

Abstract
If untreated at early stages, melanoma becomes a highly aggressive cancer with rapid metastasis to distant sites. Although cell biologic analyses have uncovered a multitude of signaling pathways involved in melanoma genesis and progression – including the MAPK, PI3K, and FAK pathways – efficacious therapies that target these cellular components have remained elusive. Genome-wide technologies such as microarray chips and array comparative genomic hybridization have generated genetic information that can identify cellular mechanisms critical for the induction and maintainence of the malignant phenotype. Thus, such data can guide the choice of a biologically relevant drug. However, these techniques have also identified melanoma as a genetically and biologically highly heterogeneous disease that likely requires individually tailored therapies based on the patient¹s individual genetic and biologic alterations. In addition, these techniques have generated a large body of data on candidate melanoma genes that await extensive functional validation to separate so called “driver” from “passenger” events. In this review, we cover several advances in melanoma therapeutics and their current limitations as well as emerging genomic, proteomic, and epigenetic strategies for the identification of critical cellular dependencies that may be tractable to therapeutic targeting.
Keywords: melanoma, growth factors, oncogenes, therapy, melanoma genome
THE DISEASE
Melanoma arises from melanocytes, specialized pigmented cells that reside in the skin, at the choroidal layer of the eye, the gastrointestinal and genitourethral mucosal surfaces and the meninges. Melanocytes produce melanins, the pigments responsible for skin and hair color. In the skin, melanocytes reside between keratinocytes in the basal layer of the epidermis and in the hair follicles and produce melanin to a variety of direct and indirect stimuli such as ultraviolet (UV) radiation. Thereby, melanocytes fulfill a key role in protection of our whole body from environmental stress factors such as damaging UV radiation, which can induce skin cancer.
There are an estimated 2–3 million cases of newly diagnosed skin cancers across the world each year and melanoma accounts for around 132,000 cases of these (World Health Organization, http://www.who.int/uv/faq/skincancer/en/index1.html). The incidence rates for cutaneous melanoma have risen faster than those of any other malignancy in the Caucasian population over the last 30 years. From 1976 to 2003, the Central Malignant Melanoma Registry of Germany has documented a 3-fold increase in the incidence of cutaneous melanoma, reaching 10.3 and 13.3 newly diagnosed melanomas for males and females, respectively, per 100,000 people per year 1. In the USA incidence rates are 17.2 (males) and 12.1 (females) per 100,000 population 2, highest incidence rates are observed in Australia with 38.5 (males) and 29.5 (females) per 100,000 people (Globocan 2002 database, http://www-dep.iarc.fr/globocan/downloads.html).
Although melanoma accounts for only 4% of all skin cancers, it is responsible for 80% of deaths from skin cancer. Nowadays, timely diagnosis and treatment of melanoma during the earliest stages of its evolution is of critical importance to patient survival. Whereas surgical resection is curative in most patients with early in situ and radial growth phase melanomas (and around 80% of melanomas can be treated this way), only 14% of patients presenting with locoregional and/or distant metastasis survive for 5 years, despite all therapeutic efforts 3.
ENVIRONMENT AND PREVENTION OF THE DISEASE
In view of the epidemiological, the clinical, and the emerging experimental evidence linking melanoma incidence to UV exposure and skin phototype, primary (sun protection) and secondary (early detection) prevention strategies have been key areas to efforts for reduction of disease incidence and severity in the last centuries.
In Queensland, Australia where secondary prevention started in the 1960s and primary prevention in the 1980s, Coory et al. found sustained increases in age-standardized incidence, but stabilization of mortality rates, with a shift to detection of more early (in situ) melanomas 4, 5. Similar results have been reported from the Central Malignant Melanoma Registry of Germany with a decrease of age-standardized mortality accompanied by a shift towards the detection of thin melanomas in patients younger than 70 years 1. These data are being regarded as reflecting the success of earlier detection of melanoma and the authors conclude that continuation of secondary preventive strategies is warranted.
In contrast, strategies to primary prevention (sun protection) in Queensland have shown at best some tentative signs that some younger-age groups may be experiencing trend improvements 5. Similar evidence comes from the USA, where sun protection campaigns have failed to substantially reduce melanoma incidence 6. These results may also be reflected by epidemiologic studies that identify UV radiation-exposure, as compared to other risk factors for developing melanoma, as a surprisingly modest one (1.7-fold) in an unselected population 7. However, the effect of UV radiation is governed by variations (polymorphisms) in particular genes that affect both the defensive response of skin (particularly its impact on cutaneous immune function) and the risk of developing melanoma, e.g. by induction of growth factors and reactive oxygen species (ROS) 8, 9. At the molecular level, exposure to UV radiation increases pigmentation through release of α-melanocyte-stimulating hormone (α-MSH), which interacts with its receptor melanocortin receptor 1 (MCR1) to induce the expression of enzymes producing melanin on the surface of melanocytes. Pigmentation as a response to UV radiation 10 is modulated by polymorphisms of MCR1, which functionally reduce the activity of this receptor 11 (as in light-skinned and red-headed individuals with skin phototype I 10) and are significantly associated with an increased risk for melanoma 12.
CLINICAL AND MOLECULAR PATHOLOGY OF CUTANEOUS MELANOMA
Cutaneous melanoma presents clinically with four different major subtypes 13. By far the most common form (around 75%) is superficial spreading melanoma. It presents clinically as a macule or variably raised plaque with distinct irregular coloration. It is the third most common cancer in young people in the USA and accounts for around 50% of melanoma cases in non-Caucasians 9, 14. In contrast, the second most common form (around 15%) of melanoma is nodular melanoma, which constitutes a raised nodule, with or without a macule in the background. Acral lentiginous melanoma (ALM) is usually found on the palms, the soles and the nail beds. It accounts for about 5% of melanomas in the Caucasian and for about 50% in the non-Caucasian population. Lentigo maligna (around 5%) is generally a flat macule occurring at chronically sun-exposed body sites of the elderly and is associated with the lifetime dose of UV radiation received.
The Clark model of the progression of melanoma emphasizes the stepwise transformation of melanocytes to melanoma. The model depicts the proliferation of melanocytes in the process of forming melanocytic nevi and the subsequent development of hyperplasia, dysplasia, invasion and metastasis 13. According to Clark’s model, the first phenotypic change in melanocytes is the development of benign nevi. Histologically, such lesions have an increased number of nested melanocytes either restricted to the epidermis along the basal layer (junctional nevus), to the dermis (dermal nevus), or to both (compound nevus).
The next step toward melanoma is the occurrence of cytological atypia such as aberrant growth and dysplastic cells, which are characteristic for so called dysplastic nevi. These lesions may occur either at a new location or in a preexisting nevus. Nevi are generally benign, but can progress rarely into radial growth phase (RGP) melanoma 13. RGP presents with a completely (in situ) or a predominantly intraepidermal proliferation of melanocytes with invasion of single cells or nests into the papillary dermis. RGP cells cannot form colonies in soft agar in vitro.
RGP cells can progress to vertical growth phase (VGP) melanoma, which is either confined to the papillary dermis with development of a tumor nodule or extends deeply into the dermis and/or the subcutaneous fat. These cells have the ability to form colonies in soft agar in vitro and tumor nodules when implanted into nude mice and have acquired metastatic potential. The distinction between RGP and VGP is the single most important pathologic observation in melanoma as most RGP melanomas are curable by surgical resection only, whereas VGP melanomas have the capacity to metastasize.
Not all melanomas strictly pass through each of these individual phases, but can directly develop from melanocytes and progress to metastatic melanoma 15.
THERAPY OF MALIGNANT MELANOMA AND ITS LIMITATIONS
Surgical excision is often curative in patients with thin primary tumors. However, the majority of patients with deeply infiltrating primary melanomas or tumors that metastasize to regional nodes will develop distant metastases later on.
Conventional Therapeutics
Over the last four decades only little, if any, progress has been made in the systemic treatment of metastatic melanoma. The alkylating agent dacarbazine (DTIC-Dome, Bayer, West Haven, CT) has been approved by the Food and Drug Administration (FDA) in the 1970s and is considered to be the reference single agent for advanced disease. DTIC has been reported to induce objective clinical responses in about 15 to 20% of patients in older trials, but with response rates of below 10% in recent multicenter trials 16, 17. Although complete clinical responses have been reported, most responses are partial and last for around 5–7 months 16, 17.
Interleukin-2 (IL-2) was approved by the FDA in 1998 based on a 6% complete response rate in phase II study data set of 270 patients 18. If induced, clinical responses may be remarkably sustained (31 patients of 270 are still alive at a 10 years follow-up) 19. In the absence of any phase III data demonstrating a clinical benefit of any dose of IL-2 in metastatic melanoma and with regard to its high and sometimes unacceptable toxicity, it is unlikely that IL-2 will be approved in Europe, particularly as long as it is not known which subgroup of patients will respond.
Targeted Therapy
Even though many if not most of the current effective therapies in cancer are targeted (e.g. taxanes are targeted to beta-tubulin, the fluoropyrimidines to thymidylate syntethase, and doxorubicin to topoisomerase-II), “targeted therapy” has become a well-worn password in clinical oncology today. This is the consequence of the remarkable efficacy of novel targeted agents such as small inhibitory molecules and antibodies in other cancers and led to a remarkable shift in the investigational paradigm for melanoma.
The concept of “oncogene addiction” argues that cancer cells rely more heavily on hyperactivated intracellular signaling pathways than do normal cells, and therefore rely critically on activated oncogenes that drive those pathways (reviewed in 21). Not all oncogenes are tractable to therapeutic targeting, but intracellular enzymes such as kinases, proteases and phosphatases and extracellular antigens such as cell surface receptors are prime targets. In the following paragraph, we want to give three prominent examples of targeted therapies that have been evaluated in advanced melanoma patients.
Based on the observation that activating mutations of a serine/threonine-specific protein kinase BRAF occur in a high proportion of melanomas (for more details see later) clinical trials in advanced melanoma patients have been initiated with sorafenib, a tyrosine kinase inhibitor that targets mutant and wild-type BRAF, vascular endothelial growth-factor receptors (VEGF-R)-2 and -3, c-KIT, and platelet-derived growth factor receptor (PDGF-R)-β. Whereas sorafenib was well tolerated, it had only little or no antitumor activity 22.
It is also known that melanoma expresses a number of growth factor receptors at the cell surface and that ligands for these receptors may be present in the tumor milieu 23. Imatinib mesylate is an oral tyrosine kinase inhibitor that targets BCR-ABL, c-KIT, PDGF-R-α and -β. As activating mutations and gene amplifications of c-KIT 24, as well as signal transduction through PDGF-R-α and -β have been described in melanoma, clinical trials have been initiated with imatinib mesylate in metastatic melanoma patients. Patients experienced significant toxicity, but no objective clinical responses 25, 26.
Another trial targeted epidermal growth factor receptor (EGFR), a type 1 receptor tyrosine kinase involved in cellular differentiation and proliferation of cancer cells including melanoma 27, 28. A phase II trial in metastatic melanoma patients with erlotinib, a small molecule EGFR kinase inhibitor, revealed mild toxicity, but no objective responses 29.
All in all, no agent or combination of agents has been shown to have an impact on survival in patients suffering from metastatic melanoma and thus, no single agent can be regarded as standard of care. The intractability of advanced melanoma painfully illustrates how much we still have to learn about the biology of this disease and the molecular changes associated with, or better, resulting in progression, metastasis and resistance to therapy. In the past, too many clinical trials have been conducted with a poor understanding of the mechanisms of action of the involved compounds and without an adequate consideration or knowledge of the biology of the disease. In other words, testing a targeted therapy when we don’t know the expression pattern and don’t understand the functional consequences of the expression of a target molecule in a disease-specific cellular context is a low-yield clinical research strategy.
THE KEY QUESTIONS
The etiology of transformation and progression of melanocytes to melanoma is not well understood. The most critical questions to melanoma cell biology that have to be answered are: (1) which genetic alterations are responsible for development, progression and maintenance of the established disease? (2) what genetic events underlie the propensity for metastasis and treatment resistance? (3) finally, what maintenance-essential biological or molecular signaling pathways/networks might prove amenable to preventive and/or therapeutic intervention in man?
Many studies conducted over the last decades on benign and malignant melanocytic lesions as well as melanoma cell lines have implicated numerous genes in melanoma development and progression. Here, we will focus on (1) validated genetic events in cell signaling pathways and growth factors, which include predisposing or somatic structural alterations in melanoma specimens on the DNA level, such as translocation, amplification/deletion and point mutations 30 and (2) how functional genomic information can contribute to the identification and validation of new candidate genes.
MELANOMA-RELEVANT CELL SIGNALING PATHWAYS
RAS, RAF and Other Activators of MAP Kinase Signaling
The RAS/RAF/MEK/ERK mitogen-activated protein (MAP) kinase pathway (Fig. 1) has been most directly linked to the development of melanoma due to its growth-promoting activities. Since self-sufficiency in growth signaling is a requisite capability acquired by all cancer cells 31, hyperactive extracellular signal-regulated kinases (ERKs) are common in many human cancers, including melanoma 32, 33. In melanocytes, this pathway can be activated by growth factors such as stem cell factor (SCF), fibroblast growth factor (FGF) and hepatocyte growth factor/scatter factor (HGF) 34, but individually these growth factors induce only weak or transient ERK activation. A sustained hyperactive state can theoretically be achieved by activating mutations of any signaling mediator upstream of ERKs and there are clear tumor-type specific patterns of mutational activation. The small G protein RAS is perhaps the most frequently activated component of this signaling cascade with a reported incidence of 15 to 30% in all human cancers 35. In the absence of gain-of-function mutations in RAS genes, BRAF alone is responsible for coupling RAS signals to MEK. However, as soon as melanoma cells acquire a mutation in RAS, cells switch their signaling from BRAF to CRAF which is accompanied by disruption of cAMP signaling, a prerequisite for CRAF signaling to MEK 36.
Figure 1.
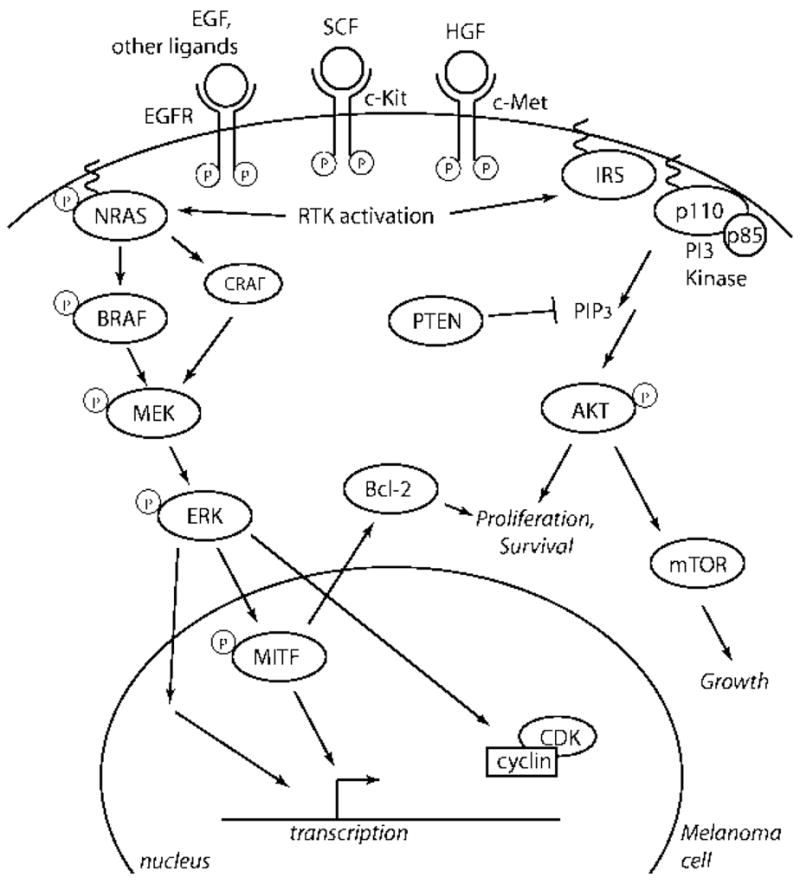
Signaling pathways important in melanoma. RTKs activate both the MAPK and PI3K pathways, which together promote mitogenesis and survival.
BRAF is also commonly targeted in human cancers with an overall mutation frequency of 7% 37, but a reported mutation frequency of up to 70% in metastatic melanoma 38–44. ERK activation can induce transcription of genes involved in melanoma cell proliferation (e.g. FGF-2, IL-8, and HIF-1a), actin organization and cell motility (e.g. PREX1, COTL1), angiogenesis (e.g. angiomotin-like 1), metastasis (TWIST1), and immune response (e.g. CD58, CD200) 45. However, ERK activation can also regulate differentiation, senescence and survival. Therefore, genotype-phenotype correlation must take into account consequences other than growth promotion by activating mutations in components of ERK signaling.
The RAS Family of Proto-Oncogenes: H-, N- and K-RAS
In contrast to other solid tumors, activating mutations of RAS are not detected with high frequency in melanoma, ranging from low to 10–15% incidence 46 (reviewed in 47). N-RAS is the most frequent RAS family member targeted in the melanocyte lineage, with activating mutations in as many as 81% of congenital nevi 48, up to 33% of primary and 26% of metastatic melanoma samples 49. Activating N-RAS mutations have been correlated with nodular lesions and sun exposure 50, 51. Interestingly, N-RAS mutations are rarely found in dysplastic nevi 50, 52, 53, which may imply their distinct evolutionary path to melanoma. H-RAS activation has occasionally been detected in melanoma, albeit more commonly associated with Spitz nevi, based on amplification of its genomic locus on 11p and oncogenic point mutations 54. K-RAS mutations have not been described in human melanocytic lesions.
These mutation patterns point to distinct biological activities of the different RAS family members in melanocyte biology. The phenotypic impact of activated H-RAS versus N-RAS transgenic mice has reinforced this view. Specifically, an activated H-RAS transgene, together with inactivating mutations in Ink4a, Arf and/or p53 promotes development of non-metastatic melanomas 55–57. In contrast, when targeted to the melanocytic compartment, an activated N-RAS transgene and Ink4a/Arf deficiency drives cutaneous melanomas with high penetrance and short latency, as well as metastatic spread to lymph nodes and other distal sites (e.g. lung and liver) in a third of the cases 58.
BRAF, a Potent Activator of ERK Protein Kinases
The most commonly mutated member of the RAS/RAF/MEK/ERK pathway is BRAF, one the of the three RAF genes (together with ARAF and CRAF). Since its discovery through a genome-wide cancer re-sequencing effort 37, mutations in BRAF have been detected in a variety of tumor types, with the highest incidence in melanoma (ranging from 27 to 70%) 38–44. BRAF mutations occur particularly in melanomas at body sites with intermittent UV exposure 59 and are associated with the occurrence of germline variants of the MC1R gene 60. The point mutations cluster in specific regions of biochemical importance, with the predominant melanoma mutation being a single phosphomimetic substitution in the kinase activation domain (V600E), which confers constitutive activation 61. BRAFV600E stimulates constitutive ERK signaling and directly and indirectly regulates expression and function of several genes critical to proliferation and survival of melanoma cells. These include transcription factors microphthalmia-associated transcription factor (MITF) 62, NF-kB 63, and the cell cycle regulators Cyclin D1 64, p16INK4A 65, and p27Kip166.
An intriguing observation is that BRAF mutations are also common in benign and dysplastic nevi 39, 42, 67, 68 suggesting a role in the earliest stages of neoplasia. It is notable, however, that most nevi remain growth-arrested for their whole lifetime and only rarely progress into melanoma. This raises the possibility that BRAFV600E-induced checkpoint mechanisms exist operating to constrain malignant transformation. Indeed, a recent study showed that human congenital nevi are positive for both p16INK4A and senescence-associated acidic beta-galactosidase (SA-beta-Gal), the classical senescence-associated marker 65. Furthermore, BRAFV600E expression alone is not sufficient to transform human melanocytes 69, but able to transform p16INK4A-deficient murine melanocytes 70. Thus, BRAFV600E alone seems not to be sufficient to induce melanoma, but induces cell cycle arrest with concomitant induction of both p16INK4A and markers of senescence 65. This phenomenon is called oncogene-induced senescence (OIS), a mechanisms known to constrain progression of early premalignant lesions 71. Together, these results argue for a model whereby p16INK4A serves as a brake to BRAFV600E-triggered melanocyte proliferation and p16INK4A pathway inactivation is required for progression to melanoma. Notably, in melanocytic nevi expression of p16INK4A is not in 100% concordance with SA-beta-Gal positivity, suggesting the presence of a non-p16INK4A dependent pathway in mediating BRAF-induced OIS 65. Therefore, it seems likely that OIS will lead to the discovery of another tumor suppressor whose importance in melanoma may rival that of p16INK4A.
The notion that BRAFV600E is not sufficient for transformation of melanocytes has also been demonstrated in other model systems. In zebrafish, it has been shown that BRAF activation leads only to development of benign nevi, while progression to frank melanoma requires p53 deficiency 72. Similarly, BRAFV600E mutation alone in TERT-immortalized RB-p53 mutant human melanocytes was found to produce only junctional nevi in the human/mouse skin graft, in contrast to activated NRAS or PI3K p110a mutants which generated invasive melanoma lesions 73. These biological outcomes indicate distinct roles for NRAS and BRAF activation in melanoma development. The mutually exclusive occurrence of either activated NRAS or B-RAF alleles in melanoma and other tumor types 37, 74, 75 may argue for some functional overlap of NRAS and BRAF activation, but may also be the result of a synthetic lethality between NRAS and BRAF activation at the single cell level 45.
PTEN, a Negative Regulator of the Phosphatidylinositol 3-Kinase (PI3K)-AKT Pathway
Another pathway promoting cell growth and survival in melanoma is the phosphoinositide-3-OH kinase (PI3K)-AKT pathway (Fig. 1). Phosphoinositides are membrane lipids that are converted to phosphatidylinositol phosphate (PIP3) second messengers through hyperphosphorylation by one of the PI3K family members 76. Integrins, extracellular matrix components such as fibronectin, and established growth factors, such as HGF and insulin-like growth factor (IGF)-1, act through this signaling pathway 77–79. In the presence of growth factor signaling, the intracellular level of PIP3 rises leading to phosphorylation of AKT, which is known to promote cell cycle progression and to inhibit apoptosis, and whose expression in its phosphorylated form is correlated adversely with patient survival 80. PIP3 second messengers are negatively regulated by the lipid and protein phosphatase phosphate and tensin homologue (PTEN) and inactivation of PTEN results in accumulation of PIP3, AKT hyperphosphorylation, and induction of expression of genes involved in enhanced cell survival/proliferation, tumor growth and metastasis such as the cell cycle promoting kinase Cyclin D3 81 and the glycophosphoprotein Osteopontin 82
Unlike the MAP kinase pathway, genetic alterations specifically targeting components of this signaling cascade do not occur at high frequency in melanoma 83. Of those that do occur, the best-known culprit is the PTEN tumor suppressor. PTEN resides on chromosome 10q, a region known to sustain LOH in many human cancers, including melanoma 84, 85. Allelic loss or altered expression of PTEN occurs in 20% and 40% of melanoma tumors, respectively 74, 86–88, although somatic point mutations and homozygous deletions are rarely observed. Functionally, ectopic expression of PTEN in PTEN-deficient melanoma cells can abolish phospho-AKT activity, induce apoptosis, and suppress growth, tumorigenicity and metastasis 89–91; reviewed in 79. Correspondingly, germline or somatic inactivation of Pten in the mouse strongly promotes tumor phenotypes in multiple cell lineages 92–95 including melanoma 96.
Most recently, additional ways of inactivation of PTEN activity have been described. PTEN can be inactivated by poly-ubiquitilation through NEDD4-1, which leads to PTEN degradation in the cytoplasm 97. By contrast, mono-ubiquitylated PTEN localizes to the cell nucleus 97, where it antagonizes the (PI3K)-AKT survival pathway and maintains chromosomal stability through physical interaction with centromeres and control of DNA repair 98. Whereas PTEN inactivation through NEDD4-1 alone is not sufficient to induce tumors, it significantly augments the efficiency of Ras-mediated transformation of mouse fibroblasts in the presence of p53 deficiency 99. This observation underlines the importance of the simultaneous activation of ERK and PI3K signaling for tumor induction, a phenomenon similarly important for melanoma development. In three-dimensional melanoma cultures as well as in the transgenic TPRas melanoma mouse model both signaling pathways must be inhibited to suppress cell growth 100, 101. In melanoma specimens, NRAS and PTEN mutations are mutually exclusive, but BRAF and PTEN mutations have been shown to coincide in about 20% of cases 59, 102.
In line with the experimental evidence supporting a melanoma suppressive role of PTEN, constitutive activation of AKT has been shown to be a potent inducer of melanocyte transformation 73 and progression of RGP into VGP melanoma in vivo 103. In addition, DNA copy gain involving the AKT3 locus has recently been described in melanoma, and selective AKT3 activation may characterize 40–60% of sporadic tumors 104. However, the complexity of this signaling cascade has not been fully understood till today. Recent data have suggested that activation of different AKT isotypes may elicit distinct effects on cell proliferation and survival. For example, one report found that targeted deletion of AKT3, whose expression correlated most strongly with melanoma tumor progression amongst the three AKT isotypes, triggered apoptotic signaling 104. On the other hand, AKT1 activation was found to inhibit the migration and invasion of certain cancer cell lines 105, including MDA-MB435, a line previously believed to derive from breast cancer but subsequently shown through transcriptional and SNP array profiling studies to be a melanoma cell line 69, 106. Thus, although the PI3K/AKT pathway clearly demonstrates enhanced activity in many melanomas, the extent to which this constitutes a critical melanoma dependency remains unresolved.
Activation of Receptor Tyrosine Kinases
Considering the prominent roles of RTKs in transmitting extracellular signals to intracellular effectors, and the importance of homotypic and heterotypic cell-cell interactions in cancers, it is not surprising that almost all of the direct signaling components of RTKs have been implicated in the development of human melanoma (Fig. 1). Several RTKs map to known regions of recurrent DNA copy number gain or amplification. Moreover, considering the example of c-KIT (see below), it is expected that systematic re-sequencing efforts will identify activating mutations in these and other RTKs in melanomas.
The c-KIT gene encodes a RTK that serves as the receptor for stem cell factor (SCF). The regulation of the KIT pathway is complex and tightly regulated. A number of isoforms are known for the receptor and its ligand, and the ligand can interact with the receptor in both soluble and membrane-bound forms 107. Binding of soluble SCF leads to KIT receptor activation, internalization and degradation, whereas binding of membrane-bound SCF leads to prolonged KIT activation. The KIT receptor can interact with multiple downstream signaling pathways including RAS/RAF/MEK/ERK, PI3K/AKT, phospholipase C, and the SRC family. The mechanisms underlying the differential activation of these pathways are not understood till today. In melanocytes, KIT plays a critical role in migration, survival, proliferation and differentiation. Mice deficient in Kit activation loose melanocytes 108 and KIT inhibition appears to drive melanocyte cell loss in human skin 109, indicating that KIT is a critical survival factor for melanocytes. KIT is also responsible for melanocyte cell proliferation. In vitro, KIT activation induces proliferation of cells of the melanocytic lineage 110 and transgenic mice overexpressing the membrane-bound form of SCF in the epidermis develop melanocytic hyperplasia 111. Most recently, the D814Y activating KIT mutation has shown to induce PI3K signaling and migration of melanocytes 112.
Numerous immunohistochemical studies have linked progressive loss of c-KIT expression with the transition from benign to primary and metastatic melanomas 113–115. Reconstitution of c-KIT in metastatic melanoma cells apparently conferred sensitivity to SCF-induced apoptosis in vitro 116. Thus, at first glance, KIT does not fit the profile of a RTK targeted for activation in melanoma. However, a recurrent L576P mutation in c-KIT has recently been reported in melanoma. Among 153 cases examined, Holden and colleagues identified four metastatic melanomas with robust expression of c-KIT on IHC. High-resolution amplicon melting analyses followed by direct DNA sequencing revealed that three of them harbored a L576P mutation with selective loss of the normal allele 117, 118. L576P is a known GIST-associated mutation that maps to the 5′ juxtamembrane domain where most activating KIT mutations cluster 119, 120. Most recently, Bastian and colleagues reported in a chort of 102 primary melanoma samples on the presence of activating KIT mutations and gene amplifications at a frequency of 28 to 39% in particular subtypes of melanomas, i.e. acral melanoma, mucosal melanoma, and melanomas from chronically sun-damaged skin. 69% of the identified KIT mutations were predicted to affect the 5′ juxta-membrane domain and 19% the kinase domain 24. The example of the EGFR mutational status as a predictor for therapeutic responses in NSCLC 121, 122 suggests the possibility of identifying a melanoma patient subpopulation that will respond to the c-KIT inhibitor imatinib mesylate based on c-KIT mutational status.
Activation of EGFR by its ligands EGF, transforming growth factor (TGF)-α, amphiregulin and heparin-binding EGF (HBEGF) has been shown to activate several downstream signaling pathways such as the MAPK pathways, the PI3K-AKT pathway, the stress-activated protein kinase C and the Janus kinase (Jak)- signal transducer and activator of transcription 16 pathway and thus critically regulates cell differentiation, proliferation, survival, and migration 123. Late stage melanomas often exhibit EGFR over-expression in association with increased copies of chromosome 7 124–126. Enforced activation of EGFR has been associated in metastatic progression in a cell-based study 127, 128. However, unlike glioblastomas or lung adenocarcinoma 129, 130, focal amplification and/or mutation of EGFR have not been reported in melanoma. The non-focal nature of chromosome 7 gains in melanoma renders it impossible to assign EGFR as a target of such genomic alterations. In an inducible HRAS-driven mouse melanoma model 131, transcriptome analysis revealed the existence of a RAS-dependent EGFR signaling loop mediated through upregulation of EGF family ligands (e.g. amphiregulin and epiregulin) 132. This EGFR signaling pathway provides important survival signals involving PI3K-dependent activation of AKT, as sustained EGFR activity is able to prolong viability of established melanoma upon inactivation of RAS. Conversely, inactivation by dominant negative EGFR abolishes tumorigenicity of RAS-driven melanoma cells, consistent with observations in other cell systems (fibroblasts, keratinocytes, and intestinal epithelial cells) that autocrine EGFR signaling is required for transformation by activated RAS 133–135. Thus, in addition to providing experimental evidence that EGFR activation is biologically relevant, the above-mentioned study in the inducible model also points out the possibility that EGFR or its ligands may constitute alternative point(s) of therapeutic intervention in RAS-activated melanoma. It should be mentioned that the contribution of EGFR signaling to melanoma development and possibly progression is evolutionarily conserved, as activating mutations in the EGFR homologue, Xmrk, increase melanoma susceptibility in Xiphophorus fish 136–138; reviewed in 139. It therefore remains possible that similar activating mutations exist in human melanoma, although systematic re-sequencing of large cohorts of melanomas from different ethnic and/or molecular subclasses will be required to uncover such examples.
The RTK c-MET is normally expressed on epithelial cells and melanocytes 140 and is activated by binding of its ligand, HGF, through a number of downstream signaling pathways including RAS/RAF/MEK/ERK, PI3K, phospolipase c-g and STAT 47, 141. c-MET is a multifaceted regulator of growth, motility and invasion in a number of cell lineages. Whereas RAS/RAF/MEK/ERK signaling may be responsible for proliferation, PI3K signaling is required for scattering and these two pathways in combination with STAT signaling may be important in morphogenesis 142–144. Although MET is normally activated in a paracrine manner, autocrine activation of HGF–MET has been described in melanoma progression 145; reviewed in 146. Accordingly, increased c-MET expression has been observed in metastatic melanoma 147, and copy number gain of the c-MET locus at 7q33 - qter seems to be a late event in melanoma progression 148. However, similar to EGFR above, neither focal MET amplifications nor activating MET point mutations have been detected in melanoma, although both have been observed in other human cancers 149–152. However, several lines of experimental and functional evidence support a causal role for MET signaling in human melanoma. For example, in explant models, it has been shown that elevated c-Met expression or Met receptor tyrosine kinase activity may correlate with metastasis 153. In genetically engineered models, constitutive and ubiquitous HGF expression establishes an autocrine loop with c-Met, leading to step-wise development and progression of cutaneous and metastatic melanomas, which cooperates with UVB, Ink4a/Arf deficiency, and activated Cdk4 154–156. Correspondingly, while enforced expression of c-Met in melanocytes provides only weak cancer-initiating activity, this mutation drives the development of metastatic disease, and such tumor lesions show concomitant activation of HGF and establishment of HGF-Met signaling loop (LC, unpublished observations). Finally, c-MET was recently shown to be a direct transcriptional target of MITF 157, the melanocytic lineage transcription factor that can be activated by focal amplification in melanoma (see below).
THE MELANOMA GENOME
In probing the cancer genome for novel targets, technological advances have enabled the rapid production of genome-wide datasets informative for changes in gene expression, DNA copy number, and loss of heterozygosity. Such assays have revealed in melanomas an increasingly complex pantheon of genetic aberrations beyond the established ones described above. Novel tumor suppressors and oncogenes are continuing to be described, but many validation steps are required before any gene can be considered bona fide therapeutic targets. Effects on tumor initiation, growth, or invasion must be experimentally demonstrable both in vitro and in vivo.
Typically, experiments in cell culture make use of overexpression vectors, knockdown vectors such as siRNA or shRNA, drug inhibitors, or competitive antibodies to modulate gene expression or product levels. Cells can then be assayed for their ability to form colonies in soft agar, indicative of anchorage-independent growth and loss of contact inhibition. Growth curves can be measured, and invasion can be tested in Boyden chambers, which simulate the initial steps of metastatic invasion through the extracellular matrix. Three- dimensional cultures, including organotypic skin rafts and collagen matrices, may better portray the conditions of the in vivo environment. Culture preparations can also be injected subcutaneously into athymic mice, and the incidence, growth, and invasiveness of the resulting xenograft tumor is considered more clinically relevant than cell culture. Tail or portal vein injections and the observation of subsequent metastasis formation can measure the intravasation and seeding steps. Finally, genetically engineered mouse models can provide strong evidence for a gene’s role in melanoma, as cell type-specific gene knockouts, knockins, and transgenics can recapitulate human mutations in the context of established mouse melanoma models (see above). A number of studies employing genome-wide assays are catalogued in Table 1, along with the extent of candidate gene validation performed in each.
Table 1.
The extent of validation for recently identified genes in melanoma. E (Expression validaton) is achieved by RT-PCR, northern, or western blots, while I (IHC or tissue microarrays) refers to histological protein analysis. O (Overexpression), K (Knockdown/dominant negative) and X (Xenograft) refer to manipulation of gene expression in cell lines. M (Mechanism) indicates studies of interactions with putative pathway members. P (Patient survival data) indicates a matched Kaplan-Meier analysis.
| Extent of validation | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Samplea | Array | Genes | Proposed | gene | E | I | O | K | X | M | P | Ref |
| Tb | aCGH | NEDD9 | Metastasis | • | • | • | • | • | • | Kim, et al, 2006 | ||
| C | SNP/cDNA | MITF | Oncogene | • | • | • | • | • | Garraway, et al, 2005 | |||
| C | cDNA | CRSP3, | Metastasis | • | • | • | • | Goldberg, 2003 | ||||
| C | cDNA | SYN, | Tumor suppressor | • | • | • | Muthusamy, et al, 2006 | |||||
| C | cDNA | CX43 | Oncogene | • | • | Su, et al, 2000 | ||||||
| C, T | cDNA | TN-C, FN | Metastasis | • | • | Kaariainen, et al, 2006 | ||||||
| T | cDNA | PTGDS, | Multiple | • | • | • | Winnepenninckx, et al, 2006 | |||||
| T | cDNA | HLA-DR, | Lymphocytic | • | • | Mandruzzato, et al, 2006 | ||||||
| C | cDNA | TWIST1, | Metastasis | • | • | Shields, et al, 2007 | ||||||
| T | cDNA | DSC1, | Multiple | • | • | Jaeger, et al, 2006 | ||||||
| C | cDNA | Mitf/Tgfb- | Progression | • | • | Hoek, et al, 2006 | ||||||
| T | aCGH | KIT | Oncogene | • | Curtin, et al, 2006 | |||||||
| C | cDNA | TSPY | Marker, | • | Gallagher, et al, 2005 | |||||||
| T | cDNA | PLAB, | Multiple | • | Talantov, et al, 2005 | |||||||
| C | SNP | PTPRDc | Multiple | Stark and Hayward, 2007 | ||||||||
| T | cDNA | TNFSF10c | Proapoptotic | Jensen, et al, 2006 | ||||||||
T = tissue, C = cell lines; all human, but see footnote b
Mouse and human tissues
Sampling of identified genes
As can be seen, the majority of gene discovery studies have utilized gene expression arrays. For example, comparisons of metastatic and non-metastatic cell lines, the latter with an engineered extra copy of chromosome 6, identified CRSP3 and TXNIP as correlated with the suppression of metastasis 158. Although overexpression of either gene did not affect tumor growth in vitro or in xenografts, they significantly and independently suppressed the formation of metastases to the lung when the cells were injected either subcutaneously or intravenously. Preliminary evidence suggested that both genes operate through the KISS1 (Kisspeptin-1) pathway, which may affect cell motility 159. Others 160 used a similar set of cell lines with or without an extra chromosome 6, with the latter displaying anchorage-independent growth in soft agar and a downregulation of the connexin gene CX43. Overexpression of CX43 significantly suppressed the soft agar colony formation. As a final example, tenascin and fibronectin were upregulated in progressed tumors, and their co-localization confirmed by IHC 161. Overexpression of either gene conferred growth and invasive behavior in three-dimensional collagen gels.
Combinations of genome-wide technologies – expression microarrays, array comparative genomic hybridization, and SNP microarrays – can together define a genomic atlas of recurrent DNA and gene regulatory aberrations. As the accumulation of genome-wide data continues to outpace the technology to validate individual genes, basic cancer research is faced with a need to isolate bona fide therapeutic targets. The brief survey of articles in Table 1 indicates that we are faced with a wealth of candidate melanoma modifiers, each with a long experimental process ahead of it (it should be noted, however, that many of the studies with a smaller extent of validation achieved goals other than gene discovery.) The extents to which these listed genes truly control melanoma behavior and thus represent promising therapeutic targets await further testing, as illustrated by the examples of NEDD9 (neural precursor cell expressed, developmentally down-regulated 9) and MITF below.
NEDD9
The power of array technology further benefits from comparisons among species, with the evolutionary conservation of genetic pathways providing a means to highlight significant changes; this is evidenced by the novel cross-species identification of Nedd9 as a regulator of melanoma metastasis. The Ink4a/Arf−/−, inducible H-ras mouse melanoma model 55, 131 provided a tractable means by which to create isogenic metastatic and non-metastatic tumors. Selective pressure for “escaper” tumors no longer dependent on the doxycycline-induced H-ras signal was generated by alternating the signal over defined time periods. Two such lines proved metastatic to distant sites in xenografts. DNA copy number profiles of these lines and a non-metastatic sister line were compared by aCGH, pinpointing an 850kb common site of amplification on chromosome 13. Only one gene in this region, Nedd9, showed a significant upregulation in mouse melanomas but not in normal melanocytes. The syntenic human region, 6p24-25, undergoes copy number gain in 36% of human metastatic, but not non-metastatic, melanomas 124, 162, indicating a common route of genetic modification between human and mouse. Indeed, human melanoma tissue microarrays (TMAs) revealed a significant correlation of Nedd9 protein levels with tumor progression.
Illustrating the power of candidate gene validation, Nedd9 showed metastasis-modulating activity at multiple levels. In Boyden chamber assays, overexpression of Nedd9 enhanced the invasiveness and growth of non-metastatic Ink4a/Arf−/− cell lines expressing activated H-Ras or B-Raf, while knockdown by shRNA inhibited invasiveness in the original metastatic escaper line. Demonstrating this bidirectional behavior revealed the dependency of the cell line on Nedd9 for invasive potential. Nedd9 also conferred a capacity to metastasize to distant sites when overexpressing cells were injected subcutaneously into SCID mice, providing more clinically relevant in vivo evidence. Finally, the mechanism of Nedd9 action was dissected with biochemical and genetic assays, which showed focal adhesion kinase (Fak) to be a critical mediator of Nedd9-induced in vitro invasion, formation of dynamic focal contacts, and attachment to matrigel. These properties are considered hallmarks of cell motility and a capacity for extravasation into the bloodstream.
The identification of Nedd9 as a cross-species melanoma metastasis gene reinforced two major concepts: that the fundamental properties of cancer genetics are conserved and thus highlighted between mouse and human and that modulation of a single genetic pathway can greatly alter tumor fate. The ongoing research into creating and testing mice genetically engineered to over- or underexpress Nedd9 moves it closer to a thorough conception for its clinical translation.
MITF
The identification of MITF as a critical melanoma survival gene took a cross-tissue approach, wherein the NCI-60 cell line panel representing nine tumor types was subjected to both gene expression and SNP array analysis 69. A recurrent gain of 3p13-14 significantly segregated melanoma from other tumor classes and was confirmed by quantitative PCR (qPCR). The combined expression and qPCR data isolated MITF as the only gene in the region with maximal amplification and overexpression. Fluorescence in situ hybridization and immunohistochemistry on melanoma TMAs revealed a correlation of increased MITF gene dosage and protein levels with malignancy and decreased survival. Correlation, however, does not discriminate between MITF upregulation as a cause or bystander effect of malignant transformation. Therefore, the effect of exogenous MITF was determined in human melanocytes engineered to be immortalized but non-transformed. Only when MITF was expressed in a melanoma-relevant signaling context (i.e. activated BRAF) cells were able to grow in the absence of otherwise essential media factors and able to form colonies in soft agar, both results suggestive of full transformation. Conversely, inhibition of MITF in cell lines showing 3p13-14 amplification reduced growth and survival and conferred sensitivity to certain anticancer drugs. This careful validation of MITF in human cells sets the stage for in vivo mouse studies.
By comparing different tumor types, genetic changes specific to the relevant lineages were highlighted. The critical role of MITF in both normal melanocyte development and melanoma survival was therefore suggested to identify, along with androgen receptor, a novel class of oncogenes termed “lineage addiction” oncogenes 163. The tumor may “hijack” extant lineage survival mechanisms in the presence of selective pressures; indeed, activated BRAF is known to target MITF for proteolytic degradation, which may select for refractory cellular variants with amplified MITF. Tellingly, MITF gene disruption leads to coat color graying in mice 164, 165 and pigmentation and hearing defects (melanocytes play a role in cochlear development) in humans, termed Waardenburg Syndrome Type 2A 166. The demonstration that a gene involved specifically in melanocyte maintenance and differentiation is dysregulated in melanomas opens the possibility that anticancer drugs can be targeted not only to specific cellular pathways, but also to specific cell types.
Epigenetics, miRNAs, and proteomics
Even with a combination of the three main avenues of genome-wide interrogation – gene expression, DNA copy number, and LOH – a full picture of the changes that take place in tumorigenesis is lacking. The intracellular milieu has many levels of regulation, both at the DNA and protein levels. Thus, a number of other large-scale technologies will build towards a more comprehensive view of melanoma: epigenetic profiling, screening for non-coding but functional DNA, and proteomic profiling (Fig 2).
Figure 2.
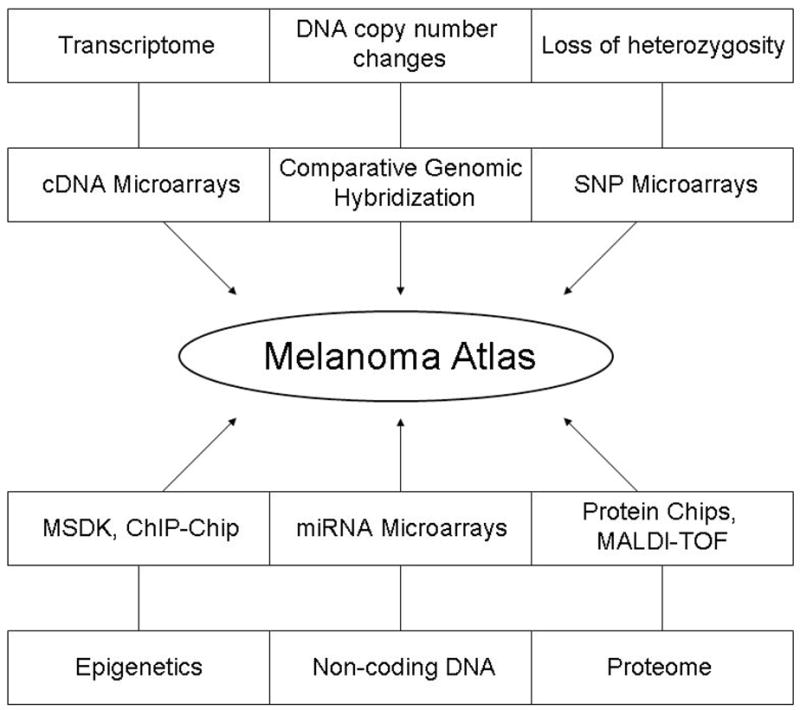
Types of cellular data and some of the respective high-throughput technologies.
Epigenetic studies in cancer have focused on the role of both DNA methylation and histone modifications to understand how genes can be aberrantly regulated in the absence of disruptive mutations. The methylation of cytosines within CpG islands upstream of gene promoters results in gene silencing, while methylation at other sites may activate genes, such as at the differentially methylated region of Igf2 (insulin-like growth factor 2) 167. In conjunction, the “histone code” represents a complex combination of modifications of specific histone tail residues, with certain methylation and acetylation (and, perhaps to a lesser extent, phosphorylation, ubiquitination, and sumoylation) patterns governing chromatin structure and hence gene expression. Though the interaction of DNA and histone modifications continues to be clarified, recent studies suggest that silencing via histone methylation may supercede the cytosine methylation status 168.
Hence, genome-wide epigenetic scans can be either DNA-specific – methylation-specific digital karyotyping 169, restriction landmark genome scanning 170, or ChIP-Chip 171 – or histone-specific, such as applying expression arrays to cells treated with histone deacetylase (HDAC) inhibitors 172. One study made use of the DNA methylation inhibitor 5-Aza-2′-deoxycytidine to compare treated and untreated melanoma cell lines 173. The genes SYN and HOXB13 were found to be significantly re-expressed upon treatment, and were also reduced in untreated cell lines compared to normal melanocytes. Overexpression of each gene individually reduced in vitro proliferation and in vivo xenograft tumor size, indicating a tumor suppressor role for both genes. Other current evidence points to a pivotal role for the epigenetic regulation of specific genes in melanoma progression: focal comparisons of primary and metastatic tumor specimens have confirmed differential methylation statuses of peroxiredoxin 2 174, skeletrophin 175, and estrogen receptor (ER)-α 176. Intriguingly, ER-α 176 and PTEN 177 methylation are increased in DNA circulating in the serum of patients with metastatic disease, suggesting that they may be useful as clinical indicators. Furthermore, HDAC inhibitors show some initial promise in cell lines and xenografts 178, supporting global epigenetic changes as relevant for tumor maintenance.
Long referred to as “junk DNA,” certain non-coding genetic elements have also recently taken a prominent spot in gene regulation. Of particular interest are miRNAs, which are short, 21-23bp RNAs that bind to homologous mRNA segments and mediate the translational blockage or argonaute-directed degradation of gene transcripts as well as initiate epigenetic gene silencing modifications. Multi-platform assays have identified an enrichment for miRNAs in various solid cancers that target known tumor suppressors and oncogenes 179. The same group discovered that miR155 can function as an oncogene in B cell lineages, with Eμ-directed overexpression resulting in the development of leukemia and lymphoma 180. Whether miRNAs play a significant functional role in melanoma remains to be seen, though initial studies on different tumor types demonstrate that melanoma can be differentiated from other tumor types based solely on miRNA expression signatures 181 and that each of 243/283 miRNAs exhibited copy number changes in at least seven of 45 melanoma cell lines 182.
Finally, gene expression levels do not necessarily correlate with protein levels, due to posttranslational modifications undetectable by gene-centric assays. Comprehensive analysis of the tumor proteome is a technically challenging process, requiring the use of low-throughput biochemical isolation techniques (2D-gel electrophoresis, protein chips) and specialized machinery for protein identification and quantification (MALDI-TOF). Nevertheless, such analysis of a mouse melanoma xenograft model identified associations of increased VEGF and cathepsin D levels with progression 183. Proteomics also provides a plausible method for non-invasive prognostic tests: the MALDI-TOF analysis of patient sera identified a protein spectrum that could retrospectively classify 55 progressive and non-progressive stage III tumors with 80% specificity 184. Such proteomic data awaits predictive testing. Continuing technology improvements will necessitate larger scale studies towards a more complete understanding of the melanoma cell.
STRATEGIES FOR TRANSLATION OF GENETIC INFORMATION INTO MELANOMA THERAPY
All in all, melanoma must be regarded as a genetically and biologically heterogeneous disease. This heterogeneity is exemplified in the clinics by the differential and until today unpredictable response to therapy. Rational drug design has significantly changed the daily practice of clinical oncology through introduction of small molecule inhibitors and antibodies, e.g. against overexpression of HER2/NEU in breast cancer, activating c-KIT mutations in gastrointestinal stromal tumors, and EGFR mutations and overexpression in non-small cell lung cancer. What could be the prospects for the paradigm of genetic targets to melanoma?
Due to the high incidence of activating mutations in genes of the RAS/RAF/MEK/ERK signaling pathway, BRAF targeting has been predicted as a promising therapeutic strategy for melanoma, but failed to accomplish meaningful clinical activity 22. A possible explanation for this could be that the agent itself may be insufficiently potent. However, with a more complete understanding of the genetic and functional data of this pathway -as outlined above- we favor explanations suggested by genetic and functional data: (1) BRAFV600E alone is not sufficient to cause melanoma, but induces cell cycle arrest with concomitant induction of oncogene induced senescence, (2) BRAF activity is modulated by the genetic background at the single cell level, e.g. the presence or absence of control mechanisms such as p16INK4A, (3) RAS/RAF/MEK/ERK signaling switches from BRAF to CRAF signaling upon the acquisition of RAS mutation, (4) pathway redundancy or digression may occur: in contrast to targets successfully targeted in other cancers, the most frequent mutation of the RAS/RAF/MEK/ERK signaling pathway lies several steps downstream of the initial receptor-ligand interaction, thus favoring the recruitment of alternative pathways of cell signaling, (5) the development of resistance mechanisms: several melanoma cell lines survive MAPK inhibition by expression of the antiapoptotic factors Mcl-1, Bcl-xL and Bcl-2 and suppression of tumor suppressor p53 through ROS 185.
Unlike the RAS/RAF/MEK/ERK pathway, genetic alterations specifically affecting components of other established signaling cascades do occur only in a small proportion of melanomas. This complex and heterogeneous genetic and biological background significantly challenges the identification of targets for drug development and should preclude the unselected enrollment of melanoma patients into clinical trials. In this context, strategies towards a genetic-based patient selection and individualized therapy have to be developed. In the last few years, we have been witnessing the establishment of first genotype-phenotype correlations. Examples are the reported association of activating N-RAS mutations with nodular lesions and sun exposure 50, 51, the occurrence of BRAF mutations particularly in melanomas from body sites with intermittent UV exposure 59 and their correlation with germline variants of the MC1R gene 60, the presence of CDK4 amplifications preferentially in acral and mucosal melanomas 59, the presence of Cyclin D1 amplifications particularly in melanomas from skin with chronic sun damage 59, and the presence of activating KIT mutations and gene amplifications in acral melanoma, mucosal melanoma, and melanomas from chronically sun-damaged skin 24.
However, these correlations can provide only a rough estimate on the presence of certain types of genetic alterations and are far from being predictive for the most appropriate avenue of personalized treatment. In this regard, individualized genomic information can provide significant contributions.
OUTLOOK: INDIVIDUAL GENETIC TYPING FOR GENOTYPE-PHENOTYPE CORRELATIONS
Success in identifying gene expression signatures predictive of survival in breast and other solid cancers 186, 187 establishes a real possibility that the molecular profiling of melanoma can inform clinical decisions. Recent descriptive, retrospective observations of gene expression in metastatic versus non metastatic tumors 188–190 provide a tantalizing glimpse into what these signatures may look like, but they must be functionally validated in order to have true clinical relevance.
Indeed, the ability to classify melanomas by particular genomic signatures suggests such a predictive capacity. For example, CGH profiling could distinguish among 126 acral, musocal, and chronically and non-chronically sun-damaged primary skin melanomas with 70% accuracy 59. BRAF and NRAS mutations were significantly associated with the non-chronically sun damaged subtype, suggesting that knowledge of common gene mutations alone could provide a degree of classifying information. In fact, a microarray analysis of cell lines with and without CDKN2A deletions, coupled with confirmatory RT-PCR on 14 cell lines, identified eight genes consistently differing in expression between the two classes 191. Conversely, although two studies supported an expression profile difference between NRAS and BRAF mutant tumors 192, 193, others employing stricter statistical parameters did not 188, 194. Overall, genome-wide profiling may provide more specific classification than single-gene sequencing.
In the farther future, the feasibility of personal whole-genome sequencing comes ever closer as sequencing costs continue to drop and novel techniques continue to be rapidly developed. Recent initiatives to exhaustively sequence annotated genes in breast and colon cancers have unearthed only a fractional minority of putatively functional aberrations 195. Given the rich diversity of cellular regulation illustrated above, any future clinically-oriented sequencing efforts may need a massive supporting network of data to have true prognostic value.
Acknowledgments
This work was supported by grants to SNW (Grant No. APP19722FW from the FWF - Fonds zur Förderung der wissenschaftlichen Forschung) and LC (NIH RO1 CA93947; UO1 CA84313 and P50 CA93683).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
BIBLIOGRAPHY
- 1.Lasithiotakis KG, Leiter U, Gorkievicz R, et al. The incidence and mortality of cutaneous melanoma in Southern Germany: trends by anatomic site and pathologic characteristics, 1976 to 2003. Cancer. 2006 Sep 15;107(6):1331–1339. doi: 10.1002/cncr.22126. [DOI] [PubMed] [Google Scholar]
- 2.Giblin AV, Thomas JM. Incidence, mortality and survival in cutaneous melanoma. J Plast Reconstr Aesthet Surg. 2007;60(1):32–40. doi: 10.1016/j.bjps.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 3.Balch CM, Buzaid AC, Soong SJ, et al. Final version of the American Joint Committee on Cancer staging system for cutaneous melanoma. J Clin Oncol. 2001 Aug 15;19(16):3635–3648. doi: 10.1200/JCO.2001.19.16.3635. [DOI] [PubMed] [Google Scholar]
- 4.Baade P, Coory M. Trends in melanoma mortality in Australia: 1950–2002 and their implications for melanoma control. Aust N Z J Public Health. 2005 Aug;29(4):383–386. doi: 10.1111/j.1467-842x.2005.tb00211.x. [DOI] [PubMed] [Google Scholar]
- 5.Coory M, Baade P, Aitken J, Smithers M, McLeod GR, Ring I. Trends for in situ and invasive melanoma in Queensland, Australia, 1982–2002. Cancer Causes Control. 2006 Feb;17(1):21–27. doi: 10.1007/s10552-005-3637-4. [DOI] [PubMed] [Google Scholar]
- 6.Weinstock MA. Sun protection and early detection: do we have the balance right? 6th World Congress on Melanoma. 2005 abstract 114. [Google Scholar]
- 7.Berwick M, Wiggins C. The current epidemiology of cutaneous malignant melanoma. Front Biosci. 2006;11:1244–1254. doi: 10.2741/1877. [DOI] [PubMed] [Google Scholar]
- 8.Thompson JF, Scolyer RA, Kefford RF. Cutaneous melanoma. Lancet. 2005 Feb 19–25;365(9460):687–701. doi: 10.1016/S0140-6736(05)17951-3. [DOI] [PubMed] [Google Scholar]
- 9.Gilchrest BA, Eller MS, Geller AC, Yaar M. The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med. 1999 Apr 29;340(17):1341–1348. doi: 10.1056/NEJM199904293401707. [DOI] [PubMed] [Google Scholar]
- 10.Valverde P, Healy E, Jackson I, Rees JL, Thody AJ. Variants of the melanocyte-stimulating hormone receptor gene are associated with red hair and fair skin in humans. Nat Genet. 1995 Nov;11(3):328–330. doi: 10.1038/ng1195-328. [DOI] [PubMed] [Google Scholar]
- 11.Frandberg PA, Doufexis M, Kapas S, Chhajlani V. Human pigmentation phenotype: a point mutation generates nonfunctional MSH receptor. Biochem Biophys Res Commun. 1998 Apr 17;245(2):490–492. doi: 10.1006/bbrc.1998.8459. [DOI] [PubMed] [Google Scholar]
- 12.Kennedy C, ter Huurne J, Berkhout M, et al. Melanocortin 1 receptor (MC1R) gene variants are associated with an increased risk for cutaneous melanoma which is largely independent of skin type and hair color. J Invest Dermatol. 2001 Aug;117(2):294–300. doi: 10.1046/j.0022-202x.2001.01421.x. [DOI] [PubMed] [Google Scholar]
- 13.Clark WH, Jr, Elder DE, Guerry Dt, Epstein MN, Greene MH, Van Horn M. A study of tumor progression: the precursor lesions of superficial spreading and nodular melanoma. Hum Pathol. 1984 Dec;15(12):1147–1165. doi: 10.1016/s0046-8177(84)80310-x. [DOI] [PubMed] [Google Scholar]
- 14.Ishihara K, Saida T, Yamamoto A. Updated statistical data for malignant melanoma in Japan. Int J Clin Oncol. 2001 Jun;6(3):109–116. doi: 10.1007/pl00012091. [DOI] [PubMed] [Google Scholar]
- 15.Miller AJ, Mihm MC., Jr Melanoma. N Engl J Med. 2006 Jul 6;355(1):51–65. doi: 10.1056/NEJMra052166. [DOI] [PubMed] [Google Scholar]
- 16.Middleton MR, Grob JJ, Aaronson N, et al. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol. 2000 Jan;18(1):158–166. doi: 10.1200/JCO.2000.18.1.158. [DOI] [PubMed] [Google Scholar]
- 17.Bedikian AY, Millward M, Pehamberger H, et al. Bcl-2 antisense (oblimersen sodium) plus dacarbazine in patients with advanced melanoma: the Oblimersen Melanoma Study Group. J Clin Oncol. 2006 Oct 10;24(29):4738–4745. doi: 10.1200/JCO.2006.06.0483. [DOI] [PubMed] [Google Scholar]
- 18.Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999 Jul;17(7):2105–2116. doi: 10.1200/JCO.1999.17.7.2105. [DOI] [PubMed] [Google Scholar]
- 19.Atkins MB, Kunkel L, Sznol M, Rosenberg SA. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: long-term survival update. Cancer J Sci Am. 2000 Feb;6(Suppl 1):S11–14. [PubMed] [Google Scholar]
- 20.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004 Sep;10(9):909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weinstein IB, Joe AK. Mechanisms of disease: Oncogene addiction--a rationale for molecular targeting in cancer therapy. Nat Clin Pract Oncol. 2006 Aug;3(8):448–457. doi: 10.1038/ncponc0558. [DOI] [PubMed] [Google Scholar]
- 22.Eisen T, Ahmad T, Flaherty KT, et al. Sorafenib in advanced melanoma: a Phase II randomised discontinuation trial analysis. Br J Cancer. 2006 Sep 4;95(5):581–586. doi: 10.1038/sj.bjc.6603291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giehl KA, Nagele U, Volkenandt M, Berking C. Protein expression of melanocyte growth factors (bFGF, SCF) and their receptors (FGFR-1, c-kit) in nevi and melanoma. J Cutan Pathol. 2007 Jan;34(1):7–14. doi: 10.1111/j.1600-0560.2006.00569.x. [DOI] [PubMed] [Google Scholar]
- 24.Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006 Sep 10;24(26):4340–4346. doi: 10.1200/JCO.2006.06.2984. [DOI] [PubMed] [Google Scholar]
- 25.Ugurel S, Hildenbrand R, Zimpfer A, et al. Lack of clinical efficacy of imatinib in metastatic melanoma. Br J Cancer. 2005 Apr 25;92(8):1398–1405. doi: 10.1038/sj.bjc.6602529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wyman K, Atkins MB, Prieto V, et al. Multicenter Phase II trial of high-dose imatinib mesylate in metastatic melanoma: significant toxicity with no clinical efficacy. Cancer. 2006 May 1;106(9):2005–2011. doi: 10.1002/cncr.21834. [DOI] [PubMed] [Google Scholar]
- 27.Ellis DL, King LE, Jr, Nanney LB. Increased epidermal growth factor receptors in melanocytic lesions. J Am Acad Dermatol. 1992 Oct;27(4):539–546. doi: 10.1016/0190-9622(92)70219-6. [DOI] [PubMed] [Google Scholar]
- 28.Sparrow LE, Heenan PJ. Differential expression of epidermal growth factor receptor in melanocytic tumours demonstrated by immunohistochemistry and mRNA in situ hybridization. Australas J Dermatol. 1999 Feb;40(1):19–24. doi: 10.1046/j.1440-0960.1999.00310.x. [DOI] [PubMed] [Google Scholar]
- 29.Wyman K, Kelley M, Puzanov I, et al. Phase II study of erlotinib given daily for patients with metastatic melanoma (MM). J Clin Oncol; 2006 ASCO Annual Meeting Proceedings Part I; June 20 Supplement; 2006. p. 18002 (abstract). [Google Scholar]
- 30.Futreal PA, Coin L, Marshall M, et al. A census of human cancer genes. Nat Rev Cancer. 2004 Mar;4(3):177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000 Jan 7;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 32.Takata M, Goto Y, Ichii N, et al. Constitutive activation of the mitogen-activated protein kinase signaling pathway in acral melanomas. J Invest Dermatol. 2005 Aug;125(2):318–322. doi: 10.1111/j.0022-202X.2005.23812.x. [DOI] [PubMed] [Google Scholar]
- 33.Zhuang L, Lee CS, Scolyer RA, et al. Activation of the extracellular signal regulated kinase (ERK) pathway in human melanoma. J Clin Pathol. 2005 Nov;58(11):1163–1169. doi: 10.1136/jcp.2005.025957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bohm M, Moellmann G, Cheng E, et al. Identification of p90RSK as the probable CREB-Ser133 kinase in human melanocytes. Cell Growth Differ. 1995 Mar;6(3):291–302. [PubMed] [Google Scholar]
- 35.Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989 Sep 1;49(17):4682–4689. [PubMed] [Google Scholar]
- 36.Dumaz N, Hayward R, Martin J, et al. In Melanoma, RAS Mutations Are Accompanied by Switching Signaling from BRAF to CRAF and Disrupted Cyclic AMP Signaling. Cancer Res. 2006 Oct 1;66(19):9483–9491. doi: 10.1158/0008-5472.CAN-05-4227. [DOI] [PubMed] [Google Scholar]
- 37.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002 Jun 27;417(6892):949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 38.Maldonado JL, Fridlyand J, Patel H, et al. Determinants of BRAF mutations in primary melanomas. J Natl Cancer Inst. 2003 Dec 17;95(24):1878–1890. doi: 10.1093/jnci/djg123. [DOI] [PubMed] [Google Scholar]
- 39.Pollock PM, Harper UL, Hansen KS, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003 Jan;33(1):19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- 40.Uribe P, Wistuba II, Gonzalez S. BRAF mutation: a frequent event in benign, atypical, and malignant melanocytic lesions of the skin. Am J Dermatopathol. 2003 Oct;25(5):365–370. doi: 10.1097/00000372-200310000-00001. [DOI] [PubMed] [Google Scholar]
- 41.Daniotti M, Oggionni M, Ranzani T, et al. BRAF alterations are associated with complex mutational profiles in malignant melanoma. Oncogene. 2004 Aug 5;23(35):5968–5977. doi: 10.1038/sj.onc.1207780. [DOI] [PubMed] [Google Scholar]
- 42.Kumar R, Angelini S, Snellman E, Hemminki K. BRAF mutations are common somatic events in melanocytic nevi. J Invest Dermatol. 2004 Feb;122(2):342–348. doi: 10.1046/j.0022-202X.2004.22225.x. [DOI] [PubMed] [Google Scholar]
- 43.Shinozaki M, Fujimoto A, Morton DL, Hoon DS. Incidence of BRAF oncogene mutation and clinical relevance for primary cutaneous melanomas. Clin Cancer Res. 2004 Mar 1;10(5):1753–1757. doi: 10.1158/1078-0432.ccr-1169-3. [DOI] [PubMed] [Google Scholar]
- 44.Libra M, Malaponte G, Navolanic PM, et al. Analysis of BRAF mutation in primary and metastatic melanoma. Cell Cycle. 2005 Oct;4(10):1382–1384. doi: 10.4161/cc.4.10.2026. [DOI] [PubMed] [Google Scholar]
- 45.Petti C, Molla A, Vegetti C, Ferrone S, Anichini A, Sensi M. Coexpression of NRASQ61R and BRAFV600E in human melanoma cells activates senescence and increases susceptibility to cell-mediated cytotoxicity. Cancer Res. 2006 Jul 1;66(13):6503–6511. doi: 10.1158/0008-5472.CAN-05-4671. [DOI] [PubMed] [Google Scholar]
- 46.Wagner SN, Ockenfels HM, Wagner C, Hofler H, Goos M. Ras gene mutations: a rare event in nonmetastatic primary malignant melanoma. J Invest Dermatol. 1995 May;104(5):868–871. doi: 10.1111/1523-1747.ep12607039. [DOI] [PubMed] [Google Scholar]
- 47.Chin L, Merlino G, DePinho RA. Malignant melanoma: modern black plague and genetic black box. Genes Dev. 1998 Nov 15;12(22):3467–3481. doi: 10.1101/gad.12.22.3467. [DOI] [PubMed] [Google Scholar]
- 48.Bauer J, Curtin JA, Pinkel D, Bastian BC. Congenital melanocytic nevi frequently harbor NRAS mutations but no BRAF mutations. J Invest Dermatol. 2007 Jan;127(1):179–182. doi: 10.1038/sj.jid.5700490. [DOI] [PubMed] [Google Scholar]
- 49.Demunter A, Stas M, Degreef H, De Wolf-Peeters C, van den Oord JJ. Analysis of N- and K-ras mutations in the distinctive tumor progression phases of melanoma. J Invest Dermatol. 2001 Dec;117(6):1483–1489. doi: 10.1046/j.0022-202x.2001.01601.x. [DOI] [PubMed] [Google Scholar]
- 50.Jafari M, Papp T, Kirchner S, et al. Analysis of ras mutations in human melanocytic lesions: activation of the ras gene seems to be associated with the nodular type of human malignant melanoma. J Cancer Res Clin Oncol. 1995;121(1):23–30. doi: 10.1007/BF01202725. [DOI] [PubMed] [Google Scholar]
- 51.van Elsas A, Zerp SF, van der Flier S, et al. Relevance of ultraviolet-induced N-ras oncogene point mutations in development of primary human cutaneous melanoma. Am J Pathol. 1996 Sep;149(3):883–893. [PMC free article] [PubMed] [Google Scholar]
- 52.Albino AP, Nanus DM, Mentle IR, et al. Analysis of ras oncogenes in malignant melanoma and precursor lesions: correlation of point mutations with differentiation phenotype. Oncogene. 1989 Nov;4(11):1363–1374. [PubMed] [Google Scholar]
- 53.Papp T, Pemsel H, Zimmermann R, Bastrop R, Weiss DG, Schiffmann D. Mutational analysis of the N-ras, p53, p16INK4a, CDK4, and MC1R genes in human congenital melanocytic naevi. J Med Genet. 1999 Aug;36(8):610–614. [PMC free article] [PubMed] [Google Scholar]
- 54.Bastian BC, LeBoit PE, Pinkel D. Mutations and copy number increase of HRAS in Spitz nevi with distinctive histopathological features. Am J Pathol. 2000 Sep;157(3):967–972. doi: 10.1016/S0002-9440(10)64609-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chin L, Pomerantz J, Polsky D, et al. Cooperative effects of INK4a and ras in melanoma susceptibility in vivo. Genes Dev. 1997 Nov 1;11(21):2822–2834. doi: 10.1101/gad.11.21.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bardeesy N, Bastian BC, Hezel A, Pinkel D, DePinho RA, Chin L. Dual inactivation of RB and p53 pathways in RAS-induced melanomas. Mol Cell Biol. 2001 Mar;21(6):2144–2153. doi: 10.1128/MCB.21.6.2144-2153.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sharpless NE, Kannan K, Xu J, Bosenberg MW, Chin L. Both products of the mouse Ink4a/Arf locus suppress melanoma formation in vivo. Oncogene. 2003 Aug 7;22(32):5055–5059. doi: 10.1038/sj.onc.1206809. [DOI] [PubMed] [Google Scholar]
- 58.Ackermann J, Frutschi M, Kaloulis K, McKee T, Trumpp A, Beermann F. Metastasizing melanoma formation caused by expression of activated N-RasQ61K on an INK4a-deficient background. Cancer Res. 2005 May 15;65(10):4005–4011. doi: 10.1158/0008-5472.CAN-04-2970. [DOI] [PubMed] [Google Scholar]
- 59.Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005 Nov 17;353(20):2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 60.Landi MT, Bauer J, Pfeiffer RM, et al. MC1R germline variants confer risk for BRAF-mutant melanoma. Science. 2006 Jul 28;313(5786):521–522. doi: 10.1126/science.1127515. [DOI] [PubMed] [Google Scholar]
- 61.Garnett MJ, Marais R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell. 2004 Oct;6(4):313–319. doi: 10.1016/j.ccr.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 62.Wellbrock C, Marais R. Elevated expression of MITF counteracts B-RAF-stimulated melanocyte and melanoma cell proliferation. J Cell Biol. 2005 Aug 29;170(5):703–708. doi: 10.1083/jcb.200505059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu J, Suresh Kumar KG, Yu D, et al. Oncogenic BRAF regulates beta-Trcp expression and NF-kappaB activity in human melanoma cells. Oncogene. 2007 Mar 22;26(13):1954–1958. doi: 10.1038/sj.onc.1209994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bhatt KV, Spofford LS, Aram G, McMullen M, Pumiglia K, Aplin AE. Adhesion control of cyclin D1 and p27Kip1 levels is deregulated in melanoma cells through BRAF-MEK-ERK signaling. Oncogene. 2005 May 12;24(21):3459–3471. doi: 10.1038/sj.onc.1208544. [DOI] [PubMed] [Google Scholar]
- 65.Michaloglou C, Vredeveld LC, Soengas MS, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005 Aug 4;436(7051):720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 66.Bhatt KV, Hu R, Spofford LS, Aplin AE. Mutant B-RAF signaling and cyclin D1 regulate Cks1/S-phase kinase-associated protein 2-mediated degradation of p27Kip1 in human melanoma cells. Oncogene. 2007 Feb 15;26(7):1056–1066. doi: 10.1038/sj.onc.1209861. [DOI] [PubMed] [Google Scholar]
- 67.Saldanha G, Purnell D, Fletcher A, Potter L, Gillies A, Pringle JH. High BRAF mutation frequency does not characterize all melanocytic tumor types. Int J Cancer. 2004 Sep 20;111(5):705–710. doi: 10.1002/ijc.20325. [DOI] [PubMed] [Google Scholar]
- 68.Yazdi AS, Palmedo G, Flaig MJ, et al. Mutations of the BRAF gene in benign and malignant melanocytic lesions. J Invest Dermatol. 2003 Nov;121(5):1160–1162. doi: 10.1046/j.1523-1747.2003.12559.x. [DOI] [PubMed] [Google Scholar]
- 69.Garraway LA, Widlund HR, Rubin MA, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005 Jul 7;436(7047):117–122. doi: 10.1038/nature03664. [DOI] [PubMed] [Google Scholar]
- 70.Wellbrock C, Ogilvie L, Hedley D, et al. V599EB-RAF is an oncogene in melanocytes. Cancer Res. 2004 Apr 1;64(7):2338–2342. doi: 10.1158/0008-5472.can-03-3433. [DOI] [PubMed] [Google Scholar]
- 71.Sharpless NE, DePinho RA. Cancer: crime and punishment. Nature. 2005 Aug 4;436(7051):636–637. doi: 10.1038/436636a. [DOI] [PubMed] [Google Scholar]
- 72.Patton EE, Widlund HR, Kutok JL, et al. BRAF mutations are sufficient to promote nevi formation and cooperate with p53 in the genesis of melanoma. Curr Biol. 2005 Feb 8;15(3):249–254. doi: 10.1016/j.cub.2005.01.031. [DOI] [PubMed] [Google Scholar]
- 73.Chudnovsky Y, Adams AE, Robbins PB, Lin Q, Khavari PA. Use of human tissue to assess the oncogenic activity of melanoma-associated mutations. Nat Genet. 2005 Jul;37(7):745–749. doi: 10.1038/ng1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Goel VK, Lazar AJ, Warneke CL, Redston MS, Haluska FG. Examination of mutations in BRAF, NRAS, and PTEN in primary cutaneous melanoma. J Invest Dermatol. 2006 Jan;126(1):154–160. doi: 10.1038/sj.jid.5700026. [DOI] [PubMed] [Google Scholar]
- 75.Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002 Aug 29;418(6901):934. doi: 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- 76.Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006 May 25;441(7092):424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- 77.Boisvert-Adamo K, Aplin AE. B-RAF and PI-3 kinase signaling protect melanoma cells from anoikis. Oncogene. 2006 Aug 10;25(35):4848–4856. doi: 10.1038/sj.onc.1209493. [DOI] [PubMed] [Google Scholar]
- 78.Meier F, Schittek B, Busch S, et al. The RAS/RAF/MEK/ERK and PI3K/AKT signaling pathways present molecular targets for the effective treatment of advanced melanoma. Front Biosci. 2005;10:2986–3001. doi: 10.2741/1755. [DOI] [PubMed] [Google Scholar]
- 79.Robertson GP. Functional and therapeutic significance of Akt deregulation in malignant melanoma. Cancer Metastasis Rev. 2005 Jun;24(2):273–285. doi: 10.1007/s10555-005-1577-9. [DOI] [PubMed] [Google Scholar]
- 80.Dai DL, Martinka M, Li G. Prognostic significance of activated Akt expression in melanoma: a clinicopathologic study of 292 cases. J Clin Oncol. 2005 Mar 1;23(7):1473–1482. doi: 10.1200/JCO.2005.07.168. [DOI] [PubMed] [Google Scholar]
- 81.Spofford LS, Abel EV, Boisvert-Adamo K, Aplin AE. Cyclin D3 expression in melanoma cells is regulated by adhesion-dependent phosphatidylinositol 3-kinase signaling and contributes to G1-S progression. J Biol Chem. 2006 Sep 1;281(35):25644–25651. doi: 10.1074/jbc.M600197200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Packer L, Pavey S, Parker A, et al. Osteopontin is a downstream effector of the PI3-kinase pathway in melanomas that is inversely correlated with functional PTEN. Carcinogenesis. 2006 Sep;27(9):1778–1786. doi: 10.1093/carcin/bgl016. [DOI] [PubMed] [Google Scholar]
- 83.Curtin JA, Stark MS, Pinkel D, Hayward NK, Bastian BC. PI3-kinase subunits are infrequent somatic targets in melanoma. J Invest Dermatol. 2006 Jul;126(7):1660–1663. doi: 10.1038/sj.jid.5700311. [DOI] [PubMed] [Google Scholar]
- 84.Bastian BC. Understanding the progression of melanocytic neoplasia using genomic analysis: from fields to cancer. Oncogene. 2003 May 19;22(20):3081–3086. doi: 10.1038/sj.onc.1206463. [DOI] [PubMed] [Google Scholar]
- 85.Wu H, Goel V, Haluska FG. PTEN signaling pathways in melanoma. Oncogene. 2003 May 19;22(20):3113–3122. doi: 10.1038/sj.onc.1206451. [DOI] [PubMed] [Google Scholar]
- 86.Mikhail M, Velazquez E, Shapiro R, et al. PTEN expression in melanoma: relationship with patient survival, Bcl-2 expression, and proliferation. Clin Cancer Res. 2005 Jul 15;11(14):5153–5157. doi: 10.1158/1078-0432.CCR-05-0397. [DOI] [PubMed] [Google Scholar]
- 87.Pollock PM, Walker GJ, Glendening JM, et al. PTEN inactivation is rare in melanoma tumours but occurs frequently in melanoma cell lines. Melanoma Res. 2002 Dec;12(6):565–575. doi: 10.1097/00008390-200212000-00006. [DOI] [PubMed] [Google Scholar]
- 88.Slipicevic A, Holm R, Nguyen MT, Bohler PJ, Davidson B, Florenes VA. Expression of activated Akt and PTEN in malignant melanomas: relationship with clinical outcome. Am J Clin Pathol. 2005 Oct;124(4):528–536. doi: 10.1309/YT58WWMTA6YR1PRV. [DOI] [PubMed] [Google Scholar]
- 89.Robertson GP, Furnari FB, Miele ME, et al. In vitro loss of heterozygosity targets the PTEN/MMAC1 gene in melanoma. Proc Natl Acad Sci U S A. 1998 Aug 4;95(16):9418–9423. doi: 10.1073/pnas.95.16.9418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stahl JM, Cheung M, Sharma A, Trivedi NR, Shanmugam S, Robertson GP. Loss of PTEN promotes tumor development in malignant melanoma. Cancer Res. 2003 Jun 1;63(11):2881–2890. [PubMed] [Google Scholar]
- 91.Stewart AL, Mhashilkar AM, Yang XH, et al. PI3 kinase blockade by Ad-PTEN inhibits invasion and induces apoptosis in RGP and metastatic melanoma cells. Mol Med. 2002 Aug;8(8):451–461. [PMC free article] [PubMed] [Google Scholar]
- 92.Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998 Aug;19(4):348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- 93.Ma X, Ziel-van der Made AC, Autar B, et al. Targeted biallelic inactivation of Pten in the mouse prostate leads to prostate cancer accompanied by increased epithelial cell proliferation but not by reduced apoptosis. Cancer Res. 2005 Jul 1;65(13):5730–5739. doi: 10.1158/0008-5472.CAN-04-4519. [DOI] [PubMed] [Google Scholar]
- 94.Podsypanina K, Ellenson LH, Nemes A, et al. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci U S A. 1999 Feb 16;96(4):1563–1568. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stambolic V, Suzuki A, de la Pompa JL, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998 Oct 2;95(1):29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 96.You MJ, Castrillon DH, Bastian BC, et al. Genetic analysis of Pten and Ink4a/Arf interactions in the suppression of tumorigenesis in mice. Proc Natl Acad Sci U S A. 2002 Feb 5;99(3):1455–1460. doi: 10.1073/pnas.022632099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Trotman LC, Wang X, Alimonti A, et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell. 2007 Jan 12;128(1):141–156. doi: 10.1016/j.cell.2006.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shen WH, Balajee AS, Wang J, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007 Jan 12;128(1):157–170. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- 99.Wang X, Trotman LC, Koppie T, et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell. 2007 Jan 12;128(1):129–139. doi: 10.1016/j.cell.2006.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bedogni B, Welford SM, Kwan AC, Ranger-Moore J, Saboda K, Powell MB. Inhibition of phosphatidylinositol-3-kinase and mitogen-activated protein kinase kinase 1/2 prevents melanoma development and promotes melanoma regression in the transgenic TPRas mouse model. Mol Cancer Ther. 2006 Dec;5(12):3071–3077. doi: 10.1158/1535-7163.MCT-06-0269. [DOI] [PubMed] [Google Scholar]
- 101.Smalley KS, Haass NK, Brafford PA, Lioni M, Flaherty KT, Herlyn M. Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases. Mol Cancer Ther. 2006 May;5(5):1136–1144. doi: 10.1158/1535-7163.MCT-06-0084. [DOI] [PubMed] [Google Scholar]
- 102.Tsao H, Goel V, Wu H, Yang G, Haluska FG. Genetic interaction between NRAS and BRAF mutations and PTEN/MMAC1 inactivation in melanoma. J Invest Dermatol. 2004 Feb;122(2):337–341. doi: 10.1046/j.0022-202X.2004.22243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Govindarajan B, Sligh JE, Vincent BJ, et al. Overexpression of Akt converts radial growth melanoma to vertical growth melanoma. J Clin Invest. 2007 Mar;117(3):719–729. doi: 10.1172/JCI30102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Stahl JM, Sharma A, Cheung M, et al. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 2004 Oct 1;64(19):7002–7010. doi: 10.1158/0008-5472.CAN-04-1399. [DOI] [PubMed] [Google Scholar]
- 105.Yoeli-Lerner M, Yiu GK, Rabinovitz I, Erhardt P, Jauliac S, Toker A. Akt blocks breast cancer cell motility and invasion through the transcription factor NFAT. Mol Cell. 2005 Nov 23;20(4):539–550. doi: 10.1016/j.molcel.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 106.Ross P, Hall L, Haff LA. Quantitative approach to single-nucleotide polymorphism analysis using MALDI-TOF mass spectrometry. Biotechniques. 2000 Sep;29(3):620–626. 628–629. doi: 10.2144/00293rr05. [DOI] [PubMed] [Google Scholar]
- 107.Ronnstrand L. Signal transduction via the stem cell factor receptor/c-Kit. Cell Mol Life Sci. 2004 Oct;61(19–20):2535–2548. doi: 10.1007/s00018-004-4189-6. [DOI] [PubMed] [Google Scholar]
- 108.Yoshida H, Kunisada T, Kusakabe M, Nishikawa S, Nishikawa SI. Distinct stages of melanocyte differentiation revealed by anlaysis of nonuniform pigmentation patterns. Development. 1996 Apr;122(4):1207–1214. doi: 10.1242/dev.122.4.1207. [DOI] [PubMed] [Google Scholar]
- 109.Grichnik JM, Burch JA, Burchette J, Shea CR. The SCF/KIT pathway plays a critical role in the control of normal human melanocyte homeostasis. J Invest Dermatol. 1998 Aug;111(2):233–238. doi: 10.1046/j.1523-1747.1998.00272.x. [DOI] [PubMed] [Google Scholar]
- 110.Sviderskaya EV, Wakeling WF, Bennett DC. A cloned, immortal line of murine melanoblasts inducible to differentiate to melanocytes. Development. 1995 May;121(5):1547–1557. doi: 10.1242/dev.121.5.1547. [DOI] [PubMed] [Google Scholar]
- 111.Kunisada T, Lu SZ, Yoshida H, et al. Murine cutaneous mastocytosis and epidermal melanocytosis induced by keratinocyte expression of transgenic stem cell factor. J Exp Med. 1998 May 18;187(10):1565–1573. doi: 10.1084/jem.187.10.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Alexeev V, Yoon K. Distinctive role of the cKit receptor tyrosine kinase signaling in mammalian melanocytes. J Invest Dermatol. 2006 May;126(5):1102–1110. doi: 10.1038/sj.jid.5700125. [DOI] [PubMed] [Google Scholar]
- 113.Isabel Zhu Y, Fitzpatrick JE. Expression of c-kit (CD117) in Spitz nevus and malignant melanoma. J Cutan Pathol. 2006 Jan;33(1):33–37. doi: 10.1111/j.0303-6987.2006.00420.x. [DOI] [PubMed] [Google Scholar]
- 114.Montone KT, van Belle P, Elenitsas R, Elder DE. Proto-oncogene c-kit expression in malignant melanoma: protein loss with tumor progression. Mod Pathol. 1997 Sep;10(9):939–944. [PubMed] [Google Scholar]
- 115.Shen SS, Zhang PS, Eton O, Prieto VG. Analysis of protein tyrosine kinase expression in melanocytic lesions by tissue array. J Cutan Pathol. 2003 Oct;30(9):539–547. doi: 10.1034/j.1600-0560.2003.00090.x. [DOI] [PubMed] [Google Scholar]
- 116.Huang S, Luca M, Gutman M, et al. Enforced c-KIT expression renders highly metastatic human melanoma cells susceptible to stem cell factor-induced apoptosis and inhibits their tumorigenic and metastatic potential. Oncogene. 1996 Dec 5;13(11):2339–2347. [PubMed] [Google Scholar]
- 117.Willmore-Payne C, Holden JA, Tripp S, Layfield LJ. Human malignant melanoma: detection of BRAF- and c-kit-activating mutations by high-resolution amplicon melting analysis. Hum Pathol. 2005 May;36(5):486–493. doi: 10.1016/j.humpath.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 118.Willmore-Payne C, Holden JA, Hirschowitz S, Layfield LJ. BRAF and c-kit gene copy number in mutation-positive malignant melanoma. Human Pathology. 2006 doi: 10.1016/j.humpath.2006.01.003. in press. [DOI] [PubMed] [Google Scholar]
- 119.Fukuda R, Hamamoto N, Uchida Y, et al. Gastrointestinal stromal tumor with a novel mutation of KIT proto-oncogene. Intern Med. 2001 Apr;40(4):301–303. doi: 10.2169/internalmedicine.40.301. [DOI] [PubMed] [Google Scholar]
- 120.Nakahara M, Isozaki K, Hirota S, et al. A novel gain-of-function mutation of c-kit gene in gastrointestinal stromal tumors. Gastroenterology. 1998 Nov;115(5):1090–1095. doi: 10.1016/s0016-5085(98)70079-4. [DOI] [PubMed] [Google Scholar]
- 121.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004 May 20;350(21):2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 122.Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004 Jun 4;304(5676):1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 123.Jost M, Kari C, Rodeck U. The EGF receptor - an essential regulator of multiple epidermal functions. Eur J Dermatol. 2000 Oct-Nov;10(7):505–510. [PubMed] [Google Scholar]
- 124.Bastian BC, LeBoit PE, Hamm H, Brocker EB, Pinkel D. Chromosomal gains and losses in primary cutaneous melanomas detected by comparative genomic hybridization. Cancer Res. 1998 May 15;58(10):2170–2175. [PubMed] [Google Scholar]
- 125.Koprowski H, Herlyn M, Balaban G, Parmiter A, Ross A, Nowell P. Expression of the receptor for epidermal growth factor correlates with increased dosage of chromosome 7 in malignant melanoma. Somat Cell Mol Genet. 1985 May;11(3):297–302. doi: 10.1007/BF01534687. [DOI] [PubMed] [Google Scholar]
- 126.Udart M, Utikal J, Krahn GM, Peter RU. Chromosome 7 aneusomy. A marker for metastatic melanoma? Expression of the epidermal growth factor receptor gene and chromosome 7 aneusomy in nevi, primary malignant melanomas and metastases. Neoplasia. 2001 May-Jun;3(3):245–254. doi: 10.1038/sj.neo/7900156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.de Wit PE, Moretti S, Koenders PG, et al. Increasing epidermal growth factor receptor expression in human melanocytic tumor progression. J Invest Dermatol. 1992 Aug;99(2):168–173. doi: 10.1111/1523-1747.ep12616793. [DOI] [PubMed] [Google Scholar]
- 128.Huang TS, Rauth S, Das Gupta TK. Overexpression of EGF receptor is associated with spontaneous metastases of a human melanoma cell line in nude mice. Anticancer Res. 1996 Nov–Dec;16(6B):3557–3563. [PubMed] [Google Scholar]
- 129.Maher EA, Furnari FB, Bachoo RM, et al. Malignant glioma: genetics and biology of a grave matter. Genes Dev. 2001 Jun 1;15(11):1311–1333. doi: 10.1101/gad.891601. [DOI] [PubMed] [Google Scholar]
- 130.Sihto H, Puputti M, Pulli L, et al. Epidermal growth factor receptor domain II, IV, and kinase domain mutations in human solid tumors. J Mol Med. 2005 Dec;83(12):976–983. doi: 10.1007/s00109-005-0699-4. [DOI] [PubMed] [Google Scholar]
- 131.Chin L, Tam A, Pomerantz J, et al. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999 Jul 29;400(6743):468–472. doi: 10.1038/22788. [DOI] [PubMed] [Google Scholar]
- 132.Bardeesy N, Kim M, Xu J, et al. Role of epidermal growth factor receptor signaling in RAS-driven melanoma. Mol Cell Biol. 2005 May;25(10):4176–4188. doi: 10.1128/MCB.25.10.4176-4188.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dlugosz AA, Hansen L, Cheng C, et al. Targeted disruption of the epidermal growth factor receptor impairs growth of squamous papillomas expressing the v-ras(Ha) oncogene but does not block in vitro keratinocyte responses to oncogenic ras. Cancer Res. 1997;57(15):3180–3188. [PubMed] [Google Scholar]
- 134.Gangarosa LM, Sizemore N, Graves-Deal R, Oldham SM, Der CJ, Coffey RJ. A raf-independent epidermal growth factor receptor autocrine loop is necessary for Ras transformation of rat intestinal epithelial cells. J Biol Chem. 1997;272(30):18926–18931. doi: 10.1074/jbc.272.30.18926. [DOI] [PubMed] [Google Scholar]
- 135.Sibilia M, Fleischmann A, Behrens A, et al. The EGF receptor provides an essential survival signal for SOS- dependent skin tumor development. Cell. 2000;102(2):211–220. doi: 10.1016/s0092-8674(00)00026-x. [DOI] [PubMed] [Google Scholar]
- 136.Wittbrodt J, Lammers R, Malitschek B, Ullrich A, Schartl M. The Xmrk receptor tyrosine kinase is activated in Xiphophorus malignant melanoma. Embo J. 1992 Nov;11(11):4239–4246. doi: 10.1002/j.1460-2075.1992.tb05518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Winnemoeller D, Wellbrock C, Schartl M. Activating mutations in the extracellular domain of the melanoma inducing receptor Xmrk are tumorigenic in vivo. Int J Cancer. 2005 Dec 10;117(5):723–729. doi: 10.1002/ijc.21232. [DOI] [PubMed] [Google Scholar]
- 138.Meierjohann S, Wende E, Kraiss A, Wellbrock C, Schartl M. The oncogenic epidermal growth factor receptor variant Xiphophorus melanoma receptor kinase induces motility in melanocytes by modulation of focal adhesions. Cancer Res. 2006 Mar 15;66(6):3145–3152. doi: 10.1158/0008-5472.CAN-05-2667. [DOI] [PubMed] [Google Scholar]
- 139.Bardeesy N, Wong KK, DePinho RA, Chin L. Animal models of melanoma: recent advances and future prospects. Adv Cancer Res. 2000;79:123–156. doi: 10.1016/s0065-230x(00)79004-x. [DOI] [PubMed] [Google Scholar]
- 140.Bottaro DP, Rubin JS, Faletto DL, et al. Identification of the hepatocyte growth factor receptor as the c- met proto-oncogene product. Science. 1991;251:802–804. doi: 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- 141.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003 Dec;4(12):915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 142.Bardelli A, Longati P, Gramaglia D, Stella MC, Comoglio PM. Gab1 coupling to the HGF/Met receptor multifunctional docking site requires binding of Grb2 and correlates with the transforming potential. Oncogene. 1997 Dec 18;15(25):3103–3111. doi: 10.1038/sj.onc.1201561. [DOI] [PubMed] [Google Scholar]
- 143.Fixman ED, Fournier TM, Kamikura DM, Naujokas MA, Park M. Pathways downstream of Shc and Grb2 are required for cell transformation by the tpr-Met oncoprotein. J Biol Chem. 1996 May 31;271(22):13116–13122. doi: 10.1074/jbc.271.22.13116. [DOI] [PubMed] [Google Scholar]
- 144.Ponzetto C, Bardelli A, Zhen Z, et al. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell. 1994 Apr 22;77(2):261–271. doi: 10.1016/0092-8674(94)90318-2. [DOI] [PubMed] [Google Scholar]
- 145.Li G, Schaider H, Satyamoorthy K, Hanakawa Y, Hashimoto K, Herlyn M. Downregulation of E-cadherin and Desmoglein 1 by autocrine hepatocyte growth factor during melanoma development. Oncogene. 2001 Dec 6;20(56):8125–8135. doi: 10.1038/sj.onc.1205034. [DOI] [PubMed] [Google Scholar]
- 146.Vande Woude GF, Jeffers M, Cortner J, Alvord G, Tsarfaty I, Resau J. Met-HGF/SF: tumorigenesis, invasion and metastasis. Ciba Found Symp. 1997;212:119–130. doi: 10.1002/9780470515457.ch8. discussion 130–112, 148–154. [DOI] [PubMed] [Google Scholar]
- 147.Natali PG, Nicotra MR, Di Renzo MF, et al. Expression of the c-Met/HGF receptor in human melanocytic neoplasms: demonstration of the relationship to malignant melanoma tumour progression. British Journal of Cancer. 1993;68:746–750. doi: 10.1038/bjc.1993.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wiltshire RN, Duray P, Bittner ML, et al. Direct visualization of the clonal progression of primary cutaneous melanoma: application of tissue microdissection and comparative genomic hybridization. Cancer Res. 1995 Sep 15;55(18):3954–3957. [PubMed] [Google Scholar]
- 149.Jeffers M, Schmidt L, Nakaigawa N, et al. Activating mutations for the met tyrosine kinase receptor in human cancer. Proc Natl Acad Sci U S A. 1997 Oct 14;94(21):11445–11450. doi: 10.1073/pnas.94.21.11445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Schmidt L, Duh FM, Chen F, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet. 1997 May;16(1):68–73. doi: 10.1038/ng0597-68. [DOI] [PubMed] [Google Scholar]
- 151.Kong-Beltran M, Seshagiri S, Zha J, et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. 2006 Jan 1;66(1):283–289. doi: 10.1158/0008-5472.CAN-05-2749. [DOI] [PubMed] [Google Scholar]
- 152.Smolen GA, Sordella R, Muir B, et al. Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752. Proc Natl Acad Sci U S A. 2006 Feb 14;103(7):2316–2321. doi: 10.1073/pnas.0508776103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Rusciano D, Lorenzoni P, Burger MM. Expression of constitutively activated hepatocyte growth factor/scatter factor receptor (c-met) in B16 melanoma cells selected for enhanced liver colonization. Oncogene. 1995;11:1979–1987. [PubMed] [Google Scholar]
- 154.Noonan FP, Recio JA, Takayama H, et al. Neonatal sunburn and melanoma in mice. Nature. 2001 Sep 20;413(6853):271–272. doi: 10.1038/35095108. [DOI] [PubMed] [Google Scholar]
- 155.Otsuka T, Takayama H, Sharp R, et al. c-Met autocrine activation induces development of malignant melanoma and acquisition of the metastatic phenotype. Cancer Res. 1998 Nov 15;58(22):5157–5167. [PubMed] [Google Scholar]
- 156.Recio JA, Noonan FP, Takayama H, et al. Ink4a/arf deficiency promotes ultraviolet radiation-induced melanomagenesis. Cancer Res. 2002 Nov 15;62(22):6724–6730. [PubMed] [Google Scholar]
- 157.McGill GG, Haq R, Nishimura EK, Fisher DE. c-met expression is regulated by mitf in the melanocyte lineage. J Biol Chem. 2006 Feb 2; doi: 10.1074/jbc.M513094200. [DOI] [PubMed] [Google Scholar]
- 158.Goldberg SF, Miele ME, Hatta N, et al. Melanoma Metastasis Suppression by Chromosome 6: Evidence for a Pathway Regulated by CRSP3 and TXNIP. Cancer Res. 2003 January 15, 2003;63(2):432–440. [PubMed] [Google Scholar]
- 159.Lee J-H, Miele ME, Hicks DJ, et al. KiSS-1, a Novel Human Malignant Melanoma Metastasis-Suppressor Gene. J Natl Cancer Inst. 1996 December 4, 1996;88(23):1731–1737. doi: 10.1093/jnci/88.23.1731. [DOI] [PubMed] [Google Scholar]
- 160.Su YA, Bittner ML, Chen Y, et al. Identification of tumor-suppressor genes using human melanoma cell lines UACC903, UACC903(+6), and SRS3 by comparison of expression profiles. Mol Carcinog. 2000 Jun;28(2):119–127. doi: 10.1002/1098-2744(200006)28:2<119::aid-mc8>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 161.Kaariainen E, Nummela P, Soikkeli J, et al. Switch to an invasive growth phase in melanoma is associated with tenascin-C, fibronectin, and procollagen-I forming specific channel structures for invasion. J Pathol. 2006 Oct;210(2):181–191. doi: 10.1002/path.2045. [DOI] [PubMed] [Google Scholar]
- 162.Namiki T, Yanagawa S, Izumo T, et al. Genomic alterations in primary cutaneous melanomas detected by metaphase comparative genomic hybridization with laser capture or manual microdissection: 6p gains may predict poor outcome. Cancer Genet Cytogenet. 2005 Feb;157(1):1–11. doi: 10.1016/j.cancergencyto.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 163.Garraway LA, Weir BA, Zhao X, et al. Lineage Addiction” in Human Cancer: Lessons from Integrated Genomics. Cold Spring Harbor Symposia on Quantitative Biology. 2005;70(1):25–34. doi: 10.1101/sqb.2005.70.016. [DOI] [PubMed] [Google Scholar]
- 164.Hemesath TJ, Steingrimsson E, McGill G, et al. microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family. Genes Dev. 1994 Nov 15;8(22):2770–2780. doi: 10.1101/gad.8.22.2770. [DOI] [PubMed] [Google Scholar]
- 165.Steingrimsson E, Moore KJ, Lamoreux ML, et al. Molecular basis of mouse microphthalmia (mi) mutations helps explain their developmental and phenotypic consequences. Nat Genet. 1994 Nov;8(3):256–263. doi: 10.1038/ng1194-256. [DOI] [PubMed] [Google Scholar]
- 166.Price ER, Fisher DE. Sensorineural Deafness and Pigmentation Genes: Melanocytes and the Mitf Transcriptional Network. Neuron. 2001;30(1):15–18. doi: 10.1016/s0896-6273(01)00259-8. [DOI] [PubMed] [Google Scholar]
- 167.Ferguson-Smith AC, Sasaki H, Cattanach BM, Surani MA. Parental-origin-specific epigenetic modification of the mouse H19 gene. Nature. 1993;362(6422):751–755. doi: 10.1038/362751a0. [DOI] [PubMed] [Google Scholar]
- 168.Stransky N, Vallot C, Reyal F, et al. Regional copy number-independent deregulation of transcription in cancer. Nat Genet. 2006;38(12):1386–1396. doi: 10.1038/ng1923. [DOI] [PubMed] [Google Scholar]
- 169.Hu M, Yao J, Cai L, et al. Distinct epigenetic changes in the stromal cells of breast cancers. Nat Genet. 2005;37(8):899–905. doi: 10.1038/ng1596. [DOI] [PubMed] [Google Scholar]
- 170.Hatada I, Hayashizaki Y, Hirotsune S, Komatsubara H, Mukai T. A Genomic Scanning Method for Higher Organisms Using Restriction Sites as Landmarks. PNAS. 1991 November 1, 1991;88(21):9523–9527. doi: 10.1073/pnas.88.21.9523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ballestar E, Paz MF, Valle L, et al. Methyl-CpG binding proteins identify novel sites of epigenetic inactivation in human cancer. Embo J. 2003 Dec 1;22(23):6335–6345. doi: 10.1093/emboj/cdg604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Shi H, Wei SH, Leu Y-W, et al. Triple Analysis of the Cancer Epigenome: An Integrated Microarray System for Assessing Gene Expression, DNA Methylation, and Histone Acetylation. Cancer Res. 2003 May 1, 2003;63(9):2164–2171. [PubMed] [Google Scholar]
- 173.Muthusamy V, Duraisamy S, Bradbury CM, et al. Epigenetic Silencing of Novel Tumor Suppressors in Malignant Melanoma. Cancer Res. 2006 December 1, 2006;66(23):11187–11193. doi: 10.1158/0008-5472.CAN-06-1274. [DOI] [PubMed] [Google Scholar]
- 174.Furuta J, Nobeyama Y, Umebayashi Y, Otsuka F, Kikuchi K, Ushijima T. Silencing of Peroxiredoxin 2 and Aberrant Methylation of 33 CpG Islands in Putative Promoter Regions in Human Malignant Melanomas. Cancer Res. 2006 June 15, 2006;66(12):6080–6086. doi: 10.1158/0008-5472.CAN-06-0157. [DOI] [PubMed] [Google Scholar]
- 175.Takeuchi T, Adachi Y, Sonobe H, Furihata M, Ohtsuki Y. A ubiquitin ligase, skeletrophin, is a negative regulator of melanoma invasion. Oncogene. 2006;25(53):7059–7069. doi: 10.1038/sj.onc.1209688. [DOI] [PubMed] [Google Scholar]
- 176.Mori T, Martinez SR, O’Day SJ, et al. Estrogen Receptor-{alpha} Methylation Predicts Melanoma Progression. Cancer Res. 2006 July 1, 2006;66(13):6692–6698. doi: 10.1158/0008-5472.CAN-06-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Mirmohammadsadegh A, Marini A, Nambiar S, et al. Epigenetic Silencing of the PTEN Gene in Melanoma. Cancer Res. 2006 July 1, 2006;66(13):6546–6552. doi: 10.1158/0008-5472.CAN-06-0384. [DOI] [PubMed] [Google Scholar]
- 178.Kato Y, Salumbides BC, Wang X-F, et al. Antitumor effect of the histone deacetylase inhibitor LAQ824 in combination with 13-cis-retinoic acid in human malignant melanoma. Mol Cancer Ther. 2007 January 1, 2007;6(1):70–81. doi: 10.1158/1535-7163.MCT-06-0125. [DOI] [PubMed] [Google Scholar]
- 179.Volinia S, Calin GA, Liu C-G, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. PNAS. 2006 February 14, 2006;103(7):2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Costinean S, Zanesi N, Pekarsky Y, et al. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E{micro}-miR155 transgenic mice. PNAS. 2006 May 2, 2006;103(18):7024–7029. doi: 10.1073/pnas.0602266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gaur A, Jewell DA, Liang Y, et al. Characterization of MicroRNA Expression Levels and Their Biological Correlates in Human Cancer Cell Lines. Cancer Res. 2007 March 15, 2007;67(6):2456–2468. doi: 10.1158/0008-5472.CAN-06-2698. [DOI] [PubMed] [Google Scholar]
- 182.Zhang L, Huang J, Yang N, et al. microRNAs exhibit high frequency genomic alterations in human cancer. PNAS. 2006 June 13, 2006;103(24):9136–9141. doi: 10.1073/pnas.0508889103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Culp WD, Neal R, Massey R, Egevad L, Pisa P, Garland D. Proteomic Analysis of Tumor Establishment and Growth in the B16-F10 Mouse Melanoma Model. J Proteome Res. 2006;5(6):1332–1343. doi: 10.1021/pr060059q. [DOI] [PubMed] [Google Scholar]
- 184.Mian S, Ugurel S, Parkinson E, et al. Serum Proteomic Fingerprinting Discriminates Between Clinical Stages and Predicts Disease Progression in Melanoma Patients. J Clin Oncol. 2005 August 1, 2005;23(22):5088–5093. doi: 10.1200/JCO.2005.03.164. [DOI] [PubMed] [Google Scholar]
- 185.Verhaegen M, Bauer JA, Martin de la Vega C, et al. A novel BH3 mimetic reveals a mitogen-activated protein kinase-dependent mechanism of melanoma cell death controlled by p53 and reactive oxygen species. Cancer Res. 2006 Dec 1;66(23):11348–11359. doi: 10.1158/0008-5472.CAN-06-1748. [DOI] [PubMed] [Google Scholar]
- 186.Minn AJ, Gupta GP, Siegel PM, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436(7050):518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33(1):49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- 188.Hoek KS, Schlegel NC, Brafford P, et al. Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment Cell Res. 2006 Aug;19(4):290–302. doi: 10.1111/j.1600-0749.2006.00322.x. [DOI] [PubMed] [Google Scholar]
- 189.Jaeger J, Koczan D, Thiesen HJ, et al. Gene expression signatures for tumor progression, tumor subtype, and tumor thickness in laser-microdissected melanoma tissues. Clin Cancer Res. 2007 Feb 1;13(3):806–815. doi: 10.1158/1078-0432.CCR-06-1820. [DOI] [PubMed] [Google Scholar]
- 190.Winnepenninckx V, Lazar V, Michiels S, et al. Gene expression profiling of primary cutaneous melanoma and clinical outcome. J Natl Cancer Inst. 2006 Apr 5;98(7):472–482. doi: 10.1093/jnci/djj103. [DOI] [PubMed] [Google Scholar]
- 191.Bloethner S, Hemminki K, Thirumaran RK, et al. Differences in global gene expression in melanoma cell lines with and without homozygous deletion of the CDKN2A locus genes. Melanoma Res. 2006 Aug;16(4):297–307. doi: 10.1097/01.cmr.0000222597.50309.05. [DOI] [PubMed] [Google Scholar]
- 192.Bloethner S, Chen B, Hemminki K, et al. Effect of common B-RAF and N-RAS mutations on global gene expression in melanoma cell lines. Carcinogenesis. 2005 Jul;26(7):1224–1232. doi: 10.1093/carcin/bgi066. [DOI] [PubMed] [Google Scholar]
- 193.Pavey S, Johansson P, Packer L, et al. Microarray expression profiling in melanoma reveals a BRAF mutation signature. Oncogene. 2004 May 20;23(23):4060–4067. doi: 10.1038/sj.onc.1207563. [DOI] [PubMed] [Google Scholar]
- 194.Shields JM, Thomas NE, Cregger M, et al. Lack of extracellular signal-regulated kinase mitogen-activated protein kinase signaling shows a new type of melanoma. Cancer Res. 2007 Feb 15;67(4):1502–1512. doi: 10.1158/0008-5472.CAN-06-3311. [DOI] [PubMed] [Google Scholar]
- 195.Sjoblom T, Jones S, Wood LD, et al. The Consensus Coding Sequences of Human Breast and Colorectal Cancers. Science. 2006 October 13, 2006;314(5797):268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]