

Abstract
Ethanol increases miniature inhibitory postsynaptic current frequency and decreases the paired-pulse ratio, which suggests that ethanol increases both spontaneous and evoked GABA release, respectively. We have shown previously that ethanol increases GABA release at the rat interneuron–Purkinje cell synapse and that this ethanol effect involves calcium release from internal stores; however, further exploration of the mechanism responsible for ethanol-enhanced GABA release was needed. We found that a cannabinoid receptor 1 (CB1) agonist, WIN-55212, and a GABAB receptor agonist, baclofen, decreased baseline spontaneous GABA release and prevented ethanol from increasing spontaneous GABA release. The CB1 receptor and GABAB receptor are Gα i–linked G protein–coupled receptors with common downstream messengers that include adenylate cyclase and protein kinase A (PKA). Adenylate cyclase and PKA antagonists blocked ethanol from increasing spontaneous GABA release, whereas a PKA antagonist limited to the postsynaptic neuron did not block ethanol from increasing spontaneous GABA release. These results suggest that presynaptic PKA plays an essential role in ethanol-enhanced spontaneous GABA release. Similar to ethanol, we found that the mechanism of the cannabinoid-mediated decrease in spontaneous GABA release involves internal calcium stores and PKA. A PKA antagonist decreased baseline spontaneous GABA release. This effect was reduced after incubating the slice with a calcium chelator, BAPTA-AM, but was unaffected when BAPTA was limited to the postsynaptic neuron. This suggests that the PKA antagonist is acting through a presynaptic, calcium-dependent mechanism to decrease spontaneous GABA release. Overall, these results suggest that PKA activation is necessary for ethanol to increase spontaneous GABA release.
INTRODUCTION
There is substantial evidence that the acute behavioral effects of alcohol (e.g., lethargy, anxiolysis, and incoordination) involve modulation of the GABAergic system (Aguayo et al. 2002; Criswell and Breese 2005; Grobin et al. 1998; Mihic 1999; Siggins et al. 2005; Weiner and Valenzuela 2006). Previous research studying the effect of ethanol on GABAergic function focused on ethanol acting directly on GABAA receptors with little emphasis on possible indirect mechanisms. One recently discovered indirect mechanism includes ethanol altering the amount of GABA released from presynaptic terminals in a number of brain regions in vitro (Criswell and Breese 2005; Siggins et al. 2005; Weiner and Valenzuela 2006). Interestingly, ethanol does not increase GABA release in every brain region (Criswell et al. 2008; Jia et al. 2008; Moriguchi et al. 2007), which is in agreement with earlier in vivo studies that showed ethanol increases GABA function in select brain regions (Bloom and Siggins 1987; Criswell et al. 1993; Givens and Breese 1990). The ability of ethanol to increase GABA release in a brain region-specific manner is consistent with alcohol-induced behaviors that are linked to certain brain regions (McCown et al. 1986). Despite this role for GABA release in the GABAergic profile of ethanol, much remains unknown about the mechanism responsible for the ethanol-induced increase in GABA release.
Progress was made toward elucidating this mechanism with the discovery that calcium release from internal stores plays an essential role in ethanol-enhanced spontaneous GABA release at the rat cerebellar interneuron–Purkinje cell synapse; moreover, this ethanol action is not dependent on the influx of extracellular calcium or on calcium-dependent retrograde messengers (Kelm et al. 2007). However, the manner in which ethanol interacts with internal calcium stores is uncertain. Although internal stores release calcium through activation of the inositol 1,4,5-trisphosphate receptors (IP3Rs) and ryanodine receptors (RyRs), there is no current evidence suggesting that ethanol interacts directly with these receptors. The amount of calcium released from the IP3Rs and RyRs is regulated by a number of factors, including calcium itself, nucleotides and protein kinases (Bardo et al. 2006; Patterson et al. 2004). Therefore one hypothesis is that ethanol indirectly modulates calcium release from internal stores to influence GABA release.
In addition to internal calcium stores, ethanol-enhanced GABA release is altered by activation of G protein–coupled receptors (GPCRs) that are linked to Gαi and Gαs G proteins. Nociceptin, which is the endogenous ligand of a Gαi-coupled GPCR (nociceptin/orphanin FQ peptide receptor), blocks ethanol from enhancing GABA release in the central nucleus of the amygdala (Roberto and Siggins 2006). Antagonists for the delta opioid receptor and the GABAB receptor, both of which are Gαi-linked GPCRs, augment the ability of ethanol to increase GABA release in the amygdala and hippocampus (Ariwodola and Weiner 2004; Kang-Park et al. 2007; Zhu and Lovinger 2006). Consistent with these results, activation of the corticotrophin-releasing factor 1 receptor, a GPCR coupled to Gαs, enhances the effect of ethanol on GABA release in the central nucleus of the amygdala (Nie et al. 2004). These results suggest that a variety of Gαi/s-coupled GPCRs can regulate ethanol-enhanced spontaneous GABA release.
Both the Gαi and Gαs subunits modulate adenylate cyclase, with Gαs activating adenylate cyclase and Gαi inhibiting it. When adenylate cyclase is activated, it converts ATP into cAMP, which can bind to protein kinase A (PKA) regulatory subunits (Hanoune and Defer 2001). The binding of cAMP to PKA frees the PKA catalytic subunits from the regulatory subunits, allowing the catalytic subunits to phosphorylate nearby targets. There are PKA phosphorylation sites on both the IP3R (Mignery et al. 1990; Patterson et al. 2004) and RyR (Sobie et al. 2006), and phosphorylation of these receptors leads to increased calcium release (Bardo et al. 2006; Bugrim 1999). In addition, there is evidence for PKA acting at the neurotransmitter release machinery to regulate synaptic transmission (Chheda et al. 2001; Seino and Shibasaki 2005; Trudeau et al. 1996). Therefore activation of adenylate cyclase and PKA could be playing a role in the ethanol-mediated increase in GABA release. This study investigates the role of the adenylate cyclase/PKA pathway in ethanol-enhanced spontaneous GABA release from the presynaptic terminals of rat cerebellar interneurons.
METHODS
Preparation of slices
Sprague-Dawley rats, 13–20 days old, were anesthetized with an intraperitoneal injection of 75% urethane (Sigma, St. Louis, MO) and decapitated after disappearance of the plantar reflex. The brain was rapidly removed and placed in a HEPES-buffered solution of the following composition (in mM): 145 NaCl, 5 KCl, 10 HEPES, 2 CaCl2, 1 MgCl2, 10 glucose, and 5 sucrose (pH to 7.4 with NaOH). The cerebella were isolated, and parasagittal slices, 350 μm thick, were cut with a vibrating microtome (Leica VT1000S, Vashaw Scientific, Norcross, GA) in a low sodium solution of the following composition (in mM): 112.5 sucrose, 63 NaCl, 3 KCl, 1.25 NaH2PO4, 24 NaHCO3, 6 MgSO4, 0.5 CaCl2, and 10 glucose, and gassed with 95% O2-5% CO2. The slices were placed in a chamber containing oxygenated artificial cerebrospinal fluid (ACSF) of the following composition (in mM): 124 NaCl, 3.25 KCl, 1.25 KH2PO4, 10 glucose, 2 MgSO4, 20 NaHCO3, and 2 CaCl2 and gassed with 95% O2-5% CO2. The slices were equilibrated at least 1 h at room temperature before starting experiments.
Whole cell voltage-clamp recordings
A slice was placed at the bottom of a chamber that was attached to the stage of a microscope (BX5OWI, Olympus) and was perfused with oxygenated ACSF (21–24°C) at a flow rate of 0.5 ml/min. The cells were visualized using infrared illumination under differential interference contrast optics with a 40× LUMPlanFl water-immersion objective (Olympus) and displayed on a monitor via a video camera (C2400, Hamamatsu). Recording electrodes were pulled from borosilicate glass (Drummond Scientific Company, Broomall, PA) and had a resistance of 2.5–3 MΩ when filled with internal solution. The internal solution consisted of the following composition (in mM):150 KCl, 3.1 MgCl2, 15 HEPES, 5 K-ATP, 5 EGTA, and 15 phosphocreatine. The internal solution pH was adjusted to 7.4 with KOH, and the osmolarity was ∼310 mOsm. The internal solution composition for the BAPTA experiments was described previously (Kelm et al. 2007). For the paired-pulse studies, 5 mM N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide (QX-314, Sigma) was added to the internal solution to block the generation of action potentials. Data were displayed on an oscilloscope (V-212, Hitachi), digitized at 5 kHz, and stored on a personal computer. The membrane potential was held at −70 mV using a patch-clamp amplifier (Axopatch 200B, Axon Instruments, Union City, CA), and data were collected with Clampex 8.1 software (Axon Instruments). The capacitance and access resistance were monitored continuously throughout the recordings, and a change of 10% or more was sufficient to exclude the recording from analysis. Only one protocol/recording was conducted per slice to avoid contamination.
Drug preparation and drug delivery system
TTX (Sigma), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, Sigma), H-89 (Sigma), d-2-amino-5-phosphonopentanoate (AP5, Sigma), CGP 52432 (Tocris, Ellisville, MO), (R)-baclofen (Tocris), cadmium chloride (CdCl2, Sigma), dibutyryl-cAMP sodium salt (dBcAMP, Tocris), and Rp-adenosine 3′,5′-cyclic monophosphorothioate triethylammonium salt hydrate (Rp-cAMP, Sigma) were made up as concentrated stock solutions (1,000×) in distilled water and stored at −20°C (except for TTX, which was stored at 4°C). 9-(Tetrahydro-2-furanyl)-9H-purin-6-amine (SQ 22536, Sigma) was made up as a concentrated stock solution (500×) in ACSF and stored at −20°C. l-(−)-noradrenaline (+)-bitartrate salt monohydrate (norepinephrine, Sigma) was made the day of use as a concentrated stock solution (1,000×) in distilled water. Protein kinase inhibitor-(6–22)-amide (PKI, Tocris) was made up as a concentrated stock solution (1,000×) in distilled water and was added to the pipette internal solution on the day of use. Thapsigargin (Tocris), 2′,3′-dideoxyadenosine (DDA, Tocris), BAPTA-AM (Sigma), and 2-aminoethoxydiphenylborate (2-APB, Tocris) were made up as concentrated stock solutions (1,000×) in dimethyl sulfoxide (DMSO) and stored at −20°C. WIN 55212-2 mesylate (WIN, Tocris) was made up as a concentrated stock solution (5,000×) in DMSO and stored at −20°C. The final concentration of DMSO used in the experiments was <0.1%, which does not alter the miniature inhibitory postsynaptic current (mIPSC) properties (Kelm et al. 2007). When BAPTA-AM, SQ 22536, DDA, and H-89 were used in an experiment, there was at least a 30-min drug preincubation period with the slice before starting the experiment. The drug stock solutions were diluted in ACSF and inserted into sealed syringes. CNQX (10 μM) and AP5 (50 μM) were added to all solutions inserted into the syringes. The sealed syringes were attached to Teflon tubing that was connected to a multi-barrel perfusion pencil (250-μm tip diam, Automate Scientific, Sarasota, FL), which was positioned 150–250 μm from the cell being tested.
Protocol and analysis for paired-pulse experiments
Platinum-iridium stimulating electrodes were lowered into the molecular layer ∼150 μm from the experimental Purkinje cell. The membrane of the cell was broken into, and the control solution was delivered through the multi-barrel perfusion pencil. After allowing the cell to stabilize, a paired-pulse (PP) stimulation (0.2-ms duration and 50-ms interstimulus interval) was delivered at 0.1 Hz, which generated a PP record of two closely spaced evoked inhibitory postsynaptic currents (eIPSCs). A maximum stimulation was applied to determine the maximal current response, and one half the maximal current response was used for the experiment. Following a minimum of six PP records obtained at 10-s intervals for the precontrol value, the ethanol solution was delivered through the multi-barrel perfusion pencil, and 5 min later, a second series of PP records was collected. Ethanol was washed out for ≥5 min, and a final series of PP records was collected. The miniAnalysis software (version 5.6.4, Synaptosoft, Decatur, GA) was used to generate an averaged PP trace from the PP records collected for each cell. The averaged PP trace was used to calculate the paired-pulse ratio (PPR), which is the ratio of the second eIPSC amplitude to the first (eIPSC2/eIPSC1). The PPR value for each cell in a given group (precontrol, ethanol, washout) were averaged together and represented as the means ± SE. The “averaged control” values were calculated from the precontrol and washout [(precontrol + washout)/2].
Protocol and analysis for mIPSC experiments
After the membrane of the cell was broken into, the control solution, which included 1 μM TTX in addition to the CNQX and AP5, was delivered through the multi-barrel perfusion pencil. Once steady state was obtained (determined from the average of at least 2 repetitive recordings), a precontrol perfusion was recorded for 60 s. For the ethanol experiments, in addition to the precontrol recording, an ethanol recording and washout recording were collected. The percent (%) change in mIPSC frequency, decay time, and amplitude were calculated as follows: 100 × (ethanol response/[(precontrol + washout)/2)] − 100. The control ethanol data points in the results went through the same protocol (precontrol, ethanol, washout) but were never subjected to ethanol. For the experiments that do not involve ethanol, there was a control recording and a drug recording with no washout. The percent change in mIPSC frequency, decay time, and amplitude for these experiments was calculated as follows: 100 × [(|drug response –control|)/control]. When different antagonists were tested against a drug effect on mIPSC frequency, a stable baseline mIPSC rate was established in the presence of the antagonist before exposure to the drug to avoid a summation of effects. Therefore for these experiments, the baseline mIPSC frequency value in the presence of the antagonist served as the precontrol or control value. All data were expressed as the means ± SE. If the control or precontrol baseline mIPSC frequency was <0.5 Hz (except for experiments with the control or precontrol value including exposure to WIN or baclofen), the experiment was excluded from analysis. The data were analyzed with miniAnalysis software. A bi-exponential fit for mIPSC decay times was determined using the miniAnalysis software, and fast (τ-fast) and slow decays (τ-slow) were analyzed separately.
Statistics
Paired Student's t-test, Student's t-test, one-way ANOVA, and Dunnett's post hoc test were performed as indicated. A two-tailed P < 0.05 was accepted as statistically significant.
RESULTS
Ethanol increases mIPSC frequency and decreases the PPR
Similar to previous results (Criswell et al. 2008; Kelm et al. 2007; Ming et al. 2006), 50 (14.2 ± 2.2%, n = 7) and 100 mM (28.7 ± 3.4%, n = 12) ethanol significantly increased mIPSC frequency compared with control (−0.53 ± 1.8%, n = 8) at the interneuron–Purkinje cell synapse as shown in Fig. 1A. In agreement with an earlier report (Ming et al. 2006), neither of the ethanol concentrations had an effect on mIPSC decay time or amplitude (data not shown). A cumulative probability curve from a representative neuron shows that 100 mM ethanol shifts the distribution of the interevent interval curve to the left, which is interpreted as ethanol increasing mIPSC frequency (Fig. 1B). These results suggest that ethanol increases spontaneous GABA release at this synapse.
FIG. 1.

Ethanol increases miniature inhibitory postsynaptic current (mIPSC) frequency and decreases the paired-pulse ratio (PPR). A: compared with control, 50 and 100 mM ethanol decreased mIPSC frequency (*P < 0.05, 1-way ANOVA, Dunnett's post hoc test). B: a cumulative frequency histogram from a representative neuron showing that the ethanol (100 mM) application shifted the distribution of the mIPSC interevent interval curve to the left, indicating that ethanol increases mIPSC frequency. C: the PPR was decreased at 50, 75, and 100 mM ethanol (*P < 0.05, paired Student's t-test). D: traces from a representative neuron showing that ethanol decreased the ratio of the amplitude of the 2nd evoked inhibitory postsynaptic current (eIPSC) to the amplitude of the 1st eIPSC compared with precontrol and washout (i.e., average control).
In addition to increasing mIPSC frequency, ethanol decreased the PPR at this same synapse. The PPR was significantly decreased by 50 mM ethanol (16.2 ± 6.3%, n = 10), 75 mM ethanol (19.8 ± 3.5%, n = 9), and 100 mM ethanol (22.5 ± 7%, n = 9), but not by 25 mM ethanol (4.2 ± 4.3%, n = 10) and 0 mM ethanol (3.9 ± 2.7%, n = 10), as shown in Fig. 1C. A decrease in the PPR by ethanol is interpreted as an increase in evoked GABA release (Siggins et al. 2005). There was a significant linear trend across concentrations for the ethanol effect on the PPR (r = −0.45, P < 0.05), which shows that ethanol dose-dependently decreased the PPR. In Fig. 1D, averaged PP traces from a representative neuron show that 100 mM ethanol decreases the ratio of the amplitude of the second eIPSCs to the first (i.e., PPR) compared with the precontrol and washout. These results suggest that ethanol increases evoked GABA release at the interneuron–Purkinje cell synapse.
Activation of Gαi-coupled GPCRs prevents ethanol from increasing mIPSC frequency
Because of previous research showing a link between Gαi/s-coupled GPCRs and ethanol-enhanced GABA release (Ariwodola and Weiner 2004; Kang-Park et al. 2007; Nie et al. 2004; Roberto and Siggins 2006; Zhu and Lovinger 2006), we determined whether this observation was also true at the interneuron–Purkinje cell synapse. Both GABAB receptors and cannabinoid receptors 1 (CB1) are located in the molecular layer of the cerebellum, where the presynaptic interneurons are located, and activation of both receptors inhibits baseline mIPSC frequency at this synapse (Harvey and Stephens 2004; Takahashi and Linden 2000; Yamasaki et al. 2006). Similar to these results, application of a GABAB receptor agonist, baclofen (5 μM), and a cannabinoid receptor agonist, WIN 55212–2 (WIN, 5 μM), caused a significant reduction in baseline mIPSC frequency (control: 2.3 ± 0.6 Hz, baclofen: 0.64 ± 0.2 Hz, n = 4; control: 2.9 ± 0.8 Hz, WIN: 1.1 ± 0.3 Hz, n = 13; Fig. 2A) with no significant effect on decay time or amplitude (data not shown). A cumulative probability curve from a representative neuron demonstrating that 5 μM WIN shifts the interevent interval curve to the right, which is interpreted as WIN decreasing mIPSC frequency, is shown in Fig. 2B. In the presence of baclofen or WIN, the ability of ethanol to increase mIPSC frequency was significantly blocked (baclofen: 3.9 ± 7.1%, n = 5; WIN: −1.2 ± 3.4%, n = 8; Fig. 2C) compared with control. Shown in Fig. 2D is a cumulative probability curve from a representative neuron showing that 5 μM WIN blocks 100 mM ethanol from shifting the curve to the left. Collectively, these data show that activation of the Gαi-coupled GPCR pathway reduces baseline spontaneous GABA release and prevents ethanol-enhanced spontaneous GABA release at the interneuron–Purkinje cell synapse.
FIG. 2.

WIN 55212-2 (WIN) and baclofen decrease baseline mIPSC frequency and prevent ethanol from increasing mIPSC frequency. A: WIN (5 μM) and baclofen (5 μM) reduced baseline mIPSC frequency (*P < 0.05, paired Student's t-test). B: a cumulative frequency histogram from a representative neuron showing that WIN caused a rightward shift of the interevent interval curve, which suggests that WIN decreases mIPSC frequency. C: WIN and baclofen prevented 100 mM ethanol from increasing mIPSC frequency compared with control (*P < 0.05, 1-way ANOVA, Dunnett's post hoc test). D: a cumulative frequency histogram from a representative neuron showing that ethanol did not shift the interevent interval curve in the presence of WIN, indicating that WIN blocks the effect of ethanol on mIPSC frequency.
Tonic activation of GABAB receptors does not alter the ability of ethanol to increase mIPSC frequency and decrease the PPR
Because activation of the Gαi-linked pathway blocks ethanol-enhanced spontaneous GABA release (Fig. 2C), we wanted to determine whether tonic activation of these Gαi-linked GPCRs was masking the extent of the ethanol effect on both spontaneous and evoked GABA release. Our laboratory has shown that blockade of CB1 receptors does not affect ethanol-enhanced spontaneous GABA release (Kelm et al. 2007); however, we have not studied GABAB receptors. Compared with control (−1.8 ± 0.8%, n = 6; Fig. 3A), 50 (7.1 ± 1.9%, n = 6) and 100 mM (22.7 ± 2.1%, n = 6) ethanol significantly increased mIPSC frequency in the presence of the GABAB receptor antagonist CGP 52432 (10 μM) to a similar extent to that seen in the absence of CGP 52432.
FIG. 3.
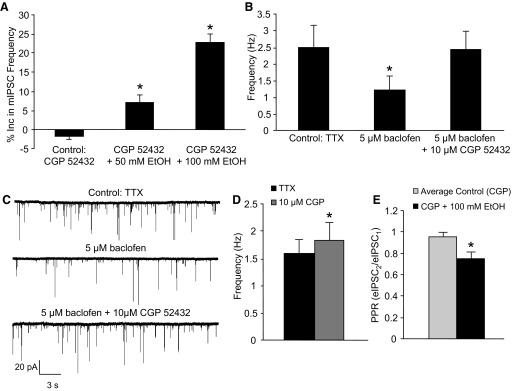
A GABAB antagonist does not affect the ethanol-induced increase in mIPSC frequency or decrease in PPR. A: CGP 52432 (CGP, 10 μM) did not alter the ability of 50 and 100 mM ethanol to increase mIPSC frequency compared with control (*P < 0.05, 1-way ANOVA, Dunnett's post hoc test). B: the reduction in mIPSC frequency by baclofen (5 μM) was reversed by CGP (*P < 0.05, paired Student's t-test). C: a trace from a representative neuron showing that baclofen inhibits mIPSC frequency and CGP reverses this effect. D: CGP increased baseline mIPSC frequency (*P < 0.05, paired Student's t-test). E: CGP did not affect the ability of 100 mM ethanol to decrease the PPR (*P < 0.05, paired Student's t-test).
To see whether an effective concentration of CGP 52432 was used, we determined whether CGP 52432 antagonized the effect of baclofen on mIPSC frequency. The baclofen (5 μM) effect on mIPSC frequency (control: 2.5 ± 0.6 Hz; baclofen: 1.2 ± 0.4 Hz, n = 6) was reversed in the presence of the GABAB receptor antagonist (CGP 52432 + baclofen: 2.4 ± 0.6 Hz, n = 6; Fig. 3B). A trace from a representative neuron showing the effect of baclofen on mIPSC frequency and the ability of CGP 52432 to block it is shown in Fig. 3C. CGP 52432 (10 μM) significantly increased baseline mIPSC frequency (control: 1.6 ± 0.2 Hz; CGP 52432: 1.8 ± 0.3 Hz, n = 14; Fig. 3D), suggesting that there is tonic activation of GABAB receptors that can affect spontaneous GABA release. In the presence of CGP 52432, 100 mM ethanol significantly decreased the PPR (20.7 ± 3.1%, n = 10; Fig. 3E) to a similar extent to that seen in the absence of CGP 52432. Overall, these results suggest that, despite tonic activation of GABAB receptors, antagonism of GABAB receptors does not alter the ability of ethanol to increase evoked and spontaneous GABA release.
Inhibition of adenylate cyclase and PKA blocks ethanol from increasing mIPSC frequency
Next we determined whether inhibiting adenylate cyclase or PKA could prevent ethanol from increasing spontaneous GABA release. To assess the role of adenylate cyclase in this ethanol mechanism, two different purine site inhibitors (SQ 22536 and DDA) were used that inhibit all isoforms of adenylate cyclase (Dessauer et al. 1999). As shown in Fig. 4A, the ability of ethanol to increase mIPSC frequency was significantly reduced in the presence of 300 μM SQ 22536 (15.0 ± 3.6%, n = 10) and 10 μM DDA (12.4 ± 2.3%, n = 9) compared with control. It has been shown previously that SQ 22536 can inhibit the norepinephrine-induced increase in mIPSC frequency at the interneuron–Purkinje cell synapse (Harvey and Stephens 2004), which activates Gαs-coupled GPCRs. Similar to these results, 10 μM norepinephrine caused a 103 ± 11.8% (n = 5) increase in mIPSC frequency, whereas in the presence of 300 μM SQ 22536, the norepinephrine effect was significantly reduced to 15.0 ± 5.8% (n = 3, P < 0.05, Student's t-test). Therefore an effective concentration of SQ 22,536 was used in these experiments.
FIG. 4.
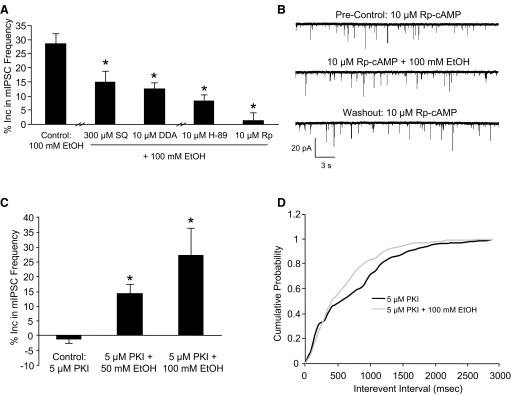
Adenylate cyclase and protein kinase A (PKA) antagonists prevent ethanol from increasing mIPSC frequency. A: SQ 22536 (SQ, 300 μM), DDA (10 μM), H-89 (10 μM), and Rp-cAMP (Rp, 10 μM) prevented 100 mM ethanol from increasing mIPSC frequency compared with control (*P < 0.05, 1-way ANOVA, Dunnett's post hoc test). B: a trace from a representative neuron showing that Rp-cAMP prevents ethanol from increasing mIPSC frequency. C: ethanol (50 and 100 mM) increased mIPSC frequency when 5 μM protein kinase inhibitor-(6–22)-amide (PKI) was in the pipette internal solution (*P < 0.05, 1-way ANOVA, Dunnett's post hoc test). D: a cumulative frequency histogram from a representative neuron showing that ethanol still shifted the curve to the left when PKI was included in the pipette internal solution.
To determine the role of PKA in ethanol-enhanced GABA release, we used two PKA antagonists, H-89 and Rp-cAMP, which have different mechanisms of action. H-89 acts at the PKA ATP-binding site, whereas Rp-cAMP binds to the cAMP binding sites to prevent the regulatory subunits from dissociating from the catalytic subunits (Lochner and Moolman 2006). Both 10 μM H-89 and 10 μM Rp-cAMP significantly reduced ethanol from increasing mIPSC frequency (8.2 ± 2.1%, n = 7 and 1.4 ± 2.5%, n = 10, respectively; Fig. 4A) compared with control. A trace from a representative neuron showing the effect of ethanol on mIPSC frequency in the presence of 10 μM Rp-cAMP is shown in Fig. 4B. A lower Rp-cAMP concentration (1 μM) did not prevent ethanol from increasing mIPSC frequency (22.5 ± 8.2%, n = 3). A higher concentration of H-89 was not tested because of known nonspecific effects that can start to occur at even 10 μM (Lochner and Moolman 2006). Overall, these results suggest that adenylate cyclase and PKA play an important role in ethanol-enhanced spontaneous GABA release.
To determine whether the PKA antagonists were acting at the presynaptic terminal and not the postsynaptic neuron, we included the membrane impermeable PKA antagonist, PKI, in the pipette internal solution, which limits the PKA antagonist to the postsynaptic neuron. With 5 μM PKI in the pipette internal solution, both 50 (14.6 ± 2.8%, n = 8) and 100 mM (27.1 ± 9.2%, n = 6) ethanol significantly increased mIPSC frequency compared with control (−1.3 ± 1.6%, n = 8; Fig. 4C). A cumulative probability curve from a representative neuron shows that 100 mM ethanol still shifts the distribution of the interevent interval curve to the left with 5 μM PKI in the pipette internal solution (Fig. 4D). Because of the lack of PKI effect, there was concern that PKI was not reaching the postsynaptic neuron; however, PKI was able to block the effect of a PKA agonist in a separate experiment (Fig. 5C). These results suggest that PKA is acting presynaptically to block ethanol-enhanced spontaneous GABA release.
FIG. 5.
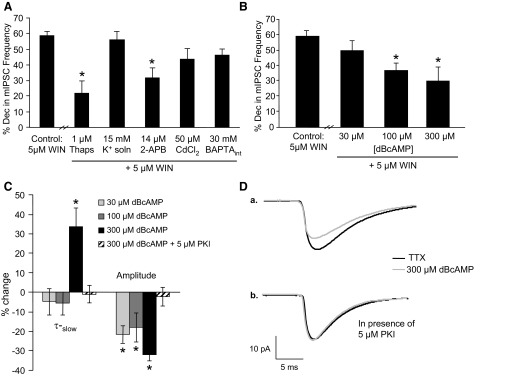
Inhibition of calcium release from internal stores and activation of PKA prevent WIN 55212-2 (WIN) from decreasing mIPSC frequency. A: thapsigargin (Thaps, 1 μM) and 2-APB (14 μM) inhibited WIN (5 μM) from decreasing mIPSC frequency, whereas the high potassium extracellular solution control (K+ soln, 15 mM), cadmium chloride (CdCl2, 50 μM), and BAPTA in the pipette internal solution (BAPTAint, 30 mM) were without effect (*P < 0.05, 1-way ANOVA, Dunnett's post hoc test). B: the ability of WIN to decrease mIPSC frequency was significantly reduced in the presence of 100 and 300 μM dBcAMP, whereas there was not a significant effect at 30 μM (*P < 0.05, 1-way ANOVA, Dunnett's post hoc test). C: baseline mIPSC τ-slow was increased in the presence of 300 μM dBcAMP, and 30, 100, and 300 μM dBcAMP decreased baseline mIPSC amplitude (*P < 0.05, paired Student's t-test). The effect of 300 μM dBcAMP on mIPSC τ-slow and amplitude was blocked when 5 μM PKI was included in the pipette internal solution. D: a trace from a representative neuron showing the effect of 300 μM dBcAMP on mIPSC τ-slow and amplitude (a.) and the ability of 5 μM PKI in the pipette internal solution to block this effect (b.).
Cannabinoids and ethanol act through similar downstream messengers to alter mIPSC frequency
A CB1 receptor agonist decreases spontaneous GABA release at the interneuron–Purkinje cell synapse (Fig. 2A), and this effect involves calcium release from RyRs (Yamasaki et al. 2006). Therefore we determined whether the downstream messengers shown to play a role in the ethanol-induced increase in spontaneous GABA release also play a role in the cannabinoid-induced decrease in spontaneous GABA release. To confirm the involvement of internal calcium stores, we used a sarcoendoplasmic reticulum calcium ATPase (SERCA) pump inhibitor, thapsigargin. The SERCA pump refills the internal stores with calcium, so inhibition of the pump leads to depletion of calcium from the internal stores. During exposure to thapsigargin, we applied a high potassium extracellular solution to deplete internal calcium stores at a faster rate by depolarizing the presynaptic terminals, which increases the rate of calcium release from the IP3Rs and RyRs (Kelm et al. 2007; Simkus and Stricker 2002). Compared with control (58.4 ± 3.0%, n = 13), 1 μM thapsigargin significantly prevented WIN from decreasing mIPSC frequency (21.7 ± 7.7%, n = 7; Fig. 5A). The high potassium (15 mM) extracellular solution in the absence of thapsigargin had no effect on the ability of WIN to decrease mIPSC frequency (55.8 ± 5.5%, n = 5; Fig. 5A). The IP3R antagonist, 2-APB (14 μM), significantly reduced the ability of WIN to decrease mIPSC frequency (31.5 ± 6.3%, n = 8; Fig. 5A). As mentioned above, a similar effect has been shown with a RyR antagonist (Yamasaki et al. 2006). The voltage-dependent calcium channel inhibitor, CdCl2 (50 μM), did not significantly prevent WIN from decreasing mIPSC frequency (43.3 ± 6.9%, n = 8; Fig. 5A). In addition, inclusion of 30 mM BAPTA in the pipette internal solution, which limits BAPTA to the postsynaptic neuron, was ineffective at blocking WIN (46.4 ± 3.8%, n = 8; Fig. 5A). Our laboratory has shown that these concentrations of CdCl2 and BAPTA are effective in this model system (Kelm et al. 2007). Overall, these results suggest that the mechanism of the cannabinoid agonist-mediated decrease in mIPSC frequency is a presynaptic, calcium-dependent process that most likely involves calcium release from internal stores with minimal involvement (if any at all) from the voltage-dependent calcium channels.
Next we determined whether PKA plays a role in the cannabinoid-induced decrease in mIPSC frequency. The ability of WIN to decrease mIPSC frequency was significantly reduced in the presence of the PKA agonist dBcAMP (100 μM: 37.1 ± 4.4%, n = 7; 300 μM: 31 ± 9.3%, n = 7; Fig. 5B) compared with control, but there was no effect at 30 μM dBcAMP (48.3 ± 8.0%, n = 6). There was a significant linear trend across concentrations for the effect of the PKA agonist on the WIN-induced decrease in mIPSC frequency (n = 7, r = −0.53, P < 0.05), which suggests that dBcAMP inhibits WIN from decreasing spontaneous GABA release in a dose-dependent manner.
At 30 and 100 μM dBcAMP, there was a significant decrease in baseline mIPSC amplitude (30 μM: 21.6 ± 4.5%, n = 7; 100 μM: 18.0 ± 7.4%, n = 7), with no change in baseline mIPSC τ-slow (Fig. 5C). With 300 μM dBcAMP, there was a significant decrease in baseline mIPSC amplitude (32 ± 2.9%, n = 6; Fig. 5C) and an increase in baseline mIPSC τ-slow (33.7 ± 9.4%, n = 6). A trace from a representative neuron showing that 300 μM dBcAMP increases mIPSC τ-slow and decreases mIPSC amplitude is shown in Fig. 5Da. There was no effect on baseline mIPSC frequency or on baseline mIPSC τ-fast at any dBcAMP concentration tested (data not shown). The change in mIPSC τ-slow and amplitude seems to be caused by a postsynaptic PKA mechanism because inclusion of 5 μM PKI in the internal solution blocked 300 μM dBcAMP from increasing mIPSC τ-slow (−0.97 ± 4.4%, n = 7) and decreasing mIPSC amplitude (2.3 ± 4.9%, n = 7; Fig. 5C). A trace from a representative neuron showing this PKI effect is in Fig. 5Db. These results suggest that the PKA agonist is having a PKA-dependent, postsynaptic effect that is manifested through a change in mIPSC τ-slow and amplitude.
Buffering presynaptic calcium prevents a PKA antagonist from decreasing baseline mIPSC frequency
Because of the established role of PKA in neurotransmitter release (Seino and Shibasaki 2005), we determined whether a PKA antagonist decreases baseline mIPSC frequency. Both 10 μM and 25 μM Rp-cAMP significantly decreased mIPSC frequency (by 23.3 ± 6.8%, n = 7 and 31.2 ± 3.6%, n = 11, respectively), whereas 1 μM Rp-cAMP was without effect (0.61 ± 4.8%, n = 4; Fig. 6A). At these concentrations of Rp-cAMP, no changes in mIPSC decay time or amplitude were observed (data not shown). In Fig. 6B, a cumulative probability curve from a representative neuron shows that 25 μM Rp-cAMP shifts the distribution of the interevent interval curve to the right, which is interpreted as Rp-cAMP decreasing mIPSC frequency. At 100 μM Rp-cAMP, there was a significant decrease in mIPSC frequency (78.9 ± 8.3%, n = 3), but there was also a significant decrease in mIPSC amplitude (29.3 ± 3.9%), making it difficult to conclude whether a presynaptic and/or postsynaptic mechanism was responsible for this change. These results suggest that presynaptic activation of PKA plays a role in the generation of spontaneous GABA release.
FIG. 6.

BAPTA-AM prevents Rp-cAMP from decreasing mIPSC frequency. A: Rp-cAMP (10 and 25 μM) was able to significantly reduce baseline mIPSC frequency, whereas 1 μM Rp-cAMP was without effect (*P < 0.05, paired Student's t-test). B: a cumulative frequency histogram from a representative neuron showing that 25 μM Rp-cAMP shifted the distribution of the interevent interval curve to the right, which indicates that Rp-cAMP decreases mIPSC frequency. C: preincubation of the slice with BAPTA-AM (BAP-AM, 100 μM) decreased the effect of 25 μM Rp-cAMP on mIPSC frequency compared with control, whereas BAPTA in the internal solution (BAPTAint, 30 mM) did not have an effect (*P < 0.05, 1-way ANOVA, Dunnett's post hoc test). D: a cumulative frequency histogram from a representative neuron showing that 100 μM BAPTA-AM prevented 25 μM Rp-cAMP from shifting the curve.
Because of the role of calcium and PKA in the ethanol and cannabinoid-induced change in spontaneous GABA release, we determined whether there is a link between PKA, calcium, and spontaneous GABA release. In the presence of BAPTA-AM, a membrane permeable calcium chelator, the ability of 25 μM Rp-cAMP to decrease mIPSC frequency was significantly reduced (5.9 ± 2.5%, n = 8; Fig. 6C) compared with control. A cumulative probability curve from a representative neuron shows that 100 μM BAPTA-AM prevents 25 μM Rp-cAMP from shifting the curve (Fig. 6D). Addition of 30 mM BAPTA to the pipette internal solution did not prevent Rp-cAMP from decreasing mIPSC frequency (31.8 ± 3.8%, n = 7; Fig. 6C). These results suggest that changes in presynaptic calcium are needed for a PKA antagonist to decrease mIPSC frequency. Thapsigargin was also able to significantly block 25 μM Rp-cAMP from decreasing mIPSC frequency (10.7 ± 5.6%, n = 6), suggesting that this Rp-cAMP mechanism involves calcium release from internal stores. However, the high potassium solution protocol also blocked the PKA antagonist effect (−2.1 ± 2.6%, n = 3). Therefore the role of internal calcium stores in the PKA antagonist-mediated suppression of spontaneous GABA release is inconclusive.
DISCUSSION
Consistent with previous results (Criswell et al. 2008; Kelm et al. 2007; Ming et al. 2006), ethanol dose-dependently increased both evoked and spontaneous GABA release at the interneuron–Purkinje cell synapse. Our laboratory has shown previously that internal calcium stores play an important role in ethanol-enhanced GABA release (Kelm et al. 2007); however, it has not been determined how ethanol interacts with these internal calcium stores. These experiments provide new insight into the mechanism responsible for ethanol-enhanced GABA release.
Involvement of the Gαi-linked GPCR pathway in ethanol-enhanced GABA release
The Gαi-coupled GPCR agonists, WIN 55212-2 and baclofen, blocked ethanol from increasing spontaneous GABA release at the interneuron–Purkinje cell synapse. Consistent with these results, it has been recently presented that WIN inhibits ethanol-enhanced GABA release in the basolateral and central nucleus of the amygdala (Roberto et al. 2008; Talani and Lovinger 2008). Similarly, baclofen prevents ethanol from increasing spontaneous GABA release in the hippocampus (Ariwodola and Weiner 2004). However, in the ventral tegmental area, baclofen does not inhibit ethanol from increasing spontaneous IPSCs, despite the fact that baclofen affects baseline GABA release (Theile et al. 2008).
Because activation of Gαi-linked GPCRs blocked ethanol from increasing spontaneous GABA release onto cerebellar Purkinje cells, we were curious to see if tonic activation of Gαi-coupled GPCRs was preventing ethanol from fully eliciting GABA release at the interneuron–Purkinje cell synapse. Previous work from our laboratory showed that blockade of CB1 receptors does not affect the ability of ethanol to increase GABA release (Kelm et al. 2007). In this study, we found that, despite tonic activation of the GABAB receptors, a GABAB receptor antagonist does not enhance the ability of ethanol to increase spontaneous and evoked GABA release at the interneuron–Purkinje cell synapse. Similar results are seen in the ventral tegmental area (Theile et al. 2008); however, a GABAB receptor antagonist enhances the ability of ethanol to increase GABA release onto basolateral amygdala neurons and CA1 hippocampal neurons (Ariwodola and Weiner 2004; Zhu and Lovinger 2006). Overall, these variable results suggest that the ability of the GABAB receptor agonist and antagonist to affect ethanol-enhanced spontaneous GABA release is brain region specific.
Adenylate cyclase and PKA play an important role in the mechanism of ethanol-enhanced GABA release
Adenylate cyclase and PKA, which are downstream messengers of the Gαi/s G proteins, were found to play an essential role in ethanol-enhanced spontaneous GABA release. Because a membrane impermeable PKA antagonist in the pipette internal solution did not prevent ethanol from increasing spontaneous GABA release, we are confident that this PKA effect is presynaptic. Although this work provides a link between ethanol-enhanced GABA release and the adenylate cyclase–PKA pathway, more research is needed to determine how ethanol is interacting with this pathway.
There is considerable evidence connecting the adenylate cyclase/PKA pathway to certain effects of ethanol (Newton and Messing 2006; Pandey 1998). Adenylate cyclase isoforms 1, 7, and 8 have all been linked to ethanol with biochemical, electrophysiological, and behavioral studies in transgenic mice (Hanoune and Defer 2001; Maas et al. 2005). Through a PKA-dependent mechanism, an in vivo exposure to ethanol induces a long-lasting potentiation of GABAergic synapses in the ventral tegmental area (Melis et al. 2002). The adenosine A2 receptor, which leads to increased activation of the adenylate cyclase/PKA pathway, mediates important ethanol effects (Mailliard and Diamond 2004). At the behavioral level, a reduction in PKA signaling affects alcohol consumption and the sensitivity to the sedative effects of alcohol (Fee et al. 2006; Lai et al. 2007; Misra and Pandey 2006; Thiele et al. 2000; Wand et al. 2001). Therefore there is evidence of the adenylate cyclase/PKA pathway playing an important role in multiple ethanol actions extending from molecular to behavioral.
CBs and ethanol act in opposite directions through similar downstream messengers to alter spontaneous GABA release
To learn more about the mechanism behind the ethanol-induced increase in GABA release, we studied the mechanism responsible for the WIN-induced decrease in GABA release. Previously, calcium release from RyRs was shown to play a role in this cannabinoid mechanism (Yamasaki et al. 2006). Our study confirmed the involvement of internal calcium stores in the cannabinoid-induced decrease in GABA release and showed that calcium release from IP3Rs is involved. Voltage-dependent calcium channels did not play a significant role in the cannabinoid-mediated suppression of spontaneous GABA release at this synapse; a similar conclusion was made previously based on data showing that CdCl2 had no significant effect on baseline mIPSC frequency (Takahashi and Linden 2000). Additionally, BAPTA in the internal solution did not significantly block the cannabinoid-induced decrease in spontaneous GABA release, which supports the idea that the calcium-dependent portion of this cannabinoid mechanism is presynaptic.
We used a PKA agonist, dBcAMP, to determine the role of PKA in the cannabinoid-mediated suppression of spontaneous GABA release. Interestingly, during the application of 300 μM dBcAMP, baseline mIPSC τ-slow and amplitude were increased and decreased, respectively. Both deceases and increases in mIPSC amplitude have been reported after application of PKA and PKA agonists (Kano and Konnerth 1992; Nusser et al. 1999; Poisbeau et al. 1999). A possible reason for this discrepancy is differences in GABAA receptor subunit composition and GABAA receptor–associated proteins (Nusser et al. 1999). Regardless, this effect seems to be a postsynaptic, PKA-specific action because the dBcAMP effect was blocked by the membrane impermeable PKA antagonist in the pipette internal solution. The PKA agonist, dBcAMP, dose-dependently reduced the ability of WIN to decrease mIPSC frequency, which suggests that the ability of the cannabinoid agonist to decrease spontaneous GABA release involves inhibition of PKA. This result is consistent with activation of Gαi-coupled GPCRs leading to reduced activation of PKA, and data showing that a PKA antagonist decreases spontaneous GABA release (Fig. 6A; Jeong et al. 2003; Lee et al. 2008). Overall, these results suggest that internal calcium stores and PKA are playing an important role in the CB-mediated decrease in spontaneous GABA release, as has been shown for the mechanism of ethanol-enhanced spontaneous GABA release.
A PKA antagonist decreases spontaneous GABA release through a calcium-dependent mechanism
The PKA antagonist, Rp-cAMP (10 μM and 25 μM), inhibited baseline spontaneous GABA release, which is consistent with the established role for PKA in neurotransmitter release (Seino and Shibasaki 2005). Additionally, similar effects of a PKA antagonist on baseline spontaneous GABA release are seen in the hippocampus and hypothalamus (Jeong et al. 2003; Lee et al. 2008). This PKA antagonist effect was not seen in the tuberomammillary nucleus, but this could be because of the low concentration of the PKA antagonist used in this study (Yum et al. 2008). At 1 μM Rp-cAMP, there was no effect on baseline spontaneous GABA release or on ethanol-enhanced spontaneous GABA release. These results suggest that the concentration of the PKA antagonist must be high enough to decrease baseline spontaneous GABA release if it is going to be effective at reducing ethanol-enhanced spontaneous GABA release.
Incubating slices with BAPTA-AM blocked Rp-cAMP from decreasing mIPSC frequency, suggesting that the ability of a PKA antagonist to decrease spontaneous GABA release involves a calcium-dependent mechanism. When BAPTA was limited to the postsynaptic neuron, Rp-cAMP still decreased GABA release. These results suggest that a presynaptic, calcium-dependent mechanism plays a role in the PKA-antagonist mediated decrease in spontaneous GABA release. Two possible calcium-dependent mechanisms related to PKA involve PKA phosphorylating the IP3R and RyR to increase calcium release from internal stores (Bardo et al. 2006; Bugrim 1999; Mignery et al. 1990; Patterson et al. 2004; Sobie et al. 2006) and/or PKA phosphorylating a protein in the neurotransmitter release machinery that is involved in calcium dependent exocytosis (Chheda et al. 2001; Trudeau et al. 1996).
We attempted to determine whether internal calcium stores are involved in the Rp-cAMP–mediated suppression of spontaneous GABA release. Even though the thapsigargin protocol was successful at blocking the PKA antagonist effect, the results are not interpretable because the high potassium extracellular solution control protocol had the same effect. One possible explanation is that the presynaptic depolarization induced by the high potassium extracellular solution altered the phosphorylation state of different proteins that are normally affected by the PKA antagonist. Therefore the role of internal calcium stores in the PKA antagonist-induced decrease in spontaneous GABA release is unknown.
Is cross-talk between PKA and PKC involved in ethanol-enhanced GABA release?
Recent data have shown that protein kinase C (PKC)ɛ is necessary for ethanol to increase GABA release in the central nucleus of the amygdala (Bajo et al. 2008). We have shown previously that internal calcium stores play an important role in ethanol-enhanced GABA release, and in this study, we found that the Gαi-coupled pathway also plays a role in this ethanol mechanism. Taken together, these data suggest that calcium release from internal stores, PKCɛ, adenylate cyclase, and PKA may all play a role in ethanol-enhanced GABA release. It is possible that ethanol is activating these components separately and/or there could be cross-talk occurring between them. Cross-talk occurs between PKA and PKC at the GABAergic nucleus basalis of Meynert synapses (Kubota et al. 2003). Moreover, there are ethanol effects that involve cross-talk between these protein kinases. One mechanism involves ethanol increasing adenylate cyclase isoform 7 (AC7) activity through PKCδ-mediated phosphorylation of AC7, which leads to activation of PKA (Tabakoff et al. 2001). Another example of an ethanol cross-talk mechanism involves ethanol inducing PKCɛ translocation to the cytosol through a PKA-dependent mechanism (Yao et al. 2008). This translocation of PKCɛ is thought to involve PKA activation of phospholipase Cβ. Therefore it is tempting to speculate that the mechanism of ethanol-enhanced GABA release depends on cross-talk occurring between PKA and PKC, but more work will be needed before any firm conclusions are reached.
Role of brain region specificity in the GABAergic actions of ethanol
Although we have presented data regarding the mechanism of ethanol-enhanced spontaneous GABA release at the interneuron–Purkinje cell synapse, it is unknown if this mechanism can be applied to every brain region where ethanol increases GABA release. When collectively looking at all of the experiments that have studied ethanol-enhanced GABA release, effects can vary by brain region. Specifically, ethanol increases GABA release in many (Carta et al. 2004; Criswell et al. 2008; Li et al. 2006; Roberto et al. 2003; Sanna et al. 2004; Sebe et al. 2003; Theile et al. 2008; Zhu and Lovinger 2006; Ziskind-Conhaim et al. 2003), but not all (Criswell et al. 2008; Jia et al. 2008; Moriguchi et al. 2007), brain regions. Additionally, activation and inhibition of GABAB receptors alters ethanol-enhanced GABA release in a brain region-specific manner (Figs. 2 and 3) (Ariwodola and Weiner 2004; Theile et al. 2008; Zhu and Lovinger 2006). Therefore these findings suggest that, depending on the brain region, multiple mechanisms can affect ethanol-enhanced GABA release. As a result, the mechanism of ethanol-enhanced GABA release should be explored in each brain region where ethanol increases GABA release. Although much remains uncertain regarding the mechanism of ethanol-enhanced GABA release, thinking in terms of the effect of ethanol in each brain region could help isolate the potentially differing mechanisms involved in this ethanol action and provide information regarding the behavioral GABAergic profile of ethanol.
GRANTS
This work was supported by National Research Service Award Predoctoral Fellowship AA 17025, National Institute on Alcohol Abuse and Alcoholism Grants R01 AA-11605, RO1 AA-14284, and RO1 AA-14949, and the Bowles Center for Alcohol Studies.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- Aguayo 2002.Aguayo LG, Peoples RW, Yeh HH, Yevenes GE. GABA(A) receptors as molecular sites of ethanol action. Direct or indirect actions? Curr Top Med Chem 2: 869–885, 2002. [DOI] [PubMed] [Google Scholar]
- Ariwodola 2004.Ariwodola OJ, Weiner JL. Ethanol potentiation of GABAergic synaptic transmission may be self-limiting: role of presynaptic GABA(B) receptors. J Neurosci 24: 10679–10686, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajo 2008.Bajo M, Cruz MT, Siggins GR, Messing R, Roberto M. Protein kinase C epsilon mediation of CRF- and ethanol-induced GABA release in central amygdala. Proc Natl Acad Sci USA 105: 8410–8415, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardo 2006.Bardo S, Cavazzini MG, Emptage N. The role of the endoplasmic reticulum Ca2+ store in the plasticity of central neurons. Trends Pharmacol Sci 27: 78–84, 2006. [DOI] [PubMed] [Google Scholar]
- Bloom 1987.Bloom FE, Siggins GR. Electrophysiological action of ethanol at the cellular level. Alcohol 4: 331–337, 1987. [DOI] [PubMed] [Google Scholar]
- Bugrim 1999.Bugrim AE Regulation of Ca2+ release by cAMP-dependent protein kinase. A mechanism for agonist-specific calcium signaling? Cell Calcium 25: 219–226, 1999. [DOI] [PubMed] [Google Scholar]
- Carta 2004.Carta M, Mameli M, Valenzuela CF. Alcohol enhances GABAergic transmission to cerebellar granule cells via an increase in Golgi cell excitability. J Neurosci 24: 3746–3751, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chheda 2001.Chheda MG, Ashery U, Thakur P, Rettig J, Sheng ZH. Phosphorylation of Snapin by PKA modulates its interaction with the SNARE complex. Nat Cell Biol 3: 331–338, 2001. [DOI] [PubMed] [Google Scholar]
- Criswell 2005.Criswell HE, Breese GR. A conceptualization of integrated actions of ethanol contributing to its GABAmimetic profile: a commentary. Neuropsychopharmacology 30: 1407–1425, 2005. [DOI] [PubMed] [Google Scholar]
- Criswell 2008.Criswell HE, Ming Z, Kelm MK, Breese GR. Brain regional differences in the effect of ethanol on gaba release from presynaptic terminals. J Pharmacol Exp Ther 326: 596–603, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criswell 1993.Criswell HE, Simson PE, Duncan GE, McCown TJ, Herbert JS, Morrow AL, Breese GR. Molecular basis for regionally specific action of ethanol on gamma-aminobutyric acidA receptors: generalization to other ligand-gated ion channels. J Pharmacol Exp Ther 267: 522–537, 1993. [PubMed] [Google Scholar]
- Dessauer 1999.Dessauer CW, Tesmer JJ, Sprang SR, Gilman AG. The interactions of adenylate cyclases with P-site inhibitors. Trends Pharmacol Sci 20: 205–210, 1999. [DOI] [PubMed] [Google Scholar]
- Fee 2006.Fee JR, Knapp DJ, Sparta DR, Breese GR, Picker MJ, Thiele TE. Involvement of protein kinase A in ethanol-induced locomotor activity and sensitization. Neuroscience 140: 21–31, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givens 1990.Givens BS, Breese GR. Site-specific enhancement of gamma-aminobutyric acid-mediated inhibition of neural activity by ethanol in the rat medial septal area. J Pharmacol Exp Ther 254: 528–538, 1990. [PMC free article] [PubMed] [Google Scholar]
- Grobin 1998.Grobin AC, Matthews DB, Devaud LL, Morrow AL. The role of GABA(A) receptors in the acute and chronic effects of ethanol. Psychopharmacology (Berl) 139: 2–19, 1998. [DOI] [PubMed] [Google Scholar]
- Hanoune 2001.Hanoune J, Defer N. Regulation and role of adenylyl cyclase isoforms. Annu Rev Pharmacol Toxicol 41: 145–174, 2001. [DOI] [PubMed] [Google Scholar]
- Harvey 2004.Harvey VL, Stephens GJ. Mechanism of GABA receptor-mediated inhibition of spontaneous GABA release onto cerebellar Purkinje cells. Eur J Neurosci 20: 684–700, 2004. [DOI] [PubMed] [Google Scholar]
- Jeong 2003.Jeong HJ, Jang IS, Nabekura J, Akaike N. Adenosine A1 receptor-mediated presynaptic inhibition of GABAergic transmission in immature rat hippocampal CA1 neurons. J Neurophysiol 89: 1214–1222, 2003. [DOI] [PubMed] [Google Scholar]
- Jia 2008.Jia F, Chandra D, Homanics GE, Harrison NL. Ethanol modulates synaptic and extrasynaptic GABAA receptors in the thalamus. J Pharmacol Exp Ther 326: 475–482, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang-Park 2007.Kang-Park MH, Kieffer BL, Roberts AJ, Siggins GR, Moore SD. Presynaptic delta opioid receptors regulate ethanol actions in central amygdala. J Pharmacol Exp Ther 320: 917–925, 2007. [DOI] [PubMed] [Google Scholar]
- Kano 1992.Kano M, Konnerth A. Potentiation of GABA-mediated currents by cAMP-dependent protein kinase. Neuroreport 3: 563–566, 1992. [DOI] [PubMed] [Google Scholar]
- Kelm 2007.Kelm MK, Criswell HE, Breese GR. Calcium release from presynaptic internal stores is required for ethanol to increase spontaneous gamma-aminobutyric acid release onto cerebellum Purkinje neurons. J Pharmacol Exp Ther 323: 356–364, 2007. [DOI] [PubMed] [Google Scholar]
- Kubota 2003.Kubota H, Katsurabayashi S, Moorhouse AJ, Murakami N, Koga H, Akaike N. GABAB receptor transduction mechanisms, and cross-talk between protein kinases A and C, in GABAergic terminals synapsing onto neurons of the rat nucleus basalis of Meynert. J Physiol 551: 263–276, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai 2007.Lai CC, Kuo TI, Lin HH. The role of protein kinase A in acute ethanol-induced neurobehavioral actions in rats. Anesth Analg 105: 89–96, 2007. [DOI] [PubMed] [Google Scholar]
- Lee 2008.Lee JJ, Hahm ET, Lee CH, Cho YW. Serotonergic modulation of GABAergic and glutamatergic synaptic transmission in mechanically isolated rat medial preoptic area neurons. Neuropsychopharmacology 33: 340–352, 2008. [DOI] [PubMed] [Google Scholar]
- Li 2006.Li Q, Wilson WA, Swartzwelder HS. Developmental differences in the sensitivity of spontaneous and miniature IPSCs to ethanol. Alcohol Clin Exp Res 30: 119–126, 2006. [DOI] [PubMed] [Google Scholar]
- Lochner 2006.Lochner A, Moolman JA. The many faces of H89: a review. Cardiovasc Drug Rev 24: 261–274, 2006. [DOI] [PubMed] [Google Scholar]
- Maas 2005.Maas JW, Vogt SK, Chan GC, Pineda VV, Storm DR, Muglia LJ. Calcium-stimulated adenylyl cyclases are critical modulators of neuronal ethanol sensitivity. J Neurosci 25: 4118–4126, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailliard 2004.Mailliard WS, Diamond I. Recent advances in the neurobiology of alcoholism: the role of adenosine. Pharmacol Ther 101: 39–46, 2004. [DOI] [PubMed] [Google Scholar]
- McCown 1986.McCown TJ, Frye GD, Breese GR. Evidence for site specific ethanol actions in the CNS. Alcohol Drug Res 6: 423–429, 1986. [PubMed] [Google Scholar]
- Melis 2002.Melis M, Camarini R, Ungless MA, Bonci A. Long-lasting potentiation of GABAergic synapses in dopamine neurons after a single in vivo ethanol exposure. J Neurosci 22: 2074–2082, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignery 1990.Mignery GA, Newton CL, Archer BT III, Sudhof TC. Structure and expression of the rat inositol 1,4,5-trisphosphate receptor. J Biol Chem 265: 12679–12685, 1990. [PubMed] [Google Scholar]
- Mihic 1999.Mihic SJ Acute effects of ethanol on GABAA and glycine receptor function. Neurochem Int 35: 115–123, 1999. [DOI] [PubMed] [Google Scholar]
- Ming 2006.Ming Z, Criswell HE, Yu G, Breese GR. Competing presynaptic and postsynaptic effects of ethanol on cerebellar purkinje neurons. Alcohol Clin Exp Res 30: 1400–1407, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra 2006.Misra K, Pandey SC. The decreased cyclic-AMP dependent-protein kinase A function in the nucleus accumbens: a role in alcohol drinking but not in anxiety-like behaviors in rats. Neuropsychopharmacology 31: 1406–1419, 2006. [DOI] [PubMed] [Google Scholar]
- Moriguchi 2007.Moriguchi S, Zhao X, Marszalec W, Yeh JZ, Narahashi T. Effects of ethanol on excitatory and inhibitory synaptic transmission in rat cortical neurons. Alcohol Clin Exp Res 31: 89–99, 2007. [DOI] [PubMed] [Google Scholar]
- Newton 2006.Newton PM, Messing RO. Intracellular signaling pathways that regulate behavioral responses to ethanol. Pharmacol Ther 109: 227–237, 2006. [DOI] [PubMed] [Google Scholar]
- Nie 2004.Nie Z, Schweitzer P, Roberts AJ, Madamba SG, Moore SD, Siggins GR. Ethanol augments GABAergic transmission in the central amygdala via CRF1 receptors. Science 303: 1512–1514, 2004. [DOI] [PubMed] [Google Scholar]
- Nusser 1999.Nusser Z, Sieghart W, Mody I. Differential regulation of synaptic GABAA receptors by cAMP-dependent protein kinase in mouse cerebellar and olfactory bulb neurones. J Physiol 521: 421–435, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey 1998.Pandey SC Neuronal signaling systems and ethanol dependence. Mol Neurobiol 17: 1–15, 1998. [DOI] [PubMed] [Google Scholar]
- Patterson 2004.Patterson RL, Boehning D, Snyder SH. Inositol 1,4,5-trisphosphate receptors as signal integrators. Annu Rev Biochem 73: 437–465, 2004. [DOI] [PubMed] [Google Scholar]
- Poisbeau 1999.Poisbeau P, Cheney MC, Browning MD, Mody I. Modulation of synaptic GABAA receptor function by PKA and PKC in adult hippocampal neurons. J Neurosci 19: 674–683, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto 2008.Roberto M, Cruz M, Bajo M, Siggins GR, Schweitzer P. Interactions of ethanol and cannabinoids on synaptic transmission in central amygdala. Alcohol Clin Exp Res 32: 48, 2008. [Google Scholar]
- Roberto 2003.Roberto M, Madamba SG, Moore SD, Tallent MK, Siggins GR. Ethanol increases GABAergic transmission at both pre- and postsynaptic sites in rat central amygdala neurons. Proc Natl Acad Sci USA 100: 2053–2058, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto 2006.Roberto M, Siggins GR. Nociceptin/orphanin FQ presynaptically decreases GABAergic transmission and blocks the ethanol-induced increase of GABA release in central amygdala. Proc Natl Acad Sci USA 103: 9715–9720, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna 2004.Sanna E, Talani G, Busonero F, Pisu MG, Purdy RH, Serra M, Biggio G. Brain steroidogenesis mediates ethanol modulation of GABAA receptor activity in rat hippocampus. J Neurosci 24: 6521–6530, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebe 2003.Sebe JY, Eggers ED, Berger AJ. Differential effects of ethanol on GABA(A) and glycine receptor-mediated synaptic currents in brain stem motoneurons. J Neurophysiol 90: 870–875, 2003. [DOI] [PubMed] [Google Scholar]
- Seino 2005.Seino S, Shibasaki T. PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol Rev 85: 1303–1342, 2005. [DOI] [PubMed] [Google Scholar]
- Siggins 2005.Siggins GR, Roberto M, Nie Z. The tipsy terminal: presynaptic effects of ethanol. Pharmacol Ther 107: 80–98, 2005. [DOI] [PubMed] [Google Scholar]
- Simkus 2002.Simkus CR, Stricker C. The contribution of intracellular calcium stores to mEPSCs recorded in layer II neurones of rat barrel cortex. J Physiol 545: 521–535, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobie 2006.Sobie EA, Guatimosim S, Gomez-Viquez L, Song LS, Hartmann H, Saleet Jafri M, Lederer WJ. The Ca 2+ leak paradox and rogue ryanodine receptors: SR Ca 2+ efflux theory and practice. Prog Biophys Mol Biol 90: 172–185, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabakoff 2001.Tabakoff B, Nelson E, Yoshimura M, Hellevuo K, Hoffman PL. Phosphorylation cascades control the actions of ethanol on cell cAMP signalling. J Biomed Sci 8: 44–51, 2001. [DOI] [PubMed] [Google Scholar]
- Takahashi 2000.Takahashi KA, Linden DJ. Cannabinoid receptor modulation of synapses received by cerebellar Purkinje cells. J Neurophysiol 83: 1167–1180, 2000. [DOI] [PubMed] [Google Scholar]
- Talani 2008.Talani G, Lovinger D. Ethanol and endocannabinoid interactions in the control of GABA release in rat basolateral amygdala. Alcohol Clin Exp Res 32: 808, 2008. [Google Scholar]
- Theile 2008.Theile JW, Morikawa H, Gonzales RA, Morrisett RA. Ethanol enhances GABAergic transmission onto dopamine neurons in the ventral tegmental area of the rat. Alcohol Clin Exp Res 32: 1040–1048, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele 2000.Thiele TE, Willis B, Stadler J, Reynolds JG, Bernstein IL, McKnight GS. High ethanol consumption and low sensitivity to ethanol-induced sedation in protein kinase A-mutant mice. J Neurosci 20: RC75, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudeau 1996.Trudeau LE, Emery DG, Haydon PG. Direct modulation of the secretory machinery underlies PKA-dependent synaptic facilitation in hippocampal neurons. Neuron 17: 789–797, 1996. [DOI] [PubMed] [Google Scholar]
- Wand 2001.Wand G, Levine M, Zweifel L, Schwindinger W, Abel T. The cAMP-protein kinase A signal transduction pathway modulates ethanol consumption and sedative effects of ethanol. J Neurosci 21: 5297–5303, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner 2006.Weiner JL, Valenzuela CF. Ethanol modulation of GABAergic transmission: the view from the slice. Pharmacol Ther 111: 533–554, 2006. [DOI] [PubMed] [Google Scholar]
- Yamasaki 2006.Yamasaki M, Hashimoto K, Kano M. Miniature synaptic events elicited by presynaptic Ca2+ rise are selectively suppressed by cannabinoid receptor activation in cerebellar Purkinje cells. J Neurosci 26: 86–95, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao 2008.Yao L, Fan P, Jiang Z, Gordon A, Mochly-Rosen D, Diamond I. Dopamine and ethanol cause translocation of epsilonPKC associated with epsilonRACK: cross-talk between cAMP-dependent protein kinase A and protein kinase C signaling pathways. Mol Pharmacol 73: 1105–1112, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yum 2008.Yum DS, Cho JH, Choi IS, Nakamura M, Lee JJ, Lee MG, Choi BJ, Choi JK, Jang IS. Adenosine A1 receptors inhibit GABAergic transmission in rat tuberomammillary nucleus neurons. J Neurochem 106: 361–371, 2008. [DOI] [PubMed] [Google Scholar]
- Zhu 2006.Zhu PJ, Lovinger DM. Ethanol potentiates GABAergic synaptic transmission in a postsynaptic neuron/synaptic bouton preparation from basolateral amygdala. J Neurophysiol 96: 433–441, 2006. [DOI] [PubMed] [Google Scholar]
- Ziskind-Conhaim 2003.Ziskind-Conhaim L, Gao BX, Hinckley C. Ethanol dual modulatory actions on spontaneous postsynaptic currents in spinal motoneurons. J Neurophysiol 89: 806–813, 2003. [DOI] [PubMed] [Google Scholar]