al. Effect of ischemic preconditioning on genomic response to cerebral ischemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. The Lancet. 2003;362:1028–1037. doi: 10.1016/S0140-6736(03)14412-1. [DOI] [PubMed] [Google Scholar]
- Stephenson J. Rising stroke rates spur efforts to identify risks, prevent disease. JAMA. 1998;279:1239–1240. doi: 10.1001/jama.279.16.1239. [DOI] [PubMed] [Google Scholar]
- Strong A, Smith S, et al. Factors influencing the frequency of fluorescence transients as markers of peri-infarct depolarizations in focal cerebral ischemia. Stroke. 2000;31:214–222. doi: 10.1161/01.str.31.1.214. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Chan P. Reactive oxygen radicals and pathogenesis of neuronal death after cerebral ischemia. Antioxidants & Redox Signaling. 2003;5:597–607. doi: 10.1089/152308603770310266. [DOI] [PubMed] [Google Scholar]
- Tarkowski E, Rosengren L, et al. Early intrathecal production of interleukin-6 predicts the size of brain lesion in stroke. Stroke. 1995;26(8):1393–8. doi: 10.1161/01.str.26.8.1393. [DOI] [PubMed] [Google Scholar]
- Tasaki K, Ruetzler CA, et al. Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontaneously hypertensive rats. Brain Research. 1997;748(12):267–70. doi: 10.1016/s0006-8993(96)01383-2. [DOI] [PubMed] [Google Scholar]
- Uematsu D, Araki N, et al. Combined therapy with MK-801 and nimodipine for protection of ischemic brain damage. Neurology. 1991;41:88–94. doi: 10.1212/wnl.41.1.88. [DOI] [PubMed] [Google Scholar]
- Wang Z, Yue T, et al. Expression of interleukin-6, c-fos, and zif268 mRNAs in rat ischemic cortex. Journal of Cerebral Blood Flow & Metabolism. 1995;15:166–171. doi: 10.1038/jcbfm.1995.18. [DOI] [PubMed] [Google Scholar]
- Wiessner C, Gehrmann J, et al. Expression of transforming growth factor beta 1 and interleukin-1 beta mRNA in rat brain following transient forebrain ischemia. Acta Neuropathol. 1993;86:439–46. doi: 10.1007/BF00228578. [DOI] [PubMed] [Google Scholar]
- Yamasaki Y, Shozuhara H, et al. Blocking of interleukin-1 activity is a beneficial approach to ischemia brain edema formation. Acta Neurochirurgica Supplementum. 1994;60:300–2. doi: 10.1007/978-3-7091-9334-1_80. [DOI] [PubMed] [Google Scholar]
- Yamasaki Y, Matsuo Y, et al. New therapeutic possibility of blocking cytokine-induced neutrophil chemoattractant on transient ischemic brain damage in rats. Brain Research. 1997;759:103–111. doi: 10.1016/s0006-8993(97)00251-5. [DOI] [PubMed] [Google Scholar]
- Yamasaki Y, Matsuura N, et al. Interleukin-1 as a pathogenetic mediator of ischemic brain damage in rats. Stroke. 1995;26:676–681. doi: 10.1161/01.str.26.4.676. [DOI] [PubMed] [Google Scholar]
- Zaremba J, Skrobanksi P, et al. Tumor necrosis factor alpha is increased in cerebrospinal fluid and serum of ischemic stroke patients and correlates to volume of evolving brain infarct. Biomedical Pharmacotherapy. 2001;55:258–263. doi: 10.1016/s0753-3322(01)00058-0. [DOI] [PubMed] [Google Scholar]
- Zausinger S, Scholler K, et al. Combination drug therapy and mild hypothermia after transient focal cerebral ischemia in rats. Stroke. 2003;34:2246–2251. doi: 10.1161/01.STR.0000083622.65684.21. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Zhang L, et al. VEGF enhances angiogenesis and promotes blood brain barrier leakage in the ischemic brain. Journal of Clinical Investigation. 2000;106:829–838. doi: 10.1172/JCI9369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Shimohata T, et al. Akt contributes to neuroprotection by hypothermia against cerebral ischemia in rats. The Journal of Neuroscience. 2005;25(42):9794–9806. doi: 10.1523/JNEUROSCI.3163-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Yenari M, et al. Bcl-2 overexpression protects against neuron loss within the ischemic margin following experimental stroke and inhibits cytochrome c translocation and caspase 3 activity. Journal of Neurochemistry. 2003;85(4):1026–36. doi: 10.1046/j.1471-4159.2003.01756.x. [DOI] [PubMed] [Google Scholar]
- Zhao H, Yenari M, et al. Mild postischemic hypothermia prolongs the time window for gene therapy by inhibiting cytochrome C release. Stroke. 2004;35:572–577. doi: 10.1161/01.STR.0000110787.42083.58. [DOI] [PubMed] [Google Scholar]
- Ziegler G, Harhausen D, et al. TLR2 has a detrimental role in mouse transient focal cerebral ischemia. Bioch Biophys Res Comm. 2007;359:574–579. doi: 10.1016/j.bbrc.2007.05.157. [DOI] [PubMed] [Google Scholar]
- d Zoppo G, Ginis I, et al. Inflammation and stroke: putative role for cytokines, adhesion molecules and iNOS in brain response to ischemia. Brain Pathol. 2000;10(1):95–112. doi: 10.1111/j.1750-3639.2000.tb00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d Zoppo GJ. Microvasculature Responses to Cerebral Ischemia/Inflammation. Ann NY Acad Sci. 1997;823:132–47. doi: 10.1111/j.1749-6632.1997.tb48386.x. [DOI] [PubMed] [Google Scholar]