

Abstract
In addition to its effects on neuronal survival and differentiation, brain-derived neurotrophic factor (BDNF) plays an important role in modulating synaptic transmission and plasticity in many brain areas, most notably the neocortex and hippocampus. These effects may underlie a role for BDNF in learning and memory as well as developmental plasticity. Consistent with localization of the tropomyosin-related kinase B receptor to both sides of the synapse, BDNF appears to have pre- and postsynaptic effects, but the underlying cellular mechanisms are unclear and it is not known whether pre- and postsynaptic modulations by BDNF occur simultaneously. To address these issues, we recorded dual-component (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid [AMPA] and N-methyl-d-aspartate [NMDA]) miniature excitatory postsynaptic currents (mEPSCs) from cortical and hippocampal pyramidal neurons and dentate gyrus granule cells from acute brain slices. BDNF had no effect on the fast component of mEPSC decay or on the peak amplitude, suggesting that BDNF did not modulate postsynaptic AMPA receptors, although BDNF rapidly modulated NMDA receptors, as seen by an enhancement of the slow component of mEPSC decay that was prevented by blocking postsynaptic NMDA receptors. At the same time, BDNF acted presynaptically to enhance mEPSC frequency. Surprisingly, the effect on frequency was also NMDA receptor dependent, but required activation of presynaptic, not postsynaptic, NMDA receptors. BDNF also enhanced action potential–dependent glutamate release via presynaptic NMDA receptors, an effect that was unmasked when voltage-gated calcium channels were partially inhibited. Our results indicate that BDNF acutely modulates presynaptic release and postsynaptic responsiveness through simultaneous effects on pre- and postsynaptic NMDA receptors.
INTRODUCTION
In addition to their well-established trophic roles in neuronal survival and differentiation, neurotrophins have rapid effects on synaptic transmission and plasticity in the CNS. In particular, brain-derived neurotrophic factor (BDNF) potently influences presynaptic and postsynaptic components of neural transmission in the cortex and hippocampus (Lu 2003), consistent with localization of the tropomyosin-related kinase B (trkB) receptor to axon terminals and dendritic spines (Aoki et al. 2000; Cabelli et al. 1996; Drake et al. 1999). Downstream targets of BDNF–trkB signaling include voltage-gated sodium channels and potassium channels as well as glutamate and γ-aminobutyric acid (GABA) receptors (Blum et al. 2002; Cheng and Yeh 2003; Kramar et al. 2004; Levine et al. 1998). Furthermore, BDNF modulates activity-dependent synaptic plasticity, including some forms of long-term potentiation (LTP) (Figurov et al. 1996; Korte et al. 1995; Patterson et al. 1996) and long-term depression (LTD) (Huber et al. 1998). These effects may underlie the proposed role for BDNF in learning and memory processes (Bekinschtein et al. 2008).
The mechanisms underlying BDNF modulation of activity-dependent plasticity are not clear. Modulation of postsynaptic N-methyl-d-aspartate (NMDA) receptors may play a part in the acute effects of BDNF–trkB signaling on some forms of LTP. BDNF rapidly enhances postsynaptic responses to exogenously applied NMDA, but not α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) (Crozier et al. 1999; Levine et al. 1998), selectively enhances synaptically evoked NMDA currents (Kolb et al. 2005) and increases NMDA single-channel open probability (Levine and Kolb 2000; Levine et al. 1998). The underlying signaling mechanism may involve BDNF enhancement of the phosphorylation of NMDA receptor subunits associated with the postsynaptic density (Lin et al. 1998; Suen et al. 1997). These data suggest that NMDA receptors are a primary postsynaptic target of the BDNF–trkB receptor signaling cascade.
On the presynaptic side, BDNF enhances glutamate release in synaptosomes (Pascual et al. 2001; Sala et al. 1998) and increases the frequency of miniature excitatory postsynaptic currents (mEPSCs) in brain slice and culture preparations (Carmignoto et al. 1997; Lessmann and Heumann 1998; Li et al. 1998a; Schinder et al. 2000; Tyler and Pozzo-Miller 2001). Although the downstream targets mediating these presynaptic effects have not been fully characterized, recent work suggests that synapsin (Jovanovic et al. 2000), rab3a (Alder et al. 2005), and a myosin VI complex (Yano et al. 2006) may contribute to the presynaptic effect of BDNF. Interestingly, presynaptic NMDA receptors have been visualized on glutamatergic axon terminals in the neocortex and hippocampus using electron microscopy (Aoki et al. 1997; Corlew et al. 2007; Jourdain et al. 2007). In the cortex, NMDA receptor antagonists decrease mEPSC frequency, suggesting that presynaptic NMDA receptors regulate spontaneous glutamate release (Berretta and Jones 1996; Brasier and Feldman 2008; Jourdain et al. 2007; Sjostrom et al. 2003; Woodhall et al. 2001). Because postsynaptic NMDA receptors are modulated by BDNF–trkB signaling, presynaptic NMDA receptors represent a potential target for BDNF modulation of transmitter release. In the present studies, we examined the potential for simultaneous pre- and postsynaptic effects of BDNF at glutamatergic synapses in the cortex and hippocampus and investigated the role of NMDA receptors at both sides of the synapse in mediating these effects.
METHODS
All animal procedures were conducted using protocols approved by the University of Connecticut Health Center Animal Care Committee. Following euthanasia of postnatal day 13 (P13) to P20 Swiss CD-1 mice (Charles River, Wilmington, MA), brains were removed and placed in ice-cold “cutting and incubating” solution composed of (in mM): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 0.5 CaCl2, 4 MgCl2, 4 MgSO4, 4 lactic acid, 2 pyruvic acid, 20 glucose, and 0.4 ascorbic acid, carboxygenated with 95% O2-5% CO2 (pH 7.3, 310 ± 5 mmol·kg−1). Transverse slices (300–400 μm) containing visual cortex were cut using a vibratome (Microslicer, Dosaka EM, Kyoto, Japan), transferred to a holding chamber, and incubated at room temperature for ≥45 min prior to recording. Slices were then individually transferred to a heated recording chamber (32–33°C) fixed to the stage of an Olympus BX51WI upright microscope fitted with a ×40 water-immersion objective lens (0.8 NA).
Pyramidal neurons (PNs) in visual cortex and the CA1 region of the hippocampus and dentate gyrus granule cells were visually identified using infrared-differential interference contrast microscopy. The recording chamber was continuously perfused with carboxygenated artificial cerebrospinal fluid (aCSF) containing (in mM): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 4 CaCl2, 17.5 glucose, 0.01 glycine, and either 0.05 or 2 MgCl2 (pH 7.3, 305 ± 4 mmol·kg−1). There was no compensation for the change in divalent cation ratio when extracellular Mg2+ was reduced. The bath solution also contained either 20 μM bicuculline or 10 μM GABAzine to block GABAA receptors. For recording miniature, action-potential (AP)–independent EPSCs, tetrodotoxin (TTX; 1 μM) was added to the bath solution to block voltage-dependent Na+ channels. Patch pipettes were pulled from borosilicate glass (3–6 MΩ) using a horizontal puller (Sutter Instruments, Novato, CA). The pipette solution contained (in mM): 20 KCl, 100 K-gluconate, 10 HEPES, 4 Mg2+-ATP, 0.3 Na+-GTP, and 10 phosphocreatine (pH 7.3, 285 ± 2 mmol·kg−1). For experiments using (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate (MK-801; 500 μM) in the pipette solution, three depolarizing pulses to −10 mV (2 s/pulse) over the course of 10–15 min were delivered to the cell to facilitate the block by MK-801. For evoked EPSCs, TTX was omitted from the perfusate and a bipolar tungsten electrode (resistance = 1 MΩ; WPI, Sarasota, FL) was positioned 100–150 μm lateral to the patched neuron. Extracellular shocks typically consisted of a single square-wave current pulse (duration 60–120 μs, amplitude = 15–100 μA) and were delivered at a frequency of 0.1 Hz. In some experiments, paired stimuli were delivered with an interstimulus interval of 50–70 ms to obtain the paired-pulse ratio (PPR). The PPR of evoked postsynaptic currents was calculated by dividing the mean peak amplitude of the second evoked response (A2) by the mean peak amplitude of the first response (A1; PPR = mean of A2/mean of A1) (Kim and Alger 2001).
Signals were recorded with an Axopatch 200B amplifier (Axon Instruments, Foster City, CA) or a HEKA EPC 9 amplifier (HEKA Elektronik, Darmstadt, Germany), sampled at ≥6 kHz and filtered at 2 kHz. Series resistance was compensated 60–70%. All drugs were delivered by bath perfusion at 2 ml·min−1. Neurons were discarded from analysis if: 1) the access resistance changed by >10% during the course of an experiment or 2) the holding current needed to keep Vm at −70 mV increased by >50 pA. Bicuculline methiodide, Ro25-6981, MK-801, d(−)-2-amino-5-phosphonopentanoic acid (AP5), and 6,7-dinitroquinoxaline-2,3-dione (DNQX) were purchased from Sigma (St. Louis, MO). BDNF, neurotrophin-3 (NT-3), and TTX were obtained from Alomone Labs (Jerusalem, Israel). GABAzine was purchased from Tocris (Ellisville, MO). K-252a was obtained from Calbiochem (La Jolla, CA) and dissolved in DMSO (final concentration = 0.01%), which by itself had no effect on synaptic transmission (Trettel and Levine 2002).
Off-line analysis of mEPSCs was performed using MiniAnalysis (v.6.0.3, Synaptosoft, Decatur, GA). The selected threshold detection level (typically 5–12 pA) was set threefold higher than the baseline root-mean-square noise and remained constant for the duration of each experiment. Overlapping events or events with poorly resolved baselines or rise times >5 ms were excluded from analysis. Changes in amplitude, frequency, and decay time were quantified during the last 120 s of each drug period relative to baseline. Decay times of events averaged over 1-min bins during the experimental time course were best fit by either a single-exponential equation, I(t) = A exp(−t/τ) or double-exponential equation, I(t) = A1 exp(−t/τ1) + A2 exp(−t/τ2), where I is the current amplitude at any given time (t), A1 and A2 are the amplitudes of the fast and slow decay components, and τ1 and τ2 are their respective decay time constants. Off-line analysis of evoked EPSCs was performed using Clampfit (v9.2, Axon Instruments, Union City, CA). Amplitudes were measured by averaging a 5- to 10-ms period around the peak and subtracting the average baseline value during a 10-ms period taken just before stimulus onset. Data were compared using Student's paired t-test and Kolmogorov–Smirnov (KS) two-sample tests. Group data are reported as means ± SE.
RESULTS
To examine the pre- and postsynaptic effects of acute exposure to BDNF at glutamatergic synapses, we recorded dual-component (AMPA and NMDA) mEPSCs from neocortical and hippocampal brain slices. The postsynaptic NMDA component was unmasked by using a very low concentration of extracellular Mg2+ (0.05 mM), which allowed for the recording of mEPSC activity at a physiological holding potential of −70 mV. Under these conditions, the decay time of individual events was best fit using two exponential time constants, where the faster time constant (τ1) predominantly reflects the AMPA receptor component and the slower time constant (τ2) reflects the NMDA receptor component (McBain and Dingledine 1992; O'Brien et al. 1997). In support of this, we found that blocking NMDA receptors with the selective antagonist (±)-3-(2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (CPP; 5 μM) or by increasing the extracellular Mg2+ concentration to 2 mM eliminated the slower component of decay. Under these conditions, the decay time was best fit with a single exponential (Fig. 1A). Blocking NMDA receptors had no significant effect on peak amplitude (baseline: 15.5 ± 1.4 vs. CPP: 14.3 ± 1.6 pA, n = 6). Addition of the non-NMDA antagonist DNQX (10 μM) in the presence of CPP completely abolished all events. Thus under low Mg2+ conditions, τ1 and peak amplitude reflected the AMPA receptor contribution and τ2 reflected the NMDA receptor contribution.
FIG. 1.
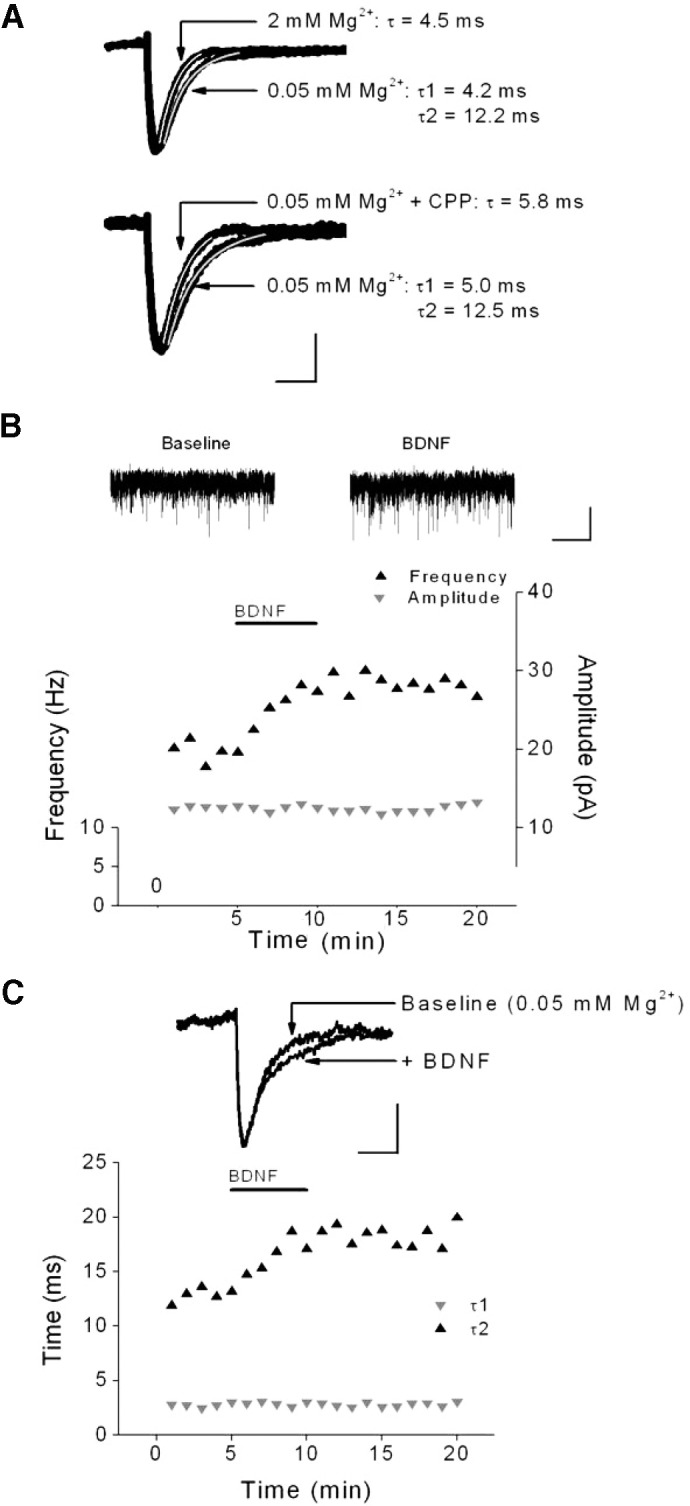
Brain-derived neurotrophic factor (BDNF) rapidly modulated miniature excitatory postsynaptic current (mEPSC) frequency and decay kinetics but not amplitude. A: representative sweeps from a single cell showing the effect of extracellular Mg2+ on mEPSC decay kinetics (top) or the effect of the N-methyl-d-aspartate (NMDA) receptor antagonist CPP (5 μM) on a different cell (bottom). Each sweep is the average of >100 events. The superimposed lines indicate the fit to single (white) or double (gray) exponentials of the decay times. Scale bars: 5 pA, 10 ms. B: effect of BDNF (0.8 nM) on the frequency and amplitude of mEPSCs from a representative layer 5 pyramidal neuron (PN) recorded in 0.05 mM extracellular Mg2+. Traces of mEPSCs prior to and during BDNF are shown above. Scale bars: 15 pA, 5 s. C: time course and example sweeps from same cell as B showing effect of BDNF on mEPSC decay times. Decay was fit using 2 exponential time constants. Scale bars: 5 pA, 10 ms.
In the first set of experiments, mEPSCs were recorded from visual cortical layer 5 PNs in ACSF containing TTX, bicuculline, and low (0.05 mM) extracellular Mg2+. As shown in the example in Fig. 1B, application of BDNF (20 ng/ml, 0.8 nM) increased mEPSC frequency within minutes of exposure, but had no effect on peak amplitude. At the same time, BDNF selectively prolonged the slow component of decay time, as reflected by an increase in τ2, but not τ1 (Fig. 1C). The effect on mEPSC parameters was not a consequence of “run-up” during recordings because prolonged recordings (>10 min) in low extracellular Mg2+ had no effect on mEPSC frequency (103.3 ± 2.5% of baseline; n = 3). As shown in the group data in Fig. 2, BDNF significantly increased mEPSC frequency (baseline: 3.7 ± 0.7 vs. BDNF: 4.8 ± 0.2 Hz, n = 16, P < 0.05) and τ2 (baseline: 13.1 ± 0.9 vs. BDNF: 18.3 ± 1.1 ms, n = 16, P < 0.05) in layer 5 PNs. The effect of BDNF on frequency and decay time typically occurred within 2 min of BDNF application, outlasted the 5-min BDNF application, and generally returned to baseline levels after 15–20 min. BDNF had no effect on τ1 (baseline: 4.4 ± 0.3 vs. BDNF: 4.5 ± 0.2 ms) or peak amplitude (baseline: 10.5 ± 0.6 vs. BDNF: 10.7 ± 0.6 pA) (Fig. 2) and did not alter the amplitude distribution (P > 0.05, KS test). This effect was not limited to neocortical PNs of the visual cortex because, as shown in Fig. 2, similar results were obtained from PNs in the hippocampal CA1 region (amplitude: baseline = 10.1 ± 0.9 vs. BDNF = 10.0 ± 0.9; frequency: baseline = 1.1 ± 0.3 vs. BDNF = 1.8 ± 0.5 Hz; τ1: baseline = 4.8 ± 0.4 vs. BDNF = 5.0 ± 0.4 ms; τ2: baseline = 16.2 ± 1.4 vs. BDNF = 23.3 ± 1.4 ms; n = 5) and granule cells in the dentate gyrus (amplitude: baseline = 14.7 ± 1.5 vs. BDNF = 14.3 ± 1.9; frequency: baseline = 0.3 ± 0.1 vs. BDNF = 0.6 ± 0.1 Hz; τ1: baseline = 5.0 ± 0.6 vs. BDNF = 4.9 ± 0.5 ms; τ2: baseline = 15.6 ± 1.4 vs. BDNF = 29.7 ± 3.4 ms; n = 5). Combining the data from all cell types, we found that BDNF enhanced mEPSC frequency (≥25% increase) in 19/26 cells tested and prolonged τ2 decay time (≥25% increase) in 21/26 cells. These results suggest that BDNF exerts simultaneous pre- and postsynaptic effects on neocortical and hippocampal glutamatergic synaptic transmission by enhancing both mEPSC frequency and decay time.
FIG. 2.

BDNF enhanced mEPSC frequency and decay kinetics but not amplitude in different brain regions. Group data showing effects of BDNF on mEPSC frequency, amplitude, and decay times in layer 5 PNs of visual cortex (n = 16), hippocampal PNs (n = 5), and dentate gyrus cells (n = 5). *P < 0.05.
Next, we wanted to determine whether the effects of BDNF were mediated by trkB receptors and explore whether activation of distinct subsets of trkB receptors were responsible for the pre- and postsynaptic effects of BDNF. As shown in Fig. 3A, bath application of K-252a, which preferentially inhibits trk tyrosine kinase activity at the concentration used (200 nM; Berg et al. 1992; Knusel and Hefti 1992), completely prevented the effects of BDNF on both mEPSC frequency and decay in L5 PNs of visual cortex. Moreover, postsynaptic loading of K-252a (200 nM) via the recording pipette, which inhibits the postsynaptic effect of BDNF on NMDA responsiveness (Kolb et al. 2005), blocked the effect of BDNF on τ2 decay time (baseline: 11.2 ± 1.2 vs. BDNF: 11.1 ± 1.2 ms; n = 5), but did not prevent the increase in mEPSC frequency (baseline: 1.3 ± 0.2 vs. BDNF: 1.9 ± 0.3 ms), indicating that activation of postsynaptic trkB receptors was required for the postsynaptic effect of BDNF on decay time, but not the presynaptic effect on mEPSC frequency. These results also indicate that intracellularly loaded K-252a did not diffuse out of the cell in sufficient amounts to block presynaptic receptors. In contrast, bath application of the membrane-impermeable analog, K-252b (400 nM), did not block any of the effects of BDNF (see Fig. 6, A and B). The related neurotrophin NT-3 (20 ng/ml), which binds to trkC receptors, had no effect on mEPSC frequency (baseline: 3.3 ± 0.2 vs. NT-3: 2.9 ± 0.2 Hz), amplitude (baseline: 13.7 ± 1.3 vs. NT-3: 13.6 ± 1.3 pA), or decay times (τ1: baseline: 4.7 ± 0.8 vs. NT-3: 4.1 ± 0.5 ms; τ2: baseline: 13.4 ± 1.4 vs. NT-3: 13.9 ± 1.2 ms), as shown in Fig. 3A. These data suggest that BDNF-induced modulation of mEPSC frequency and decay time depends on selective activation of presynaptic and postsynaptic trkB receptors, respectively.
FIG. 3.

BDNF modulation required tropomyosin-related kinase B (trkB) receptors and NMDA receptors. A: effect of BDNF with either bath-applied K-252a (200 nM; n = 4) or intracellular K-252a (200 nM; n = 5) or bath-applied neurotrophin 3 (NT-3) alone (20 ng/ml; n = 7). B: lack of effect of BDNF on mEPSC decay time in the presence of CPP (5 μM), 2 mM extracellular Mg2+, or intracellular MK-801 (500 μM). Under these conditions, decay time was fit to a single exponential. In this and all subsequent figures, numbers in parentheses indicate number of experiments. C: lack of effect of BDNF on mEPSC frequency in the presence of CPP (n = 6). All recordings were from layer 5 PNs. *P < 0.05.
FIG. 6.

The effect of BDNF on mEPSC frequency required presynaptic trkB receptors and did not involve retrograde signaling. A: individual example time courses for the effect of BDNF (0.8 nM) with vehicle, bath-applied K-252a, or postsynaptic K-252a. B: group data for effect of BDNF on mEPSC frequency in the presence of vehicle, bath-applied K-252a (200 nM), or K-252b (400 nM), or intracellular K-252a (200 nM). C: group data for effect of BDNF in the presence of the mGluR I/II antagonist MCPG (500 μM), the nitric oxide synthase inhibitor L-NAME (500 μM), the GABAB receptor antagonist CGP 35348 (CGP; 100 μM), or the adenosine A2A antagonist ZM 241385 (ZM, 50 nM). Recordings were from layer 5 PNs and were conducted in the presence of 2 mM extracellular Mg2+. *P < 0.05.
We hypothesized that the increase in τ2 decay time reflected BDNF modulation of postsynaptic NMDA receptors. BDNF has been shown to selectively enhance postsynaptic NMDA, but not AMPA, responses (Kolb et al. 2005; Levine et al. 1998) and BDNF increases NMDA receptor single-channel activity in both cell-attached and -excised patches (Levine and Kolb 2000; Levine et al. 1998). Consistent with these previous findings, the NMDA receptor antagonist CPP (5 μM) completely blocked the effect of BDNF on decay time in neocortical PNs (CPP baseline τ = 6.0 ± 0.4 vs. BDNF + CPP τ = 5.7 ± 0.4 ms, n = 6, Fig. 3B). Increasing extracellular Mg2+ to 2 mM, which causes a voltage-dependent block of NMDA receptors, also completely prevented the effect of BDNF on decay time (Mg2+ baseline: 5.8 ± 0.4 vs. Mg2+ + BDNF: 6.1 ± 0.4 ms, n = 8, Fig. 3B). In addition, the intracellular application of the noncompetitive NMDA receptor antagonist MK-801 selectively blocks postsynaptic NMDA receptors (Bender et al. 2006; Berretta and Jones 1996; Corlew et al. 2007; Garraway et al. 2003; Woodhall et al. 2001). As shown in Fig. 3B, including MK-801 (500 μM) in the patch pipette prevented the effect of BDNF on decay time (baseline: 6.5 ± 0.4 vs. BDNF: 6.7 ± 0.4 ms). As noted earlier, decay times were best fit to a single exponential under conditions where postsynaptic NMDA receptors were blocked. Taken together, the selective effect of BDNF on τ2 but not τ1 or peak amplitude, and the complete block of the effect by NMDA receptor antagonists or extracellular Mg2+, indicates that BDNF selectively modulates postsynaptic NMDA, but not AMPA, receptors.
Surprisingly, we found that CPP not only blocked the postsynaptic effect of BDNF on mEPSC decay time (Fig. 3B), but this NMDA receptor antagonist also prevented the presynaptic effect of BDNF on mEPSC frequency in layer 5 PNs (Fig. 3C), as well as in CA1 PNs and dentate gyrus granule cells (n = 7). Because the BDNF-induced increase in mEPSC frequency appeared to be dependent on presynaptic trkB receptor activation, we examined the possibility that this effect required presynaptic NMDA receptors. A number of reports have demonstrated the existence of presynaptic NMDA receptors in the neocortex and hippocampus (Aoki et al. 1994; Brasier and Feldman 2008; Charton et al. 1999; Corlew et al. 2007; DeBiasi et al. 1996; Rodriguez-Moreno and Paulsen 2008). These receptors have been shown to enhance presynaptic glutamate release probability (Bender et al. 2006; Brasier and Feldman 2008; Sjostrom et al. 2003; Woodhall et al. 2001).
We tested this hypothesis by using several established procedures to selectively block postsynaptic NMDA receptors. First, we elevated extracellular Mg2+ from 0.05 to 2 mM while recording at a holding potential of −70 mV. It has been shown in several reports that maintaining a hyperpolarized membrane potential in the presence of Mg2+ selectively blocks postsynaptic, but not presynaptic, NMDA receptors (Bender et al. 2006; Corlew et al. 2007; Woodhall et al. 2001; Yang et al. 2006). As shown earlier, we found that the postsynaptic effect of BDNF on decay time was prevented in the presence of 2 mM Mg2+ (see group data in Fig. 3B). In the presence of 2 mM Mg2+, the NMDA receptor antagonist CPP (5 μM) decreased the baseline mEPSC frequency and completely blocked the effect of BDNF (see example in Fig. 4A and group data in Fig. 4B; baseline: 1.5 ± 0.1; CPP: 1.2 ± 0.1; CPP + BDNF: 1.2 ± 0.1 Hz). To rule out potential nonspecific effects of CPP, we repeated these experiments using several structurally different NMDA receptor antagonists and obtained the same results. The NMDA receptor antagonist AP5 (100 μM, baseline: 2.3 ± 0.2; AP5: 1.4 ± 0.1; AP5 + BDNF: 1.4 ± 0.1 Hz), the selective NR2B antagonist Ro25-6981 (Ro25, 1 μM, baseline: 1.7 ± 0.1; Ro25: 1.2 ± 0.1; Ro25 + BDNF: 1.3 ± 0.1 Hz), and the selective NR2B antagonist ifenprodil (3 μM, baseline: 2.4 ± 0.3; ifenprodil: 1.6 ± 0.2; ifenprodil + BDNF: 1.5 ± 0.2 Hz) each decreased mEPSC frequency, suggesting that presynaptic NMDA receptors tonically enhance the frequency of spontaneous glutamate release. Furthermore, presynaptic NMDA receptors could be activated by application of the NMDA agonist homoquinolinic acid (HQA, 5 μM) in the presence of 2 mM extracellular Mg2+ as suggested by the increase mEPSC frequency (baseline: 3.4 ± 1.3 vs. HQA: 5.2 ± 1.1 Hz, n = 5, P < 0.05; 205 ± 54.3% of baseline). There was no effect of HQA on mEPSC amplitude (baseline: 10.7 ± 1.0 vs. HQA: 9.5 ± 1.0 pA; 90.1 ± 5.7% of baseline). Moreover, the presence of these NMDA receptor antagonists completely prevented the effect of BDNF (Fig. 4B) without altering mEPSC amplitude (101.1 ± 3.0% of baseline). These results suggest that the effect of BDNF on mEPSC frequency was mediated by presynaptic NMDA receptors that contain the NR2B subunit.
FIG. 4.

NMDA receptor antagonists decreased mEPSC frequency and blocked the effect of BDNF in the presence of 2 mM Mg2+. A: representative traces from 2 layer 5 PNs showing the effect of BDNF (0.8 nM) on mEPSC frequency in the absence or presence of CPP (5 μM, scale bars: 20 pA, 1 s). B: effects of vehicle (n = 7), CPP (5 μM; n = 12), AP5 (100 μM; n = 5), Ro25-6981 (Ro25; 1 μM; n = 5), and ifenprodil (3 μM; n = 8) alone on baseline mEPSC frequency (white bars) or in the presence of BDNF (shaded bars). C: effect of raising extracellular Mg2+ from 0.05 to 2 mM on the evoked postsynaptic NMDA current (scale bars: 25 pA, 100 ms) and the postsynaptic response to puff-applied NMDA (50 μM NMDA + 3 μM glycine; scale bars: 50 pA, 500 ms). Average of 5 sweeps per condition. All recordings were from layer 5 PNs. *P < 0.05.
It is possible that rather than acting on presynaptic NMDA receptors, NMDA antagonists were blocking a subpopulation of Mg2+-insensitive postsynaptic NMDA receptors. However, as shown in Fig. 4C, increasing Mg2+ from 0.05 to 2 mM blocked electrically evoked postsynaptic NMDA currents (baseline peak amplitude: 37.1 ± 6.5 vs. 2 mM Mg2+ peak: 3.0 ± 0.4 pA, n = 6, P < 0.05) and also blocked postsynaptic responses to puff-applied exogenous NMDA (50 μM; baseline peak NMDA response: 69.8 ± 5.6 vs. 2 mM Mg2+ NMDA response: 4.5 ± 0.6 pA, n = 4, P < 0.05). Activation of AMPA receptors could provide sufficient depolarization to relieve the voltage-dependent block of postsynaptic NMDA receptors by Mg2+; however, CPP had no effect on mEPSC decay time in the presence of 2 mM extracellular Mg2+ (baseline 2 mM Mg2+: 5.7 ± 0.3 ms vs. 2 mM Mg2+ + CPP: 5.6 ± 0.9 ms; n = 5). These results indicate that maintaining a hyperpolarized membrane potential in the presence of 2 mM extracellular Mg2+ completely blocked postsynaptic NMDA receptors.
An alternative method for selectively blocking postsynaptic NMDA receptors is to include the NMDA open channel blocker MK-801 (500 μM) in the recording pipette (Bender et al. 2006; Berretta and Jones 1996; Brasier and Feldman 2008; Corlew et al. 2007; Yang et al. 2006). As shown in Fig. 5, intracellular MK-801 did not prevent the effects of either BDNF (baseline: 0.9 ± 0.1 vs. BDNF: 1.2 ± 0.1 Hz, n = 9, P < 0.05) or CPP (baseline 3.2 ± 0.1 vs. BDNF: 2.6 ± 0.1 Hz, n = 5, P < 0.05) on mEPSC frequency in the presence of 0.05 mM Mg2+. Intracellular MK-801 did block the effects of BDNF and CPP on decay time, however, confirming its effectiveness in blocking postsynaptic NMDA receptors. Thus under conditions where postsynaptic NMDA receptors were selectively blocked by MK-801, both BDNF and CPP exerted a presynaptic but not postsynaptic effect.
FIG. 5.

Selectively blocking postsynaptic NMDA receptors does not prevent the presynaptic effects of BDNF or CPP. Hatched bars (left) show the effect of BDNF (0.8 nM) or CPP (5 μM) on mEPSC frequency with MK-801 (500 μM) in the recording pipette, whereas shaded bars (right) confirm that the effects of BDNF or CPP on decay time were blocked by MK-801. *P < 0.05.
We next examined whether the effect of BDNF on mEPSC frequency required activation of presynaptic trkB receptors. As shown earlier (Fig. 3A), in the presence of 0.05 mM Mg2+, bath application of K-252a, but not postsynaptic application, prevented the effect of BDNF on mEPSC frequency. Similar results were obtained in the presence of 2 mM Mg2+. As shown in Fig. 6, A and B, BDNF significantly increased mEPSC frequency in the presence of either the DMSO vehicle (baseline: 1.0 ± 0.1 vs. BDNF: 1.3 ± 0.1 Hz) or the membrane-impermeant analog K-252b (baseline: 1.7 ± 1.0 vs. BDNF: 2.4 ± 1.2 Hz). Bath application of K-252a prevented the effect of BDNF (baseline: 0.9 ± 0.1 vs. BDNF: 0.9 ± 0.1 Hz), whereas postsynaptic dialysis of K-252a did not prevent the effect of BDNF on mEPSC frequency (baseline: 0.9 ± 0.1 vs. BDNF: 1.2 ± 0.1 Hz). It is conceivable that BDNF could activate trkB receptors on neighboring neuronal or nonneuronal cells and induce the release of a retrograde signal that altered release probability via presynaptic NMDA receptors. We therefore examined the effect of BDNF in the presence of antagonists to potential retrograde signals, including glutamate, nitric oxide, GABA, and adenosine. As shown in Fig. 6C, however, BDNF significantly increased mEPSC frequency in the presence of the nonselective mGluR antagonist MCPG (baseline: 0.7 ± 0.03 vs. BDNF: 1.0 ± 0.05 Hz), the nitric oxide synthase inhibitor L-NAME (baseline: 1.4 ± 0.1 vs. BDNF: 1.7 ± 0.1 Hz), the GABAB antagonist CGP 35348 (baseline: 1.2 ± 0.1 vs. BDNF: 1.7 ± 0.1 Hz), or the adenosine A2A receptor inhibitor ZM 241385 (baseline: 1.7 ± 0.3 vs. BDNF: 2.2 ± 0.2 Hz). Although baseline mEPSC frequency varied across experiments, all analyses were based on within-cell comparisons and there was no correlation between the baseline frequency and the effect, or lack of effect, of BDNF.
Although BDNF reliably increased mEPSC frequency, changes in spontaneous release do not necessarily translate to changes in evoked AP-dependent release. We investigated this issue by recording from L5 PNs and evoking EPSCs using a bipolar stimulating electrode located 100–150 microns lateral to the patched neuron. As shown in the example in Fig. 7A and the group data in Fig. 7C, BDNF had no effect on the peak amplitude of evoked EPSCs under basal conditions, suggesting that calcium influx through presynaptic NMDA receptors does not contribute to AP-dependent release of glutamate under these conditions. We hypothesized that the role of presynaptic NMDA receptors on evoked release may be unmasked under conditions of reduced calcium influx through voltage-dependent calcium channels (VDCCs). To decrease the contribution of calcium influx through VDCCs, we supplemented the normal aCSF perfusate with a low concentration of cadmium (Cd2+; 5 μM), a nonspecific VDCC blocker. As shown in the example in Fig. 7B, Cd2+ decreased the size of the evoked EPSC by about 50%. Application of Cd2+ significantly increased the paired-pulse ratio (baseline PPR: 1.3 vs. Cd2+ PPR: 1.9, n = 7, P < 0.05) consistent with a presynaptic effect. Moreover, in the presence of Cd2+, BDNF significantly enhanced evoked EPSC amplitude (128.3 ± 7.9% of Cd2+ baseline, n = 7, P < 0.05; Fig. 7C). Similar to the effect of BDNF on mEPSC frequency, the potentiation of evoked EPSCs by BDNF typically occurred within 2 min, but variability in recording depths of patched neurons precluded finer resolution of onset time. This potentiation was completely blocked by bath-applied K-252a (96.8 ± 7.2% of Cd2+ baseline; Fig. 7C), suggesting that the effect was mediated by trkB receptor activation. BDNF also significantly decreased the PPR of evoked EPSCs (Fig. 7D), suggesting a presynaptic site of action. Thus under conditions of reduced calcium influx through VDCCs, BDNF significantly enhanced AP-dependent evoked glutamate release.
FIG. 7.

Differential effects of BDNF on evoked EPSCs. A: lack of effect of BDNF (0.8 nM) on evoked EPSCs from a layer 5 PN under basal conditions (artificial cerebrospinal fluid [aCSF]. For A and B, representative sweeps (average of 5 consecutive sweeps) are shown above; stimulus artifacts have been blanked for clarity; scale bars: 50 pA, 10 ms. B: effect of BDNF on evoked EPSCs from a layer 5 PN in the presence of 5 μM extracellular Cd2+. C: group data showing effect of BDNF on evoked EPSC amplitude recorded in normal aCSF and in the presence of 5 μM extracellular Cd2+. Pretreatment with 200 nM K-252a in the bath blocked the effect of BDNF. D: effect of BDNF on paired-pulse ratio (PPR) in the presence of 5 μM Cd2+. E: group data showing effect of the NMDA receptor antagonist CPP (6 μM) on evoked EPSC amplitude in normal aCSF and in the presence of 5 μM extracellular Cd2+. BDNF had no effect on evoked EPSCs in the presence of CPP and Cd2+. *P < 0.05.
Finally, we wanted to determine whether the effect of BDNF on evoked EPSCs was dependent on presynaptic NMDA receptors, similar to the effect of BDNF on mEPSC frequency. Because the postsynaptic neuron was held at a hyperpolarized membrane potential (Vh = −90 mV) in the presence of 2 mM Mg2+, this effect was most likely not due to modulation of postsynaptic NMDA receptors. In addition, under baseline conditions, CPP had no effect on evoked EPSC amplitude, but in the presence of 5 μM Cd2+, CPP decreased the size of evoked EPSCs (Fig. 7E), suggesting a role for presynaptic NMDA receptors. Importantly, in a separate set of experiments, the effect of BDNF on evoked EPSCs in the presence of Cd2+ was completely blocked by CPP (97.2 ± 6.9% of CPP baseline, Fig. 7E). Thus these data suggest that the effect of BDNF on AP-dependent evoked glutamate release required presynaptic NMDA receptor activity.
DISCUSSION
The present report demonstrates that acute application of BDNF has simultaneous pre- and postsynaptic effects at glutamatergic synapses onto neocortical layer 5 PNs, hippocampal CA1 PNs, and dentate gyrus granule cells in acute brain slices. By recording dual-component mEPSCs, we found that BDNF significantly enhanced activation of postsynaptic NMDA, but not AMPA, receptors and also increased presynaptic glutamate release probability. In contrast to a recent study in cultured embryonic hippocampal neurons (Alder et al. 2005), we found that both the pre- and postsynaptic effects of BDNF occurred within minutes of application and generally outlasted a brief BDNF application. Both the pre- and postsynaptic effects were mediated by activation of trkB receptors because these effects were completely blocked by bath application of the tyrosine kinase inhibitor K-252a. The modulation of postsynaptic NMDA receptor decay time was mediated by activation of postsynaptic trkB receptors because this effect was blocked by inclusion of K-252a in the postsynaptic cell. Blockade of postsynaptic trkB receptors, however, did not block the effect of BDNF on presynaptic release probability, suggesting an additional role for presynaptic trkB receptors. These results are consistent with a study using cultured hippocampal neurons that also showed a BDNF-induced increase in mEPSC frequency that was dependent on presynaptic, and not postsynaptic, trkB activation (Li et al. 1998b). Furthermore, trkB receptors have been localized to axon terminals (Cabelli et al. 1996; Drake et al. 1999). It is also possible that activation of trkB receptors located on nearby neuronal or nonneuronal cells could induce the release of a retrograde messenger that in turn affects presynaptic glutamate release. This is unlikely, however, because we found that the effect of BDNF on mEPSC frequency persisted in the presence of antagonists for metabotropic glutamate receptors, nitric oxide synthase, GABAB receptors, or adenosine receptors.
Surprisingly, the presynaptic effect of BDNF also required NMDA receptor activation becauses it was blocked by bath application of several structurally distinct NMDA receptor antagonists. Furthermore, the effect of BDNF on mEPSC frequency specifically depended on presynaptic NMDA receptors because it was not altered when postsynaptic NMDA receptors were blocked by hyperpolarization/elevated Mg2+ or by postsynaptic loading of the NMDA open channel blocker MK-801. Anatomical evidence supports the expression of NMDA receptors on glutamatergic terminals (Aoki et al. 1997; Corlew et al. 2007; Jourdain et al. 2007) and decreases in mEPSC frequency in the presence of NMDA receptor antagonists suggest that these presynaptic NMDA receptors contribute to spontaneous transmitter release. Presynaptic NMDA receptors have also been shown to mediate neurosteroid modulation of glutamate release probability in the CA1 area of rat hippocampus (Mameli et al. 2005). Interestingly, this NR2D-mediated effect was observed only in animals younger than P6. In the present studies we observed functional presynaptic NMDA receptors at P13–P20 in mouse hippocampal slices. Although Mameli et al. (2005) did not examine slices from rats older than P10, this possible discrepancy may be due to species differences in NMDA receptor expression or developmental changes in subunit expression that alter neurosteroid sensitivity of NMDA receptors.
It has been consistently shown, both in the present results and previous studies (Bender et al. 2006; Brasier and Feldman 2008; Corlew et al. 2007; Sjostrom et al. 2003; Woodhall et al. 2001), that presynaptic NMDA receptors are relatively insensitive to extracellular Mg2+. Although the mechanisms are unclear, relief from the voltage-dependent block by Mg2+ may be due to spontaneous depolarization of axon terminals or a higher resting membrane potential at presynaptic terminals. It is also possible that presynaptic NMDA receptors have decreased Mg2+ sensitivity due to their subunit composition; NR2C, NR2D, and NR3A subunits have been shown to confer decreased sensitivity to voltage-dependent Mg2+ block (Cull-Candy et al. 2001; Monyer et al. 1992; Sasaki et al. 2002). Interestingly, the NR2D receptor subunit is expressed in cortex and possibly forms ternary NR1/NR2B/NR2D subunit complexes (Dunah et al. 1996).
An important question is whether BDNF-induced changes in mEPSC frequency translate to changes in evoked, AP-dependent release. Under baseline conditions, we found that neither BDNF nor CPP had an effect on evoked EPSCs mediated by postsynaptic AMPA receptors. This finding is in agreement with several earlier studies (Figurov et al. 1996; Gottschalk et al. 1998; Huber et al. 1998; Patterson et al. 1996). We hypothesized that under basal conditions, calcium influx through VDCC may occlude the contribution of presynaptic NMDA receptors to evoked release. In support of this idea, partial inhibition of Ca2+ influx through VDCCs by cadmium unmasked a significant effect of BDNF as well as CPP on evoked glutamate release. The effect of BDNF under these conditions, where postsynaptic NMDA receptors were blocked by 2 mM Mg2+, was prevented by an NMDA receptor antagonist. Furthermore, BDNF decreased the PPR, consistent with a presynaptic effect of BDNF on evoked glutamate release.
The downstream targets of BDNF–trkB receptor signaling in the presynaptic terminal are uncertain. Previous studies have suggested that BDNF can increase the number of docked vesicles at axon terminals and/or enhance the phosphorylation of proteins involved in transmitter release such as synapsins (Lu 2003). More recent studies suggest that BDNF may also enhance transmission through the presynaptic vesicle protein rab3A (Alder et al. 2005) or through a myosin VI-GIPC 1 complex (Yano et al. 2006). An intriguing possibility is that modulation of presynaptic NMDA receptors by BDNF lies upstream of the targets suggested by these previous studies. BDNF–trkB modulation of presynaptic NMDA receptors may dynamically influence calcium influx at axon terminals leading to subsequent changes in the number of docked vesicles and/or the phosphorylation states of vesicle proteins and transmitter release machinery.
Most studies to date have focused on the role of postsynaptic NMDA receptors in mediating different forms of activity-dependent synaptic plasticity (Malenka and Bear 2004). Evidence from several different brain regions, however, suggests a role for presynaptic NMDA receptors in certain forms of plasticity. One study suggests presynaptic NMDA receptors are required for the induction of LTP in the amygdala (Humeau et al. 2003), whereas other studies in visual and somatosensory cortex suggest presynaptic NMDA receptors are involved in the induction of LTD (Bender et al. 2006; Corlew et al. 2007; Rodriguez-Moreno and Paulsen 2008; Sjostrom et al. 2003). Yet it is unclear how activation of presynaptic NMDA receptors, which facilitate spontaneous glutamate transmission as shown in these and other studies (Berretta and Jones 1996; Jourdain et al. 2007; Li and Han 2007; Woodhall et al. 2001), may ultimately lead to depression of synaptic transmission in these studies. The present results suggest that BDNF-induced enhancement of both pre- and postsynaptic NMDA receptors may play an important role in regulating NMDA-dependent synaptic plasticity, including LTP. In fact, BDNF has been shown to be crucial for some forms of hippocampal LTP (Lu 2003), but it is unclear whether this is primarily due to the pre- or postsynaptic actions of BDNF (Kovalchuk et al. 2002; Xu et al. 2000; Zakharenko et al. 2003). Our findings suggest that BDNF could influence the induction threshold and/or the magnitude of NMDA receptor-dependent plasticity via both pre- and postsynaptic effects. Indeed, recent studies suggest burgeoning evidence for coordinate pre- and postsynaptic mechanisms underlying the role of BDNF in synaptic plasticity (Alder et al. 2005; Gartner et al. 2006; Li and Keifer 2008). From our findings, it is possible, for example, that calcium influx through presynaptic NMDA receptors could enhance glutamate release during bouts of high-frequency stimulation used to induce LTP. Interestingly, previous work has shown that BDNF attenuates synaptic fatigue through a presynaptic locus (Figurov et al. 1996; Pozzo-Miller et al. 1999). Moreover, a recent study suggests a possible role for endogenously released BDNF in blocking the induction of LTD at murine visual cortical synapses (Huang et al. 2008). Whether endogenous BDNF may block LTD induction through effects on presynaptic NMDA receptors was not explored in this study. Modulation of postsynaptic NMDA receptors, on the other hand, could directly facilitate calcium influx required for LTP induction. The simultaneous and synergistic modulation by BDNF of presynaptic and postsynaptic NMDA receptors could therefore represent an important mechanism for increasing the fidelity of high-frequency synaptic transmission and the subsequent induction of long-term synaptic plasticity.
GRANTS
This study was supported by National Institute of Neurological Disorders and Stroke Grant NS-39167 to E. S. Levine.
Acknowledgments
We thank Drs. J. Trettel and D. Fortin for comments on the manuscript.
Present address of J. C. Madara: The Scripps Research Institute, Department of Cell Biology, 10550 North Torrey Pines Road, La Jolla, CA 92037.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- Alder et al. 2005.Alder J, Thakker-Varia S, Crozier RA, Shaheen A, Plummer MR, Black IB. Early presynaptic and late postsynaptic components contribute independently to brain-derived neurotrophic factor-induced synaptic plasticity. J Neurosci 25: 3080–3085, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki et al. 1997.Aoki C, Rhee J, Lubin M, Dawson TM. NMDA-R1 subunit of the cerebral cortex co-localizes with neuronal nitric oxide synthase at pre- and postsynaptic sites and in spines. Brain Res 750: 25–40, 1997. [DOI] [PubMed] [Google Scholar]
- Aoki et al. 1994.Aoki C, Venkatesan C, Go CG, Mong JA, Dawson TM. Cellular and subcellular localization of NMDA-R1 subunit immunoreactivity in the visual cortex of adult and neonatal rats. J Neurosci 14: 5202–5222, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki et al. 2000.Aoki C, Wu K, Elste A, Len G, Lin S, McAuliffe G, Black IB. Localization of brain-derived neurotrophic factor and TrkB receptors to postsynaptic densities of adult rat cerebral cortex. J Neurosci Res 59: 454–463, 2000. [DOI] [PubMed] [Google Scholar]
- Bekinschtein et al. 2008.Bekinschtein P, Cammarota M, Izquierdo I, Medina JH. BDNF and memory formation and storage. Neuroscientist 14: 147–156, 2008. [DOI] [PubMed] [Google Scholar]
- Bender et al. 2006.Bender VA, Bender KJ, Brasier DJ, Feldman DE. Two coincidence detectors for spike timing-dependent plasticity in somatosensory cortex. J Neurosci 26: 4166–4177, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg et al. 1992.Berg MM, Sternberg DW, Parada LF, Chao MV. K-252a inhibits nerve growth factor-induced trk proto-oncogene tyrosine phosphorylation and kinase activity. J Biol Chem 267: 13–16, 1992. [PubMed] [Google Scholar]
- Berretta and Jones 1996.Berretta N, Jones RS. Tonic facilitation of glutamate release by presynaptic N-methyl-D-aspartate autoreceptors in the entorhinal cortex. Neuroscience 75: 339–344, 1996. [DOI] [PubMed] [Google Scholar]
- Blum et al. 2002.Blum R, Kafitz KW, Konnerth A. Neurotrophin-evoked depolarization requires the sodium channel Na(V)1.9. Nature 419: 687–693, 2002. [DOI] [PubMed] [Google Scholar]
- Brasier and Feldman 2008.Brasier DJ, Feldman DE. Synapse-specific expression of functional presynaptic NMDA receptors in rat somatosensory cortex. J Neurosci 28: 2199–2211, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabelli et al. 1996.Cabelli RJ, Allendoerfer KL, Radeke MJ, Welcher AA, Feinstein SC, Shatz CJ. Changing patterns of expression and subcellular localization of TrkB in the developing visual system. J Neurosci 16: 7965–7980, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmignoto et al. 1997.Carmignoto G, Pizzorusso T, Tia S, Vicini S. Brain-derived neurotrophic factor and nerve growth factor potentiate excitatory synaptic transmission in the rat visual cortex. J Physiol 498: 153–164, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charton et al. 1999.Charton JP, Herkert M, Becker CM, Schroder H. Cellular and subcellular localization of the 2B-subunit of the NMDA receptor in the adult rat telencephalon. Brain Res 816: 609–617, 1999. [DOI] [PubMed] [Google Scholar]
- Cheng and Yeh 2003.Cheng Q, Yeh HH. Brain-derived neurotrophic factor attenuates mouse cerebellar granule cell GABA(A) receptor-mediated responses via postsynaptic mechanisms. J Physiol 548: 711–721, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corlew et al. 2007.Corlew R, Wang Y, Ghermazien H, Erisir A, Philpot BD. Developmental switch in the contribution of presynaptic and postsynaptic NMDA receptors to long-term depression. J Neurosci 27: 9835–9845, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozier et al. 1999.Crozier RA, Black IB, Plummer MR. Blockade of NR2B-containing NMDA receptors prevents BDNF enhancement of glutamatergic transmission in hippocampal neurons. Learn Mem 6: 257–266, 1999. [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy et al. 2001.Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol 11: 327–335, 2001. [DOI] [PubMed] [Google Scholar]
- DeBiasi et al. 1996.DeBiasi S, Minelli A, Melone M, Conti F. Presynaptic NMDA receptors in the neocortex are both auto- and heteroreceptors. Neuroreport 7: 2773–2776, 1996. [DOI] [PubMed] [Google Scholar]
- Drake et al. 1999.Drake CT, Milner TA, Patterson SL. Ultrastructural localization of full-length trkB immunoreactivity in rat hippocampus suggests multiple roles in modulating activity-dependent synaptic plasticity. J Neurosci 19: 8009–8026, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunah et al. 1996.Dunah AW, Yasuda RP, Wang YH, Luo J, Davila-Garcia M, Gbadegesin M, Vicini S, Wolfe BB. Regional and ontogenic expression of the NMDA receptor subunit NR2D protein in rat brain using a subunit-specific antibody. J Neurochem 67: 2335–2345, 1996. [DOI] [PubMed] [Google Scholar]
- Figurov et al. 1996.Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature 381: 706–709, 1996. [DOI] [PubMed] [Google Scholar]
- Garraway et al. 2003.Garraway SM, Petruska JC, Mendell LM. BDNF sensitizes the response of lamina II neurons to high threshold primary afferent inputs. Eur J Neurosci 18: 2467–2476, 2003. [DOI] [PubMed] [Google Scholar]
- Gartner et al. 2006.Gartner A, Polnau DG, Staiger V, Sciarretta C, Minichiello L, Thoenen H, Bonhoeffer T, Korte M. Hippocampal long-term potentiation is supported by presynaptic and postsynaptic tyrosine receptor kinase B-mediated phospholipase Cgamma signaling. J Neurosci 26: 3496–3504, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschalk et al. 1998.Gottschalk W, Pozzo-Miller LD, Figurov A, Lu B. Presynaptic modulation of synaptic transmission and plasticity by brain-derived neurotrophic factor in the developing hippocampus. J Neurosci 18: 6830–6839, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang et al. 2008.Huang Y, Yasuda H, Sarihi A, Tsumoto T. Roles of endocannabinoids in heterosynaptic long-term depression of excitatory synaptic transmission in visual cortex of young mice. J Neurosci 28: 7074–7083, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber et al. 1998.Huber KM, Sawtell NB, Bear MF. Brain-derived neurotrophic factor alters the synaptic modification threshold in visual cortex. Neuropharmacology 37: 571–579, 1998. [DOI] [PubMed] [Google Scholar]
- Humeau et al. 2003.Humeau Y, Shaban H, Bissiere S, Luthi A. Presynaptic induction of heterosynaptic associative plasticity in the mammalian brain. Nature 426: 841–845, 2003. [DOI] [PubMed] [Google Scholar]
- Jourdain et al. 2007.Jourdain P, Bergersen LH, Bhaukaurally K, Bezzi P, Santello M, Domercq M, Matute C, Tonello F, Gundersen V, Volterra A. Glutamate exocytosis from astrocytes controls synaptic strength. Nat Neurosci 10: 331–339, 2007. [DOI] [PubMed] [Google Scholar]
- Jovanovic et al. 2000.Jovanovic JN, Czernik AJ, Fienberg AA, Greengard P, Sihra TS. Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nat Neurosci 3: 323–329, 2000. [DOI] [PubMed] [Google Scholar]
- Kim and Alger 2001.Kim J, Alger BE. Random response fluctuations lead to spurious paired-pulse facilitation. J Neurosci 21: 9608–9618, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knusel and Hefti 1992.Knusel B, Hefti F. K-252 compounds: modulators of neurotrophin signal transduction. J Neurochem 59: 1987–1996, 1992. [DOI] [PubMed] [Google Scholar]
- Kolb et al. 2005.Kolb JE, Trettel J, Levine ES. BDNF enhancement of postsynaptic NMDA receptors is blocked by ethanol. Synapse 55: 52–57, 2005. [DOI] [PubMed] [Google Scholar]
- Korte et al. 1995.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA 92: 8856–8860, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalchuk et al. 2002.Kovalchuk Y, Hanse E, Kafitz KW, Konnerth A. Postsynaptic induction of BDNF-mediated long-term potentiation. Science 295: 1729–1734, 2002. [DOI] [PubMed] [Google Scholar]
- Kramar et al. 2004.Kramar EA, Lin B, Lin CY, Arai AC, Gall CM, Lynch G. A novel mechanism for the facilitation of theta-induced long-term potentiation by brain-derived neurotrophic factor. J Neurosci 24: 5151–5161, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessmann and Heumann 1998.Lessmann V, Heumann R. Modulation of unitary glutamatergic synapses by neurotrophin-4/5 or brain-derived neurotrophic factor in hippocampal microcultures: presynaptic enhancement depends on pre-established paired-pulse facilitation. Neuroscience 86: 399–413, 1998. [DOI] [PubMed] [Google Scholar]
- Levine et al. 1998.Levine ES, Crozier RA, Black IB, Plummer MR. Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-D-aspartic acid receptor activity. Proc Natl Acad Sci USA 95: 10235–10239, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine and Kolb 2000.Levine ES, Kolb JE. Brain-derived neurotrophic factor increases activity of NR2B-containing N-methyl-D-aspartate receptors in excised patches from hippocampal neurons. J Neurosci Res 62: 357–362, 2000. [DOI] [PubMed] [Google Scholar]
- Li and Keifer 2008.Li W, Keifer J. Coordinate action of pre- and postsynaptic brain-derived neurotrophic factor is required for AMPAR trafficking and acquisition of in vitro classical conditioning. Neuroscience 155: 686–697, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li and Han 2007.Li YH, Han TZ. Glycine binding sites of presynaptic NMDA receptors may tonically regulate glutamate release in the rat visual cortex. J Neurophysiol 97: 817–823, 2007. [DOI] [PubMed] [Google Scholar]
- Li et al. 1998b.Li YX, Xu Y, Ju D, Lester HA, Davidson N, Schuman EM. Expression of a dominant negative TrkB receptor, T1, reveals a requirement for presynaptic signaling in BDNF-induced synaptic potentiation in cultured hippocampal neurons. Proc Natl Acad Sci USA 95: 10884–10889, 1998b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li et al. 1998a.Li YX, Zhang Y, Lester HA, Schuman EM, Davidson N. Enhancement of neurotransmitter release induced by brain-derived neurotrophic factor in cultured hippocampal neurons. J Neurosci 18: 10231–10240, 1998a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin et al. 1998.Lin SY, Wu K, Levine ES, Mount HT, Suen PC, Black IB. BDNF acutely increases tyrosine phosphorylation of the NMDA receptor subunit 2B in cortical and hippocampal postsynaptic densities. Brain Res Mol Brain Res 55: 20–27, 1998. [DOI] [PubMed] [Google Scholar]
- Lu 2003.Lu B BDNF and activity-dependent synaptic modulation. Learn Mem 10: 86–98, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka and Bear 2004.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron 44: 5–21, 2004. [DOI] [PubMed] [Google Scholar]
- Mameli et al. 2005.Mameli M, Carta M, Partridge LD, Valenzuela CF. Neurosteroid-induced plasticity of immature synapses via retrograde modulation of presynaptic NMDA receptors. J Neurosci 25: 2285–2294, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBain and Dingledine 1992.McBain C, Dingledine R. Dual-component miniature excitatory synaptic currents in rat hippocampal CA3 pyramidal neurons. J Neurophysiol 68: 16–27, 1992. [DOI] [PubMed] [Google Scholar]
- Monyer et al. 1992.Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, Burnashev N, Sakmann B, Seeburg PH. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science 256: 1217–1221, 1992. [DOI] [PubMed] [Google Scholar]
- O'Brien et al. 1997.O'Brien JA, Isaacson JS, Berger AJ. NMDA and non-NMDA receptors are co-localized at excitatory synapses of rat hypoglossal motoneurons. Neurosci Lett 227: 5–8, 1997. [DOI] [PubMed] [Google Scholar]
- Pascual et al. 2001.Pascual M, Climent E, Guerri C. BDNF induces glutamate release in cerebrocortical nerve terminals and in cortical astrocytes. Neuroreport 12: 2673–2677, 2001. [DOI] [PubMed] [Google Scholar]
- Patterson et al. 1996.Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron 16: 1137–1145, 1996. [DOI] [PubMed] [Google Scholar]
- Pozzo-Miller et al. 1999.Pozzo-Miller LD, Gottschalk W, Zhang L, McDermott K, Du J, Gopalakrishnan R, Oho C, Sheng ZH, Lu B. Impairments in high-frequency transmission, synaptic vesicle docking, and synaptic protein distribution in the hippocampus of BDNF knockout mice. J Neurosci 19: 4972–4983, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Moreno and Paulsen 2008.Rodriguez-Moreno A, Paulsen O. Spike timing-dependent long-term depression requires presynaptic NMDA receptors. Nat Neurosci 11: 744–745, 2008. [DOI] [PubMed] [Google Scholar]
- Sala et al. 1998.Sala R, Viegi A, Rossi FM, Pizzorusso T, Bonanno G, Raiteri M, Maffei L. Nerve growth factor and brain-derived neurotrophic factor increase neurotransmitter release in the rat visual cortex. Eur J Neurosci 10: 2185–2191, 1998. [DOI] [PubMed] [Google Scholar]
- Sasaki et al. 2002.Sasaki YF, Rothe T, Premkumar LS, Das S, Cui J, Talantova MV, Wong HK, Gong X, Chan SF, Zhang D, Nakanishi N, Sucher NJ, Lipton SA. Characterization and comparison of the NR3A subunit of the NMDA receptor in recombinant systems and primary cortical neurons. J Neurophysiol 87: 2052–2063, 2002. [DOI] [PubMed] [Google Scholar]
- Schinder et al. 2000.Schinder AF, Berninger B, Poo M. Postsynaptic target specificity of neurotrophin-induced presynaptic potentiation. Neuron 25: 151–163, 2000. [DOI] [PubMed] [Google Scholar]
- Sjostrom et al. 2003.Sjostrom PJ, Turrigiano GG, Nelson SB. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 39: 641–654, 2003. [DOI] [PubMed] [Google Scholar]
- Suen et al. 1997.Suen PC, Wu K, Levine ES, Mount HT, Xu JL, Lin SY, Black IB. Brain-derived neurotrophic factor rapidly enhances phosphorylation of the postsynaptic N-methyl-D-aspartate receptor subunit 1. Proc Natl Acad Sci USA 94: 8191–8195, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trettel and Levine 2002.Trettel J, Levine ES. Cannabinoids depress inhibitory synaptic inputs received by layer 2/3 pyramidal neurons of the neocortex. J Neurophysiol 88: 534–539, 2002. [DOI] [PubMed] [Google Scholar]
- Tyler and Pozzo-Miller 2001.Tyler WJ, Pozzo-Miller LD. BDNF enhances quantal neurotransmitter release and increases the number of docked vesicles at the active zones of hippocampal excitatory synapses. J Neurosci 21: 4249–4258, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhall et al. 2001.Woodhall G, Evans DI, Cunningham MO, Jones RS. NR2B-containing NMDA autoreceptors at synapses on entorhinal cortical neurons. J Neurophysiol 86: 1644–1651, 2001. [DOI] [PubMed] [Google Scholar]
- Xu et al. 2000.Xu B, Gottschalk W, Chow A, Wilson RI, Schnell E, Zang K, Wang D, Nicoll RA, Lu B, Reichardt LF. The role of brain-derived neurotrophic factor receptors in the mature hippocampus: modulation of long-term potentiation through a presynaptic mechanism involving TrkB. J Neurosci 20: 6888–6897, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang et al. 2006.Yang J, Woodhall GL, Jones RS. Tonic facilitation of glutamate release by presynaptic NR2B-containing NMDA receptors is increased in the entorhinal cortex of chronically epileptic rats. J Neurosci 26: 406–410, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano et al. 2006.Yano H, Ninan I, Zhang H, Milner TA, Arancio O, Chao MV. BDNF-mediated neurotransmission relies upon a myosin VI motor complex. Nat Neurosci 9: 1009–1018, 2006. [DOI] [PubMed] [Google Scholar]
- Zakharenko et al. 2003.Zakharenko SS, Patterson SL, Dragatsis I, Zeitlin SO, Siegelbaum SA, Kandel ER, Morozov A. Presynaptic BDNF required for a presynaptic but not postsynaptic component of LTP at hippocampal CA1-CA3 synapses. Neuron 39: 975–990, 2003. [DOI] [PubMed] [Google Scholar]