

Abstract
Mu opioid receptor agonists are clinically valuable as analgesics; however, their use is limited by high abuse liability. Kappa opioid agonists also produce antinociception, but they do not produce mu agonist-like abuse-related effects, suggesting that they may enhance the antinociceptive effects and/or attenuate the abuse-related effects of mu agonists. To evaluate this hypothesis, the present study examined interactions between the mu agonist fentanyl and the kappa agonist U69,593 in three behavioral assays in rhesus monkeys. In an assay of schedule-controlled responding, monkeys responded under a fixed-ratio 30 (FR 30) schedule of food presentation. Fentanyl and U69,593 each produced rate-decreasing effects when administered alone, and mixtures of 0.22:1, 0.65:1 and 1.96:1 U69,593/fentanyl usually produced subadditive effects. In an assay of thermal nociception, tail withdrawal latencies were measured from water heated to 50°C. Fentanyl and U69,593 each produced dose-dependent antinociception, and effects were additive for all mixtures. In an assay of drug self-administration, rhesus monkeys responded for i.v. drug injection, and both dose and FR values were manipulated. Fentanyl maintained self-administration, whereas U69,593 did not. Addition of U69,593 to fentanyl produced a proportion-dependent decrease in both rates of fentanyl self-administration and behavioral economic measures of the reinforcing efficacy of fentanyl. Taken together, these results suggest that simultaneous activation of mu and kappa receptors, either with a mixture of selective drugs or with a single drug that targets both receptors, may reduce abuse liability without reducing analgesic effects relative to selective mu agonists administered alone.
Keywords: mu opioid agonist, kappa opioid agonist, antinociception, self-administration, drug interaction
Introduction
Mu opioid agonists such as morphine, fentanyl, hydrocodone and oxycodone are clinically valuable as analgesics, but their use is limited by undesirable effects including sedation and high abuse potential (Gutstein & Akil, 2005). One strategy to address this issue has been to develop drugs or drug mixtures that act both at mu opioid receptors and at other receptor targets to produce enhanced analgesia and/or reduced undesirable effects relative to treatment with a mu agonist alone. For example, many analgesic preparations currently include both a mu agonist and a non-steroidal anti-inflammatory agent (NSAID) (e.g. mixtures of hydrocodone and acetaminophen in medications with trade names such as Vicodin and Lortab). Mu agonist/NSAID combinations have been shown to produce synergistic antinociceptive effects under some conditions in preclinical studies (Malmberg & Yaksh, 1993; Miranda, Puig, Dursteler, Prieto, & Pinardi, 2007), but these mixtures still possess high abuse potential and are among the most widely abused prescription drugs (Manchikanti, 2007).
Like mu agonists, agonists at kappa opioid receptors also produce antinociceptive effects in rodents and non-human primates (Dykstra, Gmerek, Winger, & Woods, 1987; Vonvoigtlander, Lahti, & Ludens, 1983). However, kappa agonists do not produce mu agonist-like abuse-related effects in preclinical assays of drug self-administration (Tang & Collins, 1985; Woods, Young, & Herling, 1982), place conditioning (Mucha & Herz, 1985), intracranial self-stimulation (Todtenkopf, Marcus, Portoghese, & Carlezon, 2004) or drug discrimination (Bertalmio & Woods, 1987). Moreover, kappa agonists produce subjective effects in humans that have been described as psychotomimetic or dysphoric (Pfeiffer, Brantl, Herz, & Emrich, 1986; Walsh, Strain, Abreu, & Bigelow, 2001). Taken together, these findings suggest that kappa agonists might simultaneously enhance the antinociceptive effects and attenuate the abuse-related effects of mu agonists. A few studies have addressed different aspects of this general proposition. For example, the kappa agonist U50,488 enhanced the thermal antinociceptive effects of the mu agonists morphine in mice and sufentanil in rats in a dose-additive manner (Narita et al., 1993; Verborgh, Camu, & Meert, 1997). However, U50,488 and other kappa agonists attenuated the antinociceptive effects of morphine in a shock titration assay in squirrel monkeys (Craft & Dykstra, 1992a, 1992b). The reasons for this apparent discrepancy are not clear, but there are significant species differences in the density, distribution and function of kappa receptors (Berger et al., 2006; Mansour, Khachaturian, Lewis, Akil, & Watson, 1988). For example, relative to rats, non-human primates possess a higher proportion of kappa receptors to other opioid receptor types, and non-human primates have higher kappa receptor densities in frontal cortex, hippocampus and cerebellum (Mansour et al., 1988). These species differences in opioid receptor neurobiology may contribute to species differences in mu/kappa antinociceptive interactions. With regard to abuse-related effects, kappa agonists reduced mu agonist self-administration and mu agonist-induced place preferences in rodents (Funada, Suzuki, Narita, Misawa, & Nagase, 1993; Xi, Fuller, & Stein, 1998); however, U50,488 did not attenuate the discriminative stimulus effects of mu agonists in either rats or squirrel monkeys (S. S. Negus, Picker, & Dykstra, 1990, 1991). Lastly, it should be noted that some opioid analgesics already exist that may produce kappa receptor-mediated effects in addition to their predominant mu agonist actions (e.g. nalbuphine and butorphanol (Butelman et al., 1998; Pick, Paul, & Pasternak, 1992; Remmers et al., 1999; Vivian et al., 1999). These compounds do appear to have lower abuse liability than more selective mu agonists (Hursh & Winger, 1995; Preston & Jasinski, 1991). However, nalbuphine and butorphanol also have relatively low efficacy at both mu and kappa receptors (Butelman, Winger, Zernig, & Woods, 1995; Dykstra, 1990; Emmerson et al., 1996; Remmers et al., 1999), and it is not clear to what degree their limited abuse liability derives from their mixed mu/kappa activity or their low mu receptor efficacy.
The purpose of the present study was to further evaluate interactions between the antinociceptive and abuse-related effects of mu and kappa agonists. Specifically, interactions between the high efficacy mu agonist fentanyl (Emmerson et al., 1996) and the high efficacy kappa agonist U69,593 (Remmers et al., 1999) were examined in rhesus monkeys using assays of schedule-controlled responding and thermal nociception that have been used previously to examine interactions between mu and delta agonists (Stevenson, Folk, Rice, & Negus, 2005; Stevenson, Linsenmayer, Folk, Rice, & Negus, 2003). Dose-addition analysis was used to characterize the nature of drug interactions within each procedure, and dose-ratio analysis was used to assess the degree to which drug interactions influenced relative potency across these two procedures. Drug interactions were also examined in an assay of drug self-administration to assess the abuse-related reinforcing effects of U69,593/fentanyl mixtures in comparison to U69,593 or fentanyl alone. Drug self-administration data were evaluated using statistical comparisons of response rates maintained by different drug solutions. In addition, a behavioral economics approach was employed to quantify the reinforcing efficacy of different drug solutions (Hursh & Winger, 1995; Winger, Hursh, Casey, & Woods, 2002), and results from this established method of demand-curve analysis were compared to results generated by an alternative strategy that involves linear rather than non-linear regression and requires fewer assumptions about the data. Our results suggest that kappa agonists can attenuate the reinforcing and rate-decreasing effects of mu agonists without attenuating their antinociceptive effects.
Methods
Subjects
Twelve adult male and female rhesus monkeys (Macaca mulatta) weighing 6–12.5 kg were used as subjects. All monkeys had prior exposure to drugs (primarily dopaminergic and opioid compounds) and to the behavioral procedures in which they were tested. The subjects were individually housed, and water was freely available. Their diet consisted of PMI Feeds Jumbo monkey diet (2–8 biscuits/day; PMI Feeds, Inc., St. Louis, MO) supplemented with fresh fruit daily. In addition, monkeys in the assays of schedule-controlled behavior could earn additional food pellets under operant schedules described below. A 12 hr light/12 hr dark cycle was in effect (lights on from 7AM–7PM).
Animal maintenance and research were conducted in accordance with the guidelines provided by the NIH Committee on Laboratory Animal Resources. The facility was licensed by the United States Department of Agriculture, and protocols were approved by the Institutional Animal Care and Use Committee. The health of the monkeys was monitored daily by the staff veterinarian. Monkeys had visual, auditory and olfactory contact with other monkeys throughout the study. Monkeys also had access to puzzle feeders, mirrors and chew toys to provide environmental enrichment. Nature videotapes or music were played daily in all housing rooms.
Behavioral and Pharmacological Procedures
Behavioral studies were conducted using three procedures: (1) an assay of schedule-controlled responding for food presentation, (2) an assay of thermal nociception using warm water as the noxious stimulus, and (3) an assay of drug self-administration. A different group of 4 monkeys was used for each procedure (12 monkeys total). These procedures have been used extensively by our laboratory to examine the effects of other opioids and of opioid agonist interactions in rhesus monkeys (Stevenson et al., 2005; Stevenson et al., 2003). In each assay, the effects of the mu agonist fentanyl were studied alone or in combination with the kappa agonist U69,593. Both in vivo and in vitro data suggest that fentanyl and U69,593 are selective and relatively high efficacy agonists at mu and kappa opioid receptors, respectively (Emmerson et al., 1996; Emmerson, Liu, Woods, & Medzihradsky, 1994; S. S. Negus, Burke, Medzihradsky, & Woods, 1993; S. S. Negus & Mello, 1999; Remmers et al., 1999). As in our previous studies of mu/delta interactions, mu/kappa interactions were examined first in the assay of schedule-controlled responding, and drug potencies in this procedure were used to define the proportional mixtures of fentanyl and U69,593 subsequently studied in the assays of thermal nociception and drug self-administration.
Assay of Schedule-Controlled Responding
Experiments were conducted in each monkey’s home cage (dimensions: 60 ×65 × 75 cm). The home cages of all monkeys were modified to include an operant response panel (28 × 28 cm) mounted on the front wall. Three square translucent response keys (6.4 × 6.4 cm) were arranged 2.54 cm apart in a horizontal row 3.2 cm from the top of the operant panel. Each key could be transilluminated by red, green or yellow stimulus lights. The operant panel also supported an externally-mounted pellet dispenser (Gerbrands, Model G5210; Arlington, MA) that delivered 1 gm banana-flavored food pellets (Purina Test Diet, Richmond, IN) to a food receptacle mounted on the cage beneath the operant response panel. The panel was controlled by a MED-PC interface and an IBM compatible computer programmed in MEDSTATE Notation (MED Associates, Inc., East Fairfield, VT).
Experiments used a multiple-cycle procedure. Each of 5 cycles was 15 min long and consisted of two components: a 10-min timeout period followed by a 5-min response period. During the timeout period, no stimulus lights were illuminated and responding had no scheduled consequences. During the response period, the center key was transilluminated yellow, and the subjects could respond for up to 10 food pellets under a fixed-ratio 30 (FR 30) schedule of reinforcement. If all 10 food pellets were earned before 5 min had elapsed, the lights were turned off, and responding had no scheduled consequences for the remainder of that response period. All monkeys were trained until they responded at rates greater than 1.0 response/sec during all five cycles for 10 consecutive days, and we have shown previously that monkeys respond at relatively stable rates across successive response periods in this procedure (S. S. Negus et al., 1993).
Sessions were conducted 5 days a week. Test sessions were usually conducted on Tuesdays and Fridays, and training sessions were conducted on Mondays, Wednesdays and Thursdays. In addition, test sessions were conducted only after a training session during which the monkeys responded at rates greater than 1.0 response/sec for all five cycles. Test sessions were identical to training sessions except that test compounds were administered using a cumulative dosing procedure, in which doses of the test drug or drug mixture were administered i.m. at the beginning of each timeout period, and each dose increased the total cumulative dose by 1/4 or 1/2 log units.
Initially, complete dose-effect curves were determined for fentanyl and U69,593 administered alone, and each drug was tested twice. Subsequently, three mixtures of fentanyl in combination with U69,593 were examined, and the proportions of the drugs in the mixtures were determined by their relative potencies to decrease response rates. Relative potency (RP) was defined as ED50 U69,593 ÷ ED50 fentanyl, and the proportions of U69,593 to fentanyl in the three mixtures were RP÷3, RP, and RPx3. Each mixture was tested once. All tests with mixtures were separated by at least one week.
Operant response rates from each cycle were converted to percent of control using the average rate from the previous training day as the control value. Subsequently, linear regression was used to determine the dose and 95% confidence limits at which response rates were decreased to 50% of control (the Effective Dose producing 50% control response rates, abbreviated ED50). All points between 20 and 80% control were included in the linear regression, as were the highest dose associated with rates greater than 80% control and the lowest dose associated with rates less than 20% control. For drug mixtures, a related quantity, Zmix, was also calculated for each monkey. Zmix was defined as the total drug dose (i.e. dose fentanyl + dose U69,593) that reduced response rates to 50% of control. ED50 and Zmix values were considered to be significantly different if confidence limits did not overlap.
Assay of Thermal Nociception
Monkeys were seated in acrylic restraint chairs so that their tails hung down freely. The bottom 10 cm of each monkey’s shaved tail was immersed in a thermal container of warm water. If the subject did not withdraw its tail within 20 sec, the tail was removed from the water by the experimenter, and a latency of 20 sec was assigned to that measurement. During each cycle of measurements, tail-withdrawal latencies were measured from water heated to 38 and 50°C. The order in which the temperatures were presented varied from one set of measurements to the next. Experiments were conducted no more than twice a week. A stopwatch was used to measure and record time intervals.
Each test session consisted of multiple 15 min cycles. Before the first cycle, baseline latencies to tail-withdrawal from 38 and 50°C water were determined. Testing continued only if tail withdrawal from 38°C water did not occur before the 20 sec cutoff, and if tail withdrawal occurred in ≤3 sec from 50°C water. This criterion was met during every session with every monkey in this study. During cumulative dosing experiments, a single dose of the test drug or drug mixture was administered i.m. at the start of each of five sequential 15 min cycles, and each dose increased the total cumulative dose by 1/4 or 1/2 log units. Starting 10 minutes after each injection, tail-withdrawal latencies were recorded as described above.
Initially, complete dose-effect curves were determined for fentanyl and U69,593 alone, and each drug was tested twice. Subsequently, three mixtures of fentanyl and U69,593 were examined, and the proportions of each drug in the mixtures were identical to those examined in the assay of schedule-controlled behavior described above. Each drug mixture was tested once. All tests with mixtures were separated by at least one week.
Drug effects were expressed as %Maximum Possible Effect (%MPE) using the following equation:
where test latency was the tail withdrawal latency from 50°C water obtained after drug administration, and control latency was the latency obtained at the beginning of the session prior to drug administration. ED50 and Zmix values for each drug or drug mixture were defined as the dose that produced 50%MPE, and these values were determined by linear regression as defined above. Individual ED50 and Zmix values were considered to be significantly different if confidence limits did not overlap.
Drug Interaction Analysis
Drug interactions were evaluated both within and across assays of schedule-controlled behavior and thermal nociception, because these assays generated monotonic dose-effect curves with clearly defined maximum effects. Moreover, data between these two assays could be readily compared because they used the same route of drug administration (i.m.).
Dose-Addition Analysis
Interactions between fentanyl and U69,593 within a given assay (schedule-controlled responding or thermal nociception) were assessed using both graphical and statistical approaches to dose-addition analysis (Tallarida, 2000; Wessinger, 1986; Woolverton, 1987) as described previously (Stevenson et al., 2005; Stevenson et al., 2003). Graphically, mean ED50 values for fentanyl administered either alone or as part of a mixture were plotted as a function of the ED50 value for U69,593 administered alone or as part of a mixture. This data presentation format is known as an isobologram, and the line in an isobologram that connects the data points for each drug alone shows predicted data points for drug mixtures assuming dose additivity. Points that fall above the line of additivity (away from the origin) are suggestive of sub-additivity, whereas points that fall below the line (toward the origin) are suggestive of super-additivity.
Statistical evaluation of drug interactions was accomplished by comparing the experimentally determined ED50 values for each mixture (Zmix) with predicted additive ED50 values (Zadd) as described by Tallarida (Tallarida, 2000). Zmix values were determined empirically as described above. Zadd values were calculated individually for each monkey from the equation Zadd=fA+(1−f)B, where A was the ED50 for fentanyl alone, B was the ED50 for U69,593 alone, and f was the fractional multiplier of A in the computation of the additive total dose. Any choice of f is related to the proportion of drug A (ρA) in a mixture according to the equation ρA = fA/Zadd. Mean Zmix and Zadd values were considered to be significantly different if 95% confidence limits did not overlap.
Dose-Ratio Analysis
To evaluate drug interactions across assays, the relative potency of each drug and drug mixture in the assays of schedule-controlled responding and thermal nociception was quantified according to the equation Dose Ratio (DR) = ED50 (or Zmix) in assay of schedule-controlled responding ÷ ED50 (or Zmix) in assay of thermal nociception. A dose ratio >1 indicates that the drug or drug mixture tended to be more potent in the assay of thermal nociception. Conversely, a dose ratio <1 indicates that the drug or drug mixture tended to be less potent in the assay of thermal nociception. The potency of each drug or drug mixture was considered to be different across the two procedures if the 95% confidence limits of the ED50 or Zmix values in the two procedures did not overlap.
Assay of Drug Self-administration
For intravenous drug administration, a double-lumen catheter was implanted into a jugular or femoral vein under aseptic conditions as described previously (Mello & Negus, 1998). One lumen of the double-lumen catheter was used for i.v. drug self-administration, and the second lumen was used for intermittent saline administration (0.1 ml every 20 min) to maintain catheter patency. Each monkey was fitted with a nylon vest attached to a flexible stainless-steel cable, and the distal end of the cable was attached to a fluid swivel (Lomir Biomedical, Montreal, Canada). This tether system protected the i.v. catheter and allowed freedom of movement for the animal. Catheter patency was monitored periodically by i.v. administration of ketamine (5 mg/kg) or the short-acting barbiturate methohexital (4 mg/kg). The catheter was considered patent if i.v. ketamine or methohexital administration resulted in loss of muscle tone within 10 s.
Experimental sessions were conducted in each monkey’s home cage (76.5 × 66 × 92 cm). The front wall of each cage was adapted for the attachment of an operant response panel identical to that described above. Two infusion pumps (Model B5P-1E, Braintree Scientific, Braintree, MA; or Model 98021, Harvard Apparatus, South Natick, MA) were mounted above each cage for delivery of saline or drug solutions through the two lumen of the intravenous catheters. The schedules of reinforcement were controlled and data were collected with an IBM-compatible computer and interface (MED Associates, Inc., Georgia, VT).
Drug self-administration sessions were conducted from 11am–1pm seven days per week, and monkeys were initially trained to respond for 32 μg/kg/inj cocaine under a FR 10 TO 60 s schedule until the total number of injections per session varied by ≤ 20% for three consecutive days. Saline was then substituted for cocaine until the number of injections per day decreased to fewer than five. Subsequently, testing was initiated using two procedures.
First, the ratio requirement was held constant at FR 10, and the dose of each drug and drug mixture was manipulated to determine self-administration dose-effect curves. Specifically, saline or a test dose of cocaine alone, fentanyl alone, U69,593 alone, or a mixture of U69,593 + fentanyl was made available for a single test session. Three U69,593/fentanyl mixtures were tested, and the proportions of fentanyl and U69,593 in the mixtures were identical to those tested in the assays of schedule-controlled responding and thermal nociception. Test sessions were separated by training sessions, during which either 32 μg/kg/inj cocaine or saline was available, and the sequence of training sessions between test sessions was determined by self-administration performance. If a given test solution maintained more than 20 injections/session, then saline was substituted for at least two days and until responding declined to fewer than 20 injections/session prior to the next test. If a given test solution maintained fewer than 20 injections/session, then 32 μg/kg/inj cocaine was reinstated for at least two days and until self-administration increased to rates greater than 20 injections/session. Subsequently, saline was substituted for at least two days and until responding declined to fewer than 20 injections/session prior to the next test. Thus, each test session was preceded by a period of self-administration at rates greater than 20 injections per session (maintained either by cocaine or by a test solution) and a period of saline substitution to extinguish self-administration to rates lower than 20 injections/session. Drugs and mixtures were tested in an irregular order across monkeys.
Second, the ratio requirement was systematically manipulated during availability of saline and selected doses of fentanyl alone (1.0 μg/kg/inj), U69,593 alone (0.65 μg/kg/inj) and a mixture of U69,593 + fentanyl + (0.65μg/kg/inj + 1.0 μg/kg/inj, respectively). This fentanyl dose was selected for study because it was on the descending limb of the fentanyl dose-effect curve and was therefore expected to function as a relatively effective reinforcer under conditions of increasing FR values. The U69,593 dose was selected to provide a 0.65:1 U69,593/fentanyl mixture (the intermediate ratio tested in all other studies). Saline and each drug or drug mixture was tested for a period of at least 21 days. Prior to each test, the maintenance dose of cocaine (32 μg/kg/in) was made available under an FR 10 schedule until self-administration exceeded 20 injections per session for at least two days. Subsequently, the test solution was made available under an FR 1 schedule for at least 3 days and until the total number of injections per session varied by ≤ 20% for three consecutive days. Once responding stabilized under the FR 1 schedule (≤7 days for all tests in all monkeys), then the FR was increased every three days to FR 3, 10, 30, 100, 300 and 1000. At the conclusion of each test, access to the maintenance dose of cocaine was reinstated under an FR 10 schedule as described above. Saline, drugs and drug mixtures were tested in an irregular order across monkeys.
For dose-effect studies, dose-effect curves for fentanyl and U69,593 self-administration were plotted to show the number of injections/session as a function of unit dose in μg/kg/inj (log scale). Rates of fentanyl and U69,593 self-administration were compared to rates of saline self-administration by one-way ANOVA. A significant ANOVA was followed by the Dunnett post hoc test to assess differences from saline self-administration. Rates of self-administration maintained by fentanyl alone and in combination with U69,593 were compared by two-way ANOVA, with dose of fentanyl and proportion of U69,593 as the two factors. A significant ANOVA was followed by the Bonferroni post hoc test to assess differences from fentanyl alone at each fentanyl dose. For FR studies, data from the last two days at each FR were averaged, and these data were plotted and analyzed in three ways. First, data were plotted as the number of injections/session as a function of FR (log scale). FR-effect curves for each test solution were compared by two-way ANOVA, with test solution and FR as the two factors. A significant ANOVA was followed by the Bonferroni post-hoc test to compare self-administration rates at each FR. For all ANOVAs, the significance level was set at p<0.05.
Second, an approach was used that is analogous to procedures used to calculate ED50 values for dose-effect curves. Specifically, rates of self-administration for each test solution at each FR were normalized as the percent baseline number of injections/session delivered for that solution at FR 1. This approach controlled for different baseline rates of self-administration and permitted assessment of FR manipulations on changes from these baseline self-administration rates. Normalized rates of self-administration were then plotted as function of log FR, and linear regression was used to determine the FR and 95% confidence limits at which self-administration decreased to 50% of baseline (the Effective FR producing 50% baseline self-administration, abbreviated EFR50). All points between 20 and 80% baseline were included in the linear regression, as were the highest FR associated with rates greater than 80% baseline and the lowest FR associated with rates less than 20% baseline. EFR50s for different test solutions were considered to be significantly different if 95% confidence limits did not overlap.
Finally, data were plotted and analyzed as normalized demand curves using analytic procedures described previously (Hursh & Winger, 1995; Winger, Hursh, Casey, & Woods, 2002). Demand curves plot log drug consumption (R*d) as a function of log price (FR÷d), where R is the number of reinforcers delivered, FR is the fixed ratio value, and d is the unit dose in mg/kg/inj. Because drugs differ in potency (which affects measures of both consumption and price), data were initially normalized using a normalized dose, q, calculated as 100 ÷ total number of reinforcers delivered for each self-administered solution at the lowest FR, (i.e. FR1). Log normalized consumption (logQ=R*q) was then plotted as a function of log normalized price (logP=FR÷q). Conditions that produced 0 consumption (i.e. no reinforcers delivered) were addressed by estimating log Q as −2, because log (0) cannot be calculated. A non-linear regression curve was then fit to the data for each subject in Graphpad Prism (Graphpad Software, San Diego, CA) using the equation Y=log(100)+b(X)−(a*(10^X)*log(e), where Y is logQ, X is logP, and “b” and “a” are floating variables indicating initial slope and slope acceleration, respectively. Initial values were set to b=−0.05 and a=0.004 as described by Winger et al. (Winger, Hursh, Casey, & Woods, 2002), and the software returned final values of b and a. These values were then used to calculate Pmax (the normalized price maintaining the maximum response output) according to the equation Pmax=(1+b)/a. Individual Pmax values for each self-administered solution were averaged to yield a mean and 95% confidence limits. and Pmax values for different test solutions were considered to be significantly different if 95% confidence limits did not overlap. A correlation between Pmax and EFR50 values was determined using standard linear regression. All statistical tests were conducted with Prism 4.0c for Macintosh (GraphPad Software, San Diego, CA).
Drugs
Fentanyl HCl (National Institute on Drug Abuse, Bethesda, MD) was dissolved in distilled water, and U69,593 (Sigma Chemical Co., St. Louis, MO) was dissolved in 0.5% lactic acid in sterile water. For the assays of schedule-controlled responding and thermal nociception, all drugs were administered i.m. in the thigh. For the assay of drug self-administration, drug solutions were filter-sterilized using a 0.22 micron Millipore filter and delivered i.v. Doses were determined based on the free base or salt forms listed above.
Results
Assay of Schedule-Controlled Responding
Figure 1 shows the effects of fentanyl and U69,593 administered alone or in combination in the assay of schedule-controlled responding for food reinforcement. When administered alone, both fentanyl and U69,593 produced dose-dependent decreases in response rates (Figure 1a). Table 1 shows that the potencies of fentanyl and U69,593 were not significantly different, and the relative potency of U69,593 to fentanyl was 0.65:1. This relative potency served as the basis for the proportions of U69,593 and fentanyl in the subsequent mixtures.
Figure 1. Effects of fentanyl alone, U69,593 alone and U69,593/fentanyl mixtures in the assay of schedule-controlled responding for food reinforcement.

(a) Effects of fentanyl and U69,593 alone. Abscissa: Dose of test drug in mg/kg (log scale). Ordinate: Percent control rate of responding. (b) Isobologram for U69,593/fentanyl interactions. Abscissa: ED50 value in mg/kg for fentanyl alone or as part of a mixture. Ordinate: ED50 value in mg/kg for U69,593 alone or as part of a mixture. * Indicates significantly different from additivity (see Table 2). (c) Effects of fentanyl alone or as part of a mixture. Abscissa: Dose fentanyl in mg/kg (log scale). Ordinate: Percent control rate of responding. (d) Effects of U69,593 alone or as part of a mixture. Abscissa: Dose of U69,593 in mg/kg (log scale). Ordinate: Percent control rate of responding. All points in (a) and (b) show mean ± SEM for 4 monkeys, and an apparent absence of error bars indicates that the SEM is contained within the point.. Only mean data are shown in (c) and (d) for clarity.
Table 1.
ED50 values in mg/kg (95% CL) for the mu agonist fentanyl and the kappa agonist U69,593 administered alone or as part of a mixture in assays of schedule-controlled responding for food reinforcement and warm-water tail withdrawal in rhesus monkeys.
| Fentanyl ED50 (mg/kg) | U69,593 ED50 (mg/kg) | |
|---|---|---|
| Schedule-Controlled Responding | ||
| Fentanyl | 0.0069 (0.0058–0.0082) | |
| U69,593 | 0.0045 (0.0033–0.0062) | |
| 0.22:1 U69/Fent | 0.0141 (0.0082–0.0242)* | 0.0031 (0.0018–0.0053) |
| 0.65:1 U69/Fent | 0.0063 (0.0053–0.0076) | 0.0041 (0.0034–0.0049) |
| 1.96:1 U69/Fent | 0.0037 (0.0020–0.0069) | 0.0073 (0.0039–0.0136) |
| Warm-Water Tail Withdrawal | ||
| Fentanyl | 0.0157 (0.0075–0.0326) | |
| U69,593 | 0.0751 (0.0614–0.0919) | |
| 0.22:1 U69/Fent | 0.0130 (0.0130–0.0130) | 0.0029 (0.0029–0.0029)* |
| 0.65:1 U69/Fent | 0.0196 (0.0116–0.0332) | 0.0128 (0.0076–0.0216)* |
| 1.96:1 U69/Fent | 0.0095 (0.0051–0.0175) | 0.0186 (0.0101–0.0342)* |
Asterisks indicate that 95%CL of the ED50 for a drug in a mixture do not overlap with 95%CL of the ED50 for that drug alone.
Each of the mixtures also produced a dose-dependent decrease in response rates (Figures 1c and 1d). With one exception, the potencies of fentanyl and U69,593 in the mixtures were not different from the potencies of fentanyl and U69,593 alone (Table 1). The one exception was that the potency of fentanyl in the 0.22:1 mixtures was lower than the potency of fentanyl alone. Table 2 and Figure 1b shows the results of dose-addition analysis of the interaction between fentanyl and U69,593. In general, there was a tendency for mixtures of fentanyl and U69,593 to produce infra-additive effects, and dose-addition analysis indicated that effects produced by the 0.22:1 and 0.65:1 U69,593/fentanyl mixtures were significantly less than additive.
Table 2.
Predicted additive ED50 values (Zadd) and experimentally determined ED50 values (Zmix) for fentanyl + U69,593 mixtures in assays of schedule-controlled responding and warm-water tail-withdrawal. Zadd and Zmix values show the total dose of fentanyl + U69,593 in the mixture in mg/kg (95% confidence limits). The proportion of U69,593 to fentanyl is shown for each mixture.
| Zadd | Zmix | |
|---|---|---|
| Schedule-Controlled Responding | ||
| 0.22:1 U69/Fent | 0.0063 (0.0052–0.0076) | 0.017 (0.010–0.029)* |
| 0.65:1 U69/Fent | 0.0057 (0.0046–0.0071) | 0.010 (0.0087–0.012)* |
| 1.96:1 U69/Fent | 0.0051 (0.0040–0.0066) | 0.011 (0.0059–0.021) |
| Warm-Water Tail Withdrawal | ||
| 0.22:1 U69/Fent | 0.019 (0.010–0.034) | 0.016 (0.016–0.016) |
| 0.65:1 U69/Fent | 0.024 (0.015–0.038) | 0.032 (0.019–0.055) |
| 1.96:1 U69/Fent | 0.034 (0.026–0.047) | 0.028 (0.015–0.052) |
Asterisks indicate an experimental Zmix value significantly different from the predicted additive Zadd value (p<0.05).
Assay of Thermal Nociception
Figure 2 shows the effects of fentanyl and U69,593 administered alone or in combination in the assay of thermal nociception. When administered alone, both fentanyl and U69,593 produced dose-dependent antinociceptive effects (Figure 2a). However, in contrast to results in the assay of schedule-controlled responding, U69,593 was significantly less potent than fentanyl (Table 1).
Figure 2. Effects of fentanyl alone, U69,593 alone and U69,593/fentanyl mixtures in the assay of thermal nociception.

(a) Effects of fentanyl and U69,593 alone. Abscissa: Dose of test drug in mg/kg (log scale). Ordinate: Percent maximum possible effect (%MPE). (b) Isobologram for U69,593/fentanyl interactions. Abscissa: ED50 value in mg/kg for fentanyl alone or as part of a mixture. Ordinate: ED50 value in mg/kg for U69,593 alone or as part of a mixture. * Indicates significantly different from additivity (see Table 2). (c) Effects of fentanyl alone or as part of a mixture. Abscissa: Dose fentanyl in mg/kg (log scale). Ordinate: Percent maximum possible effect (%MPE). (d) Effects of U69,593 alone or as part of a mixture. Abscissa: Dose of U69,593 in mg/kg (log scale). Ordinate: Percent maximum possible effect (%MPE). All points in (a) and (b) show mean ± SEM for 4 monkeys, and an apparent absence of error bars indicates that the SEM is contained within the point. Only mean data are shown in (c) and (d) for clarity.
Each of the mixtures also produced dose-dependent antinociception (Figures 2c and 2d). The potencies of fentanyl in the mixtures were not different from the potencies of fentanyl administered alone; however, the potency of U69,593 in each of the mixtures was significantly higher than the potency of U69,593 alone (Table 1). Table 2 and Figure 2b shows the results of dose-addition analysis of the interaction between fentanyl and U69,593. Effects were additive for all three mixtures.
Dose -Ratio Analysis
Figure 3 shows the dose ratios for fentanyl alone, U69,593 alone and each of the mixtures in the assays of schedule-controlled responding and thermal nociception. These dose ratios provide a measure of behavioral selectivity, with dose ratios >1 indicating selectivity to produce thermal nociception and dose ratios <1 indicating selectivity to decrease rates of schedule-controlled responding. The dose-ratio for fentanyl was 0.43, and confidence limits for the fentanyl ED50 values in the two procedures overlapped, indicating that fentanyl displayed similar potencies across the two procedures. Conversely, the dose ratio for U69,593 was 0.06, and ED50 confidence limits did not overlap, indicating that U69,593 was significantly more potent in reducing rates of food-maintained responding than in producing thermal nociception. For the mixtures, the highest dose ratio (1.06) was achieved with the 0.22:1 U69.593/fentanyl mixture. However, Zmix confidence limits overlapped across the two procedures for all three mixtures, indicating that none of the mixtures was significantly more potent in one procedure than the other.
Figure 3. Dose ratios for fentanyl alone, U69,593 alone and U69.593/fentanyl mixtures.
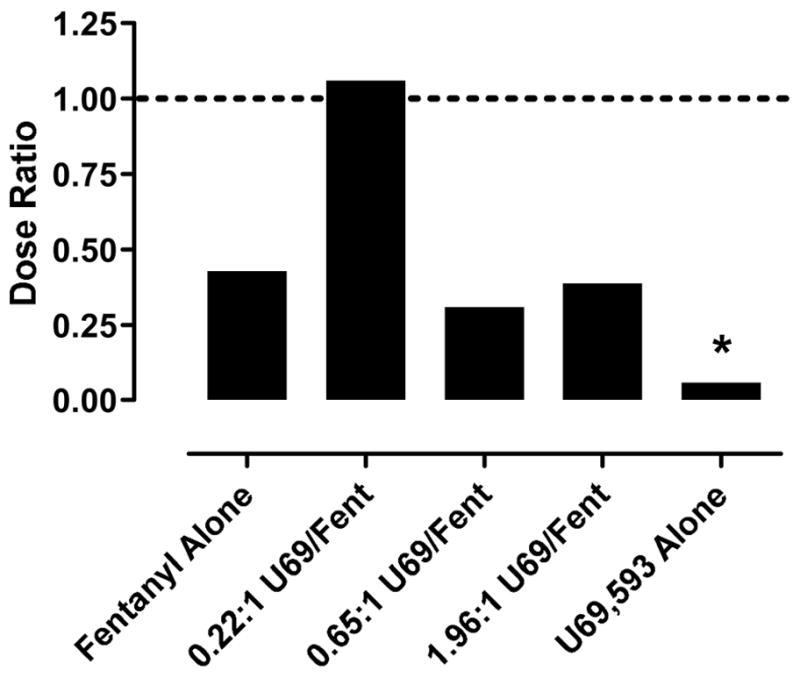
Dose ratios were determined as ED50 or Zmix in the assay of thermal nociception ÷ ED50 or Zmix in the assay of schedule-controlled responding for food reinforcement. * Asterisks indicate the confidence limits for the ED50 or Zmix values in the two procedures did not overlap.
Drug Self-Administration
Figure 4a shows drug self-administration dose-effect curves for fentanyl and U69,593 alone. The dose-effect curve for fentanyl alone displayed the typical inverted-U shape, and a dose of 0.32 μg/kg/inj fentanyl maintained a rate of self-administration significantly greater than that maintained by saline. Conversely, U69,593 did not maintain self-administration at any dose, and higher doses of 0.32–1.0 μg/kg/inj tended to maintain self-administration rates lower than the rate maintained by saline. Figure 4b compares self-administration dose-effect curves for fentanyl alone and fentanyl in combination with different proportions of U69,593. The addition of U69,593 decreased rates of self-administration relative to fentanyl alone. At a unit dose of 0.32 μg/kg/inj fentanyl, all three proportions of U69,593 reduced rates of self-administration below those maintained by fentanyl alone. At a higher unit dose of 1 μg/kg/inj fentanyl, the 1.96:1 U69,593/fentanyl mixture maintained significantly lower rates than fentanyl alone.
Figure 4. Self-administration dose-effect curves for fentanyl alone, U69,593 alone, and U69,593/fentanyl mixtures.

(a) Abscissa: Unit dose of test drug in μg/kg/inj (log scale). Points above “S” and “C” show rates of responding maintained by saline and by 0.01 mg/kg/inj cocaine, respectively. Ordinate: Number of injections/session. All points show mean data ± SEM for three monkeys, and an apparent absence of error bars indicates that the SEM is contained within the point. One-way ANOVA indicated a significant effect of fentanyl dose (p=0.0276), and asterisks in the graph indicate points significantly different from saline (p<0.05) as determined by the Dunnett post hoc test. ANOVA also indicated a significant effect of U69,593 dose (p<0.0053); however no U69,593 points were significantly different from saline. Rather, rates maintained by high U69,593 doses were significantly lower than rates maintained by low U69,593 doses. (b) Abscissa: Unit dose fentanyl in μg/kg/inj (log scale). Ordinate: Number of injections/session. All points show mean data ± SEM for three monkeys. Two-way ANOVA indicated not significant main effect of fentanyl dose (p>0.05), but there was a significant main effect of U69,593 proportion (p=0.0011) and a significant interaction between U69,593 proportion and fentanyl dose (p=0.0091). Symbols indicate a significant difference between fentanyl alone and effect of a mixture, p<0.05. * 0.22:1 U69,593/Fentanyl, $ 0.65:1 U69,593/Fentanyl, † 1.96:1 U69,593/Fentanyl. Differences between the mixtures were not significant at any dose.
Figure 5a compares the effects of varying the FR on self-administration maintained by saline, fentanyl (1.0 μg/kg/inj), U69,593 (0.65 μg/kg/inj) and the U69,593/fentanyl mixture. Fentanyl maintained significantly higher rates of self-administration than saline at FR values of 3–30, and higher rates than either U69,593 or the U69.593/fentanyl mixture at FR values of 1–30. U69,593 maintained significantly lower rates of self-administration than saline at the lowest FR (FR 1). The U69,593/fentanyl mixture did not maintain rates different from saline or from U69,593 at any FR.
Figure 5. Effects of fixed-ratio manipulations on responding maintained by saline, 1.0 μg/kg/inj fentanyl, 0.65 μg/kg/inj U69,593 or a U69,593/fentanyl mixture.

(a) Abscissa: Fixed ratio value (log scale). Ordinate: Total number of injections delivered per session. Two-way ANOVA for data in (a) indicated significant main effects of FR (p<0.0001) and self-administered solution (p<0.0094) and a significant interaction (p<0.0001). Symbols indicate a significant difference between values for saline and drug solutions at a given FR using the Bonferroni post hoc test, p<0.05. $-- fentanyl vs. saline; *--fentanyl vs. U69,593, †--fentanyl vs. the U69,593/fentanyl mixture, #--U69,593 vs. saline. (b) Regression lines on normalized self-administration data used to calculate EFR50 values. Abscissa: Fixed ratio value (log scale). Ordinate: Normalized self-administration expressed as percent baseline number of injections delivered at FR 1. (c) Normalized demand curves used to calculate Pmax values. Abscissa: Log normalized price (FR÷q). Ordinate: Normalized consumption (R*q). (d) Correlation of EFR50 and Pmax values. All points show mean data from 3 monkeys (saline) or 4 monkeys (fentanyl, U69,593 and the mixture). Error bars in (a) and (b) show the SEM. Error bars are not shown in (c) and (d) for clarity. Fentanyl alone (filled triangles, solid black line), U69,593 alone (inverted open triangles, solid black line), fentanyl + U69,593 (gray diamond, gray line), and saline (open circle, dotted black line).
Figures 5b and 5c show results of the EFR50 and demand curve analyses, respectively. Both the EFR50 and the Pmax values for fentanyl were significantly greater than the corresponding values for either saline or U69,593, which were not different from each other. The mean EFR50 and Pmax values for the mixture were intermediate between the corresponding values for fentanyl alone and U69,593 alone, but 95% confidence limits for the mixture overlapped with 95% confidence limits for each drug alone and for saline. Overall, the results of EFR50 and demand curve analyses were similar, and figure 5d shows that the correlation between EFR50 and Pmax values approached significance.
Discussion
The use of morphine-like opioid analgesics is limited by high abuse liability. The main finding of the present study was that addition of the kappa agonist U69,593 to the mu agonist fentanyl produced a proportion-dependent decrease in fentanyl self-administration without reducing the antinociceptive effects of fentanyl. The lowest proportion of U69,593 (0.22:1 U69,593/fentanyl) also attenuated the rate-decreasing effects of fentanyl in the assay of schedule-controlled responding. Taken together, these results show that kappa agonists can differentially modulate different effects of mu agonists. A potential implication of these results is that either mixtures of mu and kappa agonists or single molecules that possess both mu and kappa agonist activity may warrant further investigation as candidate analgesics with reduced abuse liability.
Interactions between fentanyl and U69,593 in the assays of schedule-controlled responding and thermal nociception
In agreement with previous studies, both the mu agonist fentanyl and the kappa agonist U69,593 produced dose-dependent rate-decreasing effects in the assay of schedule-controlled responding for food reinforcement and increases in tail-withdrawal latencies in the assay of thermal nociception (S. S. Negus et al., 1993; S S Negus, Zuzga, & Mello, 2002; Stevenson et al., 2003). Mixtures of these compounds also produced rate-decreasing and antinociceptive effects. However, the nature of the interaction between fentanyl and U69,593 varied as a function of the experimental endpoint. In the assay of schedule-controlled responding for food reinforcement, interactions between fentanyl and U69,593 were less than additive for two of the mixtures (0.22:1 and 0.65:1 U69,593/fentanyl), and effects of the 1.95:1 mixture approached subadditivity. Moreover, addition of the lowest proportion of U69,593 to fentanyl (0.22:1) actually produced a significant reduction in the potency of fentanyl to reduce response rates in this procedure.
Conversely, the combination of U69,593 and fentanyl produced dose-additive effects in the assay of thermal nociception. These additive antinociceptive effects of mu and kappa agonists in rhesus monkeys agree with previous reports of additive effects between mu and kappa agonists in assays of thermal nociception in mice and rats (Narita et al., 1993; Verborgh et al., 1997). However, these findings contrast with the finding that kappa agonists attenuated the antinociceptive effects of mu agonists in a shock-titration assay in squirrel monkeys (Craft & Dykstra, 1992a, 1992b). The finding that additive mu/kappa antinociceptive effects in rhesus monkeys resembled those found in mice and rats suggests that species may not be a critical determinant of these interactions despite the existence of species differences in opioid receptor density, distribution and function (Berger et al., 2006; Mansour et al., 1988). Rather, these findings suggest that the experimental procedure used to assess nociception may be important. Thus, additive mu/kappa effects have been observed in all studies that assessed antinociception by measuring unconditioned withdrawal responses from noxious heat stimuli (Narita et al., 1993; Verborgh et al., 1997), present study), whereas an antagonistic interaction was observed in the shock-titration procedure, which uses both a different modality of noxious stimulus and a different behavioral measure. In particular, shock titration relies on operant conditioning and can be considered a type of schedule-controlled behavior (Dykstra, 1985). From this perspective, the antagonistic mu/kappa interaction observed in the squirrel monkey shock-titration procedure may be related to the subadditive/antagonistic mu/kappa interaction observed in the present study in the assay of schedule-controlled responding for food reinforcement. At the very least, these results suggest that a more comprehensive understanding of mu/kappa antinociceptive interactions will require assessment of these interactions in a broader array of assays related to pain and analgesia.
The different interactions between fentanyl and U69,593 within the assays of schedule-controlled responding and thermal nociception resulted in only modest changes in the dose-ratios for production of these two effects across assays. The mixture with the lowest proportion of U69,593 (0.22:1 U69,593/fentanyl) produced the highest dose ratio, which was suggestive of a marginal increase in the selectivity of this mixture to produce thermal nociception vs. rate suppression relative to fentanyl alone. However, neither fentanyl nor any of the mixtures displayed significant selectivity for production of antinociception. This contrasts with the finding that some mixtures of mu and delta agonists can produce selective antinociception vs. rate suppression in these same procedures (Negus, unpublished observations).
Interactions between fentanyl and U69,593 in the assay of drug self-administration
In agreement with previous studies of mu and kappa agonists, fentanyl maintained self-administration in rhesus monkeys whereas U69,593 did not (Tang & Collins, 1985; Woods et al., 1982). Indeed, 0.65 μg/kg/inj U69,593 maintained significantly lower self-administration than saline under an FR 1 schedule, which may be consistent with the putative aversive effects of kappa agonists in place conditioning and intracranial self-stimulation procedures (Mucha & Herz, 1985; Todtenkopf et al., 2004). The addition of U69,593 to fentanyl produced a proportion-dependent decrease in fentanyl self-administration, and this finding is consistent with previous reports that kappa agonists decreased mu agonist self-administration or mu agonist-induced place preferences in rodents (Funada et al., 1993; Xi et al., 1998). Taken together, these findings suggest that U69,593 proportions that produce additive antinociceptive effects with fentanyl also produce a decrease in the abuse-related reinforcing effects of fentanyl.
Reductions in self-administration could reflect an ability of U69,593 to produce non-selective rate-suppression rather than a reduction of reinforcing effects. More specifically, it is possible that self-administered doses of U69,593/fentanyl mixtures produced more prominent or longer-acting rate-decreasing effects than fentanyl alone, and these greater rate-decreasing effects may have prevented the emission of high response rates under the conditions of this study. Two findings argue against this possibility. First, as noted above, fentanyl and U69,593 produced subadditive rate-suppressing effects in the assay of schedule-controlled responding for food reinforcement, and the lowest proportion of U69,593 (0.22:1 U69,593/fentanyl) significantly attenuated the rate-decreasing effects of fentanyl. Although drug interactions from the assay of schedule-controlled responding cannot be directly extrapolated to the self-administration procedure because the routes of administration were different (i.m. vs. i.v.), this finding suggests that lower rates of self-administration cannot be attributed simply to the rate-decreasing effects of U69,593.
Second, the effects of FR manipulations also provide evidence to suggest that U69,593 reduced the reinforcing effects of fentanyl. Manipulations of the response requirement are central to the use of progressive ratio schedules (Arnold & Roberts, 1997), behavioral economic strategies for assessment of drug reinforcing efficacy (Hursh & Winger, 1995), and evaluations of the relative persistence of drug self-administration (Meisch, 2000). A shared principle of all these approaches is that reinforcing efficacy can be inferred from the sensitivity of drug self-administration to increases in the ratio requirement. A drug (or drug dose) is considered to have higher reinforcing efficacy than another drug (or drug dose) if it is less sensitive to increases in ratio value. In the present study, sensitivity to increases in ratio requirement was quantified primarily as the EFR50, the FR that reduced self-administration to 50% of baseline levels observed at the lowest FR (FR 1). The EFR50 for 1.0 μg/kg/inj fentanyl was significantly greater than the EFR50s for either saline or U69,593. This finding provides additional evidence to suggest that fentanyl functioned as an effective reinforcer under these conditions, while also demonstrating that this experimental and analytic strategy can be used to differentiate the reinforcing efficacy of different stimuli. The mixture of U69,593/fentanyl produced an EFR50 intermediate between that of fentanyl and U69,593, suggesting a trend for U69,593 to decrease this measure of fentanyl’s reinforcing efficacy. In addition, the EFR50 values for the U69,593/fentanyl mixture and for U69,593 alone were not different from the EFR50 for saline, indicating that these solutions did not produce significant reinforcing effects by this measure.
This is the first application of EFR50 analysis to compare the reinforcing efficacy of different consequent stimuli. Advantages of this approach include (a) homology to commonly used approaches for calculating ED50 values from dose-effect curves, (b) use of linear rather than non-linear regression to calculate derived values, (c) R2 values >0.95, indicating an excellent fit of the data, and (d) well-established methods for calculation of confidence limits, which facilitates statistical comparison of derived values. Moreover, EFR50 analysis yielded results comparable to those obtained with more established demand curve analysis, in which Pmax served as the primary measure of reinforcing efficacy. Demand curve analysis has acknowledged strengths as a conceptual approach to drug self-administration data (Hursh & Winger, 1995; Winger et al., 2002), especially insofar as demand curves can incorporate data obtained with multiple unit doses and FR values into a unitary curve that yields a single metric of reinforcing efficacy (e.g. the Pmax value). However, this analysis also has some weaknesses. For example, non-linear regression requires a somewhat arbitrary selection of starting values for some variables, and the equations used to generate non-linear regression curves are still evolving (Hursh & Silberberg, 2008). Moreover, conditions that reduce consumption to “0” may be of practical or experimental interest, but as illustrated by the present study, such data must be either estimated or excluded in demand curve analysis. Consequently, strategies such as EFR50 analysis may provide a useful complement to demand curve analysis for some applications.
Implications for analgesic drug development
The finding that mu/kappa interactions were different in the assays of thermal nociception, schedule-controlled responding and drug self-administration contributes to a growing literature on the degree to which drug interactions may vary across experimental endpoints (e.g. (O’Neill, Collins, Pettit, McNutt, & Chang, 1997; Stevenson et al., 2003). This is perhaps not surprising. Drugs that target different binding sites can interact in many different ways. For example, drugs could produce interacting effects by targeting allosteric binding sites on a common protein, binding sites on adjacent proteins physically coupled in a common receptor oligomer, physically separated proteins that modulate convergent signal cascades in a common cell, or receptors located on different cells located in a common neural circuit. These mechanisms (and others, e.g. pharmacokinetic interactions) likely contribute to different degrees in different biological systems mediating different drug effects. This variety of potential mechanisms underscores both the importance of examining drug interactions across a range of endpoints and the risk of generalizing drug interactions from any one endpoint to any other endpoint. However, this complexity also provides the basis for an opportunity to develop drug mixtures or mixed-actions drugs that produce enhanced desirable effects and/or reduced undesirable effects relative to highly selective drugs administered alone (Sivachenko, Kalinin, & Yuryev, 2006).
Drug interactions depend not only on the endpoint under investigation, but also on the relative proportions of the drugs in a mixture and the relative efficacy of the drugs at their respective targets (Stevenson et al., 2003). In view of these considerations, the present study provides some insight into the characteristics that might be useful for simultaneous activation of mu and kappa receptors. For example, this study employed agonists with relatively high efficacy at mu and kappa opioid receptors, and the results provide evidence that high efficacy stimulation of mu and kappa opioid receptors can produce robust antinociception with reduced abuse liability. Several mixed-action mu/kappa opioids are already available clinically (e.g. nalbuphine and butorphanol), and these compounds are generally considered to have lower abuse liability than many selective mu agonists (Hursh & Winger, 1995; Preston & Jasinski, 1991); however, these compounds have low efficacy at both mu and kappa opioid receptors (Emmerson et al., 1996; Remmers et al., 1999) and also produce relatively weak antinociception in animals and analgesia in humans (Butelman et al., 1995; Gerak, Butelman, Woods, & France, 1994; Gutstein & Akil, 2005). The present results suggest that high efficacy activation of mu and kappa receptors could produce greater antinociceptive effects than existing mixed-action but low-efficacy mu/kappa opioids while retaining low abuse liability.
The present study also examined the effects of different proportions of mu and kappa agonists, and an argument could be made that the most favorable effects were produced by the 0.22:1 U69,593/fentanyl mixture. It is well-established that selective kappa agonists produce psychotomimetic effects in humans (Pfeiffer et al., 1986; Walsh et al., 2001), and attempts to develop selective kappa agonists as analgesics have foundered in part because patients find these psychotomimetic effects to be aversive. Accordingly, the present results would be consistent with the proposition that a relatively low proportion of kappa to mu activity might be sufficient to reduce abuse liability without producing sufficiently profound aversive effects to impede compliance.
One final comment is warranted regarding the potential abuse liability of kappa agonists alone or in combination with mu agonists. Although kappa agonists generally do not produce preclinical abuse-related effects such as maintenance of self-administration, these compounds do produce pronounced discriminative stimulus effects in animals and subjective effects in humans, and these effects may be predictive of, or contribute to, some level of misuse. For example, the highly selective kappa agonist salvinorin is the principle psychoactive constituent of the plant salvia divinorum, and recreational use of this plant has been reported (Gonzalez, Riba, Bouso, Gomez-Jarabo, & Barbanoj, 2006; Prisinzano, 2005). The role of kappa receptor-mediated discriminative/subjective effects to salvia use remains to be determined, as does the degree to which these subjective effects might be altered by co-activation of mu receptors. However, selective mu and kappa agonists did not alter each other’s discriminative stimulus effects in animals (S. S. Negus et al., 1990, 1991), which suggests that mu and kappa agonists might not alter each other’s subjective effects in humans. The implications of this finding for abuse liability of mu/kappa mixtures remains to be determined.
Table 3.
EFR50 and Pmax values (95% confidence limits) obtained for saline, 1.0 μg/kg/inj fentanyl alone, 0.65 μg/kg U69,593 alone, and a mixture of 1.0 fentanyl and 0.65 U69,593.
| Self-Administered Solution | EFR50 | Pmax |
|---|---|---|
| Fentanyl Alone | 42.5 (23.4–93.3) | 45.2 (21.6–94.3) |
| Fentanyl + U69,593 | 24.9 (13.5–49.8) | 29.9 (8.3–107.7) |
| U69,593 Alone | 15.7 (11.1–22.2)* | 5.9 (1.9–18.3)* |
| Saline | 9.6 (6.0–15.5)* | 10.2 (6.3–16.4)* |
Indicates significantly different from fentanyl as determined by non-overlapping confidence limits.
Acknowledgments
This work was supported by grant R01-DA11460 from NIDA, NIH. The authors would like to thank Ashley Bear for expert technical assistance.
References
- Arnold JM, Roberts DC. A critique of fixed and progressive ratio schedules used to examine the neural substrates of drug reinforcement. Pharmacol Biochem Behav. 1997;57(3):441–447. doi: 10.1016/s0091-3057(96)00445-5. [DOI] [PubMed] [Google Scholar]
- Berger B, Rothmaier AK, Wedekind F, Zentner J, Feuerstein TJ, Jackisch R. Presynaptic opioid receptors on noradrenergic and serotonergic neurons in the human as compared to the rat neocortex. Br J Pharmacol. 2006;148(6):795–806. doi: 10.1038/sj.bjp.0706782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertalmio AJ, Woods JH. Differentiation between mu and kappa receptor-mediated effects in opioid drug discrimination: apparent pA2 analysis. J Pharmacol Exp Ther. 1987;243(2):591–597. [PubMed] [Google Scholar]
- Butelman ER, Ko MC, Sobczyk-Kojiro K, Mosberg HI, Van Bemmel B, Zernig G, et al. kappa-Opioid receptor binding populations in rhesus monkey brain: relationship to an assay of thermal antinociception. J Pharmacol Exp Ther. 1998;285(2):595–601. [PubMed] [Google Scholar]
- Butelman ER, Winger G, Zernig G, Woods JH. Butorphanol: characterization of agonist and antagonist effects in rhesus monkeys. J Pharmacol Exp Ther. 1995;272(2):845–853. [PubMed] [Google Scholar]
- Craft RM, Dykstra LA. Agonist and antagonist activity of kappa opioids in the squirrel monkey: I. Antinociception and urine output. J Pharmacol Exp Ther. 1992a;260(1):327–333. [PubMed] [Google Scholar]
- Craft RM, Dykstra LA. Agonist and antagonist activity of kappa opioids in the squirrel monkey: II. Effect of chronic morphine treatment. J Pharmacol Exp Ther. 1992b;260(1):334–342. [PubMed] [Google Scholar]
- Dykstra LA. Behavioral and pharmacological factors in opioid analgesia. In: Seiden LS, Balster RL, editors. Behavioral Pharmacology: The Current Status. New York: Alan r. Liss, Inc; 1985. pp. 111–129. [Google Scholar]
- Dykstra LA. Butorphanol, levallorphan, nalbuphine and nalorphine as antagonists in the squirrel monkey. J Pharmacol Exp Ther. 1990;254(1):245–252. [PubMed] [Google Scholar]
- Dykstra LA, Gmerek DE, Winger G, Woods JH. Kappa opioids in rhesus monkeys. I. Diuresis, sedation, analgesia and discriminative stimulus effects. J Pharmacol Exp Ther. 1987;242(2):413–420. [PubMed] [Google Scholar]
- Emmerson PJ, Clark MJ, Mansour A, Akil H, Woods JH, Medzihradsky F. Characterization of opioid agonist efficacy in a C6 glioma cell line expressing the mu opioid receptor. J Pharmacol Exp Ther. 1996;278(3):1121–1127. [PubMed] [Google Scholar]
- Emmerson PJ, Liu MR, Woods JH, Medzihradsky F. Binding affinity and selectivity of opioids at mu, delta and kappa receptors in monkey brain membranes. J Pharmacol Exp Ther. 1994;271(3):1630–1637. [PubMed] [Google Scholar]
- Funada M, Suzuki T, Narita M, Misawa M, Nagase H. Blockade of morphine reward through the activation of kappa-opioid receptors in mice. Neuropharmacology. 1993;32(12):1315–1323. doi: 10.1016/0028-3908(93)90026-y. [DOI] [PubMed] [Google Scholar]
- Gerak LR, Butelman ER, Woods JH, France CP. Antinociceptive and respiratory effects of nalbuphine in rhesus monkeys. J Pharmacol Exp Ther. 1994;271(2):993–999. [PubMed] [Google Scholar]
- Gonzalez D, Riba J, Bouso JC, Gomez-Jarabo G, Barbanoj MJ. Pattern of use and subjective effects of Salvia divinorum among recreational users. Drug Alcohol Depend. 2006;85:157–162. doi: 10.1016/j.drugalcdep.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Gutstein H, Akil H. Opioid analgesics. In: Brunton L, Lazo J, Parker K, editors. The Pharmacological Basis of Therapeutics. Vol. 11. New York: McGraw-Hill; 2005. pp. 547–590. [Google Scholar]
- Hursh SR, Winger G. Normalized demand for drugs and other reinforcers. J Exp Anal Behav. 1995;64(3):373–384. doi: 10.1901/jeab.1995.64-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hursh SR, Silberberg A. Economic demand and essential value. Psychological Review. 2008;115(1):186–198. doi: 10.1037/0033-295X.115.1.186. [DOI] [PubMed] [Google Scholar]
- Malmberg AB, Yaksh TL. Pharmacology of the spinal action of ketorolac, morphine, ST-91, U50488H, and L-PIA on the formalin test and an isobolographic analysis of the NSAID interaction. Anesthesiology. 1993;79(2):270–281. doi: 10.1097/00000542-199308000-00012. [DOI] [PubMed] [Google Scholar]
- Manchikanti L. National drug control policy and prescription drug abuse: facts and fallacies. Pain Physician. 2007;10(3):399–424. [PubMed] [Google Scholar]
- Mansour A, Khachaturian H, Lewis ME, Akil H, Watson SJ. Anatomy of CNS opioid receptors. Trends Neurosci. 1988;11(7):308–314. doi: 10.1016/0166-2236(88)90093-8. [DOI] [PubMed] [Google Scholar]
- Meisch RA. Relative persistence of behavior: a fundamental measure of relative reinforcing effects. Exp Clin Psychopharmacol. 2000;8(3):333–349. doi: 10.1037//1064-1297.8.3.333. [DOI] [PubMed] [Google Scholar]
- Mello NK, Negus SS. Effects of kappa opioid agonists on cocaine- and food-maintained responding by rhesus monkeys. J Pharmacol Exp Ther. 1998;286(2):812–824. [PubMed] [Google Scholar]
- Miranda HF, Puig MM, Dursteler C, Prieto JC, Pinardi G. Dexketoprofen-induced antinociception in animal models of acute pain: synergy with morphine and paracetamol. Neuropharmacology. 2007;52(2):291–296. doi: 10.1016/j.neuropharm.2006.07.025. [DOI] [PubMed] [Google Scholar]
- Mucha RF, Herz A. Motivational properties of kappa and mu opioid receptor agonists studied with place and taste preference conditioning. Psychopharmacology (Berl) 1985;86(3):274–280. doi: 10.1007/BF00432213. [DOI] [PubMed] [Google Scholar]
- Narita M, Takahashi Y, Takamori K, Funada M, Suzuki T, Misawa M, et al. Effects of kappa-agonist on the antinociception and locomotor enhancing action induced by morphine in mice. Jpn J Pharmacol. 1993;62(1):15–24. doi: 10.1254/jjp.62.15. [DOI] [PubMed] [Google Scholar]
- Negus SS, Burke TF, Medzihradsky F, Woods JH. Effects of opioid agonists selective for mu, kappa and delta opioid receptors on schedule-controlled responding in rhesus monkeys: antagonism by quadazocine. J Pharmacol Exp Ther. 1993;267(2):896–903. [PubMed] [Google Scholar]
- Negus SS, Mello NK. Opioid antinociception in ovariectomized monkeys: comparison with antinociception in males and effects of estradiol replacement. J Pharmacol Exp Ther. 1999;290(3):1132–1140. [PubMed] [Google Scholar]
- Negus SS, Picker MJ, Dykstra LA. Interactions between mu and kappa opioid agonists in the rat drug discrimination procedure. Psychopharmacology (Berl) 1990;102(4):465–473. doi: 10.1007/BF02247126. [DOI] [PubMed] [Google Scholar]
- Negus SS, Picker MJ, Dykstra LA. Interactions between the discriminative stimulus effects of mu and kappa opioid agonists in the squirrel monkey. J Pharmacol Exp Ther. 1991;256(1):149–158. [PubMed] [Google Scholar]
- Negus SS, Zuzga DS, Mello NK. Sex differences in opioid antinociception in rhesus monkeys: antagonism of fentanyl and U50,488 by quadazocine. J Pain. 2002;3:218–226. doi: 10.1054/jpai.2002.124734. [DOI] [PubMed] [Google Scholar]
- O’Neill SJ, Collins MA, Pettit HO, McNutt RW, Chang KJ. Antagonistic modulation between the delta opioid agonist BW373U86 and the mu opioid agonist fentanyl in mice. J Pharmacol Exp Ther. 1997;282(1):271–277. [PubMed] [Google Scholar]
- Pfeiffer A, Brantl V, Herz A, Emrich HM. Psychotomimesis mediated by kappa opiate receptors. Science. 1986;233(4765):774–776. doi: 10.1126/science.3016896. [DOI] [PubMed] [Google Scholar]
- Pick CG, Paul D, Pasternak GW. Nalbuphine, a mixed kappa 1 and kappa 3 analgesic in mice. J Pharmacol Exp Ther. 1992;262(3):1044–1050. [PubMed] [Google Scholar]
- Preston KL, Jasinski DR. Abuse liability studies of opioid agonist-antagonists in humans. Drug Alcohol Depend. 1991;28(1):49–82. doi: 10.1016/0376-8716(91)90053-2. [DOI] [PubMed] [Google Scholar]
- Prisinzano TE. Psychopharmacology of the hallucinogenic sage Salvia divinorum. Life Sci. 2005;78(5):527–531. doi: 10.1016/j.lfs.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Remmers AE, Clark MJ, Mansour A, Akil H, Woods JH, Medzihradsky F. Opioid efficacy in a C6 glioma cell line stably expressing the human kappa opioid receptor. J Pharmacol Exp Ther. 1999;288(2):827–833. [PubMed] [Google Scholar]
- Sivachenko A, Kalinin A, Yuryev A. Pathway analysis for design of promiscuous drugs and selective drug mixtures. Curr Drug Discov Technol. 2006;3(4):269–277. doi: 10.2174/157016306780368117. [DOI] [PubMed] [Google Scholar]
- Stevenson GW, Folk JE, Rice KC, Negus SS. Interactions between delta and mu opioid agonists in assays of schedule-controlled responding, thermal nociception, drug self-administration, and drug versus food choice in rhesus monkeys: studies with SNC80 [(+)-4-[({alpha}R)-{alpha}-((2S,5R)-4-Allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide] and heroin. J Pharmacol Exp Ther. 2005;314(1):221–231. doi: 10.1124/jpet.104.082685. [DOI] [PubMed] [Google Scholar]
- Stevenson GW, Linsenmayer DC, Folk JE, Rice KC, Negus SS. Opioid interactions in rhesus monkeys: effects of delta + mu and delta + kappa agonists on schedule-controlled responding and thermal nociceptionn. J Pharmacol Exp Ther. 2003;307(3):1054–1064. doi: 10.1124/jpet.103.056515. [DOI] [PubMed] [Google Scholar]
- Tallarida RJ. Drug synergism and dose-effect data analysis. Boca Raton, FL: Chapman and Hall/CRC; 2000. [Google Scholar]
- Tang AH, Collins RJ. Behavioral effects of a novel kappa opioid analgesic, U-50488, in rats and rhesus monkeys. Psychopharmacology (Berl) 1985;85(3):309–314. doi: 10.1007/BF00428193. [DOI] [PubMed] [Google Scholar]
- Todtenkopf MS, Marcus JF, Portoghese PS, Carlezon WA., Jr Effects of kappa-opioid receptor ligands on intracranial self-stimulation in rats. Psychopharmacology (Berl) 2004;172(4):463–470. doi: 10.1007/s00213-003-1680-y. [DOI] [PubMed] [Google Scholar]
- Verborgh CM, Camu F, Meert TF. Interaction between sufentanil and U-50488H with respect to antinociception and respiratory depression in rats. Acta Anaesthesiol Scand. 1997;41(7):895–902. doi: 10.1111/j.1399-6576.1997.tb04806.x. [DOI] [PubMed] [Google Scholar]
- Vivian JA, Deyoung MB, Sumpter TL, Traynor JR, Lewis JW, Woods JH. k-opioid receptor effects of butorphanol in rhesus monkeys. J Pharmacol Exp Ther. 1999;290:259–265. [PubMed] [Google Scholar]
- Vonvoigtlander PF, Lahti RA, Ludens JH. U-50,488: a selective and structurally novel non-Mu (kappa) opioid agonist. J Pharmacol Exp Ther. 1983;224(1):7–12. [PubMed] [Google Scholar]
- Walsh SL, Strain EC, Abreu ME, Bigelow GE. Enadoline, a selective kappa opioid agonist: comparison with butorphanol and hydromorphone in humans. Psychopharmacology (Berl) 2001;157(2):151–162. doi: 10.1007/s002130100788. [DOI] [PubMed] [Google Scholar]
- Wessinger WD. Approaches to the study of drug interactions in behavioral pharmacology. Neurosci Biobehav Rev. 1986;10(2):103–113. doi: 10.1016/0149-7634(86)90021-7. [DOI] [PubMed] [Google Scholar]
- Winger G, Hursh SR, Casey KL, Woods JH. Relative reinforcing strength of three N-methyl-D-aspartate antagonists with different onsets of action. J Pharmacol Exp Ther. 2002;301(2):690–697. doi: 10.1124/jpet.301.2.690. [DOI] [PubMed] [Google Scholar]
- Woods JH, Young AM, Herling S. Classification of narcotics on the basis of their reinforcing, discriminative, and antagonist effects in rhesus monkeys. Fed Proc. 1982;41(2):221–227. [PubMed] [Google Scholar]
- Woolverton WL. Analysis of drug interactions in behavioral pharmacology. In: Thompson T, Dews PB, Barrett JE, editors. Neurobehavioral Pharmacology. Hillsdale, NJ: Lawrence Erlbaum Associates; 1987. pp. 275–302. [Google Scholar]
- Xi ZX, Fuller SA, Stein EA. Dopamine release in the nucleus accumbens during heroin self-administration is modulated by kappa opioid receptors: an in vivo fast-cyclic voltammetry study. J Pharmacol Exp Ther. 1998;284(1):151–161. [PubMed] [Google Scholar]