

Summary
Mixed lineage kinase 3 (MLK3) is a MAP3K that activates the JNK-dependent MAPK pathways. Here we show that MLK3 is required for cell migration in a manner independent of its role as a MAP3K or MLK3 kinase activity. Rather, MLK3 functions in a regulated way to limit levels of the activated GTPase, Rho, by binding to the Rho activator, p63RhoGEF/GEFT, which, in turn, prevents its activation by Gαq. These findings demonstrate a scaffolding role for MLK3 in controlling the extent of Rho activation that modulates cell migration. Moreover, they suggest that MLK3 functions as a network hub that links a number of signaling pathways.
Introduction
MLK3 (mixed-lineage kinase 3) is a ubiquitously expressed mammalian Ser/Thr kinase, best known for its function as a mitogen-activated protein kinase kinase kinase (MAP3K) that activates multiple MAPK pathways (Gallo and Johnson, 2002). Over-expression and depletion of MLK3 by RNA interference implicated it in activation of the JNK, p38 and ERK pathways (Chadee and Kyriakis, 2004; Chadee et al., 2006, and for review see Gallo and Johnson, 2002). In addition, cells from MLK3 knock-out mice are deficient in JNK activation in response to TNFα (Brancho et al., 2005) or saturated free fatty acids (Jaeschke and Davis, 2007) and the single Drosophila MLK gene, Slpr, is required for the JNK activation essential to dorsal closure (Stronach and Perrimon, 2002).
That MLK3 might have important cellular functions distinct from its signaling role as a MAP3K was suggested by sequence similarities to the fungal mitotic kinase NIMA and our demonstration that MLK3 activity was enhanced during mitosis, in the absence of JNK activation, and increased MLK3 caused a loss of central microtubules. We suggested that MLK3 might function during M phase, outside of its role as a JNK activator, to promote microtubule instability (Swenson et al., 2003). However, since MLK3 is present in many tissues and cells, including those that are non-dividing, we wondered whether its cytoskeletal effects might extend to cellular processes of microtubule remodeling such as cell migration. The requirement for Slpr in dorsal closure is consistent with such a role for MLK3 (Stronach and Perrimon, 2002). Moreover, targeted-disruption of the MLK3 gene in mice also generates a dorsal phenotype, although it is not known whether this is due to defective cell migration (Brancho et al., 2005). If MLK3 does participate in cell migration, it is unclear whether this function depends on its ability to act as a MAP3K. In Drosophila, epistasis analysis of slpr showed only a partial rescue of dorsal closure by the downstream JNK pathway components, raising the possibility that Slpr might contribute to migration in ways both dependent on and independent of its role as a JNK MAPK activator.
MLK3 function is modulated by multiple molecular interactions with other signaling proteins (Gallo, K. Mlk3. UCSD-Nature Molecule Pages, 2006;doi:10.1038/mp.a001551.01). Two of the best characterized interactions of MLK3 are with the Rho family GTPases, Cdc42 and Rac1 The GTP-bound forms of these proteins bind to the CRIB domain of MLK3 and potentiate its ability to activate JNK. In addition, co-expression of activated Cdc42 with MLK3 promotes its oligomerization and phosphorylation (for review, see Gallo and Johnson, 2002). Activated Cdc42 promotes the translocation of MLK3 from the Golgi area (Cha et al., 2004; Swenson et al., 2003) to the plasma membrane and other membrane compartments (Du et al., 2005).
Interestingly, while the interaction of MLK3 with Cdc42 and Rac1 is clear, MLK3 binding to RhoA has not been detected (Burbeloet al., 1995; Jaffe et al., 2005; Wennerberg et al., 2002). We report here that acute depletion of MLK3 blocks efficient directed cell migration. Phenotypic analyses of siRNA knock-down cells showed a spectrum of cytoarchitectural alterations strikingly similar to those observed following Rho activation. Surprisingly, given the previous failure to detect interaction between MLK3 and RhoA, we report that MLK3 can regulate Rho activity. Unlike its direct interactions with Cdc42 and Rac1, which alter the signaling/localization of MLK3, MLK3 acts on Rho to negatively regulate its activity via interaction with the Rho activator p63RhoGEF. Through this physical interaction, which is independent of its protein kinase activity, MLK3 compromises the specific activation of p63RhoGEF by Gαq, thereby uncoupling Gαq and Rho activation. The MLK3/p63RhoGEF interaction and the consequential effects on Rho activation are positively regulated by phosphorylation and activated Rac, and negatively regulated by feed-back from the activated JNK pathway. These findings show that MLK3 modulates Rho activation to affect directed cell migration via protein-protein interaction with a specific Rho GEF.
Results
MLK3 is required for efficient directed cell migration
To examine the contribution of MLK3 to directed cell migration, we designed 3 MLK3 specific siRNAs, 903, UTR497 and UTR36, and tested their ability to knockdown MLK3 protein in a human lung carcinoma cell line (A549). To assess potential non-target effects, we also examined cell cycle profiles of the transfected cells, reasoning that different siRNAs targeted to the same gene, should yield identical effects on fundamental processes such as the cell cycle if the siRNA-mediated RNAi is specific (Scacheri et al., 2004). Control cells were transfected with siRNAs containing sequences with no significant homology to a known human gene (GL2). Transfection with each of the 3 MLK3-specific siRNAs resulted in >90% knockdown of MLK3 protein (Figure 1A) and, for two of these siRNAs, UTR497 and UTR36, there was no change in cell cycle profile relative to control siRNA-transfected cells (Supplemental Figure 1A). Transfection with siRNA, 903, which is identical to that used by others to study MLK3 function (Chadee and Kyriakis, 2004; Chadee et al., 2006; Du et al., 2005) produced a marked increase in the % cells containing less than-G1 content of DNA, characteristic of apoptotic cells (Supplemental Figure 1A). Because of these apparent non-target effects, use of the 903 siRNA was discontinued.
Figure 1. MLK3 is required for directed cell migration.
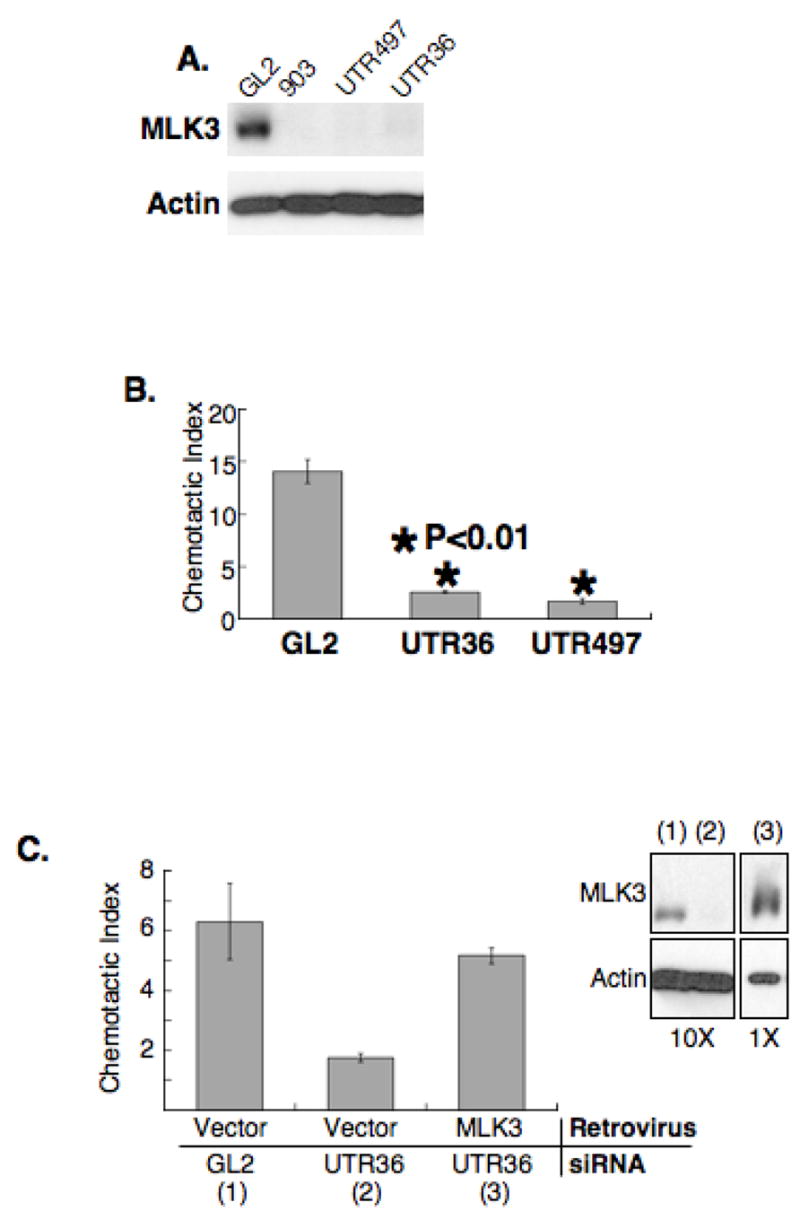
(A) Depletion of MLK3 with siRNAs. A549 cells were transfected with GL2 luciferase siRNA (nonspecific siRNA control) or one of each MLK3-specific siRNA, 903, UTR497 or UTR36. Samples from transfected cell populations were immuno-blotted with antibodies for MLK3 and actin.
(B) MLK3 depletion inhibits directed cell migration of A549 cells. A549 cells were transfected with the MLK3-specific siRNAs, UTR36 or UTR497, or with GL2 siRNA and subjected to Transwell migration assays for 11 h. Shown is the chemotactic index (fold stimulation of chemotaxis/chemokinesis) for each sample. *, statistically significant difference (P< 0.01) compared to GL2 control. Data are presented as mean (n = 3) +/−SEM.
(C) Inhibition of directed cell migration due to MLK3 depletion is reversed by expression of a non-depletable MLK3. A549 cells were infected with a GFP bicistronic retroviral expression vector encoding MLK3, or vector alone, sorted and GFP-positive cells selected and transfected with GL2 or UTR36 siRNA. Samples were prepared for immuno-blot (right panel) and Transwell migration assays. The chemotactic index is shown as mean (n = 3) +/− SEM. The top portion of the immuno-blot was probed with anti-MLK3 and the bottom portion with anti-actin. For visual clarity, 1/10th of the protein amount loaded for samples (1) and (2) was loaded for sample (3).
We first examined effects of MLK3 depletion on directed cell migration. UTR36, UTR497 or control GL2 siRNAs were transfected separately into A549 cells, which were evaluated by transwell assays to assess directed cell migration in response to serum. While the control transfected cells responded with an average chemotactic index of 14 +/− 1, cells transfected with UTR36 or UTR497 had chemotactic indices of less than 3 (Figure 1B). Although these assays were carried out for only 11 h, it was possible that negative effects on cell proliferation may have contributed to the apparent blockade of directed cell migration. To test this, 5 × 104 cells from each transfected population were cultured and cell number was determined after 23 h and in no case had the cell population doubled by this time (Supplemental Figure 1B). Thus, the inefficiency of directed cell migration measured in MLK3-depleted cells was not due to inhibition of cell proliferation. Similar effects were observed in PC3 and HEK293T cells (Supplemental Figure 2).
We next evaluated whether the defects in directed cell migration could be rescued by ectopic expression of non-depletable MLK3. A549 cells were infected either with a retrovirus co-expressing only the coding region of MLK3 along with GFP, or with control virus expressing GFP only. GFP-positive cells of both populations were selected using FACS and transfected with UTR36 siRNA to deplete endogenous MLK3, or GL2 control siRNA. Transfected cells were assessed for MLK3 protein and subjected to the cell migration assay (Figure 1D). As shown by immuno-blot, retrovirally-infected cells were as efficiently depleted of endogenous MLK3 by transfection of UTR36 (Figure 1D, compare (2) to (1) in immuno-blot) as uninfected cells (compare to immuno-blot in Figure 1A). As for uninfected cells, retrovirally-infected cells depleted of endogenous MLK3 were deficient in directed cell migration. This defect was rescued by ectopically expressed, non-depletable MLK3 (Figure 1D, graph). These results strengthen our contention that MLK3 is required for efficient directed cell migration.
Cells depleted of MLK3 have altered adhesion structures, stress fibers and lamellipodial protrusions
To assess if MLK3-depletion elicits changes in the actin cytoskeleton and/or cell adhesion structures (focal adhesions) that might account for the migration defect of these cells, we examined these cytoskeletal elements in siRNA-transfected A549 cells using immunofluorescence staining with phalloidin (specific for F-actin) or antibodies to vinculin (a marker of focal complexes/adhesions). In control cells, actin stress fibers and focal adhesions were visible and, for migrating cells on the edge of a scratch wound, extending lamellipodia were evident (Figure 2A, lower panels). However, in cells depleted of MLK3, there was a marked increase in the thickness and number of stress fibers, as well as enlarged focal adhesions. Additionally, in cells on the edge of a scratch wound, lamellipodial protrusions were absent (Figure 2A, upper panels and Figure 2B). Since enlarged focal adhesions are involved in passive anchorage rather than generation of propulsive forces (Beningo et al., 2001), and protrusions are required for cell movement (Ridley et al., 2003), we suggest that the enlarged focal adhesions and decreased protrusions of MLK3-depleted cells may account for the defects in directed cell migration.
Figure 2. MLK3 depletion induces changes in focal adhesions/stress fibers and the level of EGF-activated p38.

(A) A549 cells were transfected either with GL2 luciferase siRNA or the MLK3-specific siRNA, UTR36, and grown to confluence prior to scrape wounding (80–93 h post-transfection) to induce directed cell migration. Loose cells were washed off and fresh media added immediately after wounding. 1.5–3 h after wounding, cells were fixed and co-stained for vinculin (green), to mark focal adhesions, F-actin (red), to label stress fibers and DNA (blue). Shown are cells on the edge of the scrape wound. Exposure times and binning (2) of GL2 and UTR36 images are identical. Scale: 10 μM.
(B) Images of 15 random high power view fields along the wound were captured and wound edge cells (~75 cells) were blindly scored as high or low for stress fiber content. The experiment was performed 3×. The average % cells with high stress fibers +/− SD for MLK3-depleted (UTR36) and control cells (GL2) are plotted.
(C,D) A549 cells transfected with GL2 luciferase siRNA or MLK3-specific siRNA, UTR36, were serum-starved for 18 h and incubated with EGF (10 ng/ml) for various times before preparing protein extracts.
(C) Immuno-blots of equal protein amounts from each sample were probed with anti-MLK3, to confirm specific-depletion by siRNA, and with antibodies to phospho-JNK (P-JNK), phospho-ERK (P-ERK) and phospho-p38 (P-p38), to assess activation. The amount of MAPK was examined by probing immuno-blots with anti-JNK,-ERK1 or -p38 MAPK. Detection and quantitation were done using fluorochrome-labeled secondary antibodies and the Odyssey® infrared imaging system (LI-COR Biosciences). Data are representative of 3 experiments.
(D) The ratios of P-MAPK/MAPK for JNK, ERK1 and p38 shown in panel B, + the data of 2 additional experiments, were averaged and plotted for MLK3-depleted and GL2 control samples. Shown are mean +/− SEM.
Effects of MLK3 depletion on EGF-mediated JNK, ERK and p38 MAPK phosphorylation
Defects in cell migration as well as the presence of enlarged focal adhesions and increased stress fibers have been noted following inhibition of activation or function of JNK, a known downstream target of MLK3 (for review, see Huang et al., 2004). Additionally, previous studies implicated MLK3 in activation of ERK (Chadee and Kyriakis, 2004; Hartkamp et al., 1999) and p38 (Chadee and Kyriakis, 2004; Rana et al., 1996), two other MAP kinases that may function in cell migration (Huang et al., 2004). To question if the defects in cell migration and alterations in cytoskeletal elements after MLK3 depletion might be due to defects in the activation of ERK, JNK and/or p38 MAPK, we evaluated MLK3-depleted and control cells in response to EGF as EGF activates MLK3-mediated signaling (Chadee and Kyriakis, 2004) and stimulates MLK3-dependent migration of A549 cells (data not shown). A549 cells were transfected with UTR36 siRNA, to deplete MLK3, or control siRNA and, following serum starvation overnight, were incubated with EGF. Samples were collected as a function of time and the MAPKs assessed using anti-phospho-specific, activation-specific antibodies (Figure 2C). Our results showed no difference between MLK3-depleted and control cells in the timing or levels of JNK or ERK phosphorylation, or in the timing of p38 phosphorylation (Figure 2C and D). However, in the MLK3 depleted cells we noted an increase, rather than decrease, in p38 phosphorylation relative to controls. Together, these results show that inhibition of directed cell migration elicited by MLK3-depletion is not due to inhibition of JNK, ERK or p38 phosphorylation.
MLK3 depletion results in increased activated Rho
The changes in cytoarchitecture resulting from MLK3-depletion are similar to those that occur following microinjection of activated Rho (Nobes and Hall, 1995; Paterson et al., 1990; for review, see Jaffe and Hall, 2005). Thus, we quantified activated Rho using the Rhotekin pull-down assay (Ren et al., 1999) in MLK3-depleted and control cells that had been either serum-starved (Figure 3A and B, 0′), or serum-starved and restimulated with serum for 5 min (Figure 3A and B, 5′). While serum increased activated Rho compared to unstimulated samples for both cell types, the level of activated Rho was markedly elevated in the MLK3-depleted cells.
Figure 3. MLK3 depletion increases Rho GTPase and inhibits Rho-dependent SRF activation stimulated by Gαq.
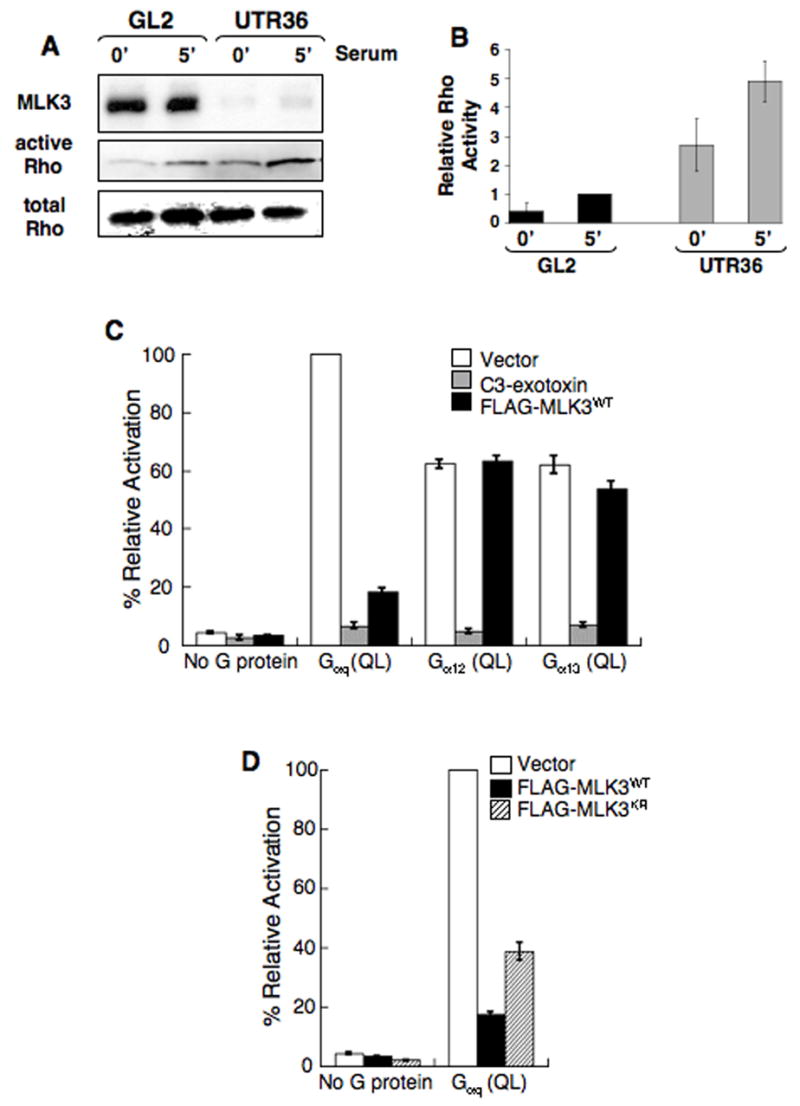
(A,B) Increased activated Rho is present in MLK3-depleted A549 cells. A549 cells transfected either with GL2 control siRNA or MLK3 UTR36 siRNA were serum-starved for 18 h then stimulated with serum for 5 min before preparating cell lysates. Activated Rho-GTP was isolated using a Rhotekin pull-down assay.
(A) Immuno-blot of samples from the pull-down assay was reacted with anti-RhoA. In parallel, immuno-blots of equal amounts of total lysate were reacted with anti-MLK3 and anti-RhoA. Data are representative of 4 experiments.
(B) Quantitation of Rho for each sample shown in panel A, + samples from 3 additional experiments (data not shown), was obtained using fluorochrome-labeled secondary antibodies and the Odyssey® infrared imaging system (LI-COR Biosciences) or densitometry and the active Rho/total Rho ratio determined (Relative Rho Activity). The ratio average from 4 experiments (+/− SEM) is plotted to compare activated Rho levels (active Rho/total Rho for the 5 min GL2 sample for each experiment is set arbitrarily at 1).
(C,D) MLK3 inhibits Rho-dependent SRF activation stimulated by Gαq.
(C) 293T cells were cotransfected with expression plasmids encoding either wild-type MLK3 (FLAG-MLK3WT), C3 exotoxin, or empty vector and constitutively active, GTPase-deficient Gαq (Q229L), Gα12 (Q229L), Gα13 (Q226L) or no G protein (empty vector alone). Transfections also included a firefly luciferase reporter plasmid that is responsive to SRF activation (pSRF-Luc) and a plasmid which expresses Renilla luciferase (phRG-TK) as an internal control. 19 h later, cell lysates were prepared and assayed for dual luciferase activities. Data represent firefly luciferase activity normalized to Renilla luciferase activity, expressed as % activation relative to that elicited by Gαq (QL) in cells transfected also with empty vector control. Total cell lysates were analyzed by immuno-blot with anti-FLAG,-Gαq,-Gα12 or -Gα13 (see Supplemental Figure 3). The data are the average of 3 experiments (+/− SEM).
(D) MLK3 inhibition of Gαq-stimulated SRF activation is not dependent on catalytic activity. 293T cells were co-transfected with expression plasmids encoding either wild-type MLK3 (FLAG-MLK3WT), an ATP-binding point mutant of MLK3 (FLAG-MLK3KR) or empty vector and GTPase-deficient Gαq (Q229L) or no G protein (empty vector alone). SRF reporter plasmids were included in these transfections, samples were analyzed for SRF activation and data expressed as described in (C). Total cell lysates were immuno-blotted with anti-FLAG or anti-Gαq (see Supplemental Figure 3). The data are the average of 3 experiments (+/− SEM).
MLK3 inhibits Gαq-specific Rho-dependent SRF activation
Since depletion of MLK3 leads to increased activated Rho, we examined if MLK3 inhibited production of activated Rho. First, we used a SRF (serum responsive factor) transcription assay commonly employed to quantitatively assess production of activated Rho (Hill et al., 1995) and evaluated the effects of MLK3 following Rho stimulation. To stimulate Rho activity, constitutively activated GTPase-deficient forms of the heterotrimeric G protein αsubunits, Gαq, Gα12, and Gα13, which have been demonstrated to stimulate Rho-dependent SRF luciferase reporter activity (Chikumi et al., 2002), were expressed following transient transfection. As shown in Figure 3C, activated Gαq, Gα12 or Gα13 stimulated SRF reporter activity relative to vector-transfected control cells. In all cases, this activity was inhibited by co-transfection of a plasmid expressing the Rho-specific inhibitor, botulinum C3 toxin (Aktories and Hall, 1989) demonstrating Rho dependence of transcription stimulation. On the other hand, C3 toxin did not affect expression of any of the Gα proteins (Supplemental Figure 3). Transfection of MLK3 (FLAG-MLK3WT) alone did not stimulate SRF-mediated transcription. This is noteworthy since activated Cdc42 and Rac, two members of the Rho family of GTPases that bind MLK3, also stimulate transcription in this assay (Hill et al., 1995) and can be ruled out as downstream mediators of MLK3 effects. In addition, MLK3 had no effect on Gα12-stimulated SRF activation and little effect on that stimulated by Gα13. However, coexpression of MLK3 caused a dramatic inhibition of SRF activation stimulated by Gαq (82% +/−1.5 reduction of the transcription activity stimulated by Gαq in absence of MLK3, Figure 3C). Importantly, immuno-blots of total protein from the samples show that MLK3 levels are similar among cells in which FLAG-MLK3WT was transfected (Supplemental Figure 3). These data suggest that MLK3 specifically regulates SRF-mediated transcription downstream of Gαq.
To test whether the inhibitory effects of MLK3 on Gαq-stimulated SRF activation were dependent on kinase activity, we compared the effects of a kinase-deficient mutant of MLK3 (FLAG-MLK3KR) with those of wild-type MLK3. As for wild-type MLK3, transfection of FLAG-MLK3KR did not result in stimulation of transcription. However, when co-expressed with activated Gαq, FLAG-MLK3KR also caused a significant inhibition of Gαq-stimulated SRF activation (61% +/− 3 reduction of transcription activity stimulated by Gαq in absence of MLK3, Figure 3D) although to a lesser extent than that elicited by wild-type MLK3. Collectively, these experiments show that MLK3 inhibits production of activated Rho in the signaling pathway downstream of active Gαq, but not Gα12 or Gα13, and that this inhibition is predominantly mediated by a mechanism independent of MLK3 kinase activity.
MLK3 and p63RhoGEF associate in mammalian cells
Activated forms of Gαq, Gα12 and Gα13 each elicit Rho activation by interacting with and stimulating subsets of Rho-specific guanosine nucleotide exchange factors (Rho GEFs, Fukuhara et al., 2000; Fukuhara et al., 1999; Hart et al., 1998; Kozasa et al., 1998; Lutz et al., 2005). Since stimulation of Rho activity occurs through Rho GEF activation, and the activity of Rho GEFs are affected by kinases as well as protein-protein interactions (for review see Schmidt and Hall, 2002), we speculated that MLK3 inhibited production of activated Rho downstream of Gαq through interaction with one or more Rho GEFs. To test this idea 293T cells were co-transfected separately with expression vectors encoding tagged versions of one of three Rho GEFs that are stimulated by activated Gα subunits (AU1-tagged PDZ-RhoGEF, HA-tagged LARG or GST-tagged p63RhoGEF), a vector encoding FLAG-MLK3WT or control vectors (Figure 4A, B and C). In anti-FLAG immune-precipitates, there was no PDZ-RhoGEF or LARG in either the presence or absence of MLK3. However, GST-p63RhoGEF, but not GST, was detected in anti-FLAG immune-precipitates only in the presence of FLAG-MLK3 (Figure 4C, right panel). Conversely, FLAG-MLK3 was detected in GST-affinity pull-downs from lysates of cells co-expressing GST-p63RhoGEF but not GST (data not shown). Additionally, endogenous MLK3, but not PAK1, was present in GST-affinity pull-downs of GST-p63RhoGEF-expressing cell lysates in a p63RhoGEF-dependent manner (Figure 4D). Conversely, endogenous GEFT, a shorter, functionally redundant p63RhoGEF isoform, was present in anti-FLAG immune-precipitates of FLAG-MLK3-expressing cell lysates but not in FLAG-tagged control protein-expressing cell lysates (Figure 4E).
Figure 4. MLK3 and p63 RhoGEF/GEFT associate in mammalian cells.
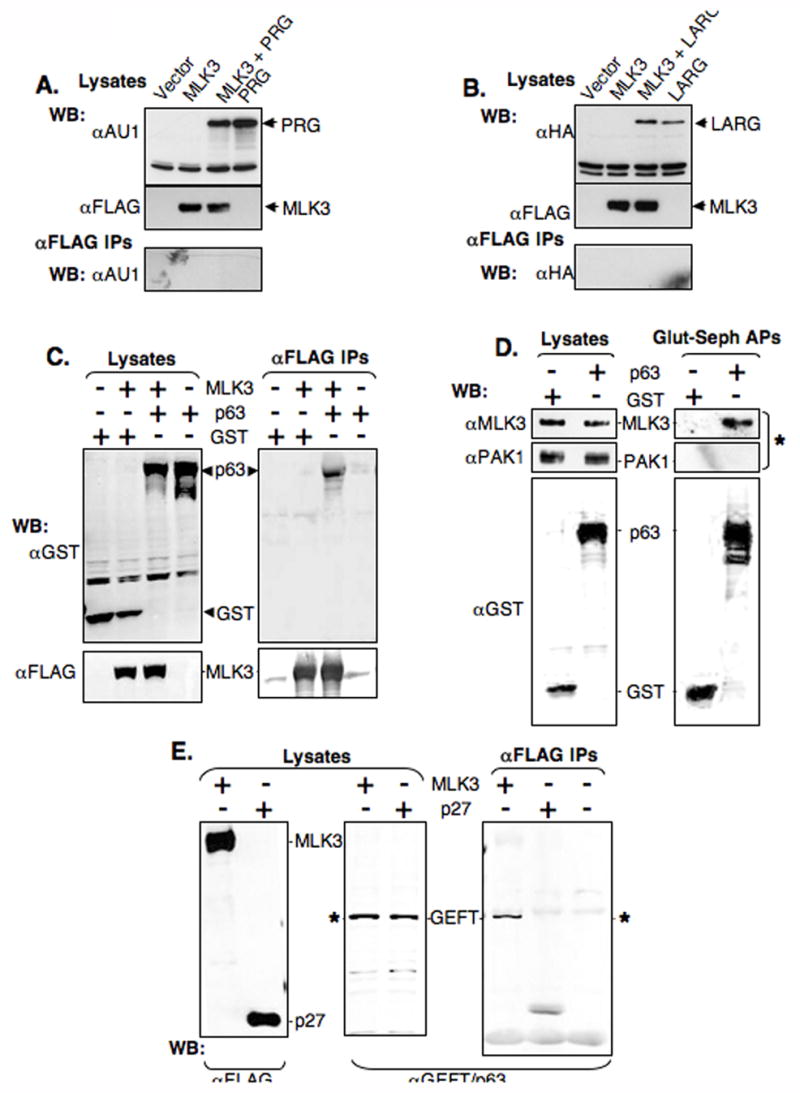
(A,B and C) HEK 293T cells were co-transfected with expression vectors for FLAG-MLK3WT (MLK3) and either AU1-tagged PDZ-RhoGEF (PRG, panel A), HA-tagged LARG (LARG panel B), GST-tagged p63 RhoGEF (p63, panel C) or control expression vectors (Vector and GST, panels A,B and C as indicated). Cell lysates were immuno-precipitated (IP) using anti-FLAG and then immuno-blotted (WB) with anti-AU1 (A), anti-HA (B) or anti-GST and anti-FLAG (C). Immuno-blots of the starting cell lysates were done in parallel to assess protein level.
(D) Endogenous MLK3, but not endogenous PAK1, is present in affinity precipitates of p63RhoGEF. 293T cells were transfected with expression vectors for GST-p63RhoGEF (p63) or GST (GST). Cell lysates were incubated with glutathione-sepharose and affinity-precipitates (Glut-Seph APs), as well as the starting lysates, were immuno-blotted (WB) with anti-MLK3, anti-PAK1 and anti-GST. * indicates endogenous proteins.
(E) Endogenous GEFT, but not p27, is present in immune-precipitates of exogenous MLK3 from mammalian cells. Cell lysates were made from 293T cells transfected with expression vectors for FLAGMLK3WT (MLK3) or FLAG-p27 (p27), immuno- precipitated (IP) using anti-FLAG and then immuno-blotted (WB) with anti-FLAG or anti-GEFT/p63.. * indicates endogenous proteins.
To identify the MLK3 domain(s) involved in p63RhoGEF interaction we created expression vectors encoding FLAG-tagged MLK3 deletion proteins and co-expressed each of these with GST-p63RhoGEF in 293T cells (Figure 5A). GST-affinity pull-downs of cell lysates were then examined by immuno-blot. We found that among the multiple interaction domains of MLK3, the kinase domain is uniquely necessary and sufficient for p63RhoGEF interaction (Figure 5A, lower panels). These experiments show that MLK3 and p63RhoGEF/GEFT can associate in mammalian cells and the kinase domain of MLK3 is sufficient for this association. In addition, the interaction of MLK3 with this Rho GEF is specific and, since p63RhoGEF signaling has been linked to Gαq, but not Gα12 or Gα13, this MLK3/RhoGEF interaction is restricted to the Gα-signaling pathways initiated by activated Gαq.
Figure 5. MLK3 binds p63RhoGEF.

(A) The kinase domain of MLK3 interacts with p63RhoGEF. MLK3WT deletion mutants were constructed and tested for interaction with p63RhoGEF. 293T cells were co-transfected with expression vectors for GST or GST-p63RhoGEF and FLAG-MLK3WT or a FLAG-MLK3WT deletion mutant (encoded MLK3 protein for each mutant is as shown and indicated by amino acid number). Cell lysates were incubated with glutathione-sepharose and the affinity-precipitates (Glut-Seph APs), as well as the starting lysates (Lysates), were immuno-blotted (WB) with anti-FLAG and anti-GST. The MLK3 domains include a Src homology 3 (SH3), kinase, leucine-zipper (LZ), Cdc42/Rac-interactive binding (CRIB) domain as well as a proline-rich carboxyl-terminal region as indicated.
(B,C) MLK3 and p63RhoGEF associate in vitro. FLAG-MLK3KR (MLK3KR) and GST-p63RhoGEF (p63) were immunopurified or affinity purified, respectively, from 293T cells following transfection of the appropriate expression vector (see also Supplemental Figure 4). Equimolar amounts of protein were incubated in buffer in duplicates for the indicated times (O.N. = over night) before addition of detergent and glutathione-sepharose to affinity-precipitate GST-p63RhoGEF complexes. The precipitates (Glut-seph APs), as well as samples of input proteins, were immuno-blotted (WB) with anti-FLAG and anti-GST (B) and proteins quantified using fluorochrome-labeled secondary antibodies and the Odyssey® infrared imaging system (LI-COR Biosciences). (C) Shown is the mean % precipitated-MLK3KR/input MLK3KR +/− SEM. (D,E) For specificity controls of the in vitro binding shown in (B,C), immunopurified FLAG-MLK3KR (MLK3KR) or FLAG-p27 (p27) was incubated with affinity purified GST-p63RhoGEF (D) or GST (E) overnight in buffer before precipitation of glutathione-sepharose affinity-precipitates and immuno-blot analysis as described for (B).
MLK3 and p63RhoGEF associate in vitro
We evaluated if MLK3 associated with p63RhoGEF in vitro to more directly study this interaction. FLAG- MLK3KR and GST-p63RhoGEF proteins were obtained following their expression in 293T cells by immune and affinity means respectively (see Supplemental Figure 4 for details). Equimolar amounts of these 2 proteins were incubated at 4°C as a function of time prior to affinity purification of GST-p63RhoGEF complexes (Figure 5B). Immuno-blots showed FLAG-MLK3KR in the GST-p63RhoGEF complexes by 2 h, with increasing amounts found at subsequent times (Figure 5B,C). To control for binding specificity, immunopurified FLAG-MLK3KR or FLAG-p27 was incubated overnight either with affinity-purified GST-p63RhoGEF (Figure 5D) or GST (Figure 5E) prior to isolation and immuno-blot analysis of the GST complexes. FLAG-MLK3KR, but not FLAG-p27, was present in the affinity complexes, and only in a GST-p63RhoGEF-dependent manner. These experiments show that the MLK3/p63RhoGEF interaction occurs in vitro.
In the experimental samples, the % associated MLK3/input MLK3 after overnight incubation was 44+/− 0.03% (Figure 5C). If a scaffold protein co-purifying specifically with FLAG MLK3KR was responsible for the MLK3/p63RhoGEF association, and assuming an equimolar ratio, then its level would correspond minimally to 44% of the molar input amount of FLAG- MLK3KR protein. Such a protein was not detected among those co-purifying proteins specific for the FLAG-MLK3KR–expressing cells (Supplemental Figure 4). These results imply that the association of MLK3 and p63RhoGEF is most likely direct.
MLK3/p63RhoGEF interaction is modulated following activation of a Gαq-coupled GPCR
Since MLK3 associates with p63RhoGEF and Rho GEF signaling has been linked to activated Gαq, we speculated that the MLK3/p63RhoGEF interaction might be modulated following acute activation of a Gαq-coupled GPCR. To test this, we used HM1 cells, which express the muscarinic type1 receptor, a Gαq-coupled GPCR that shows a Gαq-dependent response following treatment with carbachol (Peralta et al., 1988). The effects of carbachol on MLK3/p63RhoGEF interaction were examined following co-transfection of expression vectors for FLAG- MLK3KR and GST-p63RhoGEF. Expression of kinase-diminished MLK3 was chosen to prevent MLK3 activation as well as activation of downstream signaling pathways prior to carbachol treatment. The transfected HM1 cells were serum-starved overnight then stimulated with 10 μM carbachol for 0, 5 or 20 min before affinity precipitation of GST-p63RhoGEF complexes. Immuno-blot analysis showed increasing FLAG- MLK3KR as a function of time following carbachol (Figure 6A). Our results indicate the MLK3/p63RhoGEF interaction to be positively modulated following acute activation of a Gαq-coupled GPCR.
Figure 6. Modulation of MLK3/p63RhoGEF interaction.
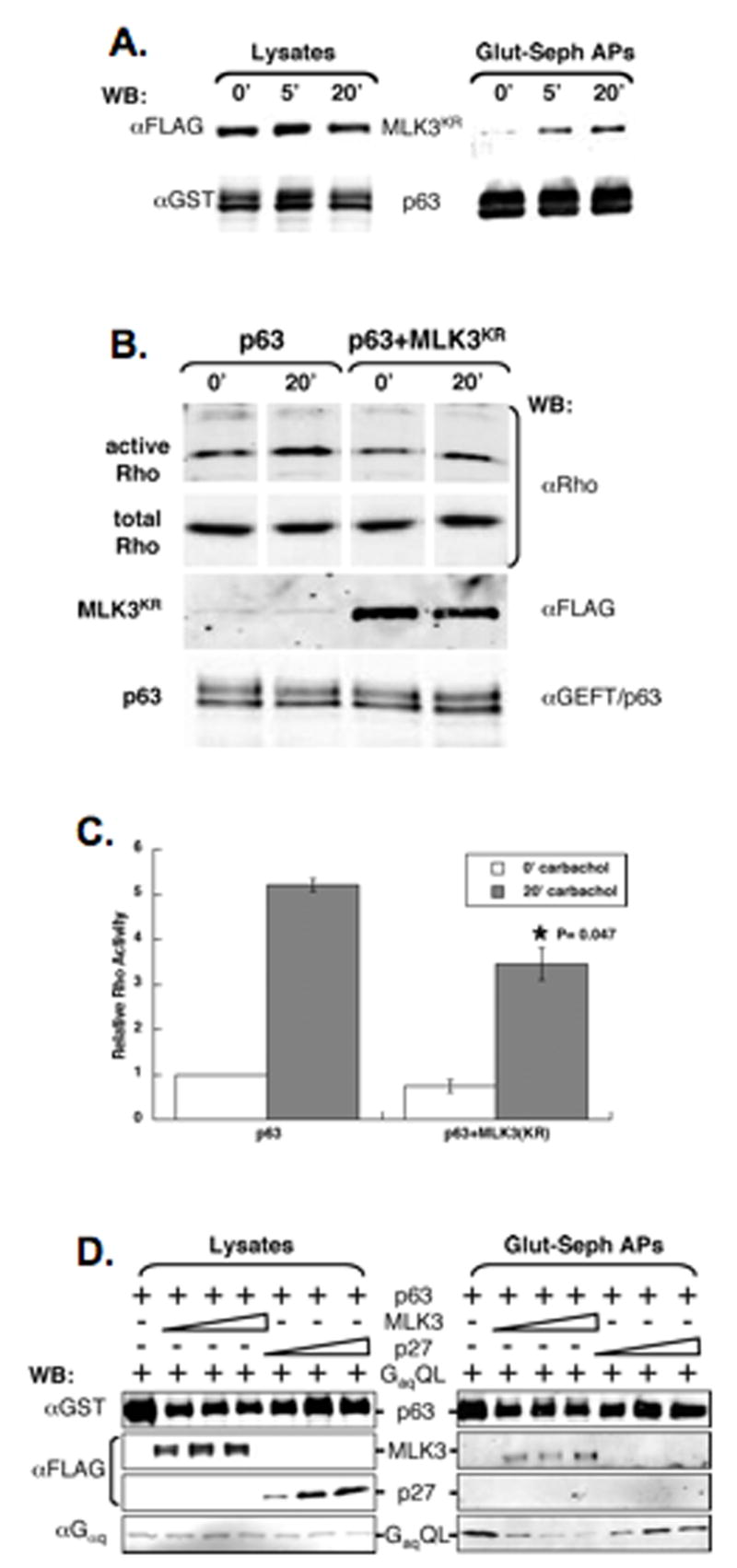
(A) The interaction of MLK3 and p63RhoGEF is modulated downstream of an activated Gq-coupled GPCR. HM1 cells co-transfected with expression vectors for FLAG-MLK3KR (MLK3KR) and GST-p63RhoGEF (p63) were serum-starved for 14.5 h then stimulated with 10 μM carbachol for 0, 5 or 20 min before preparing lysates. Lysates were incubated with glutathione-sepharose and the affinity-precipitates (Glut-seph APs) as well as the starting lysates were immuno-blotted (WB) with anti-FLAG and anti-GST. (B) Rho activation stimulated acutely by a Gq-coupled GPCR/p63RhoGEF pathway is inhibited by MLK3. HM1 cells transfected with the expression vector for GST-p63RhoGEF (p63) alone or together with the expression vector for FLAG-MLK3KR (MLK3KR) were serum-starved for 14.5 h then stimulated with 10 μM carbachol for 0 or 20 min before preparing lysates. Activated Rho-GTP was isolated using a Rhotekin pull-down assay and immuno-blotted using anti-Rho. Immuno-blots were reacted with anti-Rho, anti-FLAG and anti-GEFT/p63 in parallel. Protein bands shown for active Rho or total Rho are from the same immuno-blots with intervening, non-relevant lanes excised.
(C) Quantitation of Rho for samples shown in (B), and for samples from an additional experiment (data not shown), were obtained using fluorochrome-labeled secondary antibodies and the Odyssey® infrared imaging system (LI-COR Biosciences) and the ratio of active Rho/total Rho determined (Relative Rho Activity). The average ratio (+/−SEM) is plotted (active Rho/total Rho for the 0 min p63 sample for each experiment is set arbitrarily at 1). *, statistically significant difference (P<0.01) compared to the p63 20min sample.
(D) MLK3 interferes with p63RhoGEF binding to activated Gαq. HEK 293T cells were co-transfected with expression vectors for GST-p63RhoGEF (p63) and activated, GTPase-deficient Gαq (GqQL) either alone or in combination with increasing amounts of expression vectors for either FLAG-MLK3WT (MLK3) or FLAG-p27 (p27). Cell lysates were incubated with glutathione-sepharose and the affinity-precipitates (Glut-Seph APs), as well as the starting lysates, were immuno-blotted with anti-GST, anti-FLAG and anti-Gαq.
MLK3 inhibits Rho activation after stimulation of Gαq-coupled GPCR/p63RhoGEF
While results from SRF transcription assays show that MLK3 inhibits production of activated Rho downstream of active Gαq, we wanted to measure the effects of MLK3 on Rho activity more directly following acute activation of a Gαq-coupled GPCR/p63RhoGEF pathway. To do this HM1 cells were transfected with expression vectors for GST-p63RhoGEF alone or in combination with FLAG- MLK3KR prior to serum-starvation and 20 min after carbachol (Figure 6A). Before and after carbachol treatment, active Rho levels were quantified as described previously (Figure 6B and C). While carbachol stimulated the level of activated Rho compared with unstimulated samples in both types of transfected cells, the level of activated Rho was significantly less in cells expressing MLK3KR. Thus, increased MLK3 inhibits Rho activation stimulated acutely via a Gαq-coupled GPCR/p63RhoGEF pathway.
MLK3 compromises the interaction of p63RhoGEF and activated Gαq
p63RhoGEF activation by Gαq occurs by direct protein-protein interaction (Lutz et al., 2005; Lutz et al., 2007; Rojas et al., 2007). Our finding that MLK3 associates with p63RhoGEF suggested that the MLK3-induced inhibition of Rho activation, as well as the MLK3-induced inhibition of Rho-dependent SRF transcription stimulated by active Gαq, might result from compromising the Gαq/p63RhoGEF interaction. To test this we co-expressed activated Gαq (GαqQL) with GST-p63RhoGEF in the presence of increasing levels of FLAG-MLK3WT, or FLAG-p27 as control, and assessed whether the level of GαqQL binding to p63RhoGEF was altered by affinity purification (Figure 6D). The amount of GαqQL in the GSTp63RhoGEF affinity-precipitates is decreased in samples containing increased levels of FLAG-MLK3, but not the control protein, FLAG-p27 (Figure 6D, right panel). These results show that MLK3 can compromise the interaction of Gαq and p63RhoGEF. Since this interaction is required for p63RhoGEF activation of Rho elicited by Gαq (Lutz et al., 2007, Rojas, 2007), decreased complex formation may be sufficient to account for the MLK3-induced inhibition of Rho activation in HM1 cells stimulated with carbachol, as well as the MLK3-induced inhibition of Rho-dependent SRF transcription stimulated by active Gαq. These data mechanistically link MLK3 with the regulation of Rho activity.
MLK3/p63RhoGEF interaction is regulated by phosphorylation and Rac binding
We suspected that the increased MLK3/p63RhoGEF binding following acute activation of a Gαq-coupled GPCR, might be accompanied by modulation in the activation of MLK3. Thus, serum-starved HM1cell were treated with carbachol and samples collected as a function of time for immuno-blot analysis using activation-specific, anti-phospho-MLK3 antibodies. Phosphorylation of endogenous MLK3 was detected by 10 min of carbachol treatment and reached a peak at 20 min before declining (Figure 7). So, coincident with increased MLK3/p63RhoGEF binding, activating MLK3 phosphorylation is induced following acute stimulation of a Gαq-coupled GPCR.
Figure 7. MLK3/p63RhoGEF interaction is regulated by MLK3 phosphorylation and Rac binding.

(A) Carbachol stimulation of HM1 cells induces phosphorylation of endogenous MLK3 on T277 and S281. HM1 cells were serum-starved overnight and then treated with 10 μM carbachol for the indicated times before preparing lysates. Immuno-blots were probed either with anti-activation specific, phospho-MLK3 or anti-MLK3.
(B,C) MLK3/p63RhoGEF binding in cells is enhanced by phosphomimetic substitution of T277/S281 and decreased by anisomycin treatment. HEK 293T cells were co-transfected with expression vectors for GST-p63RhoGEF and either FLAG-MLK3KR or FLAG-MLK3KR/T277D,S281D and then treated, or not, with 10μM anisomycin for 35 min to stimulate JNK activity. Cell lysates were incubated with glutathione-sepharose and the affinity-precipitates (Glut-Seph APs), as well as the starting lysates, were immuno-blotted (WB) with anti-FLAG and anti-GEFT/p63.
(D) Detection and quantitation of MLK3 and p63 bands shown in (B, C) were done using fluorochrome-labeled secondary antibodies and the Odyssey® infrared imaging system (LI-COR Biosciences). The ratio of MLK3/p63 in the glutathione-sepharose affinity-precipitates shown in (B,C), and from data of 2 additional experiments (not shown), were averaged and plotted (MLK3/p63 for FLAG-MLK3KR-transfected sample for each experiment is set arbitrarily at 1). Shown is the mean +/− SEM.
(E,F) The in vitro binding of purified p63RhoGEF to MLK3 protein obtained from anisomycin-treated cells is decreased. FLAG-MLK3KR protein (50ng), immuno-purifed from cells untreated (MLK3KR) or treated with 10 μM anisomycin for 35 min (MLK3KRx Anis), was incubated with affinity-purified GST-p63RhoGEF (50ng) for 1.5 h before affinity-precipitation of GSTp63RhoGEF complexes. The precipitates (Glut-seph APs), as well as samples of the input proteins, were immuno-blotted (WB) with anti-FLAG and anti-GEFT/p63 (E) and quantified. The data shown are the mean precipitated MLK3KR/p63 (+/− SEM) (F).
(G,H) Preincubation of active Rac with MLK3 protein enhances binding to p63RhoGEF in vitro. Affinity-purified Rac (480ng, see also Supplemental Figures 4 and 5) or a buffer control, was loaded with GTP-γ-S and gel-filtered before pre-incubating with immuno-purified FLAG-MLK3KR (50ng) for 1 h. Affinity-purified p63RhoGEF (50ng) was then added for another 2 h before affinity precipitation. After precipitation, the GST-p63RhoGEF complexes (Glut-seph APs) were immuno-blotted (WB) with anti-FLAG, anti-Rac and anti-GEFT/p63 (G) and quantified. Data shown are the mean precipitated-MLK3KR/p63 (+/−SEM) (H).
(I) MLK3 limits activated Gαq signaling to Rho by binding to p63RhoGEF.
Stimulation of GPCRs with agonists leads to exchange of GDP for GTP bound to the αsubunit of the coupled heterotrimeric G protein, and release of the βγsubunit. The GTP- bound form of Gαq directly interacts with p63RhoGEF to promote Rho activation. The extent of p63RhoGEF-stimulated Rho activation is kept in check by MLK3, which interacts specifically with this RhoGEF and compromises its binding, and thus, stimulation by Gαq. MLK3 is activated by an unknown mechanism downstream of activated Gq-coupled GPCRs, enhancing its binding to p63RhoGEF. In addition, activated MLK3 induces the JNK pathway, which is triggered also in multiple ways in response to activated Gαq-coupled GPCR signaling (Chan and Wong, 2004; Marinissen et al., 2004). Activation of the JNK pathway results in JNK-phosphorylated forms of MLK3 resulting in decreasd MLK3 binding to p63RhoGEF. Thus, the activation of MLK3 both promotes p63RhoGEF binding to inhibit Rho activation, and initiates a negative feed-back loop to limit this interaction.
(J) In cells depleted of MLK3, the activation of p63RhoGEF by GTP-bound Gαq proceeds in an unchecked manner leading to a corresponding increase in the level of activated Rho.
In addition to the two activating phosphorylations of MLK3 within its kinase domain at residues T277 and S281, phosphorylation of additional sites occurs following activation of this kinase. For example, activated MLK3 triggers the JNK pathway, and JNK feeds back to phosphorylate MLK3 on resulting in the stabilization of MLK3 activity (Schachter et al., 2006). To assess the effects of the activating MLK3 phosphorylation on MLK3/p63RhoGEF binding an expression vector encoding FLAG- MLK3KR, as well as one in which a phosphomimetic amino acid (D) was encoded at these sites (FLAG- MLK3KR/T277D,S281D), was produced. FLAG- MLK3KR/T277D,S281D, or the parent FLAG-MLK3KR, was co-expressed in 293T cells with GST-p63RhoGEF, and the amount of these two proteins present in isolated GST-p63RhoGEF complexes was compared (Figure 7B and D). FLAG-MLK3KR/T277D,S281D was present in these complexes at almost twice the amount of FLAG-MLK3KR (Figure 7D). These results suggest that phosphorylation of MLK3 on T277/S281 promotes its interaction with p63RhoGEF and, since these phosphorylations are induced in HM1 cells following acute stimulation of a Gαq-coupled GPCR, explain the initial, positive modulation of the MLK3/p63RhoGEF association.
The effects of activating the JNK pathway on MLK3/p63RhoGEF binding were examined by acute anisomycin treatment of 293T cells co-expressing FLAG-MLK3KR, or FLAG-MLK3KR/T277D,S281D, and GST-p63RhoGEF prior to isolation of the GST-p63RhoGEF complexes. The amount of either MLK3 mutant protein in these complexes was similar and, in both cases, was less that that present in the absence of anisomycin (Figure 7B, C and D). To confirm that the negative effects of anisomycin on MLK3/p63Rho binding is due to modulating effects on MLK3, FLAG-MLK3KR was immunopurified from cells following anisomycin (Supplemental Figure 4) and incubated in vitro with p63RhoGEF (Figure 7E). Compared with FLAG-MLK3KR immunopurified from untreated cells, the p63RhoGEF binding of FLAG-MLK3KR from anisomycin-treated cells was decreased (Figure 7E and F). These results show that anisomycin-treatment modulates MLK3, contributing to decreased MLK3/p63RhoGEF interaction. While anisomycin activates the p38 MAPK pathways as well, JNK is the only MAPK that can phosphorylate MLK3 (Schachter et al., 2006). We surmise that JNK phosphorylation of MLK3 decreases binding to p63RhoGEF, therefore limiting further MLK3/p63RhoGEF interaction.
In cells, MLK3 can associate with activated forms of Rac and Cdc42, which promotes kinase activation (for review see Gallo and Johnson, 2002). To assess effects of Rac on MLK3/p63RhoGEF association in the absence of MLK3 modifications that Rac might induce in cells, we examined the interaction in vitro (Supplemental Figure 4). For quality control, we confirmed that purified Rac bound MLK3 selectively, and that an activated, GTP-γ-S-loaded form of Rac preferentially bound MLK3 compared to an inactive GDP-loaded form (Supplemental Figure 5). To examine the effects of Rac on MLK3/p63RhoGEF binding in vitro, purified, activated Rac was preincubated with immuno-purified FLAG-MLK3KR prior to the addition of GST-p63RhoGEF and isolation of GST-p63RhoGEF complexes (Figure 7G). Compared to GST-p63RhoGEF complexes from control samples, those preincubated with activated Rac for 2 h contained twice the amount of FLAG-MLK3KR (Figure 7G and H). Similar experiments in the absence of preincubation showed no effect on the amount of FLAG-MLK3KR in GST-p63RhoGEF complexes (not shown). These results show that activated Rac enhances binding of MLK3 to p63RhoGEF. Collectively, these data link two MLK3 activating events (Rac binding and phosphorylation on T277/S281) in promoting p63RhoGEF interaction and effects of the JNK pathway, which temporally follow MLK3 activation, to decreasing this interaction by exerting feed-back effects on MLK3.
Discussion
Most previous studies of MLK3 focused on its role as a MAP3K We show here that MLK3 can function in a kinase activity-independent way to negatively regulate Rho activation. Based on phenotypic similarities between MLK3-depleted cells and cells expressing activated RhoA, we speculated that MLK3 might play a role in modulating Rho function. We show that MLK3 binds p63RhoGEF, thereby impeding its activation by Gαq and preventing Gαq-mediated Rho activation (See Figure 7I and J for model). These data link MLK3, p63RhoGEF and RhoA to provide a mechanism by which MLK3 alters cell migration.
Common phenotypes due to reciprocal changes in MLK3 and RhoA activation
The amount of activated Rho is increased in cells depleted of MLK3, indicating that MLK3 must function as a negative regulator of Rho. Indeed, ectopic expression of MLK3 inhibited Rho activation induced by treatment with a cell agonist. We suspect the increase in active Rho resulting from MLK3 depletion may fully account for the phenotypes observed. First, the effects on the actin cytoskeleton, focal adhesions, protrusions and migration observed in MLK3-depleted cells, are also phenotypes resulting from increased Rho (for review see Jaffe and Hall, 2005). Increased active Rho results in increased microtubule stability (Palazzo et al., 2001), a phenotype also observed following MLK3 depletion (Swenson et al., 2003). Furthermore, inhibition of Rho can result in loss of astral microtubules as well as increased dynamic instability of microtubules (Bakal et al., 2005). Notably, these results are similar to those observed following over-expression of MLK3, which, in mitotic cells, also results in loss of astral microtubules and, in interphase cells, loss of central microtubules (Swenson et al., 2003). Although it is possible that some phenotypes observed upon MLK3 depletion may occur by a mechanism other than increased Rho, our findings indicate that MLK3 functions to limit the level of activated Rho.
MLK3 modulates activated Gαq signaling to Rho via regulated binding to p63RhoGEF
MLK3 specifically inhibits activated Gαq stimulation of Rho-dependent SRF transcription indicating that the effects of MLK3 must occur upstream of Rho activation in a Gαq-specific manner. We show that MLK3 associates with p63RhoGEF but not with other RhoGEFS like LARG or PDZ-RhoGEF. Conversely, endogenous GEFT, a functionally redundant isoform of p63RhoGEF, also specifically associates with MLK3. p63RhoGEF/GEFT is unique among the Rho GEFs activated by heterotrimeric G proteins in that it acts specifically to link Gαq/11 proteins and Gq/11-coupled receptors to Rho activation. Importantly, the activation of p63RhoGEF requires direct binding by Gαq, an interaction compromised by MLK3. Regardless of other possible functions of MLK3, blocking the Gαq/p63RhoGEF interaction would prevent full activation of Rho downstream of Gαq. The effects of MLK3 on Gαq-Rho-dependent signaling are primarily independent of its catalytic activity as a kinase-deficient mutant of MLK3, MLK3KR, interacts with p63RhoGEF and also inhibits Gαq stimulation of Rho-dependent SRF transcription, albeit with reduced effectiveness. A reduced affinity of MLK3KR for p63RhoGEF binding could explain the reduced interference in Gαq signaling. Regardless of the mechanism, these results are consistent with observations that an MLK inhibitor, CEP11004, has no effect on MLK3-inhibition of Gαq-stimulated SRF transcription or cell migration (data not shown). A non-catalytic function of MLK3 also has been reported for the maintenance of the B-Raf/Raf-1 complex which signals downstream from activated Ras (Chadee et al., 2006).
The interaction of MLK3 with p63RhoGEF is influenced both positively and negatively by modulatory factors and events affecting MLK3 directly. Within the kinase domain of MLK3, which is sufficient for p63RhoGEF binding, are two sites of phosphorylation/activation, T277 and S281, which, when substituted with phosphomimetic residues (Ds) promotes the MLK3/p63RhoGEF interaction. Thus, activating phosphorylation would be expected to enhance the MLK3/p63RhoGEF association, which occurs in HM1 cells treated with the Gαq/GPCR agonist carbachol and leads to the activating phosphorylation of MLK3 in concert with increased MLK3/p63RhoGEF interaction. MLK3/p63RhoGEF association is also promoted by activated Rac, which interacts with the MLK3 CRIB domain and promotes its full activation. The increase of MLK3/p63RhoGEF binding and resulting negative effects on Rho activation provide a mechanistic link coupling the activation of Rac (and possibly Cdc42) to the inhibition of Rho (Burridge and Wennerberg, 2004; Sander et al., 1999). The MLK3/p63RhoGEF interaction is inhibited by effects of the JNK pathway on MLK3. Since JNK phosphorylates MLK3 on multiple sites, such phosphorylation may be responsible for decreasing the interaction of MLK3 with p63RhoGEF. Regardless, activation of the JNK pathway, which is induced by activated MLK3, feeds back to negatively affect MLK3 binding to p63RhoGEF. Thus, inherent to the activation of MLK3, which promotes MLK3/p63RhoGEF binding, is the downstream activation of JNK, which deceases this binding providing an intrinsic feed-back pathway to limit negative effects on Rho activation (See Figure 7I and J for model).
Effects of MLK3 depletion on EGF-stimulated activation of MAPK pathways
The effects of MLK3 depletion on the activation of MAPK pathways in this study differ from previous siRNA-based studies (Chadee and Kyriakis, 2004). In those studies MLK3 was implicated in the activation of JNK, ERK and p38 in serum-starved cells following EGF stimulation, whereas we find no significant effect of MLK3 depletion on the extent of EGF-stimulated JNK or ERK activation, and, rather than decreased p38 activation, we find it to be increased. It is possible that differences in the cell types and/or the siRNAs used account for these discrepancies, although we found that the 903 siRNA, used in previous studies, had off-target effects resulting in apoptosis.
Our observed effects of MLK3 depletion on MAPK pathway activation are similar to those obtained using primary (MLK3−/−) mouse embryo fibroblasts (Brancho et al., 2005). EGF treatment of MLK3−/− cells showed no difference in JNK activation compared to wild-type cells, an observation similar to our studies. Furthermore, the activation of p38 appeared to be slightly enhanced in cells null for MLK3 as we observed upon acute MLK3 knock-down (see Figure 9B in Brancho et al., 2005). Interestingly, activated Rho has been found to elicit the phosphorylation and activation of p38γ(Marinissen et al., 2001) and p38α(Semenova et al., 2007). Thus, hyper-stimulation of p38 in MLK3-depleted cells following EGF treatment may have been a consequence of the elevated active Rho in these cells. Alternatively, MLK3 may negatively influence the activation of p38 more directly. Analysis of the possible regulatory relationship between MLK3 and p38 awaits future experiments.
MLK3 as a signaling hub
In the 3-kinase phospho-relay system that constitutes MAPK modules (Garrington and Johnson, 1999), MLK3 is one of at least 20 known kinases that operates as a MAP3K (Gallo and Johnson, 2002). From the analysis of the varied molecular domains of the MAP3Ks as well as the identification of their myriad protein interactions and the functional consequences of these interactions, it is likely that the MAP3Ks act individually as critical network nodes, which link a number of signaling pathways (Uhlik et al., 2004). Our study shows that MLK3 negatively influences Rho activity by blocking the interaction of Gαq with p63RhoGEF, thereby preventing Rho activation. These results as well as those of others (Chadee and Kyriakis, 2004; Chadee et al., 2006), extend our understanding of the molecular interactions and cellular processes in which MLK3 participates and present compelling evidence that, as for other MAP3Ks, MLK3 acts as a hub linking multiple signaling pathways.
Experimental Procedures
In vitro binding
Equal-molar amounts of purified proteins were incubated in filtered binding buffer (10 mM Tris-HCl, pH7.5, 150 mM NaCl, 20 mM β-glycerophosphate, 5 mM EDTA, 10% glycerol, 1% caesamino acids, 1 mM DTT, 10 μg/ml aprotinin, 5μg/ml leupeptin, 1 μg/ml pepstatin, 1 μM microcystin) in sialinized tubes at 4°C. To inhibit further binding, equal volumes of binding buffer containing 1% Igepal were added. GST complexes were isolated following 1 hr incubation with glutathione-sepharose at 4°C. Complexes were washed 2× with binding buffer containing 0.5% Igepal and 2× with TBS prior to suspension in SDS sample buffer. For in vitro binding reactions, purified Rac or binding buffer control was loaded with GTP-γ-S (or GDP) by incubating in binding buffer containing 20 mM EDTA, 100 μM GTP-γS (or 1 mM GDP) for 15 min at 30×C prior to gel filtration spin-column removal of nucleotides.
Migration assays
Cell migration assays were performed under sterile conditions using Millipore 96-well transwell plates with 8 μm pore size uncoated polycarbonate filters. For HEK293T cells, the undersides of the transwells were coated with 0.01% fibronectin. Lower wells contained 150 μl of media to which either 0.1% serum (to measure chemokinesis) or 10% serum (to measure chemotaxis) had been added, whereas upper wells contained 100 μl of cells, suspended to a concentration of 5 × 105, in media containing 0.1% serum. After incubation for 11 h, cells/media in the upper wells were discarded and the migrated cells, on the underside of the transwell membranes, were collected by incubating the transwells in 150 μl of trypsin for 45 min at 37°C and quantified using the CyQuant Cell proliferation kit (Molecular Probes). Values for the total number of cells seeded were obtained by quantifying in parallel pelleted, frozen 100 μl aliquots of the original cell suspension.
SRF Reporter assays
For reporter assays, HEK293T cells were seeded in 6-well plates at 9 ×105 cells/well 1 day prior to transfection of the expression plasmids together with 0.4 μg/well of the SRF reporter plasmid (pSRF-luc) and 0.02μg/well of ph-RGTK, which expresses Renilla luciferase from Renilla reniformis as an internal control. Expression plasmids encoding GqQL, G12QL, G13QL and C3 exotoxin were each tranfected at 0.5 μg/well, whereas pcDNA3-FLAG-MLK3WT and pcDNA3-FLAG-MLK3KR were transfected at 0.4 μg/well and 1.0 μg/well, respectively, to obtain similar expression levels. The total amount of plasmid DNA was adjusted to 2 μg/well with pcDNA3 for each transfection. Lysates were cleared by centrifugation and then assayed by luminometry for firefly and Renilla luciferase activities using the Dual-Luciferase Assay System (Promega). Light emission was quantified using a TD-20/20 luminometer (Turner Designs, Sunnyvale, CA). Standard procedures as well as oligo/siRNA design are described in Supplemental Experimental Procedures.
Supplementary Material
Acknowledgments
We thank Timothy Fields and Patrick Kelly for insightful discussions and Sally Kornbluth for critically reading the manuscript. We also thank David Loiselle and Tim Haystead for mass spectrometry analysis of MLK3-associated proteins (supported by the Duke Cancer Center Core grant, P30 CA014236). This research was supported by NIH grant R01 CA082845 to ARM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aktories K, Hall A. Botulinum ADP-ribosyltransferase C3: a new tool to study low molecular weight GTP-binding proteins. Trends Pharmacol Sci. 1989;10:415–418. doi: 10.1016/0165-6147(89)90191-0. [DOI] [PubMed] [Google Scholar]
- Bakal CJ, Finan D, LaRose J, Wells CD, Gish G, Kulkarni S, DeSepulveda P, Wilde A, Rottapel R. The Rho GTP exchange factor Lfc promotes spindle assembly in early mitosis. Proc Natl Acad Sci USA. 2005;102:9529–9534. doi: 10.1073/pnas.0504190102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beningo KA, Dembo M, Kaverina I, Small JV, Wang YL. Nascent focal adhesions are responsible for the generation of strong propulsive forces in migrating fibroblasts. J Cell Biol. 2001;153:881–888. doi: 10.1083/jcb.153.4.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancho D, Ventura JJ, Jaeschke A, Doran B, Flavell RA, Davis RJ. Role of MLK3 in the regulation of mitogen-activated protein kinase signaling cascades. Mol Cell Biol. 2005;25:3670–3681. doi: 10.1128/MCB.25.9.3670-3681.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbelo PD, Drechsel D, Hall A. A conserved binding motif defines numerous candidate target proteins for both Cdc42 and Rac GTPases. J Biol Chem. 1995;270:29071–29074. doi: 10.1074/jbc.270.49.29071. [DOI] [PubMed] [Google Scholar]
- Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–179. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- Cha H, Smith BL, Gallo K, Machamer CE, Shapiro P. Phosphorylation of golgin-160 by mixed lineage kinase 3. J Cell Sci. 2004;117:751–760. doi: 10.1242/jcs.00897. [DOI] [PubMed] [Google Scholar]
- Chadee DN, Kyriakis JM. MLK3 is required for mitogen activation of B-Raf, ERK and cell proliferation. Nat Cell Biol. 2004;6:770–776. doi: 10.1038/ncb1152. [DOI] [PubMed] [Google Scholar]
- Chadee DN, Xu D, Hung G, Andalibi A, Lim DJ, Luo Z, Gutmann DH, Kyriakis JM. Mixed-lineage kinase 3 regulates B-Raf through maintenance of the B-Raf/Raf-1 complex and inhibition by the NF2 tumor suppressor protein. Proc Natl Acad Sci USA. 2006;103:4463–4468. doi: 10.1073/pnas.0510651103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AS, Wong YH. Gbetagamma signaling and Ca2+ mobilization cooperate synergistically in a Sos and Rac-dependent manner in the activation of JNK by Gq-coupled receptors. Cell Signal. 2004;16:823–836. doi: 10.1016/j.cellsig.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Chikumi H, Vazquez-Prado J, Servitja JM, Miyazaki H, Gutkind JS. Potent activation of RhoA by Galpha q and Gq-coupled receptors. J Biol Chem. 2002;277:27130–27134. doi: 10.1074/jbc.M204715200. [DOI] [PubMed] [Google Scholar]
- Du Y, Bock BC, Schachter KA, Chao M, Gallo KA. Cdc42 induces activation loop phosphorylation and membrane targeting of mixed lineage kinase 3. J Biol Chem. 2005;280:42984–42993. doi: 10.1074/jbc.M502671200. [DOI] [PubMed] [Google Scholar]
- Fukuhara S, Chikumi H, Gutkind JS. Leukemia-associated Rho guanine nucleotide exchange factor (LARG) links heterotrimeric G proteins of the G(12) family to Rho. FEBS Lett. 2000;485:183–188. doi: 10.1016/s0014-5793(00)02224-9. [DOI] [PubMed] [Google Scholar]
- Fukuhara S, Murga C, Zohar M, Igishi T, Gutkind JS. A novel PDZ domain containing guanine nucleotide exchange factor links heterotrimeric G proteins to Rho. J Biol Chem. 1999;274:5868–5879. doi: 10.1074/jbc.274.9.5868. [DOI] [PubMed] [Google Scholar]
- Gallo KA, Johnson GL. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat Rev Mol Cell Biol. 2002;3:663–672. doi: 10.1038/nrm906. [DOI] [PubMed] [Google Scholar]
- Garrington TP, Johnson GL. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr Opin Cell Biol. 1999;11:211–218. doi: 10.1016/s0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- Hart MJ, Jiang X, Kozasa T, Roscoe W, Singer WD, Gilman AG, Sternweis PC, Bollag G. Direct stimulation of the guanine nucleotide exchange activity of p115 RhoGEF by Galpha13. Science. 1998;280:2112–2114. doi: 10.1126/science.280.5372.2112. [DOI] [PubMed] [Google Scholar]
- Hartkamp J, Troppmair J, Rapp UR. The JNK/SAPK activator mixed lineage kinase 3 (MLK3) transforms NIH 3T3 cells in a MEK-dependent fashion. Cancer Res. 1999;59:2195–2202. [PubMed] [Google Scholar]
- Hill CS, Wynne J, Treisman R. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell. 1995;81:1159–1170. doi: 10.1016/s0092-8674(05)80020-0. [DOI] [PubMed] [Google Scholar]
- Huang C, Jacobson K, Schaller MD. MAP kinases and cell migration. J Cell Sci. 2004;117:4619–4628. doi: 10.1242/jcs.01481. [DOI] [PubMed] [Google Scholar]
- Jaeschke A, Davis RJ. Metabolic stress signaling mediated by mixed-lineage kinases. Mol Cell. 2007;27:498–508. doi: 10.1016/j.molcel.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- Jaffe AB, Hall A, Schmidt A. Association of CNK1 with Rho guanine nucleotide exchange factors controls signaling specificity downstream of Rho. Curr Biol. 2005;15:405–412. doi: 10.1016/j.cub.2004.12.082. [DOI] [PubMed] [Google Scholar]
- Kozasa T, Jiang X, Hart MJ, Sternweis PM, Singer WD, Gilman AG, Bollag G, Sternweis PC. p115 RhoGEF, a GTPase activating protein for Galpha12 and Galpha13. Science. 1998;280:2109–2111. doi: 10.1126/science.280.5372.2109. [DOI] [PubMed] [Google Scholar]
- Lutz S, Freichel-Blomquist A, Yang Y, Rumenapp U, Jakobs KH, Schmidt M, Wieland T. The guanine nucleotide exchange factor p63RhoGEF, a specific link between Gq/11-coupled receptor signaling and RhoA. J Biol Chem. 2005;280:11134–11139. doi: 10.1074/jbc.M411322200. [DOI] [PubMed] [Google Scholar]
- Lutz S, Shankaranarayanan A, Coco C, Ridilla M, Nance MR, Vettel C, Baltus D, Evelyn CR, Neubig RR, Wieland T, et al. Structure of Galphaq-p63RhoGEF-RhoA complex reveals a pathway for the activation of RhoA by GPCRs. Science. 2007;318:1923–1927. doi: 10.1126/science.1147554. [DOI] [PubMed] [Google Scholar]
- Marinissen MJ, Chiariello M, Gutkind JS. Regulation of gene expression by the small GTPase Rho through the ERK6 (p38 gamma) MAP kinase pathway. Genes Dev. 2001;15:535–553. doi: 10.1101/gad.855801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinissen MJ, Chiariello M, Tanos T, Bernard O, Narumiya S, Gutkind JS. The small GTP-binding protein RhoA regulates c-jun by a ROCK-JNK signaling axis. Mol Cell. 2004;14:29–41. doi: 10.1016/s1097-2765(04)00153-4. [DOI] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- Palazzo AF, Cook TA, Alberts AS, Gundersen GG. mDia mediates Rho-regulated formation and orientation of stable microtubules. Nat Cell Biol. 2001;3:723–729. doi: 10.1038/35087035. [DOI] [PubMed] [Google Scholar]
- Paterson HF, Self AJ, Garrett MD, Just I, Aktories K, Hall A. Microinjection of recombinant p21Rho induces rapid changes in cell morphology. J Cell Biol. 1990;111:1001–1007. doi: 10.1083/jcb.111.3.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peralta EG, Ashkenazi A, Winslow JW, Ramachandran J, Capon DJ. Differential regulation of PI hydrolysis and adenylyl cyclase by muscarinic receptor subtypes. Nature. 1988;334:434–437. doi: 10.1038/334434a0. [DOI] [PubMed] [Google Scholar]
- Rana A, Gallo K, Godowski P, Hirai S, Ohno S, Zon L, Kyriakis JM, Avruch J. The mixed lineage kinase SPRK phosphorylates and activates the stress- activated protein kinase activator, SEK-1. J Biol Chem. 1996;271:19025–19028. doi: 10.1074/jbc.271.32.19025. [DOI] [PubMed] [Google Scholar]
- Ren XD, Kiosses WB, Schwartz MA. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. Embo J. 1999;18:578–585. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- Rojas RJ, Yohe ME, Gershburg S, Kawano T, Kozasa T, Sondek J. Galphaq directly activates p63RhoGEF and Trio via a conserved extension of the Dbl homology-associated pleckstrin homology domain. J Biol Chem. 2007;282:29201–29210. doi: 10.1074/jbc.M703458200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander EE, ten Klooster JP, van Delft S, van der Kammen RA, Collard JG. Rac downregulates Rho activity: reciprocal balance between both GTPases determines cellular morphology and migratory behavior. J Cell Biol. 1999;147:1009–1022. doi: 10.1083/jcb.147.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scacheri PC, Rozenblatt-Rosen O, Caplen NJ, Wolfsberg TG, Umayam L, Lee JC, Hughes CM, Shanmugam KS, Bhattacharjee A, Meyerson M, et al. Short interfering RNAs can induce unexpected and divergent changes in the levels of untargeted proteins in mammalian cells. Proc Natl Acad Sci USA. 2004;101:1892–1897. doi: 10.1073/pnas.0308698100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schachter KA, Du Y, Lin A, Gallo KA. Dynamic positive feedback phosphorylation of mixed lineage kinase 3 by JNK reversibly regulates its distribution to Triton-soluble domains. J Biol Chem. 2006;281:19134–19144. doi: 10.1074/jbc.M603324200. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Hall A. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 2002;16:1587–1609. doi: 10.1101/gad.1003302. [DOI] [PubMed] [Google Scholar]
- Semenova MM, Maki-Hokkonen AM, Cao J, Komarovski V, Forsberg KM, Koistinaho M, Coffey ET, Courtney MJ. Rho mediates calcium-dependent activation of p38alpha and subsequent excitotoxic cell death. Nat Neurosci. 2007;10:436–443. doi: 10.1038/nn1869. [DOI] [PubMed] [Google Scholar]
- Stronach B, Perrimon N. Activation of the JNK pathway during dorsal closure in Drosophila requires the mixed lineage kinase, slipper. Genes Dev. 2002;16:377–387. doi: 10.1101/gad.953002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swenson KI, Winkler KE, Means AR. A new identity for MLK3 as an NIMA-related, cell cycle-regulated kinase that is localized near centrosomes and influences microtubule organization. Mol Biol Cell. 2003;14:156–172. doi: 10.1091/mbc.E02-02-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlik MT, Abell AN, Cuevas BD, Nakamura K, Johnson GL. Wiring diagrams of MAPK regulation by MEKK1, 2, and 3. Biochem Cell Biol. 2004;82:658–663. doi: 10.1139/o04-114. [DOI] [PubMed] [Google Scholar]
- Wennerberg K, Ellerbroek SM, Liu RY, Karnoub AE, Burridge K, Der CJ. RhoG signals in parallel with Rac1 and Cdc42. J Biol Chem. 2002;277:47810–47817. doi: 10.1074/jbc.M203816200. [DOI] [PubMed] [Google Scholar]
- Zhang H, Wu W, Du Y, Santos SJ, Conrad SE, Watson JT, Grammatikakis N, Gallo KA. Hsp90/p50cdc37 is required for mixed-lineage kinase (MLK) 3 signaling. J Biol Chem. 2004;279:19457–19463. doi: 10.1074/jbc.M311377200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.