

Abstract
A possible role of cellular uptake and re-secretion of apoA-I in the mechanism of cholesterol efflux induced by apoA-I was investigated using a novel experimental approach. Incubation of adipocytes with a recombinant human apoA-I containing a consensus PKA phosphorylation site, pka-ApoA-I, leads to the appearance of phosphorylated protein in the cell culture medium unambiguously proving cellular uptake and re-secretion of pka-ApoA-I. Phosphorylation of apoA-I is abolished by PKA inhibitors and enhanced by PKA activators demonstrating the specific involvement of PKA. Studies on the concentration dependence of pka-apoA-I phosphorylation and competition experiments with human apoA-I suggest that apolipoprotein uptake is a receptor mediated process. A possible role of apoA-I recycling in the mechanism of cholesterol efflux was investigated by determining the rates of apoA-I induced cholesterol efflux and apoA-I recycling in the presence and in the absence of Brefeldin A (BFA). The studies showed that BFA strongly inhibits cholesterol efflux without affecting the rate of apoA-I recycling. Since BFA affects vesicular trafficking of ABCA1, this study suggests that the interaction of apoA-I with ABCA1 does not mediate apolipoprotein uptake and re-secretion. This result suggests that lipidation of apoA-I and apolipoprotein uptake/re-secretion are independent processes.
Plasma apolipoprotein A-1 (ApoA-I) plays an important role in the removal of cholesterol from peripheral tissues and, consequently, in the prevention of atherosclerosis dependent cardiovascular disease [1]. The rate of cellular cholesterol removal by apoA-I depends on the plasma membrane levels of cholesterol [2,3] and ATP binding cassette transporter A1, ABCA1 [1,4,5]. The importance of ABCA1 in the process of apoA-I lipidation and the concurrent formation of nascent HDL particles has been demonstrated in numerous studies and is strongly supported by the fact that nonfunctional ABCA1 molecules lead to the development of Tangier’s disease [6]. A direct interaction of apoA-I with ABCA1 is in general accepted as a main step required in the transference of cellular lipids to apoA-I [7-10]. Some studies suggest that ABCA1 promotes the transference of phospholipids alone to apoA-I [7,10,11] whereas other studies suggest that phospholipids and cholesterol are loaded into apoA-I in a concurrent manner [9]. In addition to this uncertainty, it is not clear whether or not the lipidation process requires cellular uptake of apoA-I. Models of ABCA1 dependent lipid loading of apoA-I assuming that lipidation takes place on the surface of the plasma membrane have been recently proposed [12-14]. On the other hand, some studies support models proposing that lipidation of apoA-I is, at least in part, an intracellular process that therefore requires apolipoprotein uptake and re-secretion [15-19]. Apolipoprotein uptake has been monitored using approaches based on the use of fluorescently labeled or radiolabeled apoA-I. These approaches although highly valuable have some limitations due to the difficulty of clearly distinguishing cellular incorporation of the protein from protein adsorption to plasma membrane and also because partial degradation followed by release of the small labeling probe leads to the unspecific labeling of cellular compartments.
One of the goals of the current study was to develop a method to study cellular uptake and re-secretion of apoA-I that would remove the some of the uncertainties associated to traditional methods used with this purpose. Accordingly, we have developed a method that provides unambiguous proof of protein uptake and re-secretion. Using this method, we investigated the role of apolipoprotein uptake in the lipidation of apoA-I by adipocytes. We are interested in this cell type because adipose tissue constitutes one of the largest reservoirs of cholesterol in vertebrates [20] and as such it could represent a significant contributor to the formation of nascent HDL. Previous studies have shown that adipocytes release cellular cholesterol to apoA-I [21,22]. However, as is the case for most cells, the mechanisms involved and their relative contribution to the overall lipid efflux process have not been fully established.
EXPERIMENTAL PROCEDURES
Materials
3T3 L-1 cells were purchased from American Type Cell Culture (Manassas, VA). Brefeldin A, Isoproterenol, bovine PKA (catalytic subunit), fatty acid free bovine serum albumin (BSA), isobutyl methyl xanthine (IBMX), dexamethasone, trypsin, biotin, sodium pyruvate, insulin, streptomycin and penicillin were purchased from Sigma Chemicals Co. (St. Louis, MO). Human apoA-I was purchased from Meridian Life Science, Inc (Cincinnati, OH). Fetal bovine serum (FBS) was obtained from Hyclone (Logan, UT). Dulbecco’s modified Eagle’s medium (DMEM) was purchased from Cellgro Mediatech, Inc (Herndon, VA). [3H]-Cholesterol (60 Ci/mmoL) was from Perkin-Elmer.
Cell Culture
3T3 L-1 pre-adipocytes were cultured at 37°C in 8% CO2 atmosphere in high glucose DMEM supplemented with 10% FBS and 0.01% antibiotics. One day after confluence, the differentiation into adipocytes was induced by addition of IBMX (111μg/mL), dexamethasone (0.46 μg/ml), and insulin (1.5 μg/ml) in the medium that was also supplemented with biotin (4 μg/ml) and sodium pyruvate (100 μg/ml) [23]. After 48 h, the cells were incubated in DMEM/10 % FBS containing insulin, biotin, and sodium pyruvate for additional 48 h. Afterwards the cells were maintained in DMEM/10% FBS. All experiments were conducted 12 days after completion of the differentiation period.
Cloning and Purification of pka-apoA-I
Full-length mature human apoA-1 was cloned into a pET33b vector (Novagen, Inc), which incorporates an N-terminal tag encoding for a six-His tag and a five amino acid recognition sequence (RRASV) for the catalytic subunit of cAMP-dependent protein kinase (PKA). A DNA sequence including the complete coding sequence for mature human apoA-I (243 residues) was cloned into the pET33b vector using the BamH I and Sal I cloning sites. The final sequence encoded a protein of 288 residues with a mass of 32.8 kDa. The protein was expressed in E. coli and purified by Ni-affinity chromatography using standard procedures. The protein size was confirmed by SDS-PAGE. The identity of the protein was confirmed by Maldi-tof peptide mass fingerprint. For this purpose Coomassie blue stained protein bands were excised, reduced, alkylated, and digested with trypsin. The masses of the eluted peptides were determined in a Voyager DE-Pro Maldi-tof mass spectrometer.
The presence of an accessible PKA phosphorylation site was confirmed by in vitro phosphorylation of the pure recombinant apoA-I with the catalytic subunit of bovine PKA and γ-[32P]-ATP as previously described for other protein [24].
ApoA-1 Uptake and Re-secretion
3T3-L1 fibroblasts derived adipocytes cultured in six well dishes were radiolabeled by incubation for 5 h at 37°C with 40μCi/well of [32P] orthophosphate (carrier free) in phosphate-free DMEM containing 0.12% BSA. At the end of the labeling period, the cell medium was replaced with fresh medium containing isoproterenol (10ug/ml final concentration) or DMSO (Solvent used to dissolve isoproterenol) were added to the appropriate wells. Pka-ApoA-1 (100μg) was also added immediately after the radiolabeling period. Aliquots of the cell media were collected at the specified intervals of time and loaded into a Ni-affinity column. The column was washed first with 20mM Tris, 0.5M NaCl, pH 7.8, and then with the same buffer containing increasing concentrations of imidazole. ApoA-I eluted in the fractions containing between 100mM and 1M imidazole. The eluted proteins were separated by SDS-PAGE on 4-20% gels. The gels were dried, and phosphorylation was visualized by autoradiography.
Concentration dependence of the rate of pka-apoA-I phosphorylation
These studies were carried out in six-well dishes in a similar fashion to the studies described above but varying the concentration of pka-apoA-I in the cell culture medium (KRBH medium containing 0.12% BSA and 80μCi/well of [32P] orthophosphate). Samples from the cell medium were taken 30 min after addition of the apolipoprotein and directly used to separate pka-apoA-I by SDS-PAGE and analyze its radiolabeling intensity by autoradiography.
The effect of human apoA-I concentration on the rate of pka-apoA-I phosphorylation was monitored from the radioactivity associated to pka-apoA-I after incubation of 32P-prelabeled adipocytes for 30 min in a medium containing different ratios of human apoA-I to pka-apoA-I. To determine the intensity of pka-apoA-I phosphorylation samples of the medium were subjected to SDS-PAGE followed by autoradiography.
Cholesterol Efflux
Adipocytes cultured in six-wells dishes were radiolabeled by incubation for 24 h with [1,2-3H]-cholesterol (2 μCi/ml) in DMEM containing 2.5% FBS [25]. The media was removed and the cells were incubated for 3 hours in serum-free DMEM containing 0.2% BSA (DMEM/BSA) and, before the beginning of the experiment, rinsed twice with DMEM/BSA. The efflux experiment was carried out at 37 °C and started by the addition of fresh DMEM/BSA (For the assessment of background cholesterol release) or DMEM/BSA supplemented with 75μg/ml of apoA-I (To assess background plus apoA-I induced cholesterol efflux). The effect of PKA activity on cholesterol efflux was determined in DMEM/BSA supplemented with 10μg/ml of isoproterenol. At the end of the incubation, the cells were washed with phosphate buffered saline (PBS) twice and the lipids extracted with isopropanol [26]. The lipid extract was used to determine the fraction of radiolabeled cholesterol remaining in the cells. ApoA-I induced cholesterol efflux was expressed as percentage of total cellular cholesterol and calculated as the difference between the percentages of cholesterol released in the presence and in absence of apoA-I.
RESULTS
ApoA-I recycling by adipocytes
The main goal of this work was to investigate whether or not adipocytes were able to internalize and resecrete apoA-I. In order to assess apoA-1 recycling in adipocytes we attempted developing a method that could provide an unambiguous proof of apolipoprotein recycling. It was hypothesized that a functional recombinant apoA-I containing a phosphorylation site would constitute an ideal tool to monitor recycling. Cellular uptake of the protein would allow its phosphorylation, and if resecreted, the phosphorylated protein would be found in the cell culture medium. Because phosphorylation is an intracellular process, the appearance of phosphorylated protein in the cell culture medium would unequivocally indicate cellular uptake and re-secretion of the protein. Thus, a pka-apoA-I construct was prepared by subcloning the full-length sequence of mature human apoA-I into a commercial vector that incorporate an N-terminal tag encoding for a six-His tag and a five amino acid recognition sequence (RRASV) for the catalytic subunit of cAMP-dependent protein kinase (PKA). The pka-apoA-I construct was expressed in E. coli and purified by Ni-affinity chromatography using standard procedures. In close agreement with its theoretical mass of 32.8kDa, pka-apoA-I migrated in SDS-PAGE as a 31kDa marker. The identity of the protein was confirmed by peptide mass fingerprint. Maldi-tof of the peptides obtained by in gel digested pka-apoA-I covered over 50% of the apoA-I sequence (data not shown). The presence of the phosphorylation site in pka-apoA-I was determined by in vitro phosphorylation using the catalytic subunit of PKA and γ-[32P]-ATP. Using as control human apoA-I, this study showed that only the recombinant protein was phosphorylated.
The experiment to investigate apoA-I uptake and re-secretion consisted in adding pka-ApoA-I to adipocytes that had been pre-incubated with 32P-phosphate to radiolabel the cellular pool of ATP. The cell culture medium was removed after different times of incubation and used to isolate pka-apoA-I by Ni-affinity chromatography. PKA-apoA-I containing fractions were subjected to SDS-PAGE followed by Coomassie Blue staining and autoradiography. The Figure 1 shows the results obtained when pka-apoA-I was incubated with adipocytes in the absence or in the presence of the specific PKA inhibitor H89, or in the absence or presence of isoproterenol, a β-adrenergic agonist that raises cAMP and activates of PKA. Activation of PKA in isoproterenol treated cells was confirmed by determining the rate of triglyceride hydrolysis through the production and release of glycerol into the cell culture medium. Isoproterenol promotes a ∼7-fold increase in the rate of glycerol production.
Figure 1. ApoA-I recycling in adipocytes.

32P-prelabeled adipocytes were incubated for 60 or 120 min in phosphate-free DMEM/0.12% BSA medium containing 40 μCi of 32P-PO4H3 and 100 μg of pka-apoA-I, in the presence of in the absence of either or both isoproterenol (to activate PKA) or the PKA inhibitor H89 (40μM) as indicated in the figure. ApoA-I was purified from the medium and analyzed by SDS-PAGE and autoradiography. Panels A and B show the autoradiography of the samples (top picture) and their corresponding Coomassie Blue stained gels (bottom picture); Panels C and D show the changes in apoA-I phosphorylation (ratio of radiolabeling intensity to protein mass) induced by the PKA inhibitor H89 in control cells (basal PKA activity) and cells stimulated with isoproterenol (activated PKA), respectively. Radiolabeling intensities and masses of apoA-I bands were determined by densitometry of the scanned gels and autoradiographies. The data of panels C and D were obtained simultaneously and, therefore, are comparable. However, note the difference in scales of panels C and D.
The results (Fig. 1A) show that phosphorylated apoA-I is recovered from the cell medium and, therefore, demonstrate the ability of adipocytes to internalize and re-secrete apoA-I into the cell medium. Confirming the integrity of the cells and specificity of the phosphorylation, β-adrenergic stimulation of the adipocytes promoted a ∼six-fold increase in the intensity of phosphorylation of purified apoA-I (Figure 1 C and D). Furthermore, the specificity of the phosphorylation by PKA was demonstrated by the fact that the PKA inhibitor H89 practically abolished the recovery of phosphorylated apoA-I. The inhibition of the phosphorylation for the samples shown in the figure 1 ranged between 85% and 99%.
Overall, this study clearly demonstrates the ability of adipocytes to internalize and re-secrete apoA-I.
Effect of Brefeldin A on the rates of cholesterol efflux and apoA-I recycling
Brefeldin A is a well characterized inhibitor of cholesterol efflux that has been used in numerous studies and cell types [27,28]. The precise mechanism of function of BFA is not known. However, the fact that BFA disturbs intracellular vesicular trafficking [28,29] and the dynamics and intracellular distribution of ABCA 1 [30] suggest that it affects the rate of cholesterol efflux through its effect on vesicular transport. Based on these properties of BFA, we hypothesized that a decrease in the rate of apoA-I recycling associated to the inhibition of cholesterol efflux promoted by BFA would suggest the involvement of apoA-I uptake and re-secretion in the mechanism of cholesterol efflux. To investigate this hypothesis, first we studied the effect of BFA on the rate of apoA-I induced cholesterol efflux in [3H]-cholesterol loaded adipocytes. This study was carried out under basal conditions of cellular PKA activity and also in the presence of isoproterenol to activate PKA. As shown in the Figure 2, activation of PKA does not affect the rate of apoA-I induced cholesterol efflux. However, BFA has a major inhibitory effect on the rate of cholesterol efflux (over 80% inhibition, P<0.001) under both basal and PKA-activated conditions.
Figure 2. Effect of Brefeldin A on the rate of apoA-I induced cholesterol efflux.

[3H]-cholesterol prelabeled adipocytes were incubated for 2 hours in DMEM with, or without, Brefeldin A (10 μg/ml) and with or without pka-apoA-1 (75 μg of protein/ml). The bars show the mean values of apoA-I induced cholesterol efflux (expressed as % of cellular radiolabeled cholesterol) and their corresponding S.E (n=11, control-basal PKA; n=8, BFA basal PKA; n=7 Control-activated PKA; n=6 BFA-activated PKA, where n represents the number of wells used). Average values of apoA-I induced cholesterol efflux were calculated as the difference between the mean values of cholesterol found in wells containing apoA-I and the corresponding values for wells without apoA-I. The SD of the differences was calculated as: SD= [SD2(apoA1) + SD2(blank)]1/2 . The statistical significance of the differences between the mean values of apoA-I induced cholesterol efflux was determined by one-way ANOVA. Significant differences (P<0.001) were obtained between control and BFA samples under either basal or elevated (in the presence of isoproterenol) cellular PKA activities.
In spite of the large inhibitory effect of BFA on cholesterol efflux, the study of apoA-I recycling, carried out under identical conditions, but using 32P-phosphate prelabeled cells, showed no effect of BFA on the rate of apoA-I uptake and re-secretion. As shown in the Figure 3 for basal levels of PKA activity the intensities of apoA-I phosphorylation, normalized for protein recovery, are nearly identical with or without BFA. As expected the intensities of phosphorylation of apoA-I are greater when PKA is activated. However, as observed under basal PKA conditions, comparison of apoA-I phosphorylation data obtained from PKA-activated cells does not show differences between control and BFA treated cells. These results suggest that the inhibitory effect of BFA on the rate of cholesterol efflux is not due to the inhibition of the cellular uptake or re-secretion of apoA-I.
Figure 3. Effect of BFA on apolipoprotein A-I uptake and re-secretion.
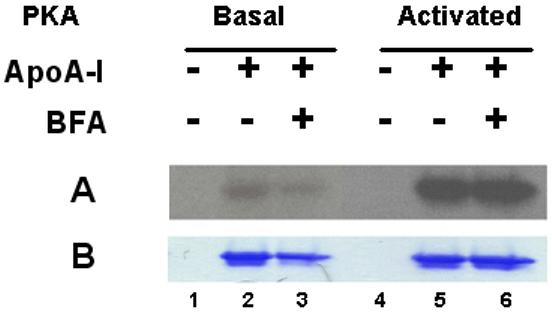
32P-prelabeled 3T3 L-1 adipocytes were incubated in phosphate-free DMEM medium containing 0.12% BSA, 80 μCi/ml of [32P]-phosphate and pka-apoA-I (75μg/ml). After incubation for 120 min, apoA-I was recovered from the cell culture media by Ni-affinity chromatography. Proteins were separated by SDS-PAGE on 4-20% gels. Panel A shows the autoradiogram and Panel B shows the corresponding apoA-I region of the Coomassie Blue-stained gel. Lane 1: Medium from adipocytes (Basal PKA) incubated without apoA-I; Lane 2: Medium from cells (Basal PKA) incubated with apoA-I; Lane 3: Medium from cells (Basal PKA activity) incubated with apoA-I and BFA (10μg/ml); Lanes 4, 5 and 6: Same as lanes 1, 2 and 3 but cellular PKA was activated with isoproterenol 10 μg/ml.
Concentration dependence of the rate of apoA-I recycling
To characterize further the process of apoA-I recycling we studied the effect of the concentration of recombinant apoA-I on the rate of appearance of phosphorylated apoA-I in the cell culture medium. As shown in the figure 4, the increase in recombinant apoA-I concentration in the cell culture medium leads to a progressive increase in the extent of apoA-I phosphorylation. At concentrations higher than 75μg/ml the rate of phosphorylation approaches a plateau suggesting that recycling of apoA-I is a receptor mediated process. The saturation observed is likely due to a rate limiting step involving binding of apoA-I to a receptor. Under this assumption the Km value inferred from the study, 1-2 μM, would be similar to the dissociation constant characterizing the apoAI/receptor interaction The figure 4 also shows that the concentration dependence of pka-apoA-I phosphorylation in the presence of BFA is nearly identical to that obtained in the absence of inhibitor. This similarity further confirms that BFA does not affect the rate limiting steps, most likely binding steps, of the process of apoA-I uptake and resecretion.
Figure 4. Concentration dependence of apoA-I recycling rate.

32P-prelabeled adipocytes were incubated for 30 min in 1ml of KRBH-BSA medium containing different amounts of recombinant pka-apoA-I. Samples of the medium were subjected to SDS-PAGE to determine the radiolabelling intensity of pka-apoA-I. Panels A and B show a representative autoradiography (A) and its corresponding Coomassie Blue stained gel (B) obtained with samples of varying pka-apoA-I concentrations. The arrows indicate the location of pka-apoA-I in the gel picture and autoradiography. C) Plots of the concentration dependence of the phosphorylation intensity of pka-apoA-I incubated with control adipocytes (mean value +/- range n=2) and BFA treated cells (n=1).
Effect of human plasma apoA-I on the rate of recycling of recombinant apoA-I
To test further the specificity of the recycling observed with pka-apoA-I, the effect of human apoA-I, purified from plasma, on the rate of phosphorylation of pka-ApoA-I was investigated. For this purpose, 32P-prelabeled adipocytes were incubated with a constant concentration of pka-ApoA-I in the presence of increasing concentrations of apoA-I isolated from human plasma. As shown in the Figure 5, human apoA-I reduces the appearance of phosphorylated pka-apoA-I in the cell culture medium in a dose dependent manner indicating a competition between human and recombinant apoA-I for binding to a common receptor. The concentration dependence of the inhibition of phosphorylation suggests that pka-apoA-I and human apoA-I have a similar affinity for the receptor (∼1-2 μM).
Figure 5. Competition between pka-apoA-I and human apoA-I.

32P-prelabeled adipocytes were incubated for 30 min in KRBH-BSA medium (1ml) containing 40 μg of pka-apoA-I and different amounts of apoA-I from human plasma, as indicated in the figure. The proteins in the medium were separated by SDS-PAGE and subjected to autoradiography. Panel A) Autoradiography; B) Coomassie Blue stained gel displaying the region containing both pka-apoA-I and human apoA-I. C) Plot of the effect of human apoA-I concentration on the phosphorylation of recombinant apoA-I.
DISCUSSION
The present study describes a novel approach to investigate cellular uptake and re-secretion of proteins that removes some of the uncertainties inherent to pulse-chase methods based on the use of radiolabeled or fluorescently-labeled proteins. Since protein phosphorylation can only occur inside the cell, the appearance of phosphorylated protein in the cell culture medium constitutes unambiguous evidence of protein uptake and subsequent exocytosis. Using this approach our study presents strong evidence supporting the ability of adipocytes to internalize and re-secrete apoA-I. No other studies have previously shown apoA-I recycling in adipocytes.
The precise mechanism/s involved in apolipoprotein mediated cholesterol removal from cells is not known. One of the models under consideration assumes that ABCA1 would facilitate the removal of cellular cholesterol by a mechanism that involves binding of apoA-I to ABCA1 [7], followed by translocation of the receptor-apoA-I complex to intracellular vesicles and subsequent secretion of lipid loaded apoA-I. This mechanism is supported by an early study that showed a correlation between apoA-I retroendocytosis and cholesterol efflux in wild type and Tangier’s cells [15]. Further support comes from more recent studies showing co-localization of apoA-I and ABCA1 in endosomal compartments [17-19,31].
To explore a possible role of apoA-I recycling in cholesterol efflux in adipocytes we investigated the effect of BFA on the rate of apoA-I recycling. The selection of BFA as inhibitor was based on the facts that it is a potent inhibitor of cholesterol efflux [27] and it has been shown to affect intracellular trafficking of ABCA1 [28,30]. As in other cells and previous studies, our results also demonstrated that BFA nearly abolishes the ability of adipocytes to promote loading of apoA-I with cholesterol. Given the high level of inhibition of apoA-I induced cholesterol efflux achieved with BFA we expected that it would also inhibit apoA-I recycling. However, as shown at different times of incubation (Fig 3) and apolipoprotein concentrations (Fig 4), BFA did not affect the rate of cellular uptake and/or re-secretion of apoA-I. These results are somehow surprising. However, the lack of effect of BFA on apoA-I recycling could be due to the fact that apoA-I uptake and re-secretion follows a BFA-insensitive path. In this regard it has been shown that in spite of its dramatic effects on the morphology of the endocytic compartments BFA does not prevent the normal recycling of several receptors, including the transferring receptor, the IgA receptor and the mannose-6-phosphate receptor [29,32,33]. It is then likely that apoA-I recycling takes place via BFA-insensitive vesicles.
Although BFA does not affect the recycling of all receptors, it has been shown to affect intracellular trafficking of ABCA1 [28,30]. Therefore, the fact that BFA did not affect the rate of apoA-I recycling suggests that ABCA1 does not mediate apoA-I uptake. Alternatively, one would conclude that the interaction between ABCA1 and apoA-I does not constitute a rate limiting factor for apolipoprotein recycling even in the presence of BFA. Further support to this suggestion arises from the fact that the estimated affinity of apoA-I for ABCA1 is nearly two orders of magnitude greater than the apparent affinity (Km ∼2000 nM) that characterizes the process of apoA-I recycling observed in our studies. The reported dissociation constant, Kd, values for apoA-I/ ABCA1 range between 23nM [34] and 74nM [35]. This analysis suggests the presence of an ABCA1 independent mechanism of apoA-I uptake and re-secretion in adipocytes.
The fact that ABCA1 plays a major role in apoA-I induced cholesterol efflux and the results of our study suggesting that ABCA1 is not involved in apoA-I recycling lead to two possible interpretations. A possible explanation of is that apoA-I lipidation is indeed confined to the plasma membrane. In this case apoA-I recycling would be a phenomenon completely unrelated to the process of cholesterol efflux and BFA would prevent cholesterol efflux by affecting the function of ABCA1 at the membrane surface. However, if lipidation of apoA-I were an intracellular process our study would indicate that BFA effectively blocks intracellular lipidation of apoA-I without affecting apolipoprotein uptake and/or secretion.
Clearly, additional studies on the relationship between apoA-I recycling and cholesterol efflux are necessary to advance on the construction of a solid mechanism of apoA-I induced cholesterol efflux. The method described to study apoA-I recycling and the results showing the possibility to decouple lipid loading from apoA-I recycling using BFA may result useful for further studies on this area.
Acknowledgments
This study was funded by Oklahoma State Experiment Station and NIH grant GM 55622.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Yokoyama S. Arterioscler Thromb Vasc Biol. 2006;26:20–7. doi: 10.1161/01.ATV.0000195789.39418.e8. [DOI] [PubMed] [Google Scholar]
- [2].Rothblat GH, de la Llera-Moya M, Atger V, Kellner-Weibel G, Williams DL, Phillips MC. J Lipid Res. 1999;40:781–96. [PubMed] [Google Scholar]
- [3].Haynes MP, Phillips MC, Rothblat GH. Biochemistry. 2000;39:4508–17. doi: 10.1021/bi992125q. [DOI] [PubMed] [Google Scholar]
- [4].Oram JF. Arterioscler Thromb Vasc Biol. 2003;23:720–7. doi: 10.1161/01.ATV.0000054662.44688.9A. [DOI] [PubMed] [Google Scholar]
- [5].Wang N, Tall AR. Arterioscler Thromb Vasc Biol. 2003;23:1178–84. doi: 10.1161/01.ATV.0000075912.83860.26. [DOI] [PubMed] [Google Scholar]
- [6].Rust S, et al. Nat Genet. 1999;22:352–5. doi: 10.1038/11921. [DOI] [PubMed] [Google Scholar]
- [7].Wang N, Silver DL, Thiele C, Tall AR. J Biol Chem. 2001;276:23742–7. doi: 10.1074/jbc.M102348200. [DOI] [PubMed] [Google Scholar]
- [8].Wang N, Silver DL, Costet P, Tall AR. J Biol Chem. 2000;275:33053–8. doi: 10.1074/jbc.M005438200. [DOI] [PubMed] [Google Scholar]
- [9].Smith JD, Le Goff W, Settle M, Brubaker G, Waelde C, Horwitz A, Oda MN. J Lipid Res. 2004;45:635–44. doi: 10.1194/jlr.M300336-JLR200. [DOI] [PubMed] [Google Scholar]
- [10].Chambenoit O, Hamon Y, Marguet D, Rigneault H, Rosseneu M, Chimini G. J Biol Chem. 2001;276:9955–60. doi: 10.1074/jbc.M010265200. [DOI] [PubMed] [Google Scholar]
- [11].Fielding PE, Nagao K, Hakamata H, Chimini G, Fielding CJ. Biochemistry. 2000;39:14113–20. doi: 10.1021/bi0004192. [DOI] [PubMed] [Google Scholar]
- [12].Vedhachalam C, Ghering AB, Davidson WS, Lund-Katz S, Rothblat GH, Phillips MC. Arterioscler Thromb Vasc Biol. 2007;27:1603–9. doi: 10.1161/ATVBAHA.107.145789. [DOI] [PubMed] [Google Scholar]
- [13].Vedhachalam C, et al. J Biol Chem. 2007;282:25123–30. doi: 10.1074/jbc.M704590200. [DOI] [PubMed] [Google Scholar]
- [14].Faulkner LE, Panagotopulos SE, Johnson JD, Woollett LA, Hui DY, Witting SR, Maiorano JN, Davidson WS. J. Lipid Res. 2008:M800048–JLR200. doi: 10.1194/jlr.M800048-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Takahashi Y, Smith JD. Proc Natl Acad Sci U S A. 1999;96:11358–63. doi: 10.1073/pnas.96.20.11358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Santamarina-Fojo S, Remaley AT, Neufeld EB, Brewer HB., Jr. J Lipid Res. 2001;42:1339–45. [PubMed] [Google Scholar]
- [17].Neufeld EB, et al. J Biol Chem. 2004;279:15571–8. doi: 10.1074/jbc.M314160200. [DOI] [PubMed] [Google Scholar]
- [18].Hassan HH, Bailey D, Lee D-YD, Iatan I, Hafiane A, Ruel I, Krimbou L, Genest J. J. Biol. Chem. 2008 doi: 10.1074/jbc.M707720200. M707720200. [DOI] [PubMed] [Google Scholar]
- [19].Smith JD, Waelde C, Horwitz A, Zheng P. J Biol Chem. 2002;277:17797–803. doi: 10.1074/jbc.M201594200. [DOI] [PubMed] [Google Scholar]
- [20].Ailhaud G, Grimaldi P, Negrel R. Annu Rev Nutr. 1992;12:207–33. doi: 10.1146/annurev.nu.12.070192.001231. [DOI] [PubMed] [Google Scholar]
- [21].Le Lay S, Robichon C, Le Liepvre X, Dagher G, Ferre P, Dugail I. J Lipid Res. 2003;44:1499–507. doi: 10.1194/jlr.M200466-JLR200. [DOI] [PubMed] [Google Scholar]
- [22].Prattes S, Horl G, Hammer A, Blaschitz A, Graier WF, Sattler W, Zechner R, Steyrer E. J Cell Sci. 2000;113(Pt 17):2977–89. doi: 10.1242/jcs.113.17.2977. [DOI] [PubMed] [Google Scholar]
- [23].Rubin CS, Hirsch A, Fung C, Rosen OM. J Biol Chem. 1978;253:7570–8. [PubMed] [Google Scholar]
- [24].Patel RT, Soulages JL, Hariharasundaram B, Arrese EL. J Biol Chem. 2005;280:22624–31. doi: 10.1074/jbc.M413128200. [DOI] [PubMed] [Google Scholar]
- [25].Verghese PB, Arrese EL, Soulages JL. Mol Cell Biochem. 2007;302:241–8. doi: 10.1007/s11010-007-9447-0. [DOI] [PubMed] [Google Scholar]
- [26].Johnson WJ, Bamberger MJ, Latta RA, Rapp PE, Phillips MC, Rothblat GH. J Biol Chem. 1986;261:5766–76. [PubMed] [Google Scholar]
- [27].Mendez AJ, Uint L. J Lipid Res. 1996;37:2510–24. [PubMed] [Google Scholar]
- [28].Zha X, Gauthier A, Genest J, McPherson R. J Biol Chem. 2003;278:10002–5. doi: 10.1074/jbc.C300024200. [DOI] [PubMed] [Google Scholar]
- [29].Lippincott-Schwartz J, Donaldson JG, Schweizer A, Berger EG, Hauri HP, Yuan LC, Klausner RD. Cell. 1990;60:821–36. doi: 10.1016/0092-8674(90)90096-w. [DOI] [PubMed] [Google Scholar]
- [30].Neufeld EB, et al. J Biol Chem. 2001;276:27584–90. doi: 10.1074/jbc.M103264200. [DOI] [PubMed] [Google Scholar]
- [31].Chen W, Wang N, Tall AR. J Biol Chem. 2005;280:29277–81. doi: 10.1074/jbc.M505566200. [DOI] [PubMed] [Google Scholar]
- [32].Wood SA, Brown WJ. J Cell Biol. 1992;119:273–85. doi: 10.1083/jcb.119.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lippincott-Schwartz J, Yuan L, Tipper C, Amherdt M, Orci L, Klausner RD. Cell. 1991;67:601–16. doi: 10.1016/0092-8674(91)90534-6. [DOI] [PubMed] [Google Scholar]
- [34].Denis M, Haidar B, Marcil M, Bouvier M, Krimbou L, Genest J., Jr. J. Biol. Chem. 2004;279:7384–7394. doi: 10.1074/jbc.M306963200. [DOI] [PubMed] [Google Scholar]
- [35].Chroni A, Liu T, Fitzgerald ML, Freeman MW, Zannis VI. Biochemistry. 2004;43:2126–39. doi: 10.1021/bi035813p. [DOI] [PubMed] [Google Scholar]