

Abstract
We sought to determine the relationship between phencyclidine (PCP)-induced alterations in behavior and NMDAR expression in the cortex by examining the effect of antischizophrenic drug treatment on both. Sprague-Dawley rat pups were pretreated with risperidone or olanzapine prior to treatment with PCP on postnatal day 7 (PN7) or sub-chronically on PN7, 9, and 11. Pre-pulse inhibition (PPI) of acoustic startle was measured on PN24–26 and following a challenge dose of 4 mg/kg PCP, locomotor activity was measured on PN28–35. PCP treatment on PN7 did not cause a deficit in PPI, but did cause locomotor sensitization. This was prevented by both antipsychotics. PCP treatment on PN7 caused an up-regulation of NR1 and NR2B, which was not affected by either antischizophrenic drug. PCP treatment on PN7, 9, and 11 caused a deficit in PPI and a sensitized locomotor response to PCP challenge as well as an up-regulation of NR1 and NR2A, all of which were prevented by both atypical antischizophrenic drugs. These data support the hypothesis that subchronic, but not single injection PCP treatment in developing rats results in behavioral alterations that are sensitive to antipsychotic drugs and these behavioral changes observed could be related to up-regulation of cortical NR1/NR2A receptors.
Keywords: phencyclidine, NMDA receptor, olanzapine, risperidone, pre-pulse inhibition, locomotor activity, behavioral sensitization
1. Introduction
Schizophrenia is a severe neuropsychiatric disorder which afflicts approximately 1% of the population worldwide and shows strong genetic tendencies, with symptoms first presenting in early adulthood (Bromet and Fennig, 1999; Lewis and Lieberman, 2000). The disease is characterized by the presence of both positive, e.g. paranoia, hallucinations, delusions, and negative symptoms, including but not limited to emotional and social withdrawal, anhedonia, and depression, as well as cognitive impairments such as memory and attention deficits (McGlashan, 1996).
Phencyclidine (PCP) intoxication in humans has been shown to mimic both the positive and negative symptoms of schizophrenia as well as exacerbate psychosis in schizophrenics. These psychotomimetic properties led researchers to examine the effects of PCP in animals (Javitt and Zukin, 1991; Luby et al., 1962). PCP administration to rats results in increased locomotor activity, stereotypy, ataxia, head-weaving, and circling and walking backwards (Braff and Geyer, 1990; Castellani and Adams, 1981; Geyer and Ellenbroek, 2003; Martinez et al., 2000; Steinpresis, 1996). In general, alterations in motoric behaviors such as these are thought to model the positive symptoms of schizophrenia.
N-methyl-D-aspartate (NMDA) receptor antagonists (PCP and MK-801) have been shown to reproducibly disrupt pre-pulse inhibition (PPI) of acoustic startle in animals and are routinely used to model the sensorimotor gating deficits of schizophrenia (Geyer et al., 2001; Rasmussen et al., 2007). It is a measure of the reduction of the startle response when a smaller non-startling acoustic stimulus (pre-pulse) is presented 80–120 ms prior to the startling stimulus (pulse) (Swerdlow et al., 1994). PPI of acoustic startle is a measure of information processing in specific pathways that are known to be abnormal in schizophrenia (Adams and Moghaddam, 1998; Bunney BG, 2000; Castellani and Adams, 1981; Ellenbroek and Cools, 2000; Wang et al., 2001). This deficit may contribute to the thought disorder and cognitive fragmentation characteristic of this disease (Braff and Geyer, 1990).
The etiology of schizophrenia has been described as a neurodevelopmental disorder (Pilowsky et al., 1993; Weinberger, 1987, 1996). Neurodegeneration during early stages of development has been shown following PCP or MK-801 treatment in the cortex, hippocampus, and striatum, all of which are regions of the brain implicated in schizophrenia (Ikonomidou et al., 1999; Wang and Johnson, 2005, 2007). In addition, PCP treatment on postnatal day (PN) 7, results in neurodegeneration (positive silver staining) in the frontal cortex, striatum, and hippocampus within 9 hours of treatment (Wang and Johnson, 2005, 2007). Accompanying increases in caspase-3 immunoreactivity and terminal dUTP nick-end labeling (TUNEL) of broken DNA show this degeneration to be apoptotic in nature. Furthermore, administration of MK-801 and/or PCP to the neonate has been shown to produce aberrant behaviors later in development (Beninger et al., 2002; Wang et al., 2001) specifically, locomotor sensitization and deficits in PPI of acoustic startle were reported in postnatal (PN24–28) rats treated with PCP on PN7, 9, and 11 (Wang et al., 2001).
While it is generally acknowledged that the schizophrenic “break” does not often occur before puberty, there is evidence that this does occur in some cases (Nicolson et al., 2000; Ross et al., 2006b). Further, when examined retrospectively, there is ample evidence of abnormalities such as poor academic performance and poor social interaction skills in children who are eventually diagnosed as schizophrenic. This laboratory and others have demonstrated that this model (PCP treatment on PN7, 9, and 11) has face validity in that it results in behavioral deficits in rats similar to those seen in schizophrenia, including deficits in spatial learning, PPI of acoustic startle and social behavior (Harich et al., 2007; Rasmussen et al., 2007; Wang et al., 2001; Wiley et al., 2003a; Wiley et al., 2003b). Further, pretreatment with olanzapine was able to prevent both behavioral and neurotoxic indices of postnatal PCP administration, suggesting that this treatment paradigm is a suitable model of schizophrenia in rats (Wang et al., 2001)
Up-regulation of the NR1 subunit of the NMDA receptor has been reported to be associated with PCP-induced behavioral changes (Wang et al., 2001); however, it is not known whether this is accompanied by commiserate increases in NR2 subunits, which would be necessary for altered receptor function (Kutsuwada et al., 1992; Monyer et al., 1994; Paoletti and Neyton, 2007). Further, it is not known whether behavioral changes following single dose PCP treatment on PN7 administration are associated with NMDAR up-regulation, and if so, whether these changes can also be modulated by antischizophrenic drugs. Therefore, in this study we determined the effect of PN7 and PN7, 9, 11 PCP administration on two behaviors thought to model different aspects of schizophrenia as well as on the levels of the three most abundant cortical and striatal NMDA receptor subunits (NR1, NR2A and NR2B). Finally, the relevance of the changes in behavior and NMDA receptor subunit protein as a model of schizophrenia was challenged by determining the effect of pretreatment with two anti-schizophrenic drugs, risperidone and olanzapine.
2. Materials and Methods
2.1 Animals
Timed, day 14 pregnant female Sprague-Dawley rats were obtained from Charles River Laboratories (Wilmington, MA). The dams were housed individually with a regular 12h light-dark cycle (lights on 0700, off at 1900) with food and water ad libitum. Following parturition, male and female pups from four dams were combined and randomly cross-fostered to one of the four lactating dams. Each litter consisted of ten to twelve pups with approximately equal numbers of each gender. All experiments were conducted in accordance with the NIH and the University of Texas Medical Branch at Galveston Institutional Animal Care and Use Committee.
2.2 Drugs
Phencyclidine was acquired from the National Institute on Drug Abuse (NIDA, Rockville, MD) and dissolved in 0.9% NaCl. Risperidone was obtained as a solution from Janssen Pharmaceutica (Titusville, NJ) and dissolved in 0.9% NaCl. Olanzapine was a generous gift from Eli Lilly and Company (Indianapolis, IN) and was dissolved in 0.1 N HCl and titrated to pH 7.0 with 0.1 N NaOH and finally diluted with 0.9% NaCl. Doses were chosen based on prior experiments that addressed PCP-induced regulation of the NMDAR (Wang et al., 2001) and our own preliminary experiments.
2.3 Experimental design
Male and female rat pups were treated on either PN7 or on PN 7, 9, and 11 (sub-chronic) with 10 mg/kg PCP or saline vehicle (s.c). Olanzapine (1 mg/kg, s.c.) or risperidone (0.25 mg/kg, s.c.) were administered 30 minutes prior to PCP or saline administration on PN7 or on PN 7, 9, 11. Pups were sacrificed by decapitation on PN7 at 24 hours following saline/vehicle (control, time=0 hours), PCP/vehicle, antagonist/vehicle, or antagonist/PCP treatment. In a separate experiment, pups were sacrificed by decapitation 24 hours following the last injection of the aforementioned drug regimens on PN7, 9 and 11 (on PN12). For biochemical studies, the frontal cortex or striatum was dissected as described below and used for Western blot analysis. We chose to investigate the frontal cortex and striatum because of the prominent role these two brain regions play in the pathophysiology of schizophrenia. Furthermore, the frontal cortex and striatum are part of the brain circuitry involved in the expression of both PPI (Ellenbroek et al., 1996; Koch and Bubser, 1994; Swerdlow et al., 1995) and locomotor sensitization (Pierce and Kalivas, 1997). In behavioral experiments, animals were assessed for PPI of acoustic startle on PN24–26 and then tested for locomotor activity following a 4 mg/kg PCP (i.p.) challenge on PN28–35.
2.4 Sub-cellular fractionation
Protein extracts were prepared from the frontal cortex or striatum as previously described with some modifications (Anastasio and Johnson, 2008; Wang et al., 2001). Briefly, 2 mm sections corresponding to 4.7 to 2.7 mm anterior to Bregma for the frontal cortex and 0.7 mm to −1.3 mm for the striatum (Paxinos and Watson, 1986) were cut with the aid of an aluminum brain mold. Cortical brain sections were homogenized in 500 µL of lysis buffer with the aid of an automatic tissue grinder (Kontes Pellet Pestle Motor, Kimble / Kontes, Vineland, New Jersey). The lysis buffer consisted of 10 mM HEPES (pH 7.4), 1 mM EDTA, 2 mM EGTA, and 500 µM DTT. Just prior to use, protease inhibitor cocktail [4-(2-aminoethyl) benzenesulfonyl fluoride (AEBSF), pepstatin A, E-64, bestatin, leupeptin, and aprotinin without metal chelators (Sigma-Aldrich, St Louis, MO)] at a concentration of 10 µL/mL was added to the lysis buffer. The homogenate was then centrifuged at 1000 × g at 4°C for 10 minutes to pellet the nuclear protein fraction (P1). The supernatant (S1) was collected and centrifuged at 8000 × g at 4°C for 30 minutes to pellet the membrane protein fraction (P2). The membrane fraction was re-suspended in homogenization buffer and centrifuged at 20,000 × g at 4°C for 30 minutes; the resultant pellet was re-suspended in lysis buffer + 1% SDS, boiled for 10 minutes and stored at −80°C.
2.5 Western blot analysis
Equal amounts of protein were separated on 10% Bis-Tris gels (Invitrogen, NY) using SDS-PAGE with a MES-SDS running buffer system, pH 7.4. Following electrophoresis (110 V for 2 hours), proteins were transferred to polyvinylidene difluoride (PVDF) membranes (0.2 µm) in a Mini Electrotransfer Unit (Bio-Rad, Hercules, CA) overnight. The membrane was blocked in 5% nonfat milk, followed by incubation with the primary antibody in 1% milk for 2 hours at room temperature. Following washes (3 × 10 minutes) in TBS+0.1% Tween 20 (TBST), the membrane was incubated with horseradish peroxidase conjugated secondary antibodies for 1 hour at room temperature. Analysis was carried out using the enhanced chemiluminescence (ECL) plus Western blotting detection reagents (Amersham Biosciences, Piscataway, NJ). The bands corresponding to the various proteins of interest were scanned and densitometrically analyzed by using an automatic imaging analysis system (Alpha Innotech Corporation, San Leandro, CA). All quantitative analyses were normalized to β-actin (after stripping [Reblot mild, Chemicon International, Temecula, CA]).
2.6 Antibodies
The monoclonal anti-NR1, anti-NR2A, and anti-NR2B were purchased from BD Biosciences (San Jose, CA). Anti-actin antibody was obtained from Chemicon International (Temecula, CA). Primary antibody dilution was 1:500–1:1000. Secondary antibodies were purchased from Zymed (Invitrogen Corporation, Carlsbad, CA) and used at a concentration of 1:5000.
2.7 Pre-pulse inhibition (PPI) of acoustic startle
Measurement of PPI of acoustic startle was performed according to previously published procedures with minor modifications (Wang et al., 2001; Wang et al., 2003). Testing was performed between 0900 and 1600 hours as described below. Male and female rat pups (PN24–26) were transferred into a small sound-attenuated, dedicated behavior room on the day of testing and allowed to acclimate to the room for 20 minutes. Animals were then placed into one of three startle chambers (SR-Lab, San Diego Instruments, San Diego, CA) with a background noise level of 65 dB. Following a 10 minute acclimation period, rats were exposed to three randomly administered stimuli: no stimulus, a 73 dB 20 ms pre-pulse 100 ms prior to a 120 dB pulse, or a 120 dB 40 ms pulse alone with a variable inter-trial interval (5–20 sec) for a total of 63 trials (21 no stimulus, 21 pulse alone, and 21 pre-pulse + pulse). % PPI of acoustic startle was calculated as the [pulse-(pre-pulse + pulse)]/pulse × 100.
2.8 Locomotor Activity
On the day of testing animals were placed in locomotor chamber boxes and allowed to habituate for 30 minutes prior to a 4 mg/kg challenge dose of PCP (i.p.). Locomotor activity was measured for an additional 90 minutes via an open-field activity system (San Diego Instruments, San Diego, CA) which consisted of a square enclosure with Plexiglas walls (40 × 40 × 40 cm). Horizontal activity was measured with a 16 × 16 photobeam matrix which recorded both central and peripheral activity in 5 min bins.
2.9 Statistical analysis
Group comparisons were specifically defined before the beginning of each experiment; therefore, planned comparisons were performed instead of an overall F test in a multifactorial ANOVA (Keppel, 1982). Statistical comparisons for each experiment were conducted using a one-way ANOVA. All values are presented as mean ± SEM. The null hypothesis was rejected at p<0.05.
3. Results
PCP administration in adult rats causes substantial impairments in normal behavior (Geyer et al., 2001; Hanania et al., 1999; Phillips et al., 2001). In perinatal rats, 10 mg/kg PCP results in significant reduction of normal behaviors, most notably, a reduction of suckling behavior. This lasts approximately 6–8 hours and causes a concomitant weight loss, especially in male pups. While females regain the lost weight within 24 hrs, the males do not; however, at the time of behavioral testing, there is no significant difference in weight between saline and PCP treated animals of either gender (Table 1). All experimental groups consisted of equal numbers of males and females.
Table 1.
| Weight (g) | |||||||
|---|---|---|---|---|---|---|---|
| PN7 | PN8 | PN9 | PN10 | PN11 | PN12 | PN28 | |
| Saline female | 14.3±0.7 | 19.7±0.2 | 17.4±1.2 | 23.7±0.5 | 20.9±1.5 | 29.1±0.7 | 81.1±2.8 |
| PCP female | 14.7±0.8 | 17.3±0.6* | 16.5±0.8 | 21.4±0.4* | 19.9±1.3 | 24.9±1.5 | 79.2±4 |
| Saline male | 16.2±0.6 | 21.2±0.4 | 20.7±0.8 | 26.4±0.6 | 25.5±1.1 | 31.5±0.6 | 91.2±3.5 |
| PCP male | 15.9±0.8 | 17.7±0.5^ | 17.5±0.8^ | 21.9±0.7^ | 20.6±1.1^ | 23.8±0.3^ | 85.6±4.6 |
Data presented as mean (g) ±SEM
p<0.05 vs saline female N=6–11/group (Student’s t-test)
p<0.05 vs saline male N=6–13/group (Student’s t-test)
3.1 Effects of PCP treatment on the development of locomotor sensitization and deficits in PPI
PCP treatment on PN7 had no significant effect on PPI (F3,31=2.264, NS) (Figure 1). Further, PCP treatment on PN7 had no effect on the startle amplitude (data not shown). However, we found that PCP treatment on PN7 caused a significantly sensitized locomotor response to PCP challenge on PN28–35 that was prevented by either olanzapine (F3,31=8.071, p<0.05) or risperidone (F3,31=10.001, p<0.05) (Figure 2).
Figure 1. Effects of PCP administration (PN7 only) on PPI of acoustic startle.
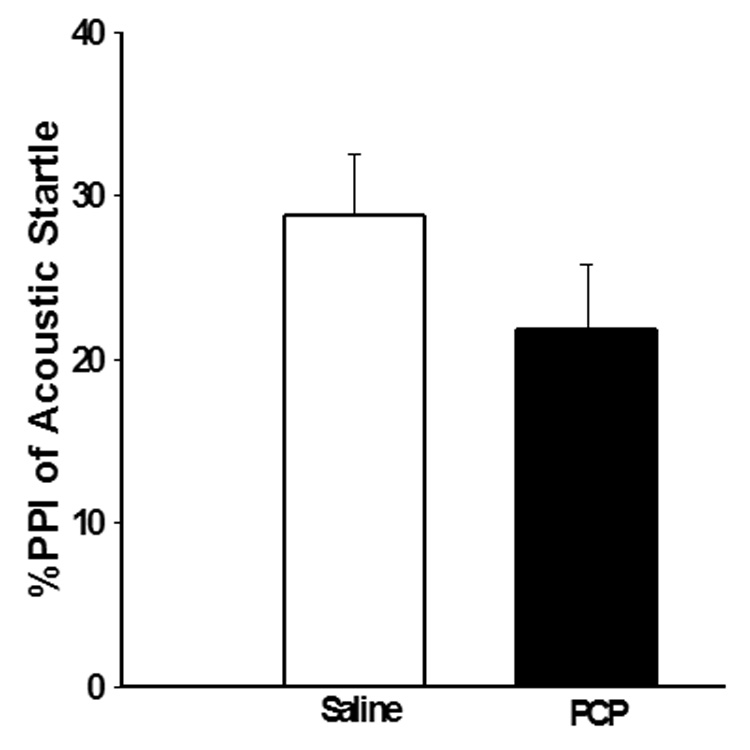
PCP (10 mg/kg) treatment on PN7 does not produce a significant deficit in PPI of acoustic startle as measured on PN24–26. N=8 in both control and experimental groups.
Figure 2. Effects of PCP administration (PN7 only) the development of locomotor sensitization.
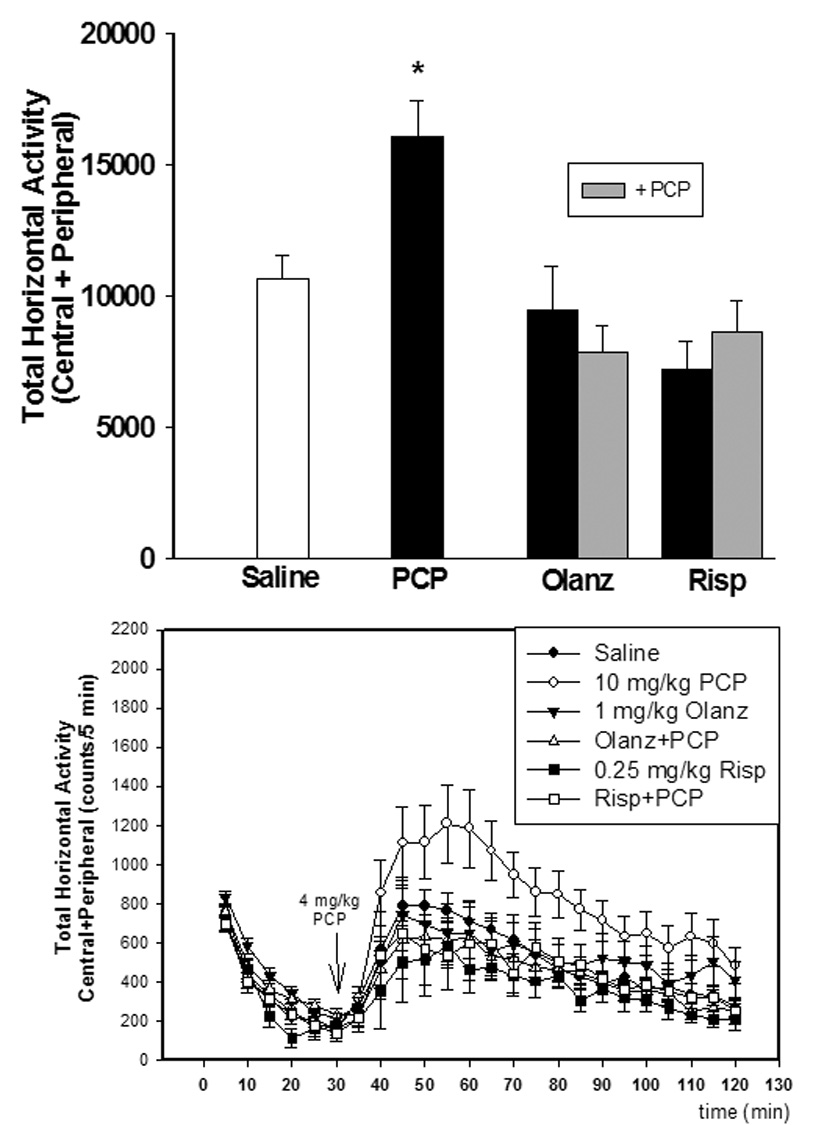
Locomotor sensitization induced by a 4 mg/kg challenge to PCP treatment on PN7 treated animals as measured on PN28–35. Pretreatment on PN7 with olanzapine (1 mg/kg) or risperidone (0.25 mg/kg) inhibits the development of locomotor sensitization N=8/group (top). Time-course of locomotor activity (bottom) *p<0.05 vs. saline (one-way ANOVA with Bonferroni’s post hoc test). N=8 in both control and experimental groups. ●-saline; ○-10 mg/kg PCP; ▼-1 mg/kg olanzapine; Δ-olanzapine + PCP; ■-0.25 mg/kg risperidone; □-risperidone + PCP
As with PCP treatment on PN7, treatment on PN7, 9, and 11 produced no significant effect on startle amplitude (data not shown). On the other hand, PCP treatment on PN7, 9, and 11 resulted in a significant deficit in PPI of acoustic startle measured on PN24–26, which was blocked by pretreatment with either olanzapine (F3,31=4.194, p<0.05) or risperidone (F3,31=2.852, p<0.05) (Figure 3). Previous studies from this laboratory reported that the atypical antipsychotic drug, olanzapine, prevented sensitization to the locomotor activating effects of PCP challenge with 2 mg/kg on PN42 as well as the inhibition of baseline pre-pulse inhibition of acoustic startle on PN24–28 in female pups (Wang et al., 2001). This experiment was repeated using the present design which also included male pups in order to exclude any gender related differences. Further, this design was extended to risperidone, which has some atypical properties at low doses. For these studies, animals treated sub-chronically with PCP and then challenged with 4 mg/kg PCP on PN28–35, showed enhanced locomotor activity compared to saline treated animals (Figure 4). Both olanzapine (F3,31=5.920, p<0.05) and risperidone (F3,31=5.044, p<0.05) pretreatment on PN7, 9, and 11 prevented PCP-induced locomotor sensitization measured on PN28–35 with no effect of their own (Figure 4).
Figure 3. Effects of subchronic PCP administration (PN7, 9 and 11) on PPI of acoustic startle.
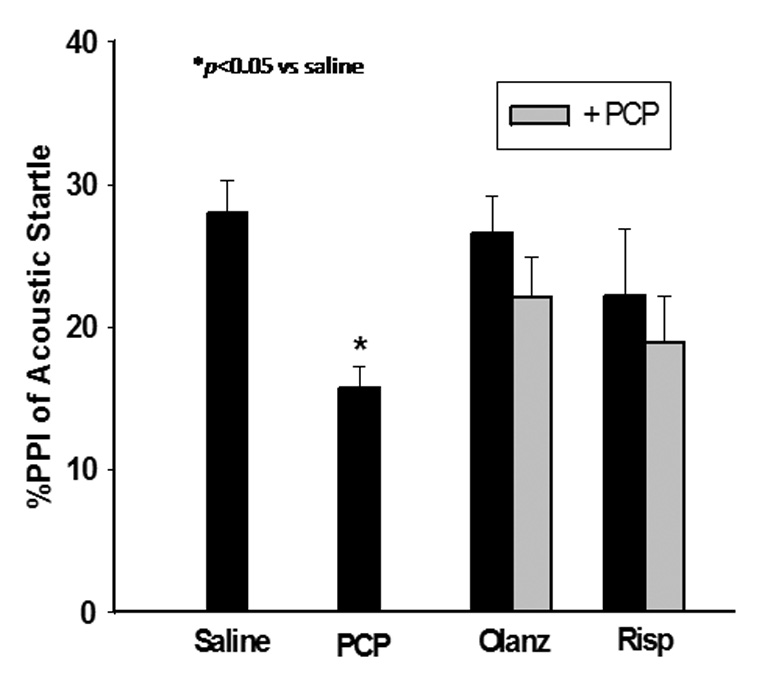
PCP treatment (10 mg/kg) treatment on PN7, 9, and 11 results in a deficit in PPI of acoustic startle as measured on PN24–26. Pretreatment on PN7, 9, and 11 with olanzapine (1 mg/kg) or risperidone (0.25 mg/kg) inhibits the PCP-induced deficit in PPI of acoustic startle. *p<0.05 vs. saline (one-way ANOVA with Bonferroni’s post hoc test). N=8 in both control and experimental groups.
Figure 4. Effects of subchronic PCP administration (PN7, 9 and 11) on the development of locomotor sensitization.
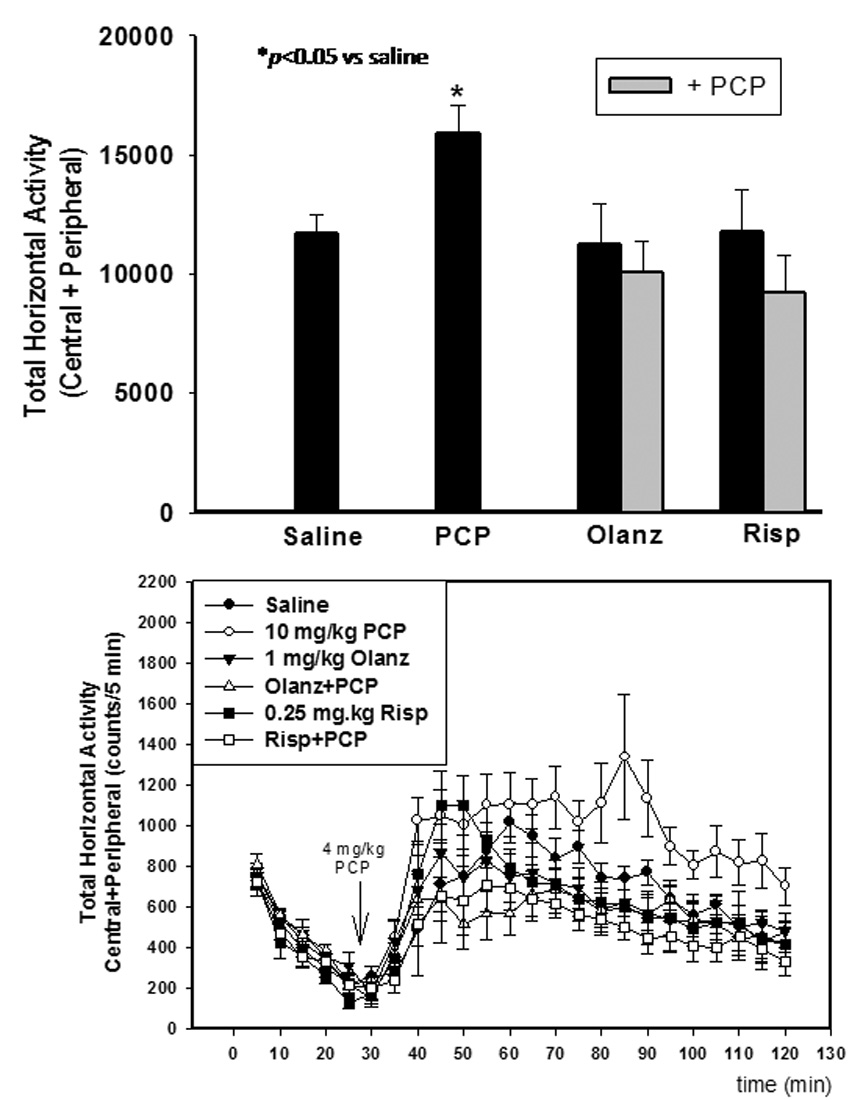
Locomotor sensitization induced by a 4 mg/kg challenge to PCP treatment on PN7, 9, and 11 treated animals as measured on PN28–35. Pretreatment on PN7, 9, and 11 with olanzapine (1 mg/kg) or risperidone (0.25 mg/kg) inhibits the development of locomotor sensitization. (top) Time course of locomotor activity (bottom) *p<0.05 vs. saline (one-way ANOVA with Bonferroni’s post hoc test). N=8 in both control and experimental groups. ●-saline; ○-10 mg/kg PCP; ▼-1 mg/kg olanzapine; Δ-olanzapine + PCP; ■-0.25 mg/kg risperidone; □-risperidone + PCP
3.2 Effects of olanzapine and risperidone on PCP-mediated regulation of NR1/2A/2B
In order to delineate one possible biochemical mechanism underlying the above mentioned behavioral changes, we determined the effects of PCP treatment on PN7 or on PN7, 9, and 11 on the density of NR1, NR2A, and NR2B in the synaptosomal membrane fraction of the frontal cortex and striatum.
Quantitative western analysis revealed that PCP treatment on PN7 produced a 3-fold increase in membrane bound NR1 and NR2B subunits in the frontal cortex 24 hours after PCP administration, while no effect on NR2A was observed (Anastasio and Johnson, 2008). In another experiment, perinatal animals were treated with either olanzapine (1 mg/kg) or risperidone (0.25 mg/kg) 30 minutes prior to PCP treatment on PN7. Twenty-four hours after the PCP injection, the rats were killed and the density of NMDA receptors was determined in the cortical membrane fraction. Pretreatment with either olanzapine or risperidone did not inhibit the up-regulation of membrane cortical NR1 or NR2B induced by PCP treatment on PN7 (Figure 5). Furthermore, treatment with olanzapine or risperidone alone had no effect on the concentration of membrane NR1 or NR2B in the frontal cortex compared to saline controls (data not shown).
Figure 5. Pharmacological antagonism of PCP treatment on PN7 effects on NR1 and NR2B in the frontal cortex.
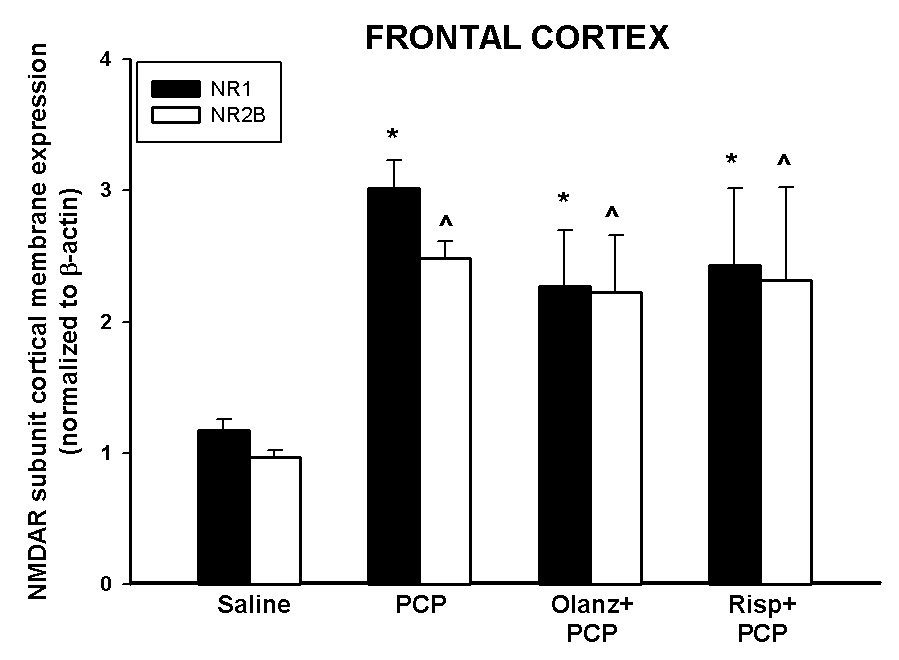
Quantitative analysis reveals that antipsychotics pretreatment has no effect on PCP treatment on PN7-induced up-regulation of NR1 or NR2B protein levels in the frontal cortex 24 hours following PCP (10 mg/kg, N=5–15/treatment). *p<0.05 vs. SAL NR1 (one-way ANOVA with Bonferroni's post hoc test) ^p<0.05 vs. SAL NR2B (one way ANOVA with Bonferroni’s post hoc test).
PCP treatment on PN7, 9, and 11 treatment caused a 3-fold increase in membrane NR1 and a 10-fold increase in NR2A protein levels in the frontal cortex with no effect on NR2B protein expression (Anastasio and Johnson, 2008). To analyze the actions of atypical antipsychotics on PCP-induced changes in the membrane protein levels of NMDAR subunits in the frontal cortex, animals were treated with olanzapine or risperidone 30 minutes prior to PCP on PN7, 9, and 11 and sacrificed 24 hours after the last of 3 injections. Each of the antipsychotics investigated inhibited PCP-induced up-regulation of the NR1 subunit in the membrane protein fraction of the frontal cortex (Figure 6), while having no effect on NR1 subunit expression on their own (data not shown). Pre-treatment with either olanzapine or risperidone also was able to completely block the increase in membrane cortical NR2A protein levels caused by PCP treatment on PN7, 9, and 11 treatment (Figure 6) with no effect on their own (data not shown).
Figure 6. Pharmacological antagonism of PCP treatment on PN7, 9, and 11-induced up-regulation on NR1 and NR2A in the frontal cortex.
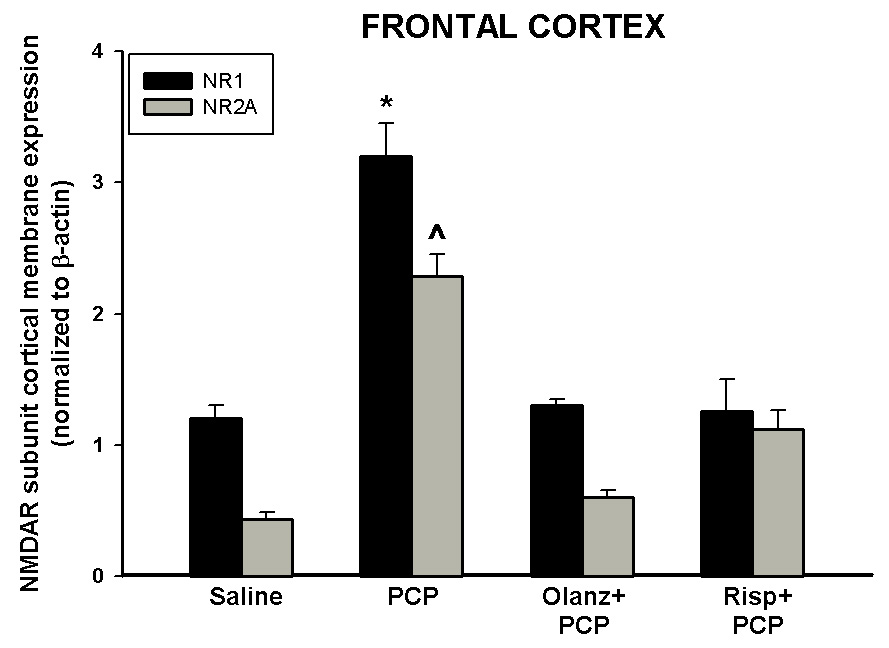
Quantitative analysis reveals that pretreatment with antipsychotics prevents PCP-induced (10 mg/kg, N=5–15/treatment) up-regulation of NR1 or NR2A protein in the frontal cortex. *p<0.05 vs. SAL NR1 (one-way ANOVA with Bonferroni's post hoc test) ^p<0.05 vs. SAL NR2A (one way ANOVA with Bonferroni’s post hoc test).
PCP treatment on PN7 had no significant effect on NR1 or NR2B protein expression levels in the striatum at 24 hours following treatment (Figure 7). The effects of pretreatment with the antipsychotics on NR1 and NR2B protein levels in the striatum were also investigated following a single injection of PCP. Treatment with olanzapine or risperidone did not significantly alter expression of NR1 or NR2B protein levels in the striatum prior to PCP on PN7 at 24 hours following treatment (Figure 7) or when administered alone (data not shown). Similarly, treatment with either PCP or atypical antipsychotics did not alter striatal NR2A protein expression levels (data not shown).
Figure 7. Effects of a single injection of PCP on PN7 on the membrane protein expression of NR1 and NR2B in the striatum.
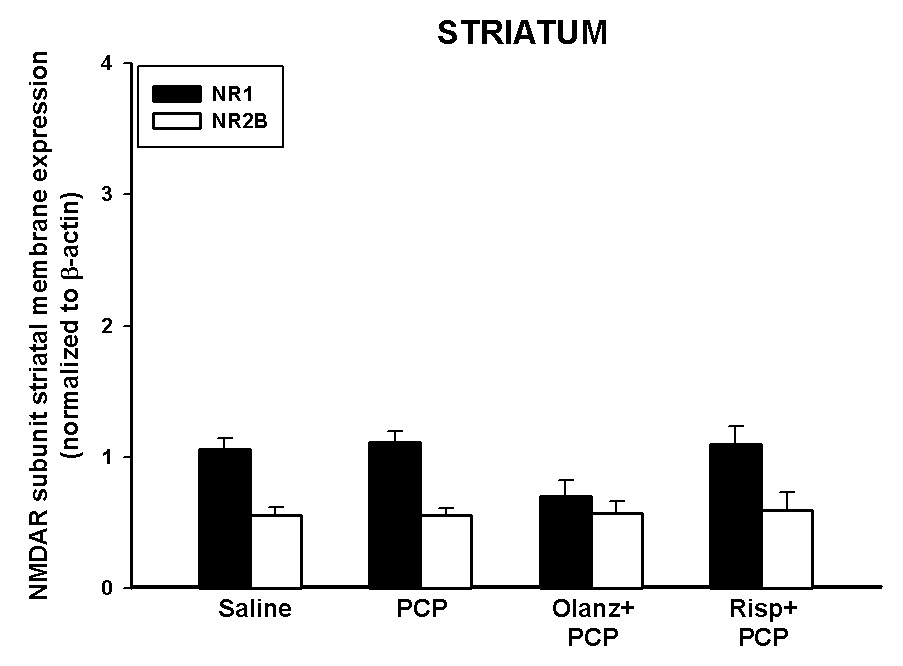
Quantitative analysis reveals that PCP treatment on PN7 has no effects on the expression of striatal NR1 or NR2B (10 mg/kg, N=5–15/treatment).
Unlike the up-regulation of NR1 observed in the frontal cortex, Western blot analysis of membrane bound protein showed that levels of striatal NR1 protein were significantly decreased following sub-chronic PCP treatment on PN7, 9, and 11 (Figure 8), with no effect on regulation of striatal NR2A (Figure 8) or NR2B (data not shown). Pretreatment with the anti-schizophrenic drug olanzapine, but not risperidone, prevented the down-regulation of NR1 protein expression in the striatum caused by sub-chronic PCP administration (Figure 8). Administration of these drugs alone had no effect on NR1 or NR2A protein levels in the striatum (data not shown).
Figure 8. Antagonism of PCP treatment on PN7, 9, and 11-induced down-regulation on NR1 in the striatum.
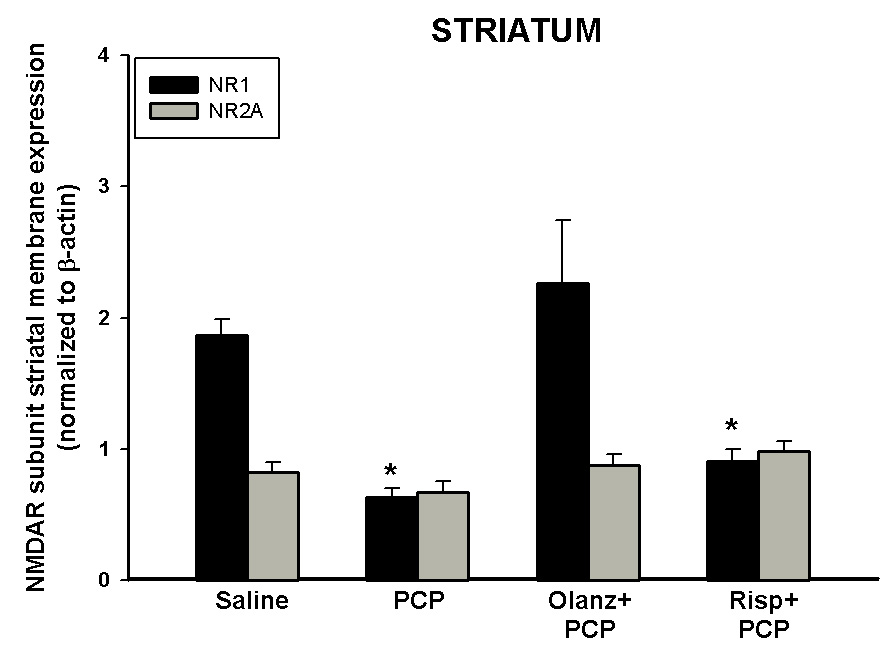
Quantitative analysis reveals that pretreatment with olanzapine prevents PCP-induced (10 mg/kg, N=5–15/treatment) down-regulation of NR1 in the striatum. PCP treatment had no effect on protein levels of NR2A *p <0.05 vs. SAL NR1 (one-way ANOVA with Bonferroni's post hoc test)
We also measured the levels of NR1, NR2A, and NR2B in the frontal cortex of animals following PCP challenge on PN28–35. No alterations in the protein levels of any of the subunits in the frontal cortex were evident in the animals treated sub-chronically with PCP (data not shown).
4. Discussion
It is well known that acute PCP treatment produces a disruption in PPI in adult rats, similar to that seen in schizophrenic patients (e.g. (Mansbach and Geyer, 1989; Martinez et al., 2000). Typical antipsychotics, such as haloperidol, are not able to reverse deficits in PPI caused by acute PCP treatment in adult rats (Geyer et al., 2001) or in pre-pubertal rats (Martinez et al., 2002), but in adult rats, they can reverse the effects of dopamine agonists (Geyer et al., 2001). Atypical antipsychotics, including clozapine, olanzapine, and quetiapine, are effective at alleviating acute PCP inhibition of PPI in adult rats (Ballmaier et al., 2001; Geyer et al., 2001; Johansson et al., 1994; Johansson et al., 1995; Martinez et al., 2002), but not in pups (PN16–19) or pre-pubertal (PN45) rats (Martinez et al., 2002). However, olanzapine is effective at preventing the deficits observed in PPI in PN24–28 pups following PCP treatment on PN7, 9 and 11 (Wang et al., 2001). In addition, both olanzapine and risperidone are able to increase PPI in NR1 −/− mice (Duncan et al., 2006). The current study shows that both olanzapine and risperidone pretreatment also blocks the PPI deficit observed in both male and female PN24–26 pups following PCP pretreatment on PN7, 9 and 11. In contrast, PCP treatment on PN7 administration did not produce a deficit in PPI in developing rat pups. It is then reasonable to postulate that PCP treatment on PN7, 9, and 11 produces a chronic deficit in NMDA receptor function compared to a single injection of PCP and that this more closely models the disease and the developmental NMDA hypofunction theory of schizophrenia (Duncan et al., 2006).
Like other psychomotor stimulants, repeated administration of PCP causes a progressive augmentation of locomotor activity (Xu and Domino, 1994), referred to as sensitization. The neuroadaptations associated with sensitization may be linked to the mechanisms underlying addiction (Robinson and Berridge, 1993). Sensitization is also thought to be an important index related to psychosis as well as movement and thought disorders in schizophrenia (Robbins, 1990). PCP-induced sensitization is blocked by haloperidol and risperidone (Kitaichi et al., 1995) and cross-sensitizes with MK-801 (Pechnick and Hiramatsu, 1994), but not with amphetamine (Balster, 1989, 1986). In the current study, PCP treatment on PN7 or on PN7, 9, and 11 produced locomotor sensitization in rats at PN28–35 that was blocked by pretreatment with either olanzapine or risperidone.
Our group and others have demonstrated that a single administration of PCP, MK-801 or ketamine to PN7 pups induces widespread neuronal apoptosis (Ikonomidou et al., 1999; Scallet et al., 2004; Wang and Johnson, 2005, 2007; Young et al., 2005). Other laboratories have also reported that transient NMDAR blockade by acute PCP, MK-801, ketamine, and ethanol to rodents during development causes behavioral, structural, and molecular abnormalities in adulthood (Fredriksson and Archer, 2003, 2004; Fredriksson et al., 2004; Harris et al., 2003; Wozniak et al., 2004). For example, Harris et al (2003) found that PN7 female rat pups administered MK-801 (0.5 mg/kg, twice, 8 hours apart, s.c.) showed PPI deficits and increased locomotor activity accompanied by a reduction of brain volume and neuronal number within the hippocampus and altered hippocampal NR1 subunit expression. Furthermore, the neurotoxicity evident in the developing frontal cortex following either a single injection or multiple injections suggests a role for altered cortical function in the development of locomotor sensitization (Wang et al., 2001; Wang and Johnson, 2005, 2007).
Several studies from this laboratory have investigated a possible mechanism by which PCP may elicit its neurotoxic effects and produce alterations in behavior in rats. In developing rats, PCP treatment on PN7, 9, and 11 of pups resulted in increased expression of NR1 mRNA in the frontal cortex, striatum, nucleus accumbens and olfactory cortex that was inhibited by pretreatment with the atypical antipsychotic olanzapine (Wang et al., 2001). These data are consistent with our previous report demonstrating that chronic PCP treatment resulted in increased NR1 immunoreactivity in the frontal cortex and striatum of adult rats treated chronically with PCP (Hanania et al., 1999). In addition, recently we reported that PCP treatment on PN7 produces an up-regulation of cortical NR1 and NR2B subunits via an increase in trafficking from intracellular compartments to the membrane, while subchronic PCP treatment results in an up-regulation of cortical NR1 and NR2A subunits by increasing new protein synthesis of these subunits (Anastasio and Johnson, 2008). We extended these experiments in this study and show that olanzapine and risperidone pre-treatment is unable to prevent the up-regulation of cortical NR1 and NR2B subunits following PCP treatment on PN7 administration. These data suggest that NMDAR up-regulation in the cortex and subsequent locomotor sensitization following a single injection of PCP are independent of each other. This suggests that expression of sensitized behavior is dependent on mechanisms requiring activation of dopamine (DA) D2 and/or serotonin (5-HT) 5-HT2A/2C receptors. Thus, even though a sensitized behavioral response to acute PCP challenge appears to involve activation of D2 and/or 5-HT2 receptors, the increase in NMDAR trafficking appears to be independent of these receptors. Furthermore, the DA and 5-HT mechanisms involved in locomotor sensitization may be downstream of receptor regulation. If so, antipsychotics would be ineffective at preventing PCP-induced changes in receptor trafficking, but still capable of preventing PCP-induced sensitization and other behaviors. In contrast, both olanzapine and risperidone are able to prevent up-regulation of NR1 and NR2A subunits in the frontal cortex following sub-chronic PCP treatment. These data suggest that up-regulation of NR1 and NR2A subunits (rather than NR1 and NR2B) may underlie the development of deficits in PPI following PCP treatment on PN7, 9, and 11, though the relative role of NMDAR subunits in locomotor behavior and PPI are not completely understood. Several studies have reported that blockade of NR2B subunits with selective antagonists (eliprodil and Ro63–1908) does not disrupt PPI (Depoortere et al., 1999; Higgins et al., 2003; Wiley, 1998) and that it is the combined blockade of NR2A and NR2B subunits which is necessary to induce deficits in PPI as well as hyperactivity in rodents (Spooren et al., 2004). Administration of the selective NR2A antagonist, NVP-AAM007, to adult rats did not disrupt PPI and resulted in hypolocomotor activity, while the NR2B antagonist, Ro 25–6981 disrupted PPI and produced a dose-dependent hyperlocomotion (Chaperon et al., 2003). Thus, the role of NR2A and NR2B subunits in the detrimental behavioral effects of PCP merits further investigation.
Neither the mechanisms underlying PCP-induced alterations in behavior nor NMDA receptor upregulation nor those responsible for the ability of olanzapine and risperidone to prevent these alterations is clear. Clozapine, olanzapine, and M100907, but not haloperidol, have been reported to prevent the blockade of NMDA responses in the medial prefrontal cortex caused by acute PCP administration (Arvanov and Wang, 1999; Wang and Liang, 1998). These data suggest a prominent role for 5-HT2A receptors in preventing the acute effect of PCP. Furthermore, clozapine has been shown to inhibit hypersensitive responses to NMDA following subchronic PCP (Arvanov and Wang, 1999), suggesting that the expected increase in NMDAR function following PCP up-regulation of NR1 and NR2A may also be prevented by olanzapine treatment. It is possible that the ability of olanzapine to inhibit PCP-induced neurotoxicity, NMDAR up-regulation and deficits in PPI following PCP treatment stems from its blockade of both DA and 5-HT2A receptors. Additionally, if PCP-induced neurotoxicity and the subsequent loss of these cortical neurons results in a hypo-glutamatergic state, then the ability of antipsychotics to prevent this loss of glutamatergic tone through blockade of DA and 5-HT receptors may be related to their effectiveness in alleviating both positive and negative symptoms of schizophrenia (Jardemark et al., 2000). Understanding the mechanism by which olanzapine and risperidone prevent PCP-induced neuronal apoptosis could provide insights into the molecular and cellular mechanisms involved in the behavioral effects of PCP in rats, which in turn could provide insight into the etiology and pharmacotherapy of schizophrenia.
PN7–11 is a critical stage in brain development, with ample evidence of neurite growth and synapse formation, and is referred to as the brain growth spurt (Olney et al., 2002). Thus, it is reasonable that even temporary changes during this critical period could result in behavioral changes as these animals mature. No variation in the expression of cortical NR1, NR2A, or NR2B was observed in either saline or sub-chronically PCP treated pups as measured immediately following PCP challenge and behavior measurement on PN28–35. Thus, it could be argued that the behavioral effects observed at this time are independent of the effects of PCP challenge on the expression of the NMDA receptor and that are more likely due to changes in NMDA receptor expression during an earlier, critical period of development and the subsequent maturation of the brain. Additionally, these results are consistent with the neurodevelopment theory of schizophrenia, which hypothesizes that damage during this early, critical stage of brain development may account for the later manifestation of mental disorders (du Bois and Huang, 2007).
PCP treatment on PN7 had no effect on the expression of either PSD-95 (data not shown) or any of the NMDA receptor subunits in the striatum. PSD-95 interacts with the intracellular tail of NR2 subunits and is thought to be responsible for anchoring the functional NR1/NR2 receptor complex in the membrane (Wenthold et al., 2003). Thus, it is possible that the lack of an observable effect on NMDA receptor expression is related to the lack of effect on PSD-95. This suggests that the striatal neurotoxicity previously reported (Wang and Johnson, 2005) is unrelated to alterations in the expression of NMDA receptor protein. Similarly, the locomotor sensitization measured following a single injection of PCP must also be due to mechanisms other than changes in striatal NMDAR expression levels. However, this does not rule the possible role that altered NMDAR expression in the frontal cortex may play in the sensitization response.
Pretreatment with olanzapine was able to prevent the down-regulation of striatal NR1 polypeptide that was caused by sub-chronic PCP administration; however, pretreatment with risperidone did not. We have recently observed that neither SCH23390 (selective D1 antagonist), sulpiride (selective D2 antagonist), nor M100907 (selective 5-HT2A antagonist) were able to prevent the down-regulation of NR1 in the striatum (Anastasio and Johnson, unpublished observations). Thus, this effect of olanzapine may require blockade of both DA and 5-HT receptors. In addition to affinity for DA and 5-HT receptors, olanzapine possesses high affinity for muscarinic ACh receptors (Ki = 1.89 nM), H1 histamine receptors (Ki=7.14 nM), as well as α1 noradrenergic receptors (Ki=19 nM) (Arnt and Skarsfeldt, 1998; Raggi et al., 2004); therefore, it is possible that in the striatum, an area rich in muscarinic ACh receptors, olanzapine’s effect of blocking down-regulation of NR1 caused by sub-chronic PCP administration to postnatal rats could also involve an action at these receptors.
Studies of postmortem schizophrenic brains have provided conflicting data on the expression of NMDA receptor subunit expression. The NR1 protein has been shown to be up-regulated in the prefrontal, parietal, and medial temporal cortices in schizophrenia (Chen et al., 1998), while radioligand binding studies showed no significant differences in NR1 in healthy versus schizophrenic brain tissue (Gao et al., 2000). Another study using in situ hybridization reported no difference between control and schizophrenic brains in either NR1 or NR2 mRNA levels (Akbarian et al., 1996), but NR2B mRNA was reportedly increased in the hippocampus while NR1 mRNA was diminished in the medial temporal cortex, superior temporal cortex, and frontal cortex (Akbarian et al., 1996; Meador-Woodruff and Healy, 2000). Although these are quite complex, this does not rule out a role for altered NMDAR function early in development in this disease. In fact, a number of genes related to glutamatergic function have been discovered that are significantly associated with schizophrenia, though in as yet undefined ways. Included in this list is NRG1 (neuregulin-1), which is known to regulate NMDAR expression (Chong et al., 2008; Craddock et al., 2005). DTNBP1 (dystrobrevin binding protein 1) has been shown to modulate glutamate function through up-regulation of presynaptic proteins and neurotrophic effects mediated by the Akt signaling pathway (Duan et al., 2007; Numakawa et al., 2004). DAOA (D-amino acid oxidase activator) activates NMDA receptors through a series of reactions involving the glycine-like molecule, D-serine (Korostishevsky et al., 2004; Ross et al., 2006a). RGS4 (regulator of G-protein signaling 4) is under-expressed in schizophrenic prefrontal cortex in postmortem microarray studies and has been reported to regulate G proteins in glutamate neurons, thereby dampening the effects of neurotransmitter interactions at G-protein coupled receptors (Chowdari et al., 2008; Chowdari et al., 2002). In addition to the glutamate related genes above found with linkage analysis, several other genes putatively associated with the NMDA synapse have been discovered to be associated with schizophrenia, including GRM3 (mGluR3 a member of the group II metabotropic receptor family, which also includes mGluR2 (Egan et al., 2004); mGluR3 is known to modulate glutamate release. Thus, genetic evidence supports a role for alterations in various glutamatergic genes including those specific to NMDA receptor function in schizophrenia. Importantly, these data are strongly supported by pharmacological data that also imply a role for altered NMDAR function. However, the complexities of these data do not allow a direct comparison between the data in this model to what may occur in the human brain in schizophrenia. Our view is that this model may reveal the gross structural features underlying behavioral alterations in rats that are similar to schizophrenia and possibly point to novel pharmacological approaches that may be helpful in treatment of the disease.
Acknowledgements
This work was supported by NIH grants F31 DA-022824 and RO1 DA-02073. The authors would like to thank Eli Lily and Co for its supply of olanzapine. We would also like to thank Michael Arriaga for his contribution to this study through his participation in Summer Undergraduate Research Program at UTMB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams B, Moghaddam B. Corticolimbic dopamine neurotransmission is temporally dissociated from the cognitive and locomotor effects of phencyclidine. J Neurosci. 1998;18:5545–5554. doi: 10.1523/JNEUROSCI.18-14-05545.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbarian S, Sucher NJ, Bradley D, Tafazzoli A, Trinh D, Hetrick WP, Potkin SG, Sandman CA, Bunney WE, Jr, Jones EG. Selective alterations in gene expression for NMDA receptor subunits in prefrontal cortex of schizophrenics. J Neurosci. 1996;16:19–30. doi: 10.1523/JNEUROSCI.16-01-00019.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasio NC, Johnson KM. Differential regulation of the NMDA receptor by acute and subchronic phencyclidine administration in the developing rat. J Neurochem. 2008;104:1210–1218. doi: 10.1111/j.1471-4159.2007.05047.x. [DOI] [PubMed] [Google Scholar]
- Arnt J, Skarsfeldt T. Do novel antipsychotics have similar pharmacological characteristics? A review of the evidence. Neuropsychopharmacology. 1998;18:63–101. doi: 10.1016/S0893-133X(97)00112-7. [DOI] [PubMed] [Google Scholar]
- Arvanov VL, Wang RY. Clozapine, but not haloperidol, prevents the functional hyperactivity of N-methyl-D-aspartate receptors in rat cortical neurons induced by subchronic administration of phencyclidine. J Pharmacol Exp Ther. 1999;289:1000–1006. [PubMed] [Google Scholar]
- Ballmaier M, Zoli M, Mazzoncini R, Gennarelli M, Spano F. Combined alpha 2-adrenergic/D2 dopamine receptor blockade fails to reproduce the ability of clozapine to reverse phencyclidine-induced deficits in prepulse inhibition of startle. Psychopharmacology (Berl) 2001;159:105–110. doi: 10.1007/s002130100905. [DOI] [PubMed] [Google Scholar]
- Balster RL. Behavioral pharmacology of PCP, NMDA and sigma receptors. NIDA Res Monogr. 1989;95:270–274. [PubMed] [Google Scholar]
- Balster RL. Clinical implications of behavioral pharmacology research on phencyclidine. NIDA Res Monogr. 1986;64:148–162. [PubMed] [Google Scholar]
- Beninger RJ, Jhamandas A, Aujla H, Xue L, Dagnone RV, Boegman RJ, Jhamandas K. Neonatal exposure to the glutamate receptor antagonist MK-801: effects on locomotor activity and pre-pulse inhibition before and after sexual maturity in rats. Neurotox Res. 2002;4:477–488. doi: 10.1080/10298420290031414. [DOI] [PubMed] [Google Scholar]
- Braff DL, Geyer MA. Sensorimotor gating and schizophrenia. Human and animal model studies. Arch Gen Psychiatry. 1990;47:181–188. doi: 10.1001/archpsyc.1990.01810140081011. [DOI] [PubMed] [Google Scholar]
- Bromet EJ, Fennig S. Epidemiology and natural history of schizophrenia. Biol Psychiatry. 1999;46:871–881. doi: 10.1016/s0006-3223(99)00153-5. [DOI] [PubMed] [Google Scholar]
- Bunney BG, BJW, Carlsson A. Schizophrenia and glutamate: an update. ACNP. 2000 [Google Scholar]
- Castellani S, Adams PM. Acute and chronic phencyclidine effects on locomotor activity, stereotypy and ataxia in rats. Eur J Pharmacol. 1981;73:143–154. doi: 10.1016/0014-2999(81)90086-8. [DOI] [PubMed] [Google Scholar]
- Chaperon F, Muller W, Auberson YP, Tricklebank MD, Neijt HC. Substitution for PCP, disruption of prepulse inhibition and hyperactivity induced by N-methyl-D-aspartate receptor antagonists: preferential involvement of the NR2B rather than NR2A subunit. Behav Pharmacol. 2003;14:477–487. doi: 10.1097/01.fbp.0000091471.79060.ed. [DOI] [PubMed] [Google Scholar]
- Chen AC, McDonald B, Moss SJ, Gurling HM. Gene expression studies of mRNAs encoding the NMDA receptor subunits NMDAR1, NMDAR2A, NMDAR2B, NMDAR2C, and NMDAR2D following long-term treatment with cis-and trans-flupenthixol as a model for understanding the mode of action of schizophrenia drug treatment. Brain Res Mol Brain Res. 1998;54:92–100. doi: 10.1016/s0169-328x(97)00326-4. [DOI] [PubMed] [Google Scholar]
- Chong VZ, Thompson M, Beltaifa S, Webster MJ, Law AJ, Weickert CS. Elevated neuregulin-1 and ErbB4 protein in the prefrontal cortex of schizophrenic patients. Schizophr Res. 2008 doi: 10.1016/j.schres.2007.12.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdari KV, Bamne M, Wood J, Talkowski ME, Mirnics K, Levitt P, Lewis DA, Nimgaonkar VL. Linkage disequilibrium patterns and functional analysis of RGS4 polymorphisms in relation to schizophrenia. Schizophr Bull. 2008;34:118–126. doi: 10.1093/schbul/sbm042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdari KV, Mirnics K, Semwal P, Wood J, Lawrence E, Bhatia T, Deshpande SN, B KT, Ferrell RE, Middleton FA, Devlin B, Levitt P, Lewis DA, Nimgaonkar VL. Association and linkage analyses of RGS4 polymorphisms in schizophrenia. Hum Mol Genet. 2002;11:1373–1380. doi: 10.1093/hmg/11.12.1373. [DOI] [PubMed] [Google Scholar]
- Craddock N, O'Donovan MC, Owen MJ. The genetics of schizophrenia and bipolar disorder: dissecting psychosis. J Med Genet. 2005;42:193–204. doi: 10.1136/jmg.2005.030718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depoortere R, Perrault G, Sanger DJ. Prepulse inhibition of the startle reflex in rats: effects of compounds acting at various sites on the NMDA receptor complex. Behav Pharmacol. 1999;10:51–62. doi: 10.1097/00008877-199902000-00005. [DOI] [PubMed] [Google Scholar]
- du Bois TM, Huang XF. Early brain development disruption from NMDA receptor hypofunction: relevance to schizophrenia. Brain Res Rev. 2007;53:260–270. doi: 10.1016/j.brainresrev.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Duan J, Martinez M, Sanders AR, Hou C, Burrell GJ, Krasner AJ, Schwartz DB, Gejman PV. DTNBP1 (Dystrobrevin binding protein 1) and schizophrenia: association evidence in the 3' end of the gene. Hum Hered. 2007;64:97–106. doi: 10.1159/000101961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan GE, Moy SS, Lieberman JA, Koller BH. Typical and atypical antipsychotic drug effects on locomotor hyperactivity and deficits in sensorimotor gating in a genetic model of NMDA receptor hypofunction. Pharmacol Biochem Behav. 2006;85:481–491. doi: 10.1016/j.pbb.2006.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan MF, Straub RE, Goldberg TE, Yakub I, Callicott JH, Hariri AR, Mattay VS, Bertolino A, Hyde TM, Shannon-Weickert C, Akil M, Crook J, Vakkalanka RK, Balkissoon R, Gibbs RA, Kleinman JE, Weinberger DR. Variation in GRM3 affects cognition, prefrontal glutamate, and risk for schizophrenia. Proc Natl Acad Sci U S A. 2004;101:12604–12609. doi: 10.1073/pnas.0405077101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellenbroek BA, Budde S, Cools AR. Prepulse inhibition and latent inhibition: the role of dopamine in the medial prefrontal cortex. Neuroscience. 1996;75:535–542. doi: 10.1016/0306-4522(96)00307-7. [DOI] [PubMed] [Google Scholar]
- Ellenbroek BA, Cools AR. Animal models for the negative symptoms of schizophrenia. Behav Pharmacol. 2000;11:223–233. doi: 10.1097/00008877-200006000-00006. [DOI] [PubMed] [Google Scholar]
- Fredriksson A, Archer T. Hyperactivity following postnatal NMDA antagonist treatment: reversal by D-amphetamine. Neurotox Res. 2003;5:549–564. doi: 10.1007/BF03033165. [DOI] [PubMed] [Google Scholar]
- Fredriksson A, Archer T. Neurobehavioural deficits associated with apoptotic neurodegeneration and vulnerability for ADHD. Neurotox Res. 2004;6:435–456. doi: 10.1007/BF03033280. [DOI] [PubMed] [Google Scholar]
- Fredriksson A, Archer T, Alm H, Gordh T, Eriksson P. Neurofunctional deficits and potentiated apoptosis by neonatal NMDA antagonist administration. Behav Brain Res. 2004;153:367–376. doi: 10.1016/j.bbr.2003.12.026. [DOI] [PubMed] [Google Scholar]
- Gao XM, Sakai K, Roberts RC, Conley RR, Dean B, Tamminga CA. Ionotropic glutamate receptors and expression of N-methyl-D-aspartate receptor subunits in subregions of human hippocampus: effects of schizophrenia. Am J Psychiatry. 2000;157:1141–1149. doi: 10.1176/appi.ajp.157.7.1141. [DOI] [PubMed] [Google Scholar]
- Geyer MA, Ellenbroek B. Animal behavior models of the mechanisms underlying antipsychotic atypicality. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:1071–1079. doi: 10.1016/j.pnpbp.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Geyer MA, Krebs-Thomson K, Braff DL, Swerdlow NR. Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology (Berl) 2001;156:117–154. doi: 10.1007/s002130100811. [DOI] [PubMed] [Google Scholar]
- Hanania T, Hillman GR, Johnson KM. Augmentation of locomotor activity by chronic phencyclidine is associated with an increase in striatal NMDA receptor function and an upregulation of the NR1 receptor subunit. Synapse. 1999;31:229–239. doi: 10.1002/(SICI)1098-2396(19990301)31:3<229::AID-SYN8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Harich S, Gross G, Bespalov A. Stimulation of the metabotropic glutamate 2/3 receptor attenuates social novelty discrimination deficits induced by neonatal phencyclidine treatment. Psychopharmacology (Berl) 2007;192:511–519. doi: 10.1007/s00213-007-0742-y. [DOI] [PubMed] [Google Scholar]
- Harris LW, Sharp T, Gartlon J, Jones DN, Harrison PJ. Long-term behavioural, molecular and morphological effects of neonatal NMDA receptor antagonism. Eur J Neurosci. 2003;18:1706–1710. doi: 10.1046/j.1460-9568.2003.02902.x. [DOI] [PubMed] [Google Scholar]
- Higgins GA, Ballard TM, Huwyler J, Kemp JA, Gill R. Evaluation of the NR2B-selective NMDA receptor antagonist Ro 63–1908 on rodent behaviour: evidence for an involvement of NR2B NMDA receptors in response inhibition. Neuropharmacology. 2003;44:324–341. doi: 10.1016/s0028-3908(02)00402-1. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vockler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–74. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- Jardemark KE, Liang X, Arvanov V, Wang RY. Subchronic treatment with either clozapine, olanzapine or haloperidol produces a hyposensitive response of the rat cortical cells to N-methyl-D-aspartate. Neuroscience. 2000;100:1–9. doi: 10.1016/s0306-4522(00)00253-0. [DOI] [PubMed] [Google Scholar]
- Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- Johansson C, Jackson DM, Svensson L. The atypical antipsychotic, remoxipride, blocks phencyclidine-induced disruption of prepulse inhibition in the rat. Psychopharmacology (Berl) 1994;116:437–442. doi: 10.1007/BF02247475. [DOI] [PubMed] [Google Scholar]
- Johansson C, Jackson DM, Zhang J, Svensson L. Prepulse inhibition of acoustic startle, a measure of sensorimotor gating: effects of antipsychotics and other agents in rats. Pharmacol Biochem Behav. 1995;52:649–654. doi: 10.1016/0091-3057(95)00160-x. [DOI] [PubMed] [Google Scholar]
- Keppel G. Design and analysis: a researcher's handbook. Englewood Cliffs, N.J.: Prentice-Hall; 1982. [Google Scholar]
- Kitaichi K, Yamada K, Yoneda Y, Ogita K, Hasegawa T, Furukawa H, Nabeshima T. Risperidone prevents the development of supersensitivity, but not tolerance, to phencyclidine in rats treated with subacute phencyclidine. Life Sci. 1995;56:531–543. doi: 10.1016/0024-3205(94)00482-8. [DOI] [PubMed] [Google Scholar]
- Koch M, Bubser M. Deficient sensorimotor gating after 6-hydroxydopamine lesion of the rat medial prefrontal cortex is reversed by haloperidol. Eur J Neurosci. 1994;6:1837–1845. doi: 10.1111/j.1460-9568.1994.tb00576.x. [DOI] [PubMed] [Google Scholar]
- Korostishevsky M, Kaganovich M, Cholostoy A, Ashkenazi M, Ratner Y, Dahary D, Bernstein J, Bening-Abu-Shach U, Ben-Asher E, Lancet D, Ritsner M, Navon R. Is the G72/G30 locus associated with schizophrenia? single nucleotide polymorphisms, haplotypes, and gene expression analysis. Biol Psychiatry. 2004;56:169–176. doi: 10.1016/j.biopsych.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Kutsuwada T, Kashiwabuchi N, Mori H, Sakimura K, Kushiya E, Araki K, Meguro H, Masaki H, Kumanishi T, Arakawa M, et al. Molecular diversity of the NMDA receptor channel. Nature. 1992;358:36–41. doi: 10.1038/358036a0. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Lieberman JA. Catching up on schizophrenia: natural history and neurobiology. Neuron. 2000;28:325–334. doi: 10.1016/s0896-6273(00)00111-2. [DOI] [PubMed] [Google Scholar]
- Luby ED, Gottlieb JS, Cohen BD, Rosenbaum G, Domino EF. Model psychoses and schizophrenia. Am J Psychiatry. 1962;119:61–67. doi: 10.1176/ajp.119.1.61. [DOI] [PubMed] [Google Scholar]
- Mansbach RS, Geyer MA. Effects of phencyclidine and phencyclidine biologs on sensorimotor gating in the rat. Neuropsychopharmacology. 1989;2:299–308. doi: 10.1016/0893-133x(89)90035-3. [DOI] [PubMed] [Google Scholar]
- Martinez ZA, Oostwegel J, Geyer MA, Ellison GD, Swerdlow NR. "Early" and "late" effects of sustained haloperidol on apomorphine- and phencyclidine-induced sensorimotor gating deficits. Neuropsychopharmacology. 2000;23:517–527. doi: 10.1016/S0893-133X(00)00147-0. [DOI] [PubMed] [Google Scholar]
- Martinez ZA, Platten A, Pollack E, Shoemaker J, Ro H, Pitcher L, Geyer MA, Swerdlow NR. "Typical" but not "atypical" antipsychotic effects on startle gating deficits in prepubertal rats. Psychopharmacology (Berl) 2002;161:38–46. doi: 10.1007/s00213-001-0977-y. [DOI] [PubMed] [Google Scholar]
- McGlashan TH. Early detection and intervention in schizophrenia: editor's introduction. Schizophr Bull. 1996;22:197–199. doi: 10.1093/schbul/22.2.197. [DOI] [PubMed] [Google Scholar]
- Meador-Woodruff JH, Healy DJ. Glutamate receptor expression in schizophrenic brain. Brain Res Brain Res Rev. 2000;31:288–294. doi: 10.1016/s0165-0173(99)00044-2. [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Nicolson R, Lenane M, Hamburger SD, Fernandez T, Bedwell J, Rapoport JL. Lessons from childhood-onset schizophrenia. Brain Res Brain Res Rev. 2000;31:147–156. doi: 10.1016/s0165-0173(99)00032-6. [DOI] [PubMed] [Google Scholar]
- Numakawa T, Yagasaki Y, Ishimoto T, Okada T, Suzuki T, Iwata N, Ozaki N, Taguchi T, Tatsumi M, Kamijima K, Straub RE, Weinberger DR, Kunugi H, Hashimoto R. Evidence of novel neuronal functions of dysbindin, a susceptibility gene for schizophrenia. Hum Mol Genet. 2004;13:2699–2708. doi: 10.1093/hmg/ddh280. [DOI] [PubMed] [Google Scholar]
- Olney JW, Wozniak DF, Jevtovic-Todorovic V, Farber NB, Bittigau P, Ikonomidou C. Drug-induced apoptotic neurodegeneration in the developing brain. Brain Pathol. 2002;12:488–498. doi: 10.1111/j.1750-3639.2002.tb00467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Curr Opin Pharmacol. 2007;7:39–47. doi: 10.1016/j.coph.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. Sydney. New York: Academic Press; 1986. The rat brain in stereotaxic coordinates. [Google Scholar]
- Pechnick RN, Hiramatsu M. The effects of MK-801 on body temperature and behavior in the rat: cross-sensitization and cross-tolerance with phencyclidine. Eur J Pharmacol. 1994;252:35–42. doi: 10.1016/0014-2999(94)90572-x. [DOI] [PubMed] [Google Scholar]
- Phillips M, Wang C, Johnson KM. Pharmacological characterization of locomotor sensitization induced by chronic phencyclidine administration. J Pharmacol Exp Ther. 2001;296:905–913. [PubMed] [Google Scholar]
- Pierce RC, Kalivas PW. A circuitry model of the expression of behavioral sensitization to amphetamine-like psychostimulants. Brain Res Brain Res Rev. 1997;25:192–216. doi: 10.1016/s0165-0173(97)00021-0. [DOI] [PubMed] [Google Scholar]
- Pilowsky LS, Kerwin RW, Murray RM. Schizophrenia: a neurodevelopmental perspective. Neuropsychopharmacology. 1993;9:83–91. doi: 10.1038/npp.1993.46. [DOI] [PubMed] [Google Scholar]
- Raggi MA, Mandrioli R, Sabbioni C, Pucci V. Atypical antipsychotics: pharmacokinetics, therapeutic drug monitoring and pharmacological interactions. Curr Med Chem. 2004;11:279–296. doi: 10.2174/0929867043456089. [DOI] [PubMed] [Google Scholar]
- Rasmussen BA, O'Neil J, Manaye KF, Perry DC, Tizabi Y. Long-term effects of developmental PCP administration on sensorimotor gating in male and female rats. Psychopharmacology (Berl) 2007;190:43–49. doi: 10.1007/s00213-006-0584-z. [DOI] [PubMed] [Google Scholar]
- Robbins TW. The case of frontostriatal dysfunction in schizophrenia. Schizophr Bull. 1990;16:391–402. doi: 10.1093/schbul/16.3.391. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev. 1993;18:247–291. doi: 10.1016/0165-0173(93)90013-p. [DOI] [PubMed] [Google Scholar]
- Ross CA, Margolis RL, Reading SA, Pletnikov M, Coyle JT. Neurobiology of schizophrenia. Neuron. 2006a;52:139–153. doi: 10.1016/j.neuron.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Ross RG, Heinlein S, Tregellas H. High rates of comorbidity are found in childhood-onset schizophrenia. Schizophr Res. 2006b;88:90–95. doi: 10.1016/j.schres.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Scallet AC, Schmued LC, Slikker W, Jr, Grunberg N, Faustino PJ, Davis H, Lester D, Pine PS, Sistare F, Hanig JP. Developmental neurotoxicity of ketamine: morphometric confirmation, exposure parameters, and multiple fluorescent labeling of apoptotic neurons. Toxicol Sci. 2004;81:364–370. doi: 10.1093/toxsci/kfh224. [DOI] [PubMed] [Google Scholar]
- Spooren W, Mombereau C, Maco M, Gill R, Kemp JA, Ozmen L, Nakanishi S, Higgins GA. Pharmacological and genetic evidence indicates that combined inhibition of NR2A and NR2B subunit containing NMDA receptors is required to disrupt prepulse inhibition. Psychopharmacology (Berl) 2004;175:99–105. doi: 10.1007/s00213-004-1785-y. [DOI] [PubMed] [Google Scholar]
- Steinpresis RE. The behavioral and neurochemical effects of phenyclidine in humans and animals: some implications for modeling psychosis. Behav Brain Res. 1996;74:45–55. doi: 10.1016/0166-4328(95)00162-x. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Braff DL, Taaid N, Geyer MA. Assessing the validity of an animal model of deficient sensorimotor gating in schizophrenic patients. Arch Gen Psychiatry. 1994;51:139–154. doi: 10.1001/archpsyc.1994.03950020063007. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Lipska BK, Weinberger DR, Braff DL, Jaskiw GE, Geyer MA. Increased sensitivity to the sensorimotor gating-disruptive effects of apomorphine after lesions of medial prefrontal cortex or ventral hippocampus in adult rats. Psychopharmacology (Berl) 1995;122:27–34. doi: 10.1007/BF02246438. [DOI] [PubMed] [Google Scholar]
- Wang C, McInnis J, Ross-Sanchez M, Shinnick-Gallagher P, Wiley JL, Johnson KM. Long-term behavioral and neurodegenerative effects of perinatal phencyclidine administration: implications for schizophrenia. Neuroscience. 2001;107:535–550. doi: 10.1016/s0306-4522(01)00384-0. [DOI] [PubMed] [Google Scholar]
- Wang C, McInnis J, West JB, Bao J, Anastasio N, Guidry JA, Ye Y, Salvemini D, Johnson KM. Blockade of phencyclidine-induced cortical apoptosis and deficits in prepulse inhibition by M40403, a superoxide dismutase mimetic. J Pharmacol Exp Ther. 2003;304:266–271. doi: 10.1124/jpet.102.041798. [DOI] [PubMed] [Google Scholar]
- Wang CZ, Johnson KM. Differential effects of acute and subchronic administration on phencyclidine-induced neurodegeneration in the perinatal rat. J Neurosci Res. 2005;81:284–292. doi: 10.1002/jnr.20559. [DOI] [PubMed] [Google Scholar]
- Wang CZ, Johnson KM. The role of caspase-3 activation in phencyclidine-induced neuronal death in postnatal rats. Neuropsychopharmacology. 2007;32:1178–1194. doi: 10.1038/sj.npp.1301202. [DOI] [PubMed] [Google Scholar]
- Wang RY, Liang X. M100907 and clozapine, but not haloperidol or raclopride, prevent phencyclidine-induced blockade of NMDA responses in pyramidal neurons of the rat medial prefrontal cortical slice. Neuropsychopharmacology. 1998;19:74–85. doi: 10.1016/S0893-133X(98)00003-7. [DOI] [PubMed] [Google Scholar]
- Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry. 1987;44:660–669. doi: 10.1001/archpsyc.1987.01800190080012. [DOI] [PubMed] [Google Scholar]
- Weinberger DR. On the plausibility of "the neurodevelopmental hypothesis" of schizophrenia. Neuropsychopharmacology. 1996;14:1S–11S. doi: 10.1016/0893-133X(95)00199-N. [DOI] [PubMed] [Google Scholar]
- Wenthold RJ, Prybylowski K, Standley S, Sans N, Petralia RS. Trafficking of NMDA receptors. Annu Rev Pharmacol Toxicol. 2003;43:335–358. doi: 10.1146/annurev.pharmtox.43.100901.135803. [DOI] [PubMed] [Google Scholar]
- Wiley JL. Nitric oxide synthase inhibitors attenuate phencyclidine-induced disruption of prepulse inhibition. Neuropsychopharmacology. 1998;19:86–94. doi: 10.1016/S0893-133X(98)00008-6. [DOI] [PubMed] [Google Scholar]
- Wiley JL, Buhler KG, Lavecchia KL, Johnson KM. Pharmacological challenge reveals long-term effects of perinatal phencyclidine on delayed spatial alternation in rats. Prog Neuropsychopharmacol Biol Psychiatry. 2003a;27:867–873. doi: 10.1016/S0278-5846(03)00146-5. [DOI] [PubMed] [Google Scholar]
- Wiley JL, Harvey SA, Balster RL, Nicholson KL. Affinity and specificity of N-methyl- D-aspartate channel blockers affect their ability to disrupt prepulse inhibition of acoustic startle in rats. Psychopharmacology (Berl) 2003b;165:378–385. doi: 10.1007/s00213-002-1297-6. [DOI] [PubMed] [Google Scholar]
- Wozniak DF, Hartman RE, Boyle MP, Vogt SK, Brooks AR, Tenkova T, Young C, Olney JW, Muglia LJ. Apoptotic neurodegeneration induced by ethanol in neonatal mice is associated with profound learning/memory deficits in juveniles followed by progressive functional recovery in adults. Neurobiol Dis. 2004;17:403–414. doi: 10.1016/j.nbd.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Xu X, Domino EF. Asymmetric cross-sensitization to the locomotor stimulant effects of phencyclidine and MK-801. Neurochem Int. 1994;25:155–159. doi: 10.1016/0197-0186(94)90034-5. [DOI] [PubMed] [Google Scholar]
- Young C, Jevtovic-Todorovic V, Qin YQ, Tenkova T, Wang H, Labruyere J, Olney JW. Potential of ketamine and midazolam, individually or in combination, to induce apoptotic neurodegeneration in the infant mouse brain. Br J Pharmacol. 2005;146:189–197. doi: 10.1038/sj.bjp.0706301. [DOI] [PMC free article] [PubMed] [Google Scholar]