

Abstract
Claudin-16 is defective in familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC). Claudin-16 knockdown (CLDN16 KD) mice show reduced cation selectivity in the thick ascending limb. The defect leads to a collapse of the lumen-positive diffusion voltage, which drives Ca2+ and Mg2+ absorption. Because of the reduced tight junction permeability ratio for Na+ over Cl−, we proposed a backleak of NaCl into the lumen. Systemic analysis had revealed lower blood pressure and a moderately increased plasma aldosterone concentration. In this study, we measured the amiloride-sensitive equivalent short-circuit current in isolated, perfused collecting ducts and found it increased by fivefold in CLDN16 KD mice compared with wild-type (WT) mice. Amiloride treatment unmasked renal Na+ loss in the thick ascending limb of the nephron. Under amiloride treatment, CLDN16 KD mice developed hyponatremia and the renal fractional excretion of Na+ was twofold higher in CLDN16 KD compared with WT mice. The loss of claudin-16 also resulted in increased urinary flow, reduced HCO3− excretion, and lower urine pH. We conclude that perturbation in salt and acid-base metabolism in CLDN16 KD mice has its origin in the defective cation permselectivity of the thick ascending limb of the nephron. This study has contributed to the still incomplete understanding of the symptoms of FHHNC patients.
Keywords: paracellular ion transport, thick ascending limb, magnesium, calcium, amiloride
tight junction properties in epithelia are key determinants of transepithelial salt and water transport. They are formed from heterogeneous protein complexes. Claudin proteins from each neighboring cell are major constituents of paracellular ion pathways and selectively control the paracellular flux of specific ions. In the thick ascending limb of Henle's loop (TAL), the epithelial cells form a water-impermeable barrier, actively transport Na+ and Cl− via the transcellular route, and provide a paracellular pathway for the selective absorption of cations. This pathway is also a second route of Na+ reabsorption (8, 9) and completely depends on a lumen-positive transepithelial voltage (Vte).
The generation of the lumen-positive Vte in TAL can be attributed to two overlapping mechanisms: 1) the polarized distribution of luminal K+ versus basolateral Cl− conductances and 2) the diffusion voltage generated by a transcellular NaCl concentration gradient (from peritubular space to lumen) through a cation-selective tight junction. The first voltage source provides around +8 mV and depends on the luminal delivery of K+; the second voltage source provides some +30 mV (10) and is higher toward the end of the TAL, where the tubular fluid is more dilute. Both mechanisms are present in parallel. At the beginning of the TAL there is no chemical gradient and therefore no diffusion voltage. At the end of the TAL, transcellular NaCl transport, with no accompanying water transport, has generated a substantial NaCl gradient across the epithelium. Owing to the ion selectivity of the tight junctions, cations can leak back into the lumen, creating +30 mV lumen-positive diffusion voltage.
Transepithelial transport in the TAL can be affected by interfering with either the transcellular or paracellular routes. The loop diuretic furosemide blocks the Na+-2Cl−-K+ cotransporter, resulting in the loss of the NaCl chemical potential across the epithelium. As a result, no diffusion potential can develop, removing the driving force for paracellular transport, and diuresis occurs, with a large increase in the excretion of Na+, Ca2+, Mg2+, and secondarily of K+ and H+ (5, 7). The diffusion voltage can also be lost when the paracellular pathway becomes leaky or loses selectivity.
A distinct defect of the paracellular pathway is caused by mutations of claudins, the major determinants of paracellular ion selectivity. One example of claudin deficiency is the hereditary disease familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC; OMIM 248250), a rare and complex renal tubular disorder with a severe defect in the ability to reabsorb divalent cations. Several mutations in the genes for the tight junction proteins claudin-16 (CLDN16) (25) and claudin-19 (CLDN19) (15, 20) are responsible for this renal transport defect. The human disease is characterized by low plasma magnesium concentrations and an intensely increased renal calcium excretion leading finally to peritubular calcification. We previously described a CLDN16 knockdown (KD) animal model for this hereditary disease (16). CLDN16 is almost exclusively expressed in the TAL (19), and it colocalizes with CLDN19 (1). CLDN16 expression has been suppressed in KD mice by transgenic small interference RNA. The phenotype mimics the symptoms of FHHNC patients. KD mice display hypomagnesemia, renal calcium loss, and nephrocalcinosis (16). In isolated, perfused TAL segments, we have shown that loss of CLDN16 did not lead to a specific loss of Mg2+ and Ca2+ permeability, but to a decrease in permeability ratio for Na+ over Cl−. Therefore we proposed that the loss of CLDN16 results in a loss of the diffusion voltage and the driving force for paracellular ion reabsorption. In addition to the symptoms of FHHNC that concern the reabsorption of the divalent cations, the CLDN16 KD mice also showed low blood pressure, 1.5-fold elevated plasma aldosterone concentration [KD 662 ± 50 pg/ml vs. wild type (WT) 402 ± 94 pg/ml; n = 5], and decreased plasma potassium. These findings would be compatible with a compensated disturbance of sodium handling.
We show here that CLDN16 KD mice have an increased Na+ absorption in the collecting duct, a partially compensated loss of acid, and an increased vulnerability toward Na+ loss. These findings could support further clinical investigations in FHHNC patients and improve the understanding of their pathophysiology.
MATERIALS AND METHODS
Animals.
CLDN16 KD mice were originally generated as described recently (16) by transgenic RNA interference. All mice were bred and maintained in the Christian-Albrechts-Universität animal research facilities. All procedures were approved by the institutional animal research and care committee. Interventions were performed under inhalative anesthesia (2.2–2.5% isoflurane). Transgenic males were crossed to B6D2F1 females, and F1 animals were analyzed as littermates. All mice were fed ad libitum and housed under a 12-h light cycle. Transgenic mice were identified by detection of green skin fluorescence with filter glasses (520-nm band pass) and illumination by a pocket torch at 480 nm (Nightsea).
Animal protocols and experimental groups.
The present study includes two experimental groups each of WT and KD mice. In the first series we investigated 5- to 9-wk-old female mice under control conditions, under water load, and under water load in the presence of amiloride. In the second series we investigated acid-base metabolism in 27-wk-old female mice.
In addition, body weight was monitored in all littermates throughout 7 wk. Five- to nine-week-old mice of either sex were used for renal tubule perfusion. These animals were killed, and the respective organs were processed at 4°C and investigated immediately within <2 h.
In the control group mice were killed, final blood samples were taken from the abdominal vena cava, and spot urine was taken directly from the urinary bladder. Water load and amiloride treatment were carried out in four groups. We designed an experimental protocol that took into account the need for a reasonable urinary flow rate within a short-term observation period, and with a minimal influence on TAL Na+ transport at the same time. The urinary bladder was voided by manual stimulation. Mice were injected with either 10 ml/kg body wt of 5% glucose or with 10 ml/kg body wt of 5% glucose plus 5 mg/kg body wt of amiloride subcutaneously (24). For urine sampling, animals were kept in metabolic cages for 2 h after injection. Animals had free access to water throughout the whole experimental period. Final blood samples were taken from the abdominal vena cava.
For investigation of acid-base metabolism, mice were kept in metabolic cages for 24 h. After 24 h, mice were killed and final blood samples were taken.
Urine and plasma analysis.
Plasma and urinary concentrations of creatinine and electrolytes were measured on a Roche Modular P analyzer (Roche Diagnostics, Germany) with an enzymatic assay for creatinine detection that is independent of mouse plasma chromogens (18). Blood and urinary acid-base analysis was performed on an Ecosys II blood gas analyzer (Eschweiler, Germany).
Experimental solutions.
Unless stated otherwise, for in vitro experiments a Ringer-type solution of the following composition was used (in mmol/l): 145 NaCl, 0.4 KH2PO4, 1.6 K2HPO4, 5 glucose, 1 MgCl2, and 1.3 Ca-gluconate; pH was adjusted to 7.4 by NaOH/HCl. Chemicals were of the highest grade of purity available and were obtained from Sigma-Aldrich. Amiloride was kindly provided by Sanofi-Aventis (Frankfurt, Germany) and utilized at 10 μmol/l in perfused tubules.
Renal tubule perfusion.
The methods for perfusion and transepithelial measurements in freshly isolated mouse cortical collecting duct (CCD) segments were described previously (8, 11). Kidney slices were stored and CCDs were dissected in sorting buffer (in mmol/l): 140 NaCl, 0.4 KH2PO4, 1.6 K2HPO4, 1 MgSO4, 10 Na-acetate, 1 α-ketoglutarate, and 1.3 Ca-gluconate; pH was adjusted to 7.4 by NaOH/HCl. Dissected tubules were transferred onto the stage of an inverted microscope (Axiovert 10, Zeiss) and monitored and measured by video imaging (Visitron Systems). The tubule was held and perfused by a concentric glass pipette system. The perfusion pipette was double barreled with an outer diameter of 10–12 μm. One barrel was used for perfusion and Vte measurement. The second barrel was used for pulsed constant current injection (13 nA). The collection side consisted of a glass pipette with an inner diameter of 45 μm. Cable equations as described previously (8) were used appropriately to calculate transepithelial resistance (Rte). Equivalent short-circuit current (Isc) was calculated from Rte and Vte according to Ohm's law. The length constant was calculated with λ = ΔV0·π·r2/(I0·ρ), where V0 is voltage deflection at the perfusion side, r is tubular radius, I0 is injection current, and ρ is resistivity of the perfusion solution; λ < 0.3 × tubular length. The perfusion rate was 10–20 nl/min. The bath was thermostated at 38°C. Continuous bath perfusion at 3–5 ml/min was obtained by gravity.
Statistical analyses.
All data are given as means ± SE. Unpaired t-test was used to test for differences between groups. A P value ≤0.05 was accepted for statistical significance. Unless stated differently, n indicates the number of mice.
RESULTS
Weight gain is impaired in CLDN16 KD mice.
CLDN16 KD mice showed normal development, but both sexes presented a delay in weight gain that persisted through the end of 7 wk (Fig. 1A). Starting from the second week of life, male and female CLDN16 KD mice weighed less compared with their respective littermates. Figure 1B gives two examples for the differences at 2 and 7 wk of age.
Fig. 1.

Growth curves. Claudin-16 (CLDN16) knockdown (KD) mice are significantly smaller compared with their counterpart wild-type (WT) litters already at the weaning age. A: weight development for each genotype and sex in the first 7 wk after birth. B: comparison between genotype and sex at 2 and 7 wk of age. Data are means ± SE; n = 5–11/group. *P < 0.05, WT vs. KD; +P < 0.05, female vs. male.
Net tubular Na+ transport was reduced in female CLDN16 KD mice under control conditions.
It is already known that mice show sex differences in Ca2+ and Mg2+ homeostasis (31). Because young female animals are more sensitive to challenges in Na+ metabolism (6), we decided to focus on female mice. To characterize NaCl handling in CLDN16 KD mice under control conditions, we therefore standardized animal groups for sex and analyzed blood and urine samples of 5- to 9-wk-old female mice. CLDN16 KD animals, as previously shown (16), suffered from hypomagnesemia and hypercalciuria. Irrespective of the high Ca2+ loss, plasma calcium concentration was elevated by 0.14 mmol/l. No obvious alterations in plasma sodium and chloride concentrations were detectable. However, CLDN16 KD mice exhibited a significant increase by twofold in the fractional excretion of Na+ (FE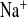 ) and Cl− (FE
) and Cl− (FE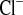 ), indicating a net transport defect for Na+ along the nephron. Plasma K+ was slightly elevated by 0.27 mmol/l in KD compared with WT mice. Plasma and FE values under control conditions are summarized in Table 1.
), indicating a net transport defect for Na+ along the nephron. Plasma K+ was slightly elevated by 0.27 mmol/l in KD compared with WT mice. Plasma and FE values under control conditions are summarized in Table 1.
Table 1.
Plasma values and fractional excretions
| Genotype | n | Mg, mmol/l | Ca, mmol/l | Na, mmol/l | Cl, mmol/l | K, mmol/l | n | FECa2+,% | FENa+, % | FECl−, % | FEK+, % |
|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | 21 | 0.88±0.02 | 2.34±0.02 | 149.7±0.6 | 112.2±0.5 | 3.93±0.08 | 10–13 | 0.4±0.1 | 0.4±0.1 | 0.6±0.1 | 28±6 |
| KD | 13 | 0.78±0.02* | 2.48±0.03* | 150.2±0.6 | 110.9±0.7 | 4.20±0.14* | 5–7 | 6.4±0.9* | 0.9±0.3* | 1.1±0.3* | 33±8 |
Values are means ± SE for n mice. WT, wild type; KD, knockdown; FE, fractional excretion.
Significant difference between KD and WT, P < 0.05.
Water load induced hyponatremia in CLDN16 KD mice.
While the diuretic experimental protocol did not affect plasma Na+ and Cl− concentrations in WT mice, CLDN16 KD mice had lower plasma Na+ concentrations 2 h after the injection of isotonic glucose solution, which is a challenge to develop hypotonic dehydration. In water-loaded CLDN16 KD mice plasma Na+ decreased by 2.5 mmol/l compared with control conditions without any treatment (Table 1, Fig. 2A). This finding might indicate that the total body Na+ content had already been reduced in KD animals, compatible with a latent Na+ deficit under control conditions and consistent with our earlier finding that CLDN16 KD mice are hypotensive (16). In parallel, water load decreased the fractional excretion of Na+, Cl−, K+, and Ca2+ in KD mice by twofold while WT mice were only affected with respect to plasma Ca2+ and FE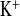 (Table 1, Figs. 2, 4, 5).
(Table 1, Figs. 2, 4, 5).
Fig. 2.

Parameters of NaCl metabolism under water load (w) and water load + amiloride (w+amil) treatment from a 2-h experimental period. A and E: plasma Na+ (P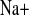 ) and Cl− (P
) and Cl− (P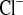 ) concentrations. B and F: urinary Na+ (U
) concentrations. B and F: urinary Na+ (U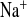 ) and Cl− (U
) and Cl− (U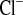 ) concentrations. C and G: Na+ (FE
) concentrations. C and G: Na+ (FE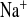 ) and Cl− (FE
) and Cl− (FE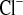 ) fractional excretions. D and H: urinary Na+ and Cl− excretion, calculated for 24 h. Mice with a defect in CLDN16 develop hyponatremia under amiloride treatment. Increase in FE
) fractional excretions. D and H: urinary Na+ and Cl− excretion, calculated for 24 h. Mice with a defect in CLDN16 develop hyponatremia under amiloride treatment. Increase in FE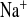 and FE
and FE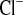 after amiloride treatment is doubled in CLDN16 KD compared with WT mice. Dashed lines indicate values in WT under control conditions. Data are means ± SE; n = 6–8/group. *P < 0.05, WT vs. KD; §P < 0.05, w vs. w+amil. BW, body weight.
after amiloride treatment is doubled in CLDN16 KD compared with WT mice. Dashed lines indicate values in WT under control conditions. Data are means ± SE; n = 6–8/group. *P < 0.05, WT vs. KD; §P < 0.05, w vs. w+amil. BW, body weight.
Fig. 4.

Parameters of K+ metabolism under water load and water load + amiloride treatment from a 2-h experimental period. A: plasma K+ concentrations. B: urinary K+ concentrations. C: FE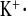 D: urinary K+ excretion, calculated for 24 h. CLDN16 KD mice showed altered potassium homeostasis. Plasma K+ concentrations were increased under water load in CLDN16 KD compared with WT mice. WT mice reduced FE
D: urinary K+ excretion, calculated for 24 h. CLDN16 KD mice showed altered potassium homeostasis. Plasma K+ concentrations were increased under water load in CLDN16 KD compared with WT mice. WT mice reduced FE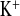 threefold under amiloride, while it did not change in CLDN16 KD mice. Dashed lines indicate values in WT mice under control conditions. Data are means ± SE; n = 6–8/group. *P < 0.05, WT vs. KD; §P < 0.05, w vs. w+amil.
threefold under amiloride, while it did not change in CLDN16 KD mice. Dashed lines indicate values in WT mice under control conditions. Data are means ± SE; n = 6–8/group. *P < 0.05, WT vs. KD; §P < 0.05, w vs. w+amil.
Fig. 5.

Parameters of Ca2+ metabolism under water load and water load + amiloride treatment from a 2-h experimental period. A: plasma Ca2+ concentrations. B: urinary Ca2+ concentrations. C: FE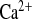 .D: urinary Ca2+ excretion, calculated for 24 h. CLDN16 KD mice display slightly elevated plasma Ca2+ concentrations and hypercalciuria. In contrast to WT, amiloride influences FE
.D: urinary Ca2+ excretion, calculated for 24 h. CLDN16 KD mice display slightly elevated plasma Ca2+ concentrations and hypercalciuria. In contrast to WT, amiloride influences FE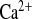 by a 2-fold increase. Dashed lines indicate values in WT mice under control conditions. Data are means ± SE; n = 6–8/group. *P < 0.05, WT vs. KD; §P < 0.05, w vs. w+amil.
by a 2-fold increase. Dashed lines indicate values in WT mice under control conditions. Data are means ± SE; n = 6–8/group. *P < 0.05, WT vs. KD; §P < 0.05, w vs. w+amil.
Hyponatremia and hypochloremia under water load and amiloride treatment in CLDN16 KD mice.
Combined treatment with water load and amiloride resulted in a marked drop in plasma Na+ and Cl− concentrations in KD but not WT animals. After 2-h treatment, plasma Na+ had dropped by a further 3.1 mmol/l and was ∼5 mmol/l below the control values (Fig. 2A and Table 1). In CLDN16 KD animals under water load and in the presence of amiloride, plasma Cl− was reduced by 2.5 and 3.5 mmol/l, respectively, compared with WT animals. In contrast, WT animals showed no significant effect of amiloride on plasma NaCl (Fig. 2E).
Effect of amiloride revealed compensatory Na+ absorption in CLDN16 KD mice.
The two groups treated with water load and amiloride showed an expected rise in fractional excretion of Na+ and Cl−. In KD mice, amiloride increased FE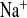 4.5-fold, in contrast to the much smaller effect in WT mice (2.7-fold, Fig. 2C). FE
4.5-fold, in contrast to the much smaller effect in WT mice (2.7-fold, Fig. 2C). FE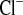 changed in parallel with FE
changed in parallel with FE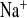 in both genotypes (Fig. 2G). This indicated upregulation of amiloride-inhibitable NaCl absorption in connecting tubules and collecting ducts, distal to the TAL containing the CLDN16 knockdown.
in both genotypes (Fig. 2G). This indicated upregulation of amiloride-inhibitable NaCl absorption in connecting tubules and collecting ducts, distal to the TAL containing the CLDN16 knockdown.
CLDN16 KD mice were not able to further increase urine flow under treatment with amiloride.
Under amiloride treatment the urine concentrations of Na+ and Cl− were higher in KD compared with WT mice (Fig. 2, B and F). However, as shown in Fig. 2, D and H, urine NaCl concentration and the fractional excretions did not translate into increased total excretion of NaCl. Indeed, KD animals excreted less NaCl in the presence of amiloride compared with WT animals under the same treatment. This suggested that in the KD mice NaCl loss in the presence of amiloride was prevented by a decrease in glomerular filtration rate (GFR) and urine flow. Figure 3 shows the data for urinary flow, creatinine, and creatinine clearance as a measurement of GFR. Water load induced a comparable diuresis in both genotypes. Addition of amiloride almost doubled the diuresis in WT animals, while it did not further increase urine flow in KD animals. Plasma creatinine concentrations were higher under water load in KD compared with WT mice and were twofold increased by amiloride in KD mice (Fig. 3B). The increase in plasma creatinine together with the limited amiloride diuresis suggested that KD mice were already at their limits with respect to body volume homeostasis and that they reduced GFR to further prevent salt and water loss. The GFR values are shown in Fig. 3D. The high creatinine clearance under water load was stable in WT, while KD mice started with reduced values and creatinine clearance further dropped in the presence of amiloride.
Fig. 3.

Renal function parameters under water load and water load + amiloride treatment from a 2-h experimental period (same experiment as in Fig. 2). A: urinary flow rate. B: plasma creatinine concentration. C: urine creatinine concentration. D: 2-h creatinine clearance. CLDN16 KD mice show limitations in water and electrolyte handling under amiloride treatment. Comparison of basic kidney parameters reveals that CLDN16 KD fails to further increase urinary flow under amiloride. Plasma creatinine was significantly higher and further increased under amiloride injection as a measure for impairment of glomerular filtration rate (GFR). Dashed lines indicate values in WT mice under control conditions. Data are means ± SE; n = 6–8/group. *P < 0.05, WT vs. KD; §P < 0.05, w vs. w+amil.
Potassium metabolism.
Because of the electrogenic coupling of Na+ reabsorption and K+ secretion in the connecting tubule and collecting duct, a fall in K+ excretion mirrored the increase in Na+ excretion. Consequently, plasma K+ concentration increased in both genotypes when treated with amiloride (Fig. 4). In WT mice, amiloride increased FE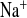 and decreased FE
and decreased FE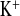 . In contrast, although FE
. In contrast, although FE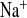 increased more in KD mice, the decrease in FE
increased more in KD mice, the decrease in FE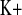 did not reach significance (Fig. 2C and 4C). Hence, the tubular handling of potassium is affected by CLDN16 knockdown.
did not reach significance (Fig. 2C and 4C). Hence, the tubular handling of potassium is affected by CLDN16 knockdown.
Ca2+ metabolism.
Amiloride treatment in WT mice under water load changed neither the plasma concentration nor the fractional or absolute excretion of Ca2+ (Table 1 and Fig. 5). The urine concentration of Ca2+ was lower, inversely proportional to the higher flow rate under amiloride diuresis. In KD animals, amiloride treatment did not affect the already elevated Ca2+ values for plasma, urine, and total excretion. Interestingly, amiloride almost doubled FE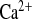 (Table 1 and Fig. 5C). CLDN16 KD mice under amiloride treatment therefore showed additional impairment in tubular Ca2+ absorption, suggesting that the amiloride effect might interfere with Ca2+-scavenging mechanisms along the tubular segments.
(Table 1 and Fig. 5C). CLDN16 KD mice under amiloride treatment therefore showed additional impairment in tubular Ca2+ absorption, suggesting that the amiloride effect might interfere with Ca2+-scavenging mechanisms along the tubular segments.
Amiloride-sensitive electrogenic sodium absorption was fivefold increased in CLDN16 KD mouse collecting ducts.
To verify the upregulation of tubular sodium uptake in CCD we performed measurements on isolated, perfused tubules. Representative recordings for each genotype are shown in Fig. 6, A and B, and illustrate that Vte was considerably higher in KD compared with WT collecting ducts (−6.6 ± 1.1 mV vs. −1.4 ± 0.5 mV; n = 6). Luminal application of amiloride nearly completely abolished Vte in both genotypes. Calculated I′sc as a measurement of electrogenic Na+ absorption is summarized in Fig. 6C. The amiloride-sensitive current ΔI′sc was fivefold increased in KD compared with WT.
Fig. 6.

Transepithelial measurement of electrogenic ion transport in isolated, perfused cortical collecting duct in vitro. A and B: original chart recordings of transepithelial voltage (Vte) in WT and CLDN16 KD mice. Scale bar, 1 min. Luminal addition of amiloride was performed at the indicated time frame. C: equivalent short-circuit current (I′sc) under control conditions (ctrl) and in the presence of amiloride (amil). Effect of amiloride on I′sc (ΔI′sc) reveals 5-fold higher electrogenic Na+ reabsorption in CLDN16 KD mice. Data are means ± SE; n = 6/group. *P < 0.05, WT vs. KD.
CLDN16 KD mice lost acid and retained HCO3−.
In addition to alteration in electrolyte homeostasis, acid/base balance was affected in CLDN16 KD mice. Although blood pH was completely compensated in both genotypes, KD blood showed significantly higher HCO3− concentrations and an increased Pco2 compared with WT (Table 2). Urine H+ concentration was sixfold higher and the urine HCO3− concentration was reduced by more than fivefold in CLDN16 KD mice (Table 2).
Table 2.
Blood and urine values of acid-base metabolism
| Genotype | n | Blood Pco2, Torr | Blood pH | Blood HCO3−, mmol/l | Urine Pco2, Torr | Urine pH | Urine HCO3−, mmol/l | Urine Flow, μl·24 h−1·g body wt−1 |
|---|---|---|---|---|---|---|---|---|
| WT | 4–8 | 35.9±2.2 | 7.42±0.02 | 22.7±1.3 | 9.6±1.0 | 6.8±0.1 | 1.47±0.35 | 55±5 |
| KD | 4–9 | 44.7±1.6* | 7.41±0.01 | 27.3±1.7* | 10.1±1.0 | 6.0±0.1* | 0.27±0.05* | 89±9* |
Values are means ± SE for n mice.
Significant difference between KD and WT, P < 0.05.
DISCUSSION
Our initial study (16) reported the generation of CLDN16-deficient (CLDN16 KD) mouse models that recapitulate human FHHNC phenotypes, including hypomagnesemia, hypercalciuria, and nephrocalcinosis. Our theory (15) of CLDN16 function as a nonselective cation channel predicted defects in renal handling of Na+ and K+ and acid/base balance, in addition to Mg2+ and Ca2+. However, the complex compensatory mechanisms in the kidney masked these defects and preclude a comprehensive understanding of CLDN16 function. In this study, we have carefully addressed this issue in animal groups standardized for sex and age and elucidated the role of CLDN16 in renal handling of salt and acid-base metabolism. Although the investigations were carried out under different housing conditions and diets, and these factors impact on behavior, hormones, and electrolyte handling (2, 32, 33), we have confirmed most of the previously reported phenotypes (16), except hypokalemia.
Defective paracellular cation selectivity in the TAL leads to salt loss.
Besides the transcellular ion transport in the TAL, the crucial mechanism for cation reabsorption in this segment is the generation of the lumen-positive diffusion voltage. This equilibrium potential is enabled on one hand by the established concentration gradient for NaCl across the tubular epithelium and on the other hand by the property of the WT tight junction to let Na+ pass and to hold Cl− back. We have shown that with the loss of CLDN16 and thereby the loss of cation selectivity without gain in epithelial tightness, the diffusion potential was markedly reduced (16), i.e., the voltage was below normal equilibrium. In consequence we would hypothesize a substantial backleak of Na+ into the lumen, now together with Cl− and no longer electrogenic. These changes will cause severe disturbances in ion transport along the TAL and most probably trigger complex compensation mechanisms. Here we report the phenotype and provide first insights into these mechanisms.
Reduced body weight and symptoms of impaired salt and water homeostasis.
In our initial analysis of body weight at one time point in nine animals of either sex the reduction in CLDN16 KD mice did not reach significance. Careful sex-specific analysis of CLDN16 KD animals and a paired control group of respective WT animals revealed lower body weight in CLDN16 KD animals. Delayed growth is a symptom that can be generally observed in diseases and animal models with chronic salt and water loss (3, 22). Therefore we hypothesize that this part of the CLDN16 KD phenotype is associated with the impairment in renal electrolyte handling and indicates that compensatory mechanisms might only be partially effective and make the animals more vulnerable.
The present data support our hypothesis. Young female CLDN16 KD mice under standard conditions already showed a higher tubular loss of NaCl. One compensatory mechanism was an upregulated Na+ absorption in the collecting duct. After the inhibition of this compensating pathway by amiloride, the additional loss of NaCl affected Na+ balance. After a 2-h diuretic period CLDN16 KD mice became hyponatremic, whereas control mice tolerated the treatment without any further side effects. Notably under amiloride treatment, FE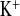 was not decreased in CLDN16 KD in contrast to WT mice. This might indicate the loss of K+ upstream of the amiloride-sensitive nephron (i.e., the TAL). Reprogramming of the transcellular pathway as a result of altered tight junction properties could be one mechanism helping to explain these observations.
was not decreased in CLDN16 KD in contrast to WT mice. This might indicate the loss of K+ upstream of the amiloride-sensitive nephron (i.e., the TAL). Reprogramming of the transcellular pathway as a result of altered tight junction properties could be one mechanism helping to explain these observations.
Homeostatic coupling of Na+ handling with extracellular fluid volume and Ca2+.
The initial analysis showed that CLDN16 KD mice were able to cope with the renal loss of calcium. They even displayed a slightly increased plasma Ca2+ concentration compared with WT mice. Plasma parathyroid hormone concentrations were normal whereas 1,25(OH)2D3 levels were threefold increased in CLDN16 KD mice (16).
In this study we show that CLDN16 KD mice lose Ca2+ and Na+ (and accompanying extracellular fluid). How could these parameters be connected in this mouse model? Recent studies (26, 27) show that Na+ homeostasis is tightly coupled to Ca2+ homeostasis and that Ca2+ is retained by the kidney when the total body Na+ content is reduced. This suggests that the extracellular fluid volume and the blood pressure might be sacrificed by CLDN16 KD animals to maintain Ca2+ balance (4) when these animals are coping with Ca2+ and Na+ loss at the same time. In addition, altered concentrations of luminal and interstitial Ca2+ along the distal nephron may increase urine volume production via Ca2+-sensing receptor signaling (12, 13, 23). In FHHNC patients this mechanism might contribute significantly to polyuria and increase their vulnerability toward restrictions in salt and water intake.
Renal handling of acid and base in CLDN16 KD mice.
An intriguing finding in this study was that CLDN16 KD mice had acidic urine. In fact, acid-base metabolism and Na+ and Mg2+/Ca2+ transport are coupled to each other in a complex manner (21, 28): a mouse model with renal Ca2+ loss, lacking the renal calcium channel TRPV5, showed acidic urine (14). Hypercalcemia, induced by chronic oral administration of 1,25(OH)2D3, was accompanied by metabolic alkalosis and urinary acidification (17). With regard to urine acidification and hypercalcemia-induced metabolic alkalosis, the outer medullary collecting duct might be the key segment, where the vacuolar H+-ATPase under the control of aldosterone plays a crucial role (29, 30).
GRANTS
This work was supported by CAU grants to M. Bleich and by National Eye Institute Grant EY-02430 to D. A. Goodenough.
Acknowledgments
We thank the department of animal care of Christian-Albrechts-Universität Kiel (CAU) for their excellent support in breeding and animal handling. We thank T. Stegmann and J. Brdon for technical assistance.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Angelow S, El-Husseini R, Kanzawa SA, Yu AS. Renal localization and function of the tight junction protein, claudin-19. Am J Physiol Renal Physiol 293: F166–F177, 2007. [DOI] [PubMed] [Google Scholar]
- 2.Argmann CA, Auwerx J. Minimizing variation due to genotype and environment. Curr Protoc Mol Biol 29: 29A.2, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Berger S, Bleich M, Schmid W, Cole TJ, Peters J, Watanabe H, Kriz W, Warth R, Greger R, Schütz G. Mineralocorticoid receptor knockout mice: pathophysiology of Na+ metabolism. Proc Natl Acad Sci USA 95: 9424–9429, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bukoski RD Linkage of Na+ and Ca2+ balance: evidence that Na+ retention preserves Ca2+ balance and limits bone wasting. J Hypertens 22: 683–685, 2004. [DOI] [PubMed] [Google Scholar]
- 5.Edwards BR, Baer PG, Sutton RA, Dirks JH. Micropuncture study of diuretic effects on sodium and calcium reabsorption in the dog nephron. J Clin Invest 52: 2418–2427, 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fraser CL, Kucharczyk J, Arieff AI, Rollin C, Sarnacki P, Norman D. Sex differences result in increased morbidity from hyponatremia in female rats. Am J Physiol Regul Integr Comp Physiol 256: R880–R885, 1989. [DOI] [PubMed] [Google Scholar]
- 7.Greger R Loop diuretics. In: Diuretics, edited by Greger RF, Knauf H, Mutschler E. Berlin: Springer, 1995, p. 221–267.
- 8.Greger R Cation selectivity of the isolated perfused cortical thick ascending limb of Henle's loop of rabbit kidney. Pflügers Arch 390: 30–37, 1981. [DOI] [PubMed] [Google Scholar]
- 9.Greger R Ion transport mechanisms in thick ascending limb of Henle's loop of mammalian nephron. Physiol Rev 65: 760–797, 1985. [DOI] [PubMed] [Google Scholar]
- 10.Greger R Principles of renal transport, concentration and dilution of urine. In: Comprehensive Human Physiology—From Cellular Mechanism to Integration, edited by Greger R, Windhorst U. Berlin: Springer, 1996, p. 1489–1516.
- 11.Greger R, Hampel W. A modified system for in vitro perfusion of isolated renal tubules. Pflügers Arch 389: 175–176, 1981. [DOI] [PubMed] [Google Scholar]
- 12.Hebert SC Calcium and salinity sensing by the thick ascending limb: a journey from mammals to fish and back again. Kidney Int Suppl S28–S33, 2004. [DOI] [PubMed]
- 13.Hebert SC, Brown EM, Harris HW. Role of the Ca2+-sensing receptor in divalent mineral ion homeostasis. J Exp Biol 200: 295–302, 1997. [DOI] [PubMed] [Google Scholar]
- 14.Hoenderop JG, van Leeuwen JP, van der Eerden BC, Kersten FF, van der Kemp AW, Merillat AM, Waarsing JH, Rossier BC, Vallon V, Hummler E, Bindels RJ. Renal Ca2+ wasting, hyperabsorption, and reduced bone thickness in mice lacking TRPV5. J Clin Invest 112: 1906–1914, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hou J, Renigunta A, Konrad M, Gomes AS, Schneeberger EE, Paul DL, Waldegger S, Goodenough DA. Claudin-16 and claudin-19 interact and form a cation-selective tight junction complex. J Clin Invest 118: 619–628, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hou J, Shan Q, Wang T, Gomes AS, Yan Q, Paul DL, Bleich M, Goodenough DA. Transgenic RNAi depletion of claudin-16 and the renal handling of magnesium. J Biol Chem 282: 17114–17122, 2007. [DOI] [PubMed] [Google Scholar]
- 17.Hulter HN, Sebastian A, Toto RD, Bonner EL Jr, Ilnicki LP. Renal and systemic acid-base effects of the chronic administration of hypercalcemia-producing agents: calcitriol, PTH, and intravenous calcium. Kidney Int 21: 445–458, 1982. [DOI] [PubMed] [Google Scholar]
- 18.Jung K, Wesslau C, Priem F, Schreiber G, Zubek A. Specific creatinine determination in laboratory animals using the new enzymatic test kit “Creatinine-PAP.” J Clin Chem Clin Biochem 25: 357–361, 1987. [DOI] [PubMed] [Google Scholar]
- 19.Kiuchi-Saishin Y, Gotoh S, Furuse M, Takasuga A, Tano Y, Tsukita S. Differential expression patterns of claudins, tight junction membrane proteins, in mouse nephron segments. J Am Soc Nephrol 13: 875–886, 2002. [DOI] [PubMed] [Google Scholar]
- 20.Konrad M, Schaller A, Seelow D, Pandey AV, Waldegger S, Lesslauer A, Vitzthum H, Suzuki Y, Luk JM, Becker C, Schlingmann KP, Schmid M, Rodriguez-Soriano J, Ariceta G, Cano F, Enriquez R, Juppner H, Bakkaloglu SA, Hediger MA, Gallati S, Neuhauss SC, Nurnberg P, Weber S. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet 79: 949–957, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nijenhuis T, Renkema KY, Hoenderop JG, Bindels RJ. Acid-base status determines the renal expression of Ca2+ and Mg2+ transport proteins. J Am Soc Nephrol 17: 617–626, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Ronzaud C, Loffing J, Bleich M, Gretz N, Grone HJ, Schutz G, Berger S. Impairment of sodium balance in mice deficient in renal principal cell mineralocorticoid receptor. J Am Soc Nephrol 18: 1679–1687, 2007. [DOI] [PubMed] [Google Scholar]
- 23.Sands JM, Naruse M, Baum M, Jo I, Hebert SC, Brown EM, Harris HW. Apical extracellular calcium/polyvalent cation-sensing receptor regulates vasopressin-elicited water permeability in rat kidney inner medullary collecting duct. J Clin Invest 99: 1399–1405, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schulz-Baldes A, Berger S, Grahammer F, Warth R, Goldschmidt I, Peters J, Schütz G, Greger R, Bleich M. Induction of the epithelial Na+ channel via glucocorticoids in mineralocorticoid receptor knockout mice. Pflügers Arch 443: 297–305, 2001. [DOI] [PubMed] [Google Scholar]
- 25.Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, Casari G, Bettinelli A, Colussi G, Rodriguez-Soriano J, McCredie D, Milford D, Sanjad S, Lifton RP. Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science 285: 103–106, 1999. [DOI] [PubMed] [Google Scholar]
- 26.Titze J, Bauer K, Schafflhuber M, Dietsch P, Lang R, Schwind KH, Luft FC, Eckardt KU, Hilgers KF. Internal sodium balance in DOCA-salt rats: a body composition study. Am J Physiol Renal Physiol 289: F793–F802, 2005. [DOI] [PubMed] [Google Scholar]
- 27.Titze J, Rittweger J, Dietsch P, Krause H, Schwind KH, Engelke K, Lang R, Kirsch KA, Luft FC, Hilgers KF. Hypertension, sodium retention, calcium excretion and osteopenia in Dahl rats. J Hypertens 22: 803–810, 2004. [DOI] [PubMed] [Google Scholar]
- 28.Wagner CA, Kovacikova J, Stehberger PA, Winter C, Benabbas C, Mohebbi N. Renal acid-base transport: old and new players. Nephron Physiol 103: p1–p6, 2006. [DOI] [PubMed] [Google Scholar]
- 29.Wang W, Praetorius J, Li C, Praetorius HA, Kwon TH, Frokiaer J, Nielsen S. Vacuolar H+-ATPase expression is increased in acid-secreting intercalated cells in kidneys of rats with hypercalcaemia-induced alkalosis. Acta Physiol (Oxf) 189: 359–368, 2007. [DOI] [PubMed] [Google Scholar]
- 30.Winter C, Schulz N, Giebisch G, Geibel JP, Wagner CA. Nongenomic stimulation of vacuolar H+-ATPases in intercalated renal tubule cells by aldosterone. Proc Natl Acad Sci USA 101: 2636–2641, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wittner M, Desfleurs E, Pajaud S, Moine G, Simeone S, de Rouffignac C, Di Stefano A. Calcium and magnesium transport in the cortical thick ascending limb of Henle's loop: influence of age and gender. Pflügers Arch 434: 451–456, 1997. [DOI] [PubMed] [Google Scholar]
- 32.Wurbel H Ideal homes? Housing effects on rodent brain and behaviour. Trends Neurosci 24: 207–211, 2001. [DOI] [PubMed] [Google Scholar]
- 33.Zhou X, Hansson GK. Effect of sex and age on serum biochemical reference ranges in C57BL/6J mice. Comp Med 54: 176–178, 2004. [PubMed] [Google Scholar]