

Abstract
Hepatitis C virus (HCV) NS5B RNA polymerase is crucial for replicating the HCV RNA genome and is an attractive target for developing anti-HCV drugs. A novel series of 2,3-diaryl-1,3-thiazolidin-4-one derivatives were evaluated for their ability to inhibit HCV NS5B. Of this series, compounds 4c, 5b, 5c and 6 emerged as more potent, displaying over 95% inhibition of NS5B RNA polymerase activity in vitro. The two most active compounds 4c and 5c exhibited an IC50 of 31.9 µM and 32.2 µM, respectively against HCV NS5B.
Hepatitis C virus (HCV) is a blood-borne pathogen belonging to the Flaviviridae1 family of viruses, which also includes the West Nile, Yellow Fever, and Dengue viruses. HCV infection is one of the most significant cause for liver cirrhosis and hepatocellular carcinoma2 leading to liver failure and as such is a growing medical problem that affects an estimated 170 million individuals worldwide.3 HCV is a positive strand RNA virus, and its genome comprises of 9600 base pairs that encode several structural and nonstructural proteins.4 Non-structural protein 5B (NS5B), encodes the viral RNA dependent RNA polymerase (RdRp), which plays a pivotal role in replicating the HCV RNA genome.5 By analogy to AIDS, most small molecule inhibitor approaches to HCV have centered on the inhibition of essential viral targets, especially the NS3-4A protease (analogous to HIV protease) and the NS5B RdRp (analogous to HIV RT), although other targets are also being followed.6 More interestingly, there is no functional counter part of this enzyme in mammalian cells thus making it an ideal drug target.7 Several classes of potent NS5B inhibitors have been reported in the past couple of years8 e.g. nucleoside NS5B inhibitors NM2839 and R-1626,10 and non-nucleoside inhibitors HCV-79611 and wedelolactone12 (Fig. 1) among others. However, despite a proliferation of pharmaceutical and academic research in the past decade, no specific antiviral agents are available for the treatment of HCV. Therefore, development of anti-HCV drugs remains an enormous unmet medical need for adequate therapeutic options.
Figure 1.

NS5B RNA polymerase inhibitors.
4-Thiazolidinone scaffold has been gaining prominence in recent years, due to the fact that its derivatives are known to possess wide spectrum of activities such as antibacterial,13,14 antifungal,15,16 anticonvulsant,17,18 antiCOX-1,19 antituberculosis,20–22 antihistaminic23 and anticancer.24 The persuasive antiviral activity of 4-thiazolidinone scaffold has been enlightened by several studies.
These include the inhibition of HIV-1 RT by 2,3-diaryl-1,3-thiazolidin-4-ones.25–27 More recently, the inhibitory potency of 4-thiazolidinone ring system against HCV NS5B polymerase has been reported by Kaushik-Basu et al.28
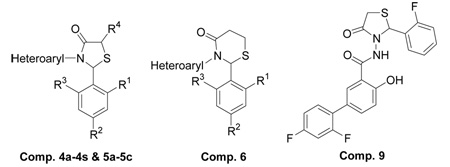
In this study, we have investigated the therapeutic potential of the 4-thiazolidinone scaffold against HCV NS5B, utilizing a series of 2,3-diaryl-1,3-thiazolidin-4-one derivatives synthesized by our group. The synthesis of all compounds reported in Table 1 except compounds 4c, 4p, 7 and 8 have been described previously.26 Our investigations have focused on building the structure–activity relationship (SAR) around 2- and 3-positions of the 4-thiazolidinone template in contrast to the recently reported 4-oxo-2-thionothiazolidines, which carry arylsulfonamido and arylidene substituents at 3- and 5-positions, respectively.28 Here we report the identification of a new series of 4-thiazolidinone derivatives as promising inhibitors of HCV NS5B polymerase. These seminal findings should assist in the development of novel 4-thiazolidinone compounds harboring potent anti-NS5B activity.
Table 1.
Physical data of 2,3-diaryl-1,3-thiazolidin-4-one derivatives.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Comp. | Heteroaryl | R1 | R2 | R3 | R4 | MWt | % yield | m.p. °C |
| 4a | Pyridin-2-yl | Quinolin-4-yl | H | 307 | 75 | - | ||
| 4b | Pyridin-2-yl | H | NMe2 | H | H | 299 | 68 | - |
| 4c | Pyridin-2-yl | H | F | H | H | 274 | 78 | - |
| 4d | 6-methyl-pyridin-2-yl | Pyridin-3-yl | H | 271 | 55 | 104–106 | ||
| 4e | 6-methyl-pyridin-2-yl | H | NMe2 | H | H | 313 | 72 | 133–135 |
| 4f | Pyridin-3-ylmethyl | Cl | H | Cl | H | 339 | 92 | - |
| 4g | 4-methyl-6-trifluoromethyl-pyrimidin-2-yl | Cl | H | Cl | H | 408 | 50 | 111–115 |
| 4h | 4-methyl-6-trifluoromethyl-pyrimidin-2-yl | Cl | H | F | H | 392 | 47 | 92–94 |
| 4i | 4-methyl-6-trifluoromethyl-pyrimidin-2-yl | Br | H | Br | H | 497 | 36 | 133–137 |
| 4j | 4-methyl-6-phenyl-pyrimidin-2-yl | Cl | H | Cl | H | 416 | 48 | 168–170 |
| 4k | 4-methyl-6-phenyl-pyrimidin-2-yl | Cl | H | F | H | 400 | 41 | 138–140 |
| 4l | 4-phenyl-6-trifluoromethyl-pyrimidin-2-yl | Cl | H | Cl | H | 470 | 46 | 206–208 |
| 4m | 4,6-diphenyl-pyrimidin-2-yl | Cl | H | Cl | H | 478 | 38 | 206–208 |
| 4n | 4,6-diphenyl-pyrimidin-2-yl | Cl | H | F | H | 462 | 30 | 176–178 |
| 4o | 4,6-diphenyl-pyrimidin-2-yl | F | H | F | H | 445 | 28 | 192–194 |
| 4p | Quinolin-2-yl | Cl | H | F | H | 358 | 56 | 144–146 |
| 4q | Furan-2-ylmethyl | Cl | H | Cl | H | 328 | 92 | 96–98 |
| 4r | Thiophen-2-ylmethyl | Br | H | Br | H | 433 | 64 | - |
| 4s | 5-ethyl-[1,3,4]-thiadiazol-2-yl | F | H | F | H | 327 | 44 | 110–114 |
| 5a | Pyridin-2-yl | Cl | H | Cl | CH3 | 339 | 80 | 85–89 |
| 5b | Pyridin-3-ylmethyl | Cl | H | Cl | CH3 | 353 | 90 | - |
| 5c | Furan-2-ylmethyl | Cl | H | Cl | CH3 | 342 | 88 | - |
| 6 | Furan-2-ylmethyl | Cl | H | Cl | H | 342 | 90 | - |
| 7 | Pyridin-2-yl | 1-benzyl-piperidinyl | H | 339 | 50 | - | ||
| 8a | Furan-2-ylmethyl | Cl | H | Cl | H | 344 | 78 | 155–158 |
| 9 | 2',4'-Difluoro-4-hydroxy-biphenyl-3-carboxylic acid [2-(2-fluoro-phenyl)-4-oxo-thiazolidin-3-yl]-amide | - | - | - | ||||
sulfoxide.
The target compounds in this study (4a–4f, 4q, 5a–5c and 6) were prepared by the multi-component DCC mediated reaction protocol29 earlier reported from this laboratory as shown in Scheme 1. In this protocol N,N-Dicyclohexylcarbodiimide (DCC) is used as a dehydrating agent to accelerate the intramolecular cyclization resulting in faster reaction and improved yields. The reactions were performed by reacting theappropriate heteroaryl amines (1), substituted benzaldehydes (2) and mercapto acids (3) in the presence of DCC at room temperature. After completion of the reaction ranging around 1.0 hr, the desired products were obtained in excellent yields and purity as confirmed by spectral data analysis. Compounds 7, 4g–4p and 4r–4s were synthesized by using the toluene reflux protocol26 in the presence of 4Å molecular sieve and p-toluene sulphonic acid (PTSA). Reaction time for these compounds varied from 18–24 hours and yielded the desired products in moderate yields and purity. Sulfoxide (8) was synthesized by using Oxone® (2 equivalents) in methanol:water (1:1) at room temperature stirring for 30 minutes. The spectral data including the elemental analysis of this compound reported in supplemental information correlates with the expected structure. Physical data for all 4-thiazolidinone derivatives are given in Table 1.
Scheme 1.

Synthesis of Compounds 4a–4s, 5a–5c, 6 and 7. (i) DCC, THF, at RT (ii) Toluene, 4 Å MS at 120 °C.
To investigate the influence of the 4-thiazolidinone compounds on the RdRp activity of NS5B, we employed the N-terminal His-tagged HCV NS5BCΔ21 (genotype 1b), lacking the C-terminal 21-amino acid membrane-spanning domain. Purification of NS5BCΔ21 and determination of its RdRp activity was carried out in accordance with previously described procedures.12,28 Primarily, the anti-NS5B activity was evaluated for all compounds (4a–4s, 5a–5c and 6–8) and their results are summarized in Table 2. The compounds showed varied pattern of inhibition of HCV NS5B RdRp ranging from moderate to good at 0.25 mM compound concentration. Importantly, compounds 4c, 5b, 5c and 6 exhibited 95% or higher inhibition of NS5B at this concentration, thus revealing somewhat higher potency than compound 9.28 Further, compounds 5b, 5c and 6 exhibited relatively poor anti-HIV-1 RT activity, in contrast to the inhibition pattern seen with HCV NS5B. More-over, examination of the inhibitory activity of the thiazolidin-4-one derivatives against SARS Co-V RdRp (nsp12) and Klenow polymerase as described previously,12,28 yielded ≤50% inhibition at 0.5 mM concentration of these compounds (data not shown), thus suggesting their specificity for HCV NS5B.
Table 2.
Anti-NS5B RdRp activity of compounds (4a–4s, 5a–5c and 6–9).
| Comp. | Anti-NS5B Activity % Inhibitiona | IC50 µM |
|---|---|---|
| 4a | 62.2±4.3 | - |
| 4b | 76.4±3.6 | - |
| 4cb | 98±0.9 | 31.9±1.2 |
| 4d | 64.2±4.1 | - |
| 4e | 84.7±4.2 | 79.4±1.1 |
| 4f | 75.6±2.1 | - |
| 4g | 72.1±1.6 | - |
| 4h | 63.6±2.5 | - |
| 4i | 76.3±2.5 | - |
| 4j | 63.6±2.1 | - |
| 4k | 65.5±2.3 | - |
| 4l | 66.4±2.9 | - |
| 4m | 73.1±3.1 | - |
| 4n | 70.6±3.8 | - |
| 4o | 73.3±2.1 | - |
| 4pb | 60.0±2.6 | - |
| 4q | 72.3±1.6 | - |
| 4r | 62.3±3.5 | - |
| 4s | 62.9±1.0 | - |
| 5a | 73.0±3.6 | - |
| 5b | 95.6±0.8 | 41.8±1.1 |
| 5c | 97.8±0.8 | 32.2±1.1 |
| 6 | 94.2±2.8 | 52.6±1.2 |
| 7b | 85.6±4.9 | 71.9±1.1 |
| 8b | 68.7±5.4 | - |
| 9 | 92.4±2.5 | 48 |
| wedelolactone | - | 36.1 |
Percentage inhibition was determined at 0.25 mM concentration of the indicated compound and represents an average of at least three independent measurements. NS5B RdRp activity in the absence of the inhibitor was taken as 100 percent after subtraction of residual background activity. The concentration of DMSO in all reactions was kept constant at 10%. The IC50 values of the compounds 4c, 4e, 5b, 5c, 6 and 7 were determined from dose–response curves using 8–12 concentrations of each compound in duplicate, in two independent experiments. Curves were fitted to data points using nonlinear regression analysis and IC50 values were interpolated from the resulting curves using GraphPad Prism 5.0 software. Wedelolactone (IC50=36.1 µM) was included as an internal reference standard.
compounds 4c, 4p, 7 & 8 are new.
It may be inferred from the biological activity data reported in Table 2 that the anti-HCV NS5B activity is sturdily dependent on the nature of the substituent at C-2, N-3 and C-5 of the 4-thiazolidinone scaffold. In particular, a high activity level was observed for compounds possessing a halophenyl and 4-dimethylaminophenyl group at C-2, substituted/unsubstituted pyridine-2-yl, pyridine-3-ylmethyl, substituted pyrimidin-2-yl and furan-2-ylmethyl at N-3 and unsubstituted or methyl substitution at C-5. In fact, the compounds with the best combination of high potency were unsubstituted pyridine-2-yl, pyridine-3-ylmethyl or furan-2-ylmethyl substituted at N-3 of 4-thiazolidinone scaffold, derivatives such as 4c, 5b, 5c and 6. The effect of 2,6-dihalosubstituent on the phenyl ring at C-2 was apparent in compound 4i. This compound was more active than the corresponding 2,6-dichloro (4g) and 2-chloro-6-fluoro substituted (4h) compounds. Furthermore the favourable effect of 2,6-dibromo was confirmed by the finding that 2,6-dichloro derivative (4g) possessed intermediate activity between 2,6-dibromo and 2-chloro-6-fluoro analogues. But the 2,6-difluoro substituted compounds (4o) possessed almost same activity as 2,6-dichloro substituted compound (4m) in case of 4,6-diphenylpyrimidin-2-yl derivatives.
The introduction of pyrimidin-2-yl substituent at the N-3 atom of the thiazolidinone ring moderated their anti-NS5B activity. Considering the effect of the substituents on the pyrimidine ring at N-3, introduction of 4-phenyl-6-trifluoromethyl pyrimidin-2-yl moiety (4l) resulted in moderate activity. Introduction of the 4-methyl-6-phenyl pyrimidin-2-yl moiety (4j–4k) led to substantial decrease in their ability to inhibit NS5B. Compounds 4m, 4n and 4o with the 4,6-diphenylpyrimidin-2-yl derivatives, compounds 4g and 4i with the 4-methyl-6-trifluoromethyl pyrimidin-2-yl derivatives exhibited near equal potency.
However, introduction of the quinolin-2-yl (4p), thiophen-2-ylmethyl (4r) and 5-ethyl-[1,3,4]-thiadiazol-2-yl (4s) at N-3 position also resulted in a substantial decrease in their activity compared to the pyridin-2yl, pyridine-3-ylmethyl and furan-2-ylmethyl containing compounds.
Compounds 4c, 4e, 5b, 5c, 6 and 7, thus emerged as the most potent compounds of this series with percentage inhibition in the range of 85 to 98%. We therefore evaluated the IC50 values of these compounds by monitoring the total incorporation of the radiolabeled UTP on poly rA/U12 as a function of inhibitor concentration. Compounds 4c, 4e, 5b, 5c, 6 and 7 yielded IC50 value ranging from 31.9–79.4 µM (Table 2). The most potent compound 4c and 5c exhibited an IC50 value of 31.9 and 32.2 µM whereas the compound 4e, 5b, 6 and 7 exhibited modest IC50 values (41.8–79.4 µM).
Earlier reports had focused mostly on exploring the substitution at C-2 and N-3 position of the 4-thiazolidinone scaffold.28 As a first step towards exploring the importance of thiazolidinone scaffold per se, we have evaluated compounds having methyl substitution at C-5, ring expansion metathiazanone and sulfoxide modification. It is apparent from the data presented in Table 2 that changes in the basic thiazolidinone moiety either by introducing a methyl group at C-5 (5a–5c), ring expansion (6) or spiro (7) has favorable effect on the anti-HCV NS5B activity as opposed to the introduction of sulfoxide (8) moiety, which decreased the activity.
In conclusion, previously synthesized novel 2,3-diaryl-1,3-thiazolidin-4-one derivatives were evaluated against HCV NS5B polymerase. Compounds 4c, 4e, 5b, 5c, 6 and 7 of this series showed promise as anti-HCV NS5B agents and exhibited over 85% inhibition. Compounds 4c and 5c were the most potent of this group with IC50 values of 31.9 and 32.2 µM, respectively. All other derivatives also exhibited greater than 60% NS5B RdRp inhibition. Taken together our data indicate that changes at C-2, N-3 and C-5 position of 4-thiazolidinone scaffold with appropriate substitution may provide compounds with improved potency. Thus 4-thiazolidinone skeleton holds promise for further activity optimization studies.
Supplementary Material
Acknowledgements
The authors thank the Director, CDRI for support and the SAIF for the spectral data. CDRI communication number: 7558. This research was partly supported by National Institute of Health (NIH) research grant DK066837 to NKB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.(a) Choo Q-L, Han J, Weiner AJ, Overby LR, Bradley DW, Kuo G, Houghton M. In: Hepatitis C virus is a distant relative of the flaviviruses and pestiviruses. Shikata T, Purcell RH, Uchida T, editors. Amsterdam, The Netherlands: Viral hepatitis C, D and E Elsevier Science Publishers B.V.; 1991. pp. 47–52. [Google Scholar]; (b) Choo Q-L, Richman KH, Han JH, Berger K, Lee C, Dong C, Gallegos C, Coit D, Medina-Selby A, Barr PJ, Weiner AJ, Bradley DW, Kuo G, Houghton M. Proc. Natl. Acad. Sci. USA. 1991;88:2451–2455. doi: 10.1073/pnas.88.6.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McHutchison JG. Am. J. Manag. Care. 2004;10:S21–S29. [PubMed] [Google Scholar]
- 3.Cohen J. Science. 1999;285:26. doi: 10.1126/science.285.5424.26. [DOI] [PubMed] [Google Scholar]
- 4.Reed KE, Rice CM. Curr. Top. Microbiol. Immunol. 2000;242:55–84. doi: 10.1007/978-3-642-59605-6_4. [DOI] [PubMed] [Google Scholar]
- 5.Beaulieu PL, Montse L-B. Curr. Med. Chem. Anti-Infective Agents. 2002;1:163–176. [Google Scholar]
- 6.(a) Gordon CP, Keller PA. J. Med. Chem. 2005;48:1–20. doi: 10.1021/jm0400101. [DOI] [PubMed] [Google Scholar]; (b) De Francesco R, Migliaccio G. Nature. 2005;436:953–960. doi: 10.1038/nature04080. [DOI] [PubMed] [Google Scholar]
- 7.(a) Wu JZ, Hong Z. Curr. Drug Targets–Infect. Dis. 2003;3:207. doi: 10.2174/1568005033481114. [DOI] [PubMed] [Google Scholar]; (b) LaPlante S, Jakalian A, Aubry N, Bousquet Y, Ferland J-M, Gillard J, Lefebvre S, Poirier M, Tsantrizos YS, Kukolj G, Beaulieu PL. Angew. Chem., Int. Ed. 2004;43:4306. doi: 10.1002/anie.200460326. [DOI] [PubMed] [Google Scholar]; (c) Tan S-L, He Y, Huang Y, Gale M., Jr Curr. Opin. Pharmacol. 2004;4:465. doi: 10.1016/j.coph.2004.07.003. [DOI] [PubMed] [Google Scholar]; (d) Beaulieu PL, Tsantrizos YS. Curr. Opin. Investig. Drugs. 2004;5:838. [PubMed] [Google Scholar]; (e) Gordon CP, Keller PA. J. Med. Chem. 2005;48:1. doi: 10.1021/jm0400101. [DOI] [PubMed] [Google Scholar]; (f) Condon SM, LaPorte MG, Herbertz T. Curr. Med. Chem.–Anti-Infective Agents. 2005;4:99. [Google Scholar]; (g) De Francesco R, Migliaccio G. Nature. 2005;436:953. doi: 10.1038/nature04080. [DOI] [PubMed] [Google Scholar]
- 8.(a) Beaulieu PL, Bousquet Y, Gauthier J, Gillard J, Marquis M, McKercher G, Pellerin C, Valois S, Kukolj G. J. Med. Chem. 2004;47:6884–6892. doi: 10.1021/jm040134d. [DOI] [PubMed] [Google Scholar]; (b) Harper S, Avolio S, Pacini B, Di Filippo M, Altamura S, Tomei L, Paonessa G, Di Marco S, Carfi A, Giuliano C, Padron J, Bonelli F, Migliaccio G, De Francesco R, Laufer R, Rowley M, Narjes F. J. Med. Chem. 2005;48:4547–4557. doi: 10.1021/jm050056+. [DOI] [PubMed] [Google Scholar]; (c) Tedesco R, Shaw AN, Bambal R, Chai D, Concha NO, Darcy MG, Dhanak D, Fitch DM, Gates A, Gerhardt WG, Halegoua DL, Han C, Hofmann GA, Johnston VK, Kaura AC, Liu N, Keenan RM, Lin-Goerke J, Sarisky RT, Wiggall KJ, Zimmerman MN, Duffy KJ. J. Med. Chem. 2006;49:971–983. doi: 10.1021/jm050855s. [DOI] [PubMed] [Google Scholar]; (d) Hirashima S, Suzuki T, Ishida T, Noji S, Yata S, Ando I, Komatsu M, Ikeda S, Hashimoto H. J. Med. Chem. 2006;49:4721–4736. doi: 10.1021/jm060269e. [DOI] [PubMed] [Google Scholar]
- 9.Afdhal N, Rodriguez-Torres M, Lawitz E, Godofsky E, Chao G, Fielman B, Knox S, Broen N. 40th EASL, Enhanced antiviral efficacy for Valopicitabine (NM283) plus Peg-interferon in hepatitis C patients with HCV genotype-1 infection: results of a phase IIa multicenter trial; April 13–17, 2005; Paris, France. [Google Scholar]
- 10.Roberts S, Cooksley G, Shaw D, Berns HK, Brandl MT, Fettner SH, Hill G, Ipe D, Klumpp K, Mannino M, O’Mara E, Tu Y, Washington CB. Abstract 731, 41st EASL, Interim results of a multiple ascending dose study of R1626, a novel nucleoside analog targeting HCV polymerase in chronic HCV patients; Vienna, Austria. [Google Scholar]
- 11.Chandra C, Raible D, Harper D, Speth J, Villano S, Bichier G. DDW 2006, Antiviral activity of the non-nucleoside polymerase inhibitor, HCV-796, in patients with chronic hepatitis C virus: preliminary results from a randomized, double-blind, placebo-controlled, ascending multiple dose study; May 20–25, 2006; Los Angeles, CA. [Google Scholar]
- 12.Kaushik-Basu N, Bopda-Waffo A, Talele TT, Basu A, Costa PR, Da Silva AJ, Sarafianos SG, Noël F. Nucleic Acids Res. 2008;36(5):1482–1496. doi: 10.1093/nar/gkm1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anders CJ, Bronson JJ, D’Andrea SV, Deshpande SM, Falk PJ, Grant-Young KA, Harte WE, Ho H, Misco PF, Robertson JG, Stock D, Sun Y, Walsh AW. Bioorg. Med. Chem. Lett. 2000;10:715–717. doi: 10.1016/s0960-894x(00)00073-1. [DOI] [PubMed] [Google Scholar]
- 14.Kucukguzel SG, Oruc EE, Rollas S, Sahin F, Ozbek A. Eur. J. Med. Chem. 2002;37:197–206. doi: 10.1016/s0223-5234(01)01326-5. [DOI] [PubMed] [Google Scholar]
- 15.Karali N, Ilhan E, Gu¨rsoy A, Kiraz M. Farmaco. 1998;53:346–349. [Google Scholar]
- 16.Fahmy HTY. Boll. Chim. Farm. 2001;140:422–427. [PubMed] [Google Scholar]
- 17.Ergenc N, Capan G. Farmaco. 1994;49:133–135. [PubMed] [Google Scholar]
- 18.Capan G, Ergenc N, Ekinci AC, Vidin A. Farmaco. 1996;51:729–732. [PubMed] [Google Scholar]
- 19.Look GC, Schullek JR, Homes CP, Chinn JP, Gordon EM, Gallop MA. Bioorg. Med. Chem. Lett. 1996;6:707–712. [Google Scholar]
- 20.Bukowski L, Janowiec M, Zwolska-Kwiek Z, Andrezejczyk Z. Pharmazie. 1998;53:373–376. [PubMed] [Google Scholar]
- 21.Ulusoy N. Arzneim.-Forsch.-Drug Res. 2002;52:565–571. doi: 10.1055/s-0031-1299931. [DOI] [PubMed] [Google Scholar]
- 22.Babaoglu K, Page MA, Jones VC, McNeil MR, Dong C, Naismith JH, Lee RE. Bioorg. Med. Chem. Lett. 2003;13:3227–3230. doi: 10.1016/s0960-894x(03)00673-5. [DOI] [PubMed] [Google Scholar]
- 23.Diurno MV, Mazzoni O, Calignano PE, Giordano F, Bolognese A. J. Med. Chem. 1992;35:2910–2912. doi: 10.1021/jm00093a025. [DOI] [PubMed] [Google Scholar]
- 24.Bhatt JJ, Shah BR, Shah HP, Trivedi PB, Undavia NK, Desai NC. Indian J. Chem. 1994;33B:189–192. [Google Scholar]
- 25.Barreca ML, Balzarini J, Chimirri A, De Clerc E, Luca LD, Holtje MH, Holtje M, Monforte AM, Monforte P, Pannecouque C, Rao A, Zappala M. J. Med. Chem. 2002;45:5410–5413. doi: 10.1021/jm020977+. [DOI] [PubMed] [Google Scholar]
- 26.(a) Rawal RK, Prabhakar YS, Katti SB, De Clercq E. Bioorg. Med. Chem. 2005;13:6771–6776. doi: 10.1016/j.bmc.2005.07.063. [DOI] [PubMed] [Google Scholar]; (b) Rawal RK, RajaSolomon V, Prabhakar YS, Katti SB, De Clercq E. Comb. Chem. High Throughput Screening. 2005;8:439–443. doi: 10.2174/1386207054546496. [DOI] [PubMed] [Google Scholar]; (c) Rawal RK, Tripathi R, Katti SB, Pannecouque C, De Clercq E. Bioorg. Med. Chem. 2007;15:1725–1731. doi: 10.1016/j.bmc.2006.12.003. [DOI] [PubMed] [Google Scholar]; (d) Rawal RK, Tripathi R, Katti SB, Pannecouque C, De Clercq E. Bioorg. Med. Chem. 2007;15:3134–3142. doi: 10.1016/j.bmc.2007.02.044. [DOI] [PubMed] [Google Scholar]; (e) Rawal RK, Tripathi R, Katti SB, Pannecouque C, De Clercq E. Med. Chem. 2007;3:355–363. doi: 10.2174/157340607781024393. [DOI] [PubMed] [Google Scholar]
- 27.Barreca ML, Chimirri A, Luca LD, Monforte AM, Monforte P, Rao A, Zappala M, Balzarini J, De Clercq E, Pannecouque C, Witvrouw M. Bioorg. Med. Chem. Lett. 2001;11:1793–1796. doi: 10.1016/s0960-894x(01)00304-3. [DOI] [PubMed] [Google Scholar]
- 28.Kaushik-Basu N, Bopda-Waffo A, Talele TT, Basu A, Chen Y, Guniz Kucukguzel S. Frontiers in Bioscience. 2008;13:3857–3868. doi: 10.2741/2974. [DOI] [PubMed] [Google Scholar]
- 29.Srivastava T, Haq W, Katti SB. Tetrahedron. 2002;58:7619–7624. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.