

Abstract
Brain-derived neurotrophic factor (BDNF), a potent modulator of synaptic transmission, is known to influence associative synaptic plasticity and refinement of neural connectivity. We now show that BDNF modulation of glutamate currents in hippocampal neurons exhibits the additional property of use dependence, a postsynaptic mechanism resulting in selective modulation of active channels. We demonstrate selectivity by varying the repetition rate of iontophoretically applied glutamate pulses during BDNF exposure. During relatively high-frequency glutamate pulses (0.1 Hz), BDNF application elicited a doubling of the glutamate current. During low-frequency pulses (0.0033 Hz), however, BDNF evoked a dramatically diminished response. This effect was apparently mediated by calcium because manipulations that prevented elevation of intracellular calcium largely eliminated the action of BDNF on glutamate currents. To confirm N-methyl-d-aspartate (NMDA) receptor involvement and assess spatial requirements, we made cell-attached single-channel recordings from somatic NMDA receptors. Inclusion of calcium in the pipette was sufficient to produce enhancement of channel activity by BDNF. Substitution of EGTA for calcium prevented BDNF effects. We conclude that BDNF modulation of postsynaptic NMDA receptors requires concurrent neuronal activity potentially conferring synaptic specificity on the neurotrophin's actions.
INTRODUCTION
A central tenet of Hebbian synaptic plasticity holds that synaptic strengthening requires concurrent pre- and postsynaptic activity. This assertion encompasses the concept of use or activity dependence and provides insight into processes ranging from refinement of synaptic connectivity during development to learning and memory. In the visual system, for example, interventions that inhibit the firing of retinal ganglion neurons can disrupt segregation of afferent fibers into cortical ocular dominance columns (Stryker and Harris 1986).
In many systems, the signal underlying activity dependence is the opening of the N-methyl-d-aspartate (NMDA) glutamate receptors. NMDA receptor antagonists can disrupt development of visual pathways (For review, see Debski and Cline 2002) and can prevent forms of associative synaptic plasticity such as hippocampal long-term potentiation (LTP) (for review, see Bliss and Collingridge 1993). Physiological features of the receptor, such as calcium permeability and voltage-dependent magnesium block (Mayer and Westbrook 1987) make it ideal to signal neuronal activity, leading to the description of this receptor as a “coincidence detector.”
NMDA receptor involvement alone, however, cannot explain the multifunctional changes observed in the nervous system. One candidate for additional modulation of synaptic plasticity is the neurotrophin brain-derived neurotrophic factor (BDNF) (for review, see Chao 2003; Lu 2004; McAllister et al. 1999; Poo 2001; Tyler et al. 2002). In slices of visual cortex, BDNF requires glutamate receptor activity to promote alterations in dendritic morphology (McAllister et al. 1996). Injection of BDNF into cat visual cortex in vivo inhibited ocular dominance column formation in the vicinity of injection but not elsewhere (Cabelli et al. 1995), and blockade of endogenous ligand signaling via TrkB, the BDNF receptor, also inhibited ocular dominance column formation (Cabelli et al. 1997). In accord with this are studies demonstrating the activity dependence of BDNF release (Balkowiec and Katz 2000, 2002; Kolarow et al. 2007; Kuczewski et al. 2008; Magby et al. 2006).
Regulation of NMDA receptor-dependent synaptic plasticity by BDNF implies a link between TrkB activation and NMDA receptor activity. Recent studies have revealed such an interaction that may be critical for manifestation of activity dependence (Arvanian and Mendell 2001; Crozier et al. 1999; Garraway et al. 2005; Jarvis et al. 1997; Kerr et al. 1999; Legrand et al. 2005; Levine et al. 1998; Song et al. 1998). We have used glutamate receptor agonists (Levine et al. 1998) and antagonists (Crozier et al. 1999) to show that the predominant postsynaptic effect of BDNF is to enhance selectively the NMDA component of glutamate current. Previous studies, however, have not addressed this critical question: will BDNF enhancement of NMDA receptor availability occur in the absence of neuronal activity? In experiments done to date, glutamate receptors were co-activated with BDNF presentation. If BDNF was instead applied in the absence of glutamate receptor activation, would subsequent testing reveal increased NMDA receptor activity or would it be unaffected? If the former, then BDNF modulation is not use-dependent and would not be categorized as contributing to Hebbian plasticity. If the latter, the conclusion would be the opposite. To address this issue, we employed two different stimulation protocols: an “activity” protocol consisting of relatively high-frequency glutamate stimulation and a “low activity” protocol with low-frequency stimulation. Our results indicate that NMDA receptor opening and elevation of intracellular calcium are critical for postsynaptic enhancement of NMDA receptor activity by BDNF and that the NMDA receptors themselves are sufficient to provide the calcium influx.
METHODS
Cell culture
High-density hippocampal cultures were grown as previously described (Levine et al. 1995). In brief, time-mated pregnant Sprague-Dawley rats were killed by CO2 asphyxiation in accordance with institutional guidelines for care and use of animals. Hippocampi were obtained at 18 days gestation and placed into cold PBS. Cells were triturated in 2 ml of Eagle's minimum essential medium (MEM) with added glucose and 10% FBS and plated on poly-d-lysine-coated Nunc Petri dishes. Cultures were maintained in serum-free medium (SFM) in a 95% air-5% CO2 humidified incubator at 37° centigrade. SFM consisted of a 1:1 (vol/vol) mixture of Ham's F-12 (Gibco) and MEM (Gibco) and supplemented with 25 μg/ml insulin, 100 μg/ml transferrin, 60 μM putrescine, 20 nM progesterone, 30 nM selenium, 6 mg/ml glucose, and 0.5 U/ml and 0.5 mg/ml of penicillin and streptomycin, respectively.
Electrophysiology
Whole cell voltage-clamp recordings were made from pyramidal-type cells after 12–16 days in vitro by standard techniques (Hamill et al. 1981). Cells were held at a resting potential of −40 mV to reduce Mg2+ blockade of NMDA receptors. For each condition tested, experiments were performed from multiple platings. The external bath solution contained (in mM) 1.67 CaCl2, 1 Mg Cl2, 5.36 KCl, 137 NaCl, 17 glucose, 10 HEPES, 0.001 TTX, and 20 sucrose. The osmolarity was 325 mOsm. For experiments with zero-added calcium in the bath solution, the osmolarity was readjusted with sucrose. For anti-BDNF antibody (Promega) experiments, a concentration of 10 μg/ml was added to the external solution. The internal pipette solution contained (in mM) 105 Cs-methanesulfonate, 15 CsCl, 10 HEPES, 0.2 EGTA, 8 NaCl, 2 Mg-ATP, 2 Na2-ATP, 0.3 Na3-GTP, 20 phosphocreatinine, and 50 creatinine phosphokinase. The pH of the pipette solution was set to 7.3 with CsOH and the osmolarity was 310 mOsm. For experiments with bis-(o-aminophenoxy)-N,N,N',N'-tetraacetic acid (BAPTA) included in the pipette solution, EGTA was removed, 10 mM BAPTA was added and Cs-methanesulfonate concentration was reduced to return the osmolarity to control; all other component concentrations remained the same. Signals were recorded with an Axopatch 200 amplifier (Axon Instruments), sampled at 2.5 kHz (INDEC IDA15125 or CED Power1401 interface) and filtered at 5 kHz. Three-barrel micropipettes were used for iontophoretic application of glutamate. One barrel, filled with 0.1 Μ NaCl, was used for current balancing. Other barrels were filled with either l-glutamate (0.2 M, pH 7.5) or saline control. During baseline recording, typical currents were elicited at 10 s intervals by 2- to 10-ms pulses yielding currents with peak amplitudes ranging from 25 to 50 pA. Experiments without exogenously applied BDNF followed the same aforementioned stimulation protocols.
For single-channel recording of NMDA receptor activity, cell-attached patches were made from neuronal somata. The intracellular potential was set to ∼0 mV with a bath solution containing (in mM) 140 potassium gluconate, 10 HEPES, and 5 EGTA, pH 7.5. To activate NMDA receptor channels, 10 μM NMDA and 3 μM glycine were added to a magnesium-free solution containing (in mM) 10 Cs-methanesulfonate, 110 Na2SO4, 25 HEPES, 33 glucose, and 1.3 CaCl2, pH 7.35. To isolate NMDA receptor activity, the membrane potential was set to −80 mV, which is below the threshold for activating most voltage-gated channels. Channel identity was confirmed as described previously (Levine et al. 1998). Patches typically contained one to three channels as judged by the presence of overlapping openings. Patches with larger numbers of channels were not used. Signals were sampled at 5 kHz and filtered at 1 kHz with an intersweep interval of 1 s. For most patches, activity was quantified by obtaining open times and number of openings from sweeps with nonoverlapping openings by idealization with the half-amplitude crossing criterion and cubic spline interpolation (Sigworth and Sine 1987). Open probability (Po) was calculated by dividing open time by total time per sweep. All sweeps, including nulls, were included in the calculation of average Po. For two recordings with a large number of overlapping openings, Po was calculated by measuring charge and dividing by maximal possible charge. For all recordings, activity was recorded for ≥10 min prior to applying BDNF (baseline). Only recordings for which baseline line activity remained stable for the 5 min before BDNF application were used. Recordings in which activity was gradually decreasing or increasing were not used.
All chemicals were from Sigma unless otherwise noted. BDNF (Peprotech) was prepared as a stock in phosphate-buffered saline with 1 μg/ml bovine serum albumin and diluted 500-fold into bath solution. K-252a/b (Calbiochem) were prepared as 1,000-fold stocks in DMSO. Kynurenate and anti-BDNF antibody were purchased from Tocris and Promega, respectively. Anti-BDNF antibody was heat inactivated by boiling the stock solution at 100°C for 15 min prior to dilution in bath solution to a final concentration of 10 μg/ml. Student's t-test was used with P < 0.05 indicating significance.
RESULTS
Frequency of glutamate stimulation affects BDNF modulation
The overall goal of this study was to determine whether the postsynaptic effect of BDNF on glutamate current required co-activation of glutamate receptors. Whole cell voltage-clamp recordings were obtained from cultured embryonic hippocampal neurons. To allow NMDA receptor openings, the holding potential was set to −40 mV, and pulses of glutamate were applied iontophoretically along the initial segment of major dendrites. In addition, presynaptic actions of BDNF on ongoing synaptic activity were eliminated by addition of TTX to the bath solution. Baseline current amplitudes ranged from 25 to 50 pA, and currents were reversed at ∼0 mV. To determine whether BDNF enhancement of glutamate-evoked currents requires concurrent neurotransmitter receptor activation, we used two types of stimulation protocols during BDNF exposure (Fig. 1A). Use of an “activity” protocol (6 glutamate pulses per minute for the duration of the recording), resulted in a doubling of total glutamate current (184 ± 13% of baseline, mean ± SE, n = 8; Fig. 1, B and C) when measured 20–25 min during BDNF (20 ng/ml) exposure, a result in accordance with previous studies (Crozier et al. 1999; Levine et al. 1998).
FIG. 1.

Brain-derived neurotrophic factor (BDNF)-induced potentiation of glutamate currents is activity-dependent. A: illustration of the recording configuration and stimulation protocols. Whole cell recordings (“patch” electrode) were made from the somata of cultured embryonic hippocampal neurons. Ionic current was elicited by iontophoretic application of glutamate to dendritic processes (“ionto” electrode) and BDNF or vehicle solution was bath-applied with a perfusion system (Ogata and Tatebayashi 1991). For the relatively high-frequency stimulation “activity” protocol, pulses of glutamate were delivered at 0.1 Hz for the duration of the recording. The diagram shows the 20-min period that begins at the onset of BDNF or vehicle perfusion. For the lower stimulation frequency “low activity” protocol, a single pulse of glutamate was delivered at 5-min intervals. After 20 min, stimulation frequency was increased to 0.1 Hz. B: example sweeps of glutamate currents during baseline period and 20 min into BDNF exposure. Repetitive glutamate application at 0.1 Hz (activity protocol) resulted in a twofold increase in total glutamate current (left). Reduction of stimulation frequency to 0.0033 Hz (low activity protocol) during BDNF exposure elicited a much smaller response (right). These and subsequent examples are the average of 3 sweeps from a single minute. C: averaged time courses of the response to BDNF during the activity (▪, n = 8) or low activity (⧫, n = 7) stimulation protocols. Peak current amplitude was normalized to baseline. Time = 0 has been set to onset of the BDNF perfusion. In this and subsequent figures, responses to 0.1-Hz stimuli were combined into 1-min bins and averaged. Responses to the 0.0033-Hz stimuli were not binned. —, the arithmetic addition of 2 time courses: BDNF during low activity, and activity alone from D (□ + ◊, see text). - - -, no response to BDNF. D: example sweeps of glutamate currents during baseline period and after 20 min of either the activity (left) or low activity (right) stimulation protocols. BDNF was not present during stimulation although cells were perfused with the BDNF vehicle solution in the same way as for BDNF. The activity protocol evoked a modest increase in glutamate current whereas the low activity protocol had no effect. E: averaged time courses of responses to activity (□, n = 10) and low activity protocols (◊, n = 7). Scale bars refer to sweeps in B and D.
The experiments in the preceding text, however, do not answer the question of whether exposure to BDNF, in the absence of concurrent neurotransmitter receptor activation, will still augment glutamate current. This is an important issue because an effect of BDNF in the absence of glutamate receptor activity would argue that BDNF acts postsynaptically in an activity-independent, and thus non-Hebbian, fashion. To address this, we used a “low activity” protocol (Fig. 1A, single pulses of glutamate delivered every 5 min for the first 20 min of BDNF exposure, stimulation at 6 pulse/min thereafter); this allowed us to periodically assess the stability of the recording while largely eliminating glutamate receptor activation. With this protocol, the effect of BDNF was significantly reduced (118 ± 11% of baseline, n = 7, P < 0.01; Fig. 1, B and C). Thus potentiation of glutamate currents by BDNF requires concurrent postsynaptic glutamate receptor activation during BDNF exposure.
Work from other investigators has shown that iontophoretic application of multiple, long pulses of glutamate (10 s) onto neurons in hippocampal slices can produce LTP by enhancing postsynaptic receptor responsiveness (Cormier and Kelly 1996; Cormier et al. 1993). To assess the use of the activity protocol on cultured neurons, we repeated our experiments but in the absence of exogenous BDNF. The activity protocol resulted in a significant increase in glutamate current measured at 20–25 min (129 ± 9% of baseline, n = 10, P < 0.05; Fig. 1, D and E). To ensure that the increase in current was stimulus-dependent, we also tested the low activity protocol in the absence of BDNF. This procedure evoked no enhancement of glutamate current at 20–25 min (101 ± 8% of baseline, n = 7; Fig. 1, D and E). Consequently, repetitive glutamate receptor activation alone can produce a small increase in glutamate current if given at a sufficiently high frequency.
These experiments identified three conditions in which glutamate current increased to varying degrees: use of the activity protocol in the presence of BDNF (condition 1), use of the activity protocol in the absence of BDNF (condition 2), and use of the low activity protocol in the presence of BDNF (condition 3). Of the three conditions, the activity protocol plus BDNF had the greatest effect. To determine whether this last response was simply an additive result of activity stimulation alone plus low activity stimulation in BDNF (i.e., condition 2 + condition 3), or whether it represented a synergistic action produced by co-activation of TrkB and glutamate receptors, the response to activity stimulation alone was added to low activity stimulation in BDNF (Fig. 1C, —). The calculated current magnitude did not reach the level of simultaneous TrkB and glutamate receptor stimulation. Therefore simultaneous TrkB and glutamate receptor activation acted synergistically to produce the largest increase in current.
Endogenous neurotrophins
To gain further insight into how the activity protocol produced the enhancement of glutamate current observed in the absence of exogenously applied BDNF (Fig. 1D), we investigated the possibility that glutamate iontophoresis stimulated release of endogenous neurotrophin that acted on glutamate receptors via “autocrine” signaling pathways (Segal and Greenberg 1996). BDNF is released by activity-dependent manipulations such as potassium depolarization or electrical stimulation (Balkowiec and Katz 2000; Goodman et al. 1996; Hartmann et al. 2001; Haubensak et al. 1998; Kojima et al. 2001; Lindholm et al. 1994). We hypothesized that the activity protocol used here increased glutamate currents by stimulating release of endogenous BDNF which then acted on the cell's intrinsic TrkB receptors. To test this hypothesis, we repeated the activity protocol in the presence of K-252a (200 nM), an inhibitor of Trk receptor tyrosine kinases (Knusel and Hefti 1992). As compared with stimulation alone, which produced a 132 ± 9% increase in current compared with baseline (n = 7; Fig. 2), stimulation in the presence of K-252a produced a significantly smaller increase in current (109 ± 6% of baseline, n = 8, P < 0.05; Fig. 2) an effect consistent with activity-dependent release of endogenous BDNF or related neurotrophins. To investigate BDNF specifically, experiments with function-blocking anti-BDNF antibody (10 μg/ml) and TrkB-Fc (2 μg/ml) were performed. Similar to K-252a, the antibody (107 ± 5% of baseline, n = 5, P < 0.05; Fig. 2) and soluble receptor (101 ± 10% of baseline, n = 6, P < 0.05; Fig. 2) reduced the increase in glutamate current. Control recordings with K-252b (200 nM), the nonmembrane-permeable analogue of K-252a (133 ± 7% of baseline, n = 7, P > 0.8; Fig. 2), and heat-inactivated anti-BNDF antibody (133 ± 8% of baseline, n = 7, P > 0.9; Fig. 2) had no effect on the increase in current. These data suggest that activity-dependent release of endogenous BDNF was predominantly responsible for the increase in glutamate current induced by the activity protocol.
FIG. 2.

Iontophoretic application of glutamate alone produces a modest increase in glutamate current via endogenously released BDNF. A: example sweeps of glutamate currents during baseline period and 20 min into the recording. Repetitive glutamate application at 0.1 Hz (activity protocol) produced an increase in glutamate current (left), which was prevented by bath application of K-252a (right). B: averaged time courses of 0.1-Hz stimulation alone + the K-252a/b vehicle (▪, n = 7) and 0.1-Hz stimulation in the presence of antagonists of neurotrophin signaling. K-252a (▿, n = 7), anti-BDNF function-blocking antibody (▵, n = 5), and TrkB-Fc (○, n = 6) reduced the increase in current seen in response to stimulation alone. The control compounds heat inactivated anti-BDNF antibody (▴, n = 7), and K-252b (▾, n = 7) had no effect. 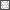 and □ (bars), application of the indicated compounds in separate sets of experiments. For all conditions, the activity protocol (0.1-Hz stimulation frequency) was used.
and □ (bars), application of the indicated compounds in separate sets of experiments. For all conditions, the activity protocol (0.1-Hz stimulation frequency) was used.
Role of calcium
Our observation that the enhancement of glutamate current produced by BDNF is activity-dependent led us to examine the mechanistic role of calcium. We employed three different manipulations to explore this issue. In each case, the activity stimulation protocol was used. First, application of BDNF during recordings performed in nominally calcium-free extracellular solution largely eliminated the increase in glutamate current (109 ± 5% of baseline, n = 6; Fig. 3). Second, we included 10 mM BAPTA, a high-affinity calcium chelator, in the recording pipette. Intracellular injection of BAPTA also strongly attenuated the action of BDNF (112 ± 7% of baseline, n = 6; Fig. 3). Finally, we employed a nonpharmacological approach by changing the membrane holding potential from −40 to −80 mV, a voltage that favors blockade of NMDA receptors by magnesium (Mayer and Westbrook 1987), thus preventing activation and calcium influx through these channels. The negative holding potential also eliminated the action of BDNF (103 ± 7% of baseline, n = 7; Fig. 3). Taken together, these results confirm the observation that BDNF enhancement of glutamate currents is activity-dependent and is dependent on calcium influx.
FIG. 3.

BDNF-induced potentiation of glutamate currents requires calcium influx and N-methyl-d-aspartate (NMDA) receptor activation. A: example sweeps of glutamate currents during baseline period and 20 min into BDNF exposure. BDNF application during repetitive glutamate application at 0.1 Hz (activity protocol) failed to produce a significant increase in glutamate current when 10 mM bis-(o-aminophenoxy)-N,N,N',N'-tetraacetic acid (BAPTA) was present in the recording pipette solution (left) or when calcium was not added to the bath solution (right). B: averaged time courses of responses to BDNF with 10 mM BAPTA included in the recording pipette solution (○, n = 6), bath solution nominally-free of calcium (•, n = 6), and cells held at –80 mV to prevent NMDA receptor openings (▿, n = 7). For comparison, a dotted line representing the time course of the normal response to BDNF during the activity protocol (from Fig. 1C) is included. For all conditions the activity protocol (0.1-Hz stimulation frequency) was used.
Role of glutamate receptors
If the effect of BDNF requires co-incident activation of glutamate receptors, then it should also be prevented during the “activity” protocol if the receptors are blocked. To test this prediction, kynurenate (1 mM) was co-applied with BDNF (Fig. 4A). During this period, the response to iontophoretically applied glutamate was substantially reduced. On washout of the kynurenate, the glutamate current returned to baseline without any indication of an effect of BDNF (104 ± 7% of baseline, n = 4), even though the BDNF had been applied for 25 min and glutamate pulses delivered at 0.1 Hz.
FIG. 4.

Glutamate receptor antagonist and change of holding potential during BDNF presentation confirms an absence of modulation when NMDA receptors are blocked. A: averaged time course of response to BDNF during co-application of kynurenate (n = 4 recordings). The holding potential was −40 mV, and the activity protocol was used. Block of the glutamate current during BDNF application prevented BDNF enhancement of current that was assessed following kynurenate washout. B: averaged time course of response to BDNF (n = 7 recordings). The holding potential was maintained at −80 mV except for the 2 indicated periods at −40 mV to assess the NMDA receptor component of current. As a result of changing the holding potential during these periods, the average amplitude of glutamate current increased slightly prior to BDNF perfusion and increased by a similar amount during BDNF perfusion. The data further support the conclusion that NMDA receptors are not being affected by BDNF in the absence of calcium influx.
To address whether NMDA receptors are critical, we again took advantage of the voltage-dependent block that affects NMDA receptors but not AMPA receptors. When neurons are held at –40 mV and the “activity” protocol is used, BDNF increased glutamate currents (Fig. 1). On the other hand, no response to BDNF was seen at a holding potential of −80 mV (Fig. 3). This manipulation prevents calcium entry through NMDA receptors but also prevents assessment of NMDA receptor activity because they are no longer available. To determine whether NMDA receptor activity is necessary during BDNF application, responses were compared at –40 mV before and after BDNF application but cells were held at −80 mV during BDNF exposure (Fig. 4B). During the baseline period when the holding potential was depolarized from −80 to −40 mV, the change in total glutamate current was somewhat variable but was on average almost unchanged (103 ± 22% of baseline, n = 7), a result expected from the decrease in driving force coupled with an increase in the NMDA component of current, which is relatively small under baseline conditions (Crozier et al. 1999). During BDNF perfusion, when the glutamate current would have been expected to increase had the holding potential been kept at −40 mV, the current amplitude was similar to that seen in the pre-BDNF period (107 ± 15% of baseline, n = 7, P > 0.8; Fig. 4). These data support the notions that NMDA receptors must be available during BDNF application to generate an increase in glutamate current and that TrkB activation alone is insufficient.
Single-channel modulation
Although previous work from our laboratory has implicated NMDA receptors as the primary target of postsynaptic BDNF enhancement of glutamate current in embryonic hippocampal neurons (Crozier et al. 1999; Levine et al. 1998), the present results led us to address directly the question of whether calcium influx through individual NMDA receptor channels is sufficient to support BDNF modulation. We therefore made cell-attached single-channel recordings of NMDA receptor channels (Fig. 5) during which the primary source of calcium was provided via the recording pipette (i.e., calcium was omitted from the external bath solution). In agreement with a prior study (Levine et al. 1998), BDNF applied outside of the recording pipette is capable of increasing NMDA receptor activity inside the pipette when calcium is included in the pipette solution (Fig. 5, A, C, and E). The effect of BDNF began within a few minutes of application and became maximal after 15 min. On average, channel activity approximately doubled (244 ± 41% above baseline, n = 10 recordings). Consistent with the results of the whole cell recordings, replacing calcium in the recording pipette with EGTA prevented the action of BDNF (Fig. 5, B, D, and E). Without calcium influx, NMDA receptor activity remained unchanged during exposure to BDNF (116 ± 27%, n = 9) and was comparable to having calcium in the pipette but exposing the cell to the BDNF vehicle solution (91 ± 14%, n = 10, Fig. 5F). These experiments suggest that sufficient calcium enters through the small number of NMDA receptor channels in the patch to allow for modulation of channel activity by BDNF.
FIG. 5.

Cell-attached single-channel recording shows that local calcium entry is sufficient for BDNF modulation of NMDA receptor activity. A: diagram illustrating the recording conditions and sample sweeps indicating modulation of NMDA receptor activity by BDNF. In addition to NMDA, glycine and other ingredients (see methods), the pipette solution contained 1.3 mM calcium. The patch contained ≥2 channels. Top: recording of baseline activity. Bottom: increased activity elicited by bath perfusion of 20 ng/ml BDNF. B: sample activity from a recording in which calcium in the pipette solution was replaced by EGTA. The patch contained ≥2 channels. Top and bottom traces: baseline activity and activity in the presence of BDNF, respectively. In this case, perfusion of BDNF had no effect on NMDA receptor activity. C: plot of open probability (Po) vs. time during for the recording illustrated in A. Time = 0 has been set to onset of the BDNF perfusion. Baseline activity is defined as the 5 min prior to BDNF perfusion. Note that Po increases with time during the BDNF perfusion. D: Po plot for the recording illustrated in B. Note that replacing Ca with EGTA in the pipette solution resulted in no effect of BDNF on NMDAR activity. E: plot in which Po values have been combined into 1-min bins and normalized to baseline activity. Experiments in which calcium was present in the pipette solution and which were exposed to BDNF show elevated activity (○, n = 10). Experiments in which the pipette contained calcium but were exposed to BDNF vehicle solution did not (□, n = 10). Experiments in which patches were exposed to BDNF but contained EGTA instead of calcium in the pipette solution also showed no increase in activity (▾, n = 9). F: summary graph in which average activity at 15–20 min during the BDNF perfusion is plotted. Addition of BDNF elicited a significant increase in activity as compared with vehicle when calcium was in the pipette solution (*, P < 0.01) but not when EGTA was in the pipette solution (P > 0.4).
The magnitude of the effect of BDNF on glutamate currents is primarily dependent on three conditions. First, simultaneous TrkB and NMDA receptor activation act synergistically to enhance glutamate current. Second, BDNF-induced enhancement of current requires elevation of intracellular calcium. Finally, the stimulation-induced increase of current results, at least in part, from endogenously released BDNF. These points are represented diagrammatically in Fig. 6, which provides an overall view of the combinations of conditions used in this study. The horizontal bars at the top of Fig. 6A indicate the presence of activity (high-frequency glutamate stimulation), elevation of intracellular calcium, and BDNF. The two black columns represent the change in glutamate current magnitude when all three manipulations were used simultaneously. It is evident that when NMDA receptors are allowed to open, the response produced exceeds that of any pairwise combination; if NMDA receptors are not allowed to open (−80 mV holding potential), then the concurrence of activity, BDNF, and extracellular calcium is without effect. Based on these data, we propose that BDNF increases the NMDA receptor component of the total glutamate current under restricted conditions (Fig. 6B). When comparing BDNF presentation to two neurons, one that is quiescent and one that is active, maintained TrkB signaling produces no change in NMDA receptor properties in the inactive cell but does so in the active cell. The reason is that TrkB activation alone is insufficient to influence NMDA receptor availability. When NMDA receptors are open and conducting current, however, the calcium influx is sufficient to support TrkB signaling and consequent upregulation of their own activity.
FIG. 6.

Concurrence of activity, BDNF and extracellular calcium produced the largest enhancement of glutamate currents. A: horizontal bars (top) indicate the 3 stimulus conditions tested: 1) activity in the form of 0.1-Hz iontophoretic application of glutamate, 2) presence of extracellular calcium (or absence of augmented buffering of intracellular calcium), and 3) activation of TrkB by binding of BDNF. Vertical columns represent the magnitude of the increase in glutamate current observed in response to the different stimuli. The number of simultaneous conditions is represented by the color of the columns: 1 = light gray, 2 = dark gray, all 3 = black. Labels beneath the columns indicate special manipulations used to achieve the stimulus conditions. If no label is present, the condition was simply omitted. For example, the 1st 3 columns show results from experiments in which activity and calcium were present, but in which BDNF was not present or activation of TrkB by BDNF was prevented. This was accomplished by not adding BDNF (column 1), adding K-252a (column 2), or adding anti-BDNF (column 3). Overall, the figure shows that simultaneous use of activity, BDNF, and calcium produced the largest effect on glutamate current but only if NMDA receptors were allowed to open (compare columns 4 and 7). The number of experiments is noted at the top of each column. *, responses significantly different from calcium alone (column 9). †, responses significantly different from activity + calcium (column 1). B: model depicting BDNF modulation of glutamate current in active, but not inactive, cells. Left: BDNF binding to TrkB alone is not sufficient to elicit an effect on NMDA receptors because the cell and NMDA receptors are inactive. However, as shown on the right,synaptic release of glutamate produces a depolarization supplied by stimulation of AMPA receptors that also results in the opening of unblocked NMDA receptors. Consequently, calcium influx through NMDA receptors, together with BDNF binding to TrkB, results in enhancement of active NMDA receptor function.
DISCUSSION
In this study, we tested the hypothesis that postsynaptic modulation of embryonic hippocampal neuron glutamate current by BDNF is activity-dependent. We examined the effect of stimulation frequency on BDNF-enhanced glutamate receptor responsiveness and found that the trophin action depended on glutamate receptor activation. In previous studies, we showed that NMDA receptor antagonists prevented the action of BDNF (Crozier et al. 1999) and that current through AMPA receptors was largely unaffected by acute application of BDNF (Levine et al. 1998). Here we show that a holding potential of −80 mV, which would prevent opening of NMDA receptors while leaving activation of non-NMDA ionotropic and metabotropic receptors intact, also prevented the effect of BDNF on glutamate current. We propose that glutamate application elevates intracellular calcium through NMDA receptors that acts synergistically with TrkB signaling to enhance NMDA receptor activity and consequently produce an increase in glutamate current. Exposure to BDNF during low-frequency stimulation, in which TrkB activation is not accompanied by repeated activation of glutamate receptors, elicits a markedly smaller response. A high spatial resolution is likely because calcium entry through individual NMDA receptor channels can support modulation. Thus in the developing mammalian nervous system, BDNF modulation is subject to a Hebbian-like condition, requiring coordinate TrkB activation and postsynaptic activity to enhance synaptic transmission. And, in common with other forms of synaptic plasticity, NMDA receptor participation is essential.
BDNF modulation of NMDA receptors has now been found in a variety of cell types and preparations. Recordings from hippocampal neurons demonstrated BDNF-potentiated transmission due to increased NMDA receptor sensitivity (Caldeira et al. 2007; Crozier et al. 1999; Kolb et al. 2005; Levine et al. 1998; Song et al. 1998). NMDA receptor single-channel recordings from cell-attached (Jarvis et al. 1997; Levine et al. 1998) or excised patches (Levine and Kolb 2000) demonstrated increased channel openings in response to BDNF application. Further, in spinal cord (Arvanian and Mendell 2001; Arvanov et al. 2000; Garraway et al. 2005; Kerr et al. 1999; Legrand et al. 2005; Slack and Thompson 2002) and suprachiasmatic nucleus neurons (Kim et al. 2006), BDNF has been shown to enhance NMDA-mediated responses. Together, these studies provide evidence for a general neurotrophin-induced enhancement of NMDA receptor function.
One potential mechanism for BDNF-induced enhancement of glutamate currents involves elevation of intracellular calcium (Li et al. 1998; Stoop and Poo 1996). Calcium influx activates multiple signaling molecules, including calcium/calmodulin-dependent protein kinase II, which was implicated in our previous work as an important link between TrkB activation and increased NMDA receptor function (Crozier et al. 1999). For the modulation described here, calcium elevation is critical. Exclusion of calcium from the external solution or intracellular injection of the calcium chelator BAPTA prevented potentiation. Moreover, in single-channel recordings with and without calcium in the pipette solution, only the former showed modulation by BDNF, indicating that calcium influx through individual NMDA receptor channels is sufficient to support modulation. On their own, these experiments do not completely rule out other potential sources of elevated calcium, including release from intracellular stores or other calcium-permeable channel types including voltage-gated calcium channels. For the modest level of depolarization (−40 mV) used here, however, we regard these possibilities as unlikely. We have shown previously that BDNF enhances the current produced by iontophoretic application of NMDA, suggesting that metabotropic receptors are not required (Levine et al. 1998). Furthermore, the response to BDNF was eliminated by application of kynurenate (Fig. 4A), which should not interfere with channel types other than glutamate receptors. Moreover, even though it has been established that BDNF can elicit an elevation of intracellular calcium (Amaral et al. 2007), our experiments indicate that this increase is not sufficient on its own to support NMDA receptor modulation because it could occur independent of glutamate application. This does not mean that other influences could not come into play, particularly at higher depolarizations, but for the NMDA receptor-dependent effect of BDNF on glutamate current, the direct source is the most likely. Regardless of the source of calcium entry, the key point is that BDNF modulation of glutamate current will only occur in active neurons, because the magnesium block of NMDA receptors must be removed and because calcium elevation must occur.
Given the dramatic effects that resulted from exogenously applied BDNF, we examined possible stimulation-dependent endogenous neurotrophin action. A previous report examining chronic NMDA treatment revealed activity-dependent release of BDNF with subsequent TrkB activation (Marini et al. 1998). Similarly, in the present study, glutamate receptor stimulation alone increased glutamate current in the absence of exogenous BDNF. This potentiation was reduced by the nonselective Trk antagonist, K-252a, suggesting neurotrophin involvement. Experiments using a function-blocking antibody to BDNF reproduced the K-252a result, suggesting that the activity protocol induced release of endogenous BDNF that then increased glutamate current. These results support the contention that BDNF can be released by glutamate receptor activity, stimulate postsynaptic TrkB receptors and enhance glutamate receptor responsiveness in an autocrine fashion in a manner strikingly similar to that of exogenously applied BDNF, albeit on a smaller scale.
Studies of BDNF-modulated LTP in the adult hippocampus have not demonstrated a requirement for NMDA receptor activation (Xu et al. 2000). Possible explanations for the apparent variation of effects on NMDA receptors may reside in the type and age of preparation used. In cultured embryonic hippocampal neurons, blockade of NR2B-containing NMDA receptors prevented BDNF enhancement of glutamate-induced currents (Crozier et al. 1999) and the increased activity of single NMDA receptor channels (Levine and Kolb 2000). NR2B-containing NMDA receptors are generally expressed at higher levels in the developing hippocampus and their relative contribution to synaptic currents subsequently diminishes in the adult (Monyer et al. 1994; Stocca and Vicini 1998; Tovar and Westbrook 1999); thus it is possible that hippocampal slices from adult animals do not reveal effects on synaptic NMDA receptors because NR2A and NR2B subunits can become associated with different sets of synaptic molecules (Al-Hallaq et al. 2007). This idea is consistent with our previous biochemical report demonstrating a BDNF-induced increase in phosphorylation of juvenile NR2B but not NR2A subunits (Lin et al. 1998).
The critical role played by NMDA receptors in the induction of synaptic plasticity has led us to focus our attention on postsynaptic actions of BDNF; work from other laboratories, however, has shown equally potent effects of this neurotrophin on presynaptic properties. For example, experiments on cultured hippocampal neurons (Berninger et al. 1999; Lessmann et al. 1994; Li et al. 1998; Schinder et al. 2000) revealed enhanced excitatory transmission arising from increased release of neurotransmitter. Experiments in hippocampal and cerebellar slices demonstrated enhancement due to increased basal excitatory transmission (Kang and Schuman 1995) or reduced inhibitory input (Cheng and Yeh 2003; Frerking et al. 1998). Activity-dependent processes such as LTP are also potentiated by BDNF, attributable to presynaptic mechanisms that do not require NMDA receptor activation (Figurov et al. 1996) but do require high-frequency stimulation (Gottschalk et al. 1998). Activity dependence has also been found in modulation of presynaptic properties by BDNF. In a nonmammalian neuromuscular synapse preparation (Boulanger and Poo 1999) brief depolarization in the presence of low levels of BDNF produced potentiation under circumstances when neither BDNF alone nor depolarization would—a result in accord with the synergistic effect of BDNF and NMDA receptor activation reported in this study. It was hypothesized in a later study (Zhang and Poo 2002) that the activity dependence and synapse specificity resulted from a localized response to BDNF that required local protein synthesis. The mechanism described in the present study characterizes a postsynaptic component of synapse specificity that does not require restricted release of BDNF. This mechanism will target active synapses selectively even in a uniform field of BDNF. Moreover, our single-channel recordings suggest that this effect could be highly spatially restricted because calcium through individual channels is sufficient to support responsiveness to BDNF.
The modulatory effects of BDNF influence both rapid and long-term synaptic and neurotrophin signaling. For example, several studies indicate that BDNF and depolarizing stimuli acutely increase TrkB localization at the cell surface in retinal ganglia and hippocampal neurons (Du et al. 2000; Meyer-Franke et al. 1998). Increased TrkB surface expression may enhance a neuron's ability to compete for limited supply of neurotrophins. Relevant to the present study, at a lower stimulation frequency (0.16 Hz) relatively little change in TrkB surface expression was observed by Du et al., indicating that this type of signaling may be operationally different from modulation of glutamate receptor function described by stimulation at 0.1 Hz used in this study. Also relevant is the finding that the combination of activity and BDNF was necessary for conversion of silent synapses to AMPA-containing synapses (Itami et al. 2003). In another study, alterations in the actual morphology of dendrites from slices of visual cortex were observed to require glutamate receptor activation that were manifested over 36 h in culture (McAllister et al. 1996). Together with the results presented here, these studies may highlight two separate phases of BDNF activity; one an early rapid phase on transmitter receptors and a later phase that may effect long-term signaling and connectivity.
For the mechanism of action, it is clear that near concomitant NMDA receptor opening and TrkB activation is critical. Although the two need not occur simultaneously, our results imply that if TrkB activation does not occur while calcium-potentiated signaling is enabled, then NMDA receptor activity will not be upregulated. In terms of the signaling pathways, the low-affinity trk receptor, p75, is not likely to be involved (Reichardt 2006). This conclusion is supported by studies which show that p75 and TrkB activation have opposite effects (Kim et al. 2003; Sandoval et al. 2007), making it less likely that p75 participates in the enhancement of NMDA receptor activity. The effect of BDNF reported here is also unlikely to be mediated by binding to the glycine site (Jarvis et al. 1997) because it can be blocked by K-252a and can be observed in cell-attached single-channel recordings (Levine et al. 1998). The requirement for calcium could be present at different levels in the many signaling cascades initiated by BDNF binding to TrkB. Our past work has implicated CaMKII (Crozier et al. 1999), which would be a good starting target for future work to elucidate the calcium dependence.
Activity dependence may confer specificity of neurotrophin synaptic action. Although the exact source of BDNF release is not yet known, it may not always be subject to precise targeting. The activity dependence described here provides a mechanism for synapse specificity; increased glutamate current depends on conjoint elevation of calcium via NMDA receptors, and TrkB stimulation in synaptic terminals. Thus exposure of active synapses to BDNF results in a reinforcement of that activity, which may consequently lead to maintenance and integration of those synapses into mature circuits.
GRANTS
This work was supported by National Institutes of Health Grants HD-23315 and NS-041310.
Acknowledgments
The authors thank Dr. Robin L. Davis for valuable discussions and a critical reading of the manuscript. We also thank G. Hale and S. Metry for some of the single-channel recordings and Q. Hsu for expert technical assistance.
Present address of R. A. Crozier, Wyeth Research, Discovery Neuroscience, CN8000, Princeton, NJ 08543.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
R. A. Crozier and C. Bi contributed equally to this work.
REFERENCES
- Al-Hallaq et al. 2007.Al-Hallaq RA, Conrads TP, Veenstra TD, Wenthold RJ. NMDA di-heteromeric receptor populations and associated proteins in rat hippocampus. J Neurosci 27: 8334–8343, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral et al. 2007.Amaral MD, Chapleau CA, Pozzo-Miller L. Transient receptor potential channels as novel effectors of brain-derived neurotrophic factor signaling: potential implications for Rett syndrome. Pharmacol Ther 113: 394–409, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvanian and Mendell 2001.Arvanian VL, Mendell LM. Acute modulation of synaptic transmission to motoneurons by BDNF in the neonatal rat spinal cord. Eur J Neurosci 14: 1800–1808, 2001. [DOI] [PubMed] [Google Scholar]
- Arvanov et al. 2000.Arvanov VL, Seebach BS, Mendell LM. NT-3 Evokes an LTP-like facilitation of AMPA/Kainate receptor-mediated synaptic transmission in the neonatal rat spinal cord. J Neurophysiol 84: 752–758, 2000. [DOI] [PubMed] [Google Scholar]
- Balkowiec and Katz 2000.Balkowiec A, Katz DM. Activity-dependent release of endogenous brain-derived neurotrophic factor from primary sensory neurons detected by ELISA in situ. J Neurosci 20: 7417–7423, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkowiec and Katz 2002.Balkowiec A, Katz DM. Cellular mechanisms regulating activity-dependent release of native brain-derived neurotrophic factor from hippocampal neurons. J Neurosci 22: 10399–10407, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berninger et al. 1999.Berninger B, Schinder AF, Poo MM. Synaptic reliability correlates with reduced susceptibility to synaptic potentiation by brain-derived neurotrophic factor. Learn Mem 6: 232–242, 1999. [PMC free article] [PubMed] [Google Scholar]
- Bliss and Collingridge 1993.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361: 31–9, 1993. [DOI] [PubMed] [Google Scholar]
- Boulanger and Poo 1999.Boulanger L, Poo MM. Presynaptic depolarization facilitates neurotrophin-induced synaptic potentiation. Nat Neurosci 2: 346–351, 1999. [DOI] [PubMed] [Google Scholar]
- Cabelli et al. 1995.Cabelli RJ, Hohn A, Shatz CJ. Inhibition of ocular dominance column formation by infusion of NT-4/5 or BDNF. Science 267: 1662–1666, 1995. [DOI] [PubMed] [Google Scholar]
- Cabelli et al. 1997.Cabelli RJ, Shelton DL, Segal RA, Shatz CJ. Blockade of endogenous ligands of trkB inhibits formation of ocular dominance columns. Neuron 19: 63–76, 1997. [DOI] [PubMed] [Google Scholar]
- Caldeira et al. 2007.Caldeira MV, Melo CV, Pereira DB, Carvalho RF, Carvalho AL, Duarte CB. BDNF regulates the expression and traffic of NMDA receptors in cultured hippocampal neurons. Mol Cell Neurosci 35: 208–219, 2007. [DOI] [PubMed] [Google Scholar]
- Chao 2003.Chao MV Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci 4: 299–309, 2003. [DOI] [PubMed] [Google Scholar]
- Cheng and Yeh 2003.Cheng Q, Yeh HH. Brain-derived neurotrophic factor attenuates mouse cerebellar granule cell GABA(A) receptor-mediated responses via postsynaptic mechanisms. J Physiol 548: 711–721, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormier and Kelly 1996.Cormier RJ, Kelly PT. Glutamate-induced long-term potentiation enhances spontaneous EPSC amplitude but not frequency. J Neurophysiol 75: 1909–1918, 1996. [DOI] [PubMed] [Google Scholar]
- Cormier et al. 1993.Cormier RJ, Mauk MD, Kelly PT. Glutamate iontophoresis induces long-term potentiation in the absence of evoked presynaptic activity. Neuron 10: 907–919, 1993. [DOI] [PubMed] [Google Scholar]
- Crozier et al. 1999.Crozier RA, Black IB, Plummer MR. Blockade of NR2B-containing NMDA receptors prevents BDNF enhancement of glutamatergic transmission in hippocampal neurons. Learn Mem 6: 257–266, 1999. [PMC free article] [PubMed] [Google Scholar]
- Debski and Cline 2002.Debski EA, Cline HT. Activity-dependent mapping in the retinotectal projection. Curr Opin Neurobiol 12: 93–99, 2002. [DOI] [PubMed] [Google Scholar]
- Du et al. 2000.Du J, Feng L, Yang F, Lu B. Activity- and Ca(2+)-dependent modulation of surface expression of brain-derived neurotrophic factor receptors in hippocampal neurons. J Cell Biol 150 :1423–1434, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figurov et al. 1996.Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature 381: 706–709, 1996. [DOI] [PubMed] [Google Scholar]
- Frerking et al. 1998.Frerking M, Malenka RC, Nicoll RA. Brain-derived neurotrophic factor (BDNF) modulates inhibitory, but not excitatory, transmission in the CA1 region of the hippocampus. J Neurophysiol 80: 3383–3386, 1998. [DOI] [PubMed] [Google Scholar]
- Garraway et al. 2005.Garraway SM, Anderson AJ, Mendell LM. BDNF-induced facilitation of afferent-evoked responses in lamina II neurons is reduced after neonatal spinal cord contusion injury. J Neurophysiol 94: 1798–1804, 2005. [DOI] [PubMed] [Google Scholar]
- Goodman et al. 1996.Goodman LJ, Valverde J, Lim F, Geschwind MD, Federoff HJ, Geller AI, Hefti F. Regulated release and polarized localization of brain-derived neurotrophic factor in hippocampal neurons. Mol Cell Neurosci 7: 222–238, 1996. [DOI] [PubMed] [Google Scholar]
- Gottschalk et al. 1998.Gottschalk W, Pozzo-Miller LD, Figurov A, Lu B. Presynaptic modulation of synaptic transmission and plasticity by brain-derived neurotrophic factor in the developing hippocampus. J Neurosci 18: 6830–6839, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill et al. 1981.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pfluegers 391: 85–100, 1981. [DOI] [PubMed] [Google Scholar]
- Hartmann et al. 2001.Hartmann M, Heumann R, Lessmann V. Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J 20: 5887–5897, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubensak et al. 1998.Haubensak W, Narz F, Heumann R, Lessmann V. BDNF-GFP containing secretory granules are localized in the vicinity of synaptic junctions of cultured cortical neurons. J Cell Sci 111 :1483–1493, 1998. [DOI] [PubMed] [Google Scholar]
- Itami et al. 2003.Itami C, Kimura F, Kohno T, Matsuoka M, Ichikawa M, Tsumoto T, Nakamura S. Brain-derived neurotrophic factor-dependent unmasking of “silent” synapses in the developing mouse barrel cortex. Proc Natl Acad Sci USA 100: 13069–13074, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis et al. 1997.Jarvis CR, Xiong ZG, Plant JR, Churchill D, Lu WY, MacVicar BA, MacDonald JF. Neurotrophin modulation of NMDA receptors in cultured murine and isolated rat neurons. J Neurophysiol 78: 2363–2371, 1997. [DOI] [PubMed] [Google Scholar]
- Kang and Schuman 1995.Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science 267: 1658–1662, 1995. [DOI] [PubMed] [Google Scholar]
- Kerr et al. 1999.Kerr BJ, Bradbury EJ, Bennett DL, Trivedi PM, Dassan P, French J, Shelton DB, McMahon SB, Thompson SW. Brain-derived neurotrophic factor modulates nociceptive sensory inputs and NMDA-evoked responses in the rat spinal cord. J Neurosci 19: 5138–5148, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim et al. 2003.Kim HJ, Hwang JJ, Behrens MM, Snider BJ, Choi DW, Koh JY. TrkB mediates BDNF-induced potentiation of neuronal necrosis in cortical culture. Neurobiol Dis 14: 110–119, 2003. [DOI] [PubMed] [Google Scholar]
- Kim et al. 2006.Kim YI, Choi HJ, Colwell CS. Brain-derived neurotrophic factor regulation of N-methyl-d-aspartate receptor-mediated synaptic currents in suprachiasmatic nucleus neurons. J Neurosci Res 84: 1512–1520, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knusel and Hefti 1992.Knusel B, Hefti F. K-252 compounds: modulators of neurotrophin signal transduction. J Neurochem 59: 1987–1996, 1992. [DOI] [PubMed] [Google Scholar]
- Kojima et al. 2001.Kojima M, Takei N, Numakawa T, Ishikawa Y, Suzuki S, Matsumoto T, Katoh-Semba R, Nawa H, Hatanaka H. Biological characterization and optical imaging of brain-derived neurotrophic factor-green fluorescent protein suggest an activity- dependent local release of brain-derived neurotrophic factor in neurites of cultured hippocampal neurons. J Neurosci Res 64: 1–10, 2001. [DOI] [PubMed] [Google Scholar]
- Kolarow et al. 2007.Kolarow R, Brigadski T, Lessmann V. Postsynaptic secretion of BDNF and NT-3 from hippocampal neurons depends on calcium calmodulin kinase II signaling and proceeds via delayed fusion pore opening. J Neurosci 27: 10350–10364, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb et al. 2005.Kolb JE, Trettel J, Levine ES. BDNF enhancement of postsynaptic NMDA receptors is blocked by ethanol. Synapse 55: 52–57, 2005. [DOI] [PubMed] [Google Scholar]
- Kuczewski et al. 2008.Kuczewski N, Porcher C, Ferrand N, Fiorentino H, Pellegrino C, Kolarow R, Lessmann V, Medina I, Gaiarsa JL. Backpropagating action potentials trigger dendritic release of BDNF during spontaneous network activity. J Neurosci 28: 7013–7023, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legrand et al. 2005.Legrand JC, Darbon P, and Streit J. Effects of brain-derived neurotrophic factor (BDNF) on activity mediated by NMDA receptors in rat spinal cord cultures. Neurosci Lett 390: 145–149, 2005. [DOI] [PubMed] [Google Scholar]
- Lessmann et al. 1994.Lessmann V, Gottmann K, Heumann R. BDNF and NT-4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurones. Neuroreport 6: 21–25, 1994. [DOI] [PubMed] [Google Scholar]
- Levine et al. 1998.Levine ES, Crozier RA, Black IB, Plummer MR. Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-d-aspartic acid receptor activity. Proc Natl Acad Sci USA 95: 10235–10239, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine et al. 1995.Levine ES, Dreyfus CF, Black IB, Plummer MR. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci USA 92: 8074–8077, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine and Kolb 2000.Levine ES, Kolb JE. Brain-derived neurotrophic factor increases activity of NR2B-containing N-methyl-d-aspartate receptors in excised patches from hippocampal neurons. J Neurosci Res 62: 357–362, 2000. [DOI] [PubMed] [Google Scholar]
- Li et al. 1998.Li YX, Zhang Y, Lester HA, Schuman EM, Davidson N. Enhancement of neurotransmitter release induced by brain-derived neurotrophic factor in cultured hippocampal neurons. J Neurosci 18: 10231–10240, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin et al. 1998.Lin SY, Wu K, Levine ES, Mount HT, Suen PC, Black IB. BDNF acutely increases tyrosine phosphorylation of the NMDA receptor subunit 2B in cortical and hippocampal postsynaptic densities. Brain Res Mol Brain Res 55: 20–27, 1998. [DOI] [PubMed] [Google Scholar]
- Lindholm et al. 1994.Lindholm D, Castren E, Berzaghi M, Blochl A, Thoenen H. Activity-dependent and hormonal regulation of neurotrophin MRNA levels in the brain—implications for neuronal plasticity. J Neurobiol 25: 1362–1372, 1994. [DOI] [PubMed] [Google Scholar]
- Lu 2004.Lu B Acute and long-term synaptic modulation by neurotrophins. Prog Brain Res 146: 137–150, 2004. [DOI] [PubMed] [Google Scholar]
- Magby et al. 2006.Magby JP, Bi C, Chen ZY, Lee FS, Plummer MR. Single-cell characterization of retrograde signaling by brain-derived neurotrophic factor. J Neurosci 26: 13531–13536, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini et al. 1998.Marini AM, Rabin SJ, Lipsky RH, and Mocchetti I. Activity-dependent release of brain-derived neurotrophic factor underlies the neuroprotective effect of N-methyl-d-aspartate. J Biol Chem 273: 29394–29399, 1998. [DOI] [PubMed] [Google Scholar]
- Mayer and Westbrook 1987.Mayer ML, Westbrook GL. Permeation and block of N-methyl-d-aspartic acid receptor channels by divalent cations in mouse cultured central neurones. J Physiol 394: 501–527, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister et al. 1996.McAllister AK, Katz LC, Lo DC. Neurotrophin regulation of cortical dendritic growth requires activity. Neuron 17: 1057–1064, 1996. [DOI] [PubMed] [Google Scholar]
- McAllister et al. 1999.McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci 22: 295–318, 1999. [DOI] [PubMed] [Google Scholar]
- Meyer-Franke et al. 1998.Meyer-Franke A, Wilkinson GA, Kruttgen A, Hu M, Munro E, Hanson MG Jr, Reichardt LF, Barres BA. Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron 21: 681–693, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer et al. 1994.Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12: 529–540, 1994. [DOI] [PubMed] [Google Scholar]
- Ogata and Tatebayashi 1991.Ogata N, Tatebayashi H. A simple and multi-purpose “concentration-clamp” method for rapid superfusion. J Neurosci Methods 39: 175–183, 1991. [DOI] [PubMed] [Google Scholar]
- Poo 2001.Poo MM Neurotrophins as synaptic modulators. Nat Rev Neurosci 2: 24–32, 2001. [DOI] [PubMed] [Google Scholar]
- Reichardt 2006.Reichardt LF Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci 361: 1545–1564, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval et al. 2007.Sandoval M, Sandoval R, Thomas U, Spilker C, Smalla KH, Falcon R, Marengo JJ, Calderon R, Saavedra V, Heumann R, Bronfman F, Garner CC, Gundelfinger ED, Wyneken U. Antagonistic effects of TrkB and p75(NTR) on NMDA receptor currents in post-synaptic densities transplanted into Xenopus oocytes. J Neurochem 101: 1672–1684, 2007. [DOI] [PubMed] [Google Scholar]
- Schinder et al. 2000.Schinder AF, Berninger B, Poo M. Postsynaptic target specificity of neurotrophin-induced presynaptic potentiation. Neuron 25: 151–163, 2000. [DOI] [PubMed] [Google Scholar]
- Segal and Greenberg 1996.Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci 19: 463–489, 1996. [DOI] [PubMed] [Google Scholar]
- Sigworth and Sine 1987.Sigworth FJ, Sine SM. Data Transformations for improved display and fitting of single-channel dwell time histograms. Biophys J 52: 1047–1054, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack and Thompson 2002.Slack SE, Thompson SW. Brain-derived neurotrophic factor induces NMDA receptor 1 phosphorylation in rat spinal cord. Neuroreport 13: 1967–1970, 2002. [DOI] [PubMed] [Google Scholar]
- Song et al. 1998.Song DK, Choe B, Bae JH, Park WK, Han IS, Ho WK, Earm YE. Brain-derived neurotrophic factor rapidly potentiates synaptic transmission through NMDA, but suppresses it through non-NMDA receptors in rat hippocampal neuron. Brain Res 799: 176–179, 1998. [DOI] [PubMed] [Google Scholar]
- Stocca and Vicini 1998.Stocca G, Vicini S. Increased contribution of NR2A subunit to synaptic NMDA receptors in developing rat cortical neurons. J Physiol 507 :13–24, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoop and Poo 1996.Stoop R, Poo MM. Synaptic msodulation by neurotrophic factors: differential and synergistic Effects Of brain-derived neurotrophic factor and ciliary neurotrophic factor. J Neurosci 16: 3256–3264, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stryker and Harris 1986.Stryker MP, Harris WA. Binocular impulse blockade prevents the formation of ocular dominance columns in cat visual cortex. J Neurosci 6: 2117–2133, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar and Westbrook 1999.Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci 19: 4180–4188, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler et al. 2002.Tyler WJ, Alonso M, Bramham CR, Pozzo-Miller LD. From acquisition to consolidation: on the role of brain-derived neurotrophic factor signaling in hippocampal-dependent learning. Learn Mem 9: 224–237, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu et al. 2000.Xu B, Gottschalk W, Chow A, Wilson RI, Schnell E, Zang K, Wang D, Nicoll RA, Lu B, Reichardt LF. The role of brain-derived neurotrophic factor receptors in the mature hippocampus: modulation of long-term potentiation through a presynaptic mechanism involving TrkB. J Neurosci 20: 6888–6897, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang and Poo 2002.Zhang X, Poo MM. Localized synaptic potentiation by BDNF requires local protein synthesis in the developing axon. Neuron 36: 675–688, 2002. [DOI] [PubMed] [Google Scholar]