

Abstract
This manuscript reports an approach to the screening of natural product extracts for compounds which are active at membrane-bound receptors, ion channels and transporters. The technique is based upon cellular membrane affinity chromatography (CMAC) columns created through the immobilization of cellular membrane fragments on liquid chromatography stationary phases. In this study a CMAC(nAChR(+)) column was created out of membranes from a transfected cell line expressing the α3β4 neuronal nicotinic acetylcholine receptor (nAChR) and the column was used to screen tobacco smoke condensates. A strategy involving parallel screening with a CMAC column created from a non-transfected form of the same cell line, CMAC(nAChR(−)) was adopted. The condensate was chromatographed on both columns, timed fractions collected and concentrated. Each fraction was analyzed on a C18 column in order to establish a chromatographic fingerprint of each fraction and a differential elution profile of each compound. Comparison of the elution profiles from the CMAC(nAChR(+)) and CMAC(nAChR(−)) columns identified patterns that could be associated with high affinity ligands and with low-affinity/non-binding compounds. Known strong ligands ((S)-Nicotine, (R,S)-Anatabine, N’-Nitrosonornicotine), weak ligands ((R,S)-Nornicotine, Anabasine) as well as known non-ligands (N-Methyl-γ-oxo-3-pyridinebutanamide, (1’S,2′S)-Nicotine 1’-oxide) have been identified in the complex extract. The results demonstrate that CMAC-based screens can be used in the identification of compounds within natural product extracts that bind to membrane-based targets.
Keywords: natural products screening, α3β4 nicotinic receptor, affinity chromatography, nicotine derivatives, tobacco smoke fingerprinting, drug discovery
1. Introduction
The screening of natural products is a historically valid and successful approach to drug discovery. Indeed, natural product extracts can be considered as smart libraries with a high occurrence of biologically relevant structures [1]. Traditional drug screens have involved the fractionation of an extract using global physicochemical properties such as acidity and hydrophobicity, followed by functional testing of the fractions using the selected drug target. The active fractions are then further fractionated and tested. Consequently, bioguided fractionation is time consuming and costly. In addition, extracts are also highly complex mixtures in which concentration differences of several orders of magnitude can bias bioactivity assays producing false positive as well as false negative results.
The difficulties associated with the screening of natural products are currently being addressed using analytical techniques that were developed for the screening of combinatorial chemistry libraries [2]. For example, dereplication, is an approach that is designed to screen extracts for known compounds. If this is accomplished in the early stages of the screening process, significant time and effort can be avoided by the identification of known components with established activities that are present in the extract. Dereplication has been approached using a variety of hyphenated techniques such as fractionation using HPLC-SPE coupled with NMR-MS structural identification [3]. However, while dereplication identifies known compounds, it does not solve the problem of the identification and isolation of previously unknown compounds that are active at the pharmacological target.
Online screening with bioactive detection is an experimental technique that has been used to identify structurally new and active components within natural product extracts. In one approach, a highly efficient chromatographic phase, such as an octadecyl (C18) reversed phase column, is used to separate the components of an extract and the target is used as a bio-detector to evaluate the pharmacological activity of the resolved components. This system can be coupled with detectors such as NMR or MS to produce highly informative structure-function screens.
This approach has been successfully applied to the detection of inhibitors of acetylcholine esterase [4–6], cytochrome P450 [7], phosphodiesterase [8], angiotensin-converting enzyme [9], glutathione-S-transferase [10], and cathepsin B [11] in a variety of extracts. In these studies, the enzymatic conversion of a target substrate is monitored and inhibiton of this activity is used to identify active components in the chromatographic eluate. This technique has also been used to screen for ligands of estrogen receptors α and β [12–14]. In these studies, the functional activity of the resolved components was detected by their ability to competitively displace coumestrol, a compound showing a fluorescence enhancement when bound to the estrogen receptor.
In addition to enzymes and nuclear receptors, membrane-bound targets have significant therapeutic importance in a wide variety of diseases comprising 30% of the therapeutic targets [15]. A number of affinity-based screening strategies have been reported for use with membrane receptors [16], as have online chromatographic screens using cellular membrane affinity chromatography (CMAC). The CMAC approach involves the immobilization of cellular membrane fragments on a silica support or the surface of a glass capillary and the use of frontal, zonal or nonlinear chromatography techniques. This approach has been recently reviewed [17,18]. Functional CMAC columns have been prepared and validated for use with a wide variety of targets including nicotinic acetylcholine receptors CMAC(nAChR) [17] and the P-glycoprotein drug transporter CMAC(PgP) [19]. CMAC(nAChR) columns have been used to screen synthetic mixtures using a two-dimensional chromatographic system [20] and the CMAC(PgP) has been used with fast frontal chromatography to identify Pgp ligands [21]. In the latter study, the CMAC(PgP) produced data and throughput comparable to the Caco-2 cell screen, the industrial standard.
While the CMAC approach presents a varied and powerful tool for binding studies, it can not be directly applied to the screening of natural product extracts. Unlike columns that contain a purified protein or that rely on a specific enzymatic conversion, CMAC columns contain cellular membrane fragments which contain multiple specific and non-specific binding sites. Thus, in a complex mixture, it is hard to identify which compound is binding to which target using a fast frontal or zonal chromatographic technique. This paper describes a simple strategy for overcoming this problem, “missing peak chromatography,” and its application to the screening of a complex extract.
In this study, two CMAC columns were used. One contained cellular membranes from a HEK-293 cell line that stably expressed the α3β4 nAChR, denoted in this paper as the CMAC(+) column. The other contained cellular membranes from the native HEK-293 cell line and is denoted as the CMAC(−) column.
The α3β4 nAChR used in this study is one of a family of ligand gated ion channels that are found in different locations of the central and peripheral nervous system and have been associated with different pharmacological functions including cognition, memory, sensory gating including pain, anxiety and depression as well as cardiovascular and gastrointestinal action [17].
The extract was obtained from tobacco smoke condensates. It is estimated that more than 4,000 compounds are present in tobacco leaves and tobacco smoke [22]. While the pharmacological activity of the majority of these compounds have not been identified, several commercially available alkaloid components of tobacco smoke have been structurally characterized [22] and their binding affinities to nAChRs established [23–25]. Thus, tobacco smoke represents a complex mixture containing pharmacologically active components, which can be used as a model for the "missing peak" chromatographic screening of medicinally active biological and chemical mixtures.
2. Materials and methods
2.1. Methodology
The experimental technique described in this work, “missing peak chromatography”, is outlined in Fig. 1. The approach is based upon the construction of a CMAC column which contains membranes from a cell line that expresses the target either natively or via transfection, the CMAC(+) column, and a CMAC column from the same cell line that does not express the target either natively or via chemical or biological knockout, the CMAC(−) column. Since the CMAC(−) and CMAC(−) columns essentially differ only in the expression of the target, any differences in the retention of a compound on the CMAC(+) column relative to the CMAC(−) column will be due to specific interactions with the target. This principle has been validated by parallel screening on CMAC(PgP(+)) and CMAC(PgP(−)) columns prepared from native and MDR1 transfected forms of the MDA435/LCC6 cell line [19,21].
Figure 1.
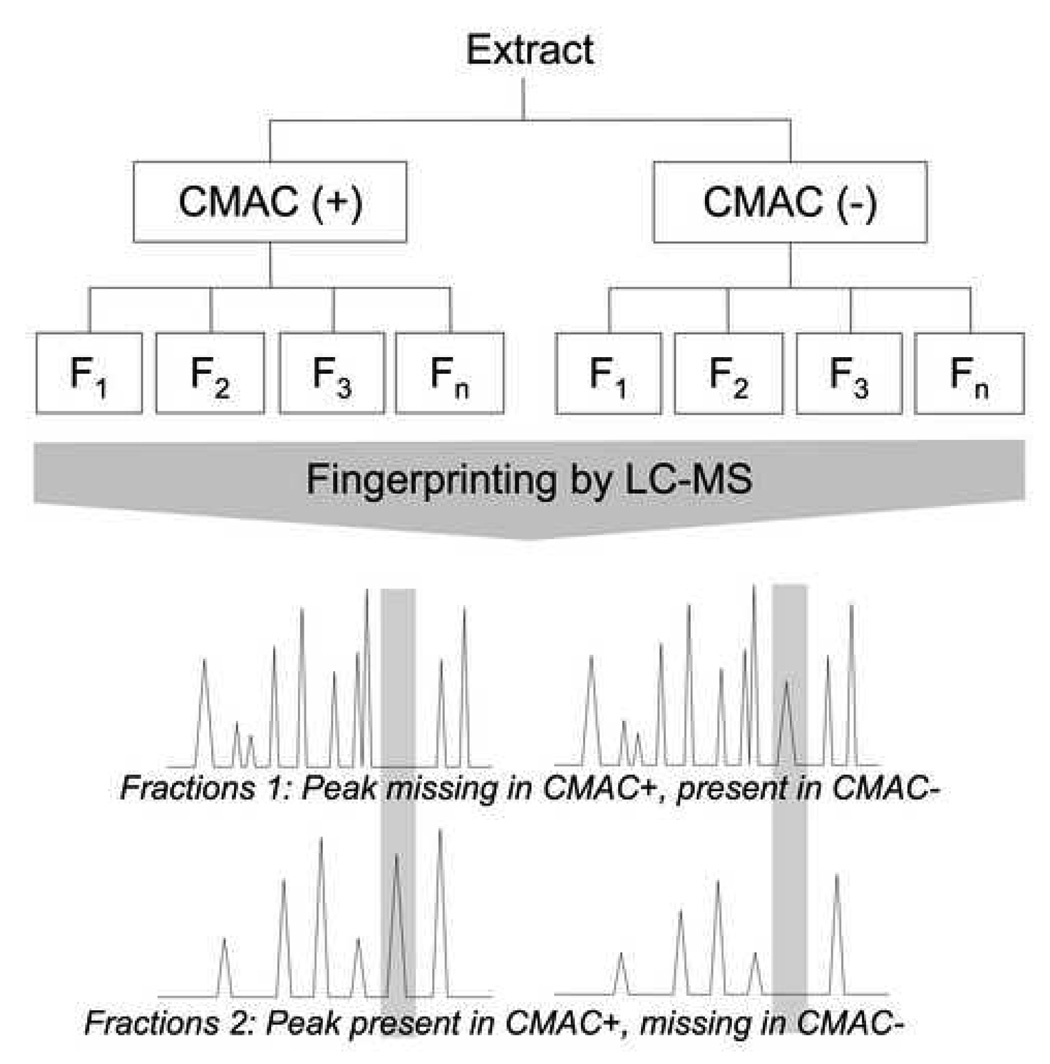
General approach of the screening of ligands using parallel injection on CMAC+ (membranes from cells expressing the receptor) and CMAC− (membranes from control cells), using missing peak chromatography analysis.
In missing peak chromatography the extract is initially analyzed on an efficient analytical column, such as a C18 column, in order to establish a “control peak fingerprint” and where possible individual components are identified. The mixture is then chromatographed on the CMAC(+) and CMAC(−) columns and timed fractions are collected and concentrated. Each fraction from the CMAC(+) and CMAC(−) columns is analyzed on the C18 column to obtain peak fingerprints and the fingerprints are compared to the control fingerprint and to each other. Peaks similarly retained by both columns result from non-specific interactions. A peak that is missing in the fingerprint of a CMAC(+) early fraction, but present in the control and corresponding CMAC(−) early fraction is assumed to bind to the immobilized target on the CMAC(+) column. The presence of this peak in a later fraction of CMAC(+), along with its absence in the corresponding fraction in CMAC(−) further confirms the affinity of this compound for the expressed receptor in CMAC(+). Once identified, the “missing peaks” can be isolated from the initial mixture using an analytical or semi-preparative C18 column, the structure determined and the compound tested for activity at the target.
2.2. Materials
MEM medium, FBS, penicilline/streptomycine and G418 were purchased from Invitrogen (Carlsbad, CA, USA). Methylene chloride, methanol, acetonitrile and isopropanol were purchased from Fisher Scientific (Fairlawn, NJ, USA). Acetic acid was purchased from Mallinckrodt (Hazelwood, MO, USA). Ammonium acetate, trifluoroacetic acid (TFA), propionic acid, (S)-nicotine hydrogen tartrate, myosmine, quinoline, (±)-nornicotine, (−)-cotinine and (±)-anabasine were purchased from Sigma-Aldrich (St Louis, MO, USA). Other nicotine derivatives including (±)-anatabine, N-nitrosonornicotine, N-methyl-γ-oxo-3-pyridinebutanamide and (1′S,2′S)-nicotine 1′-oxide were obtained from Toronto Research Chemicals (North York, ON, Canada). [3H]-Epibatidine ([3H]-EB) and [3H]-(S)-nicotine were purchased from PerkinElmer (Boston, MA, USA). Ecoscint A scintillation liquid was purchased from National Diagnostics (Atlanta, GA, USA).
2.3. Cell culture
HEK 293 KXα3β4R2 cells were kindly provided by Dr K. Kellar (Georgetown University Medical School, Washington, DC, USA) [26], and were cultured in MEM medium supplemented with 10% FBS, 1% penicillin/streptomycin and 1 mM G418. HEK control cells (ATCC, Manassas, VA, USA) were cultured in the same medium without G418.
2.4. Column preparation
Columns containing immobilized cellular membranes obtained from the HEK 293 KXα3β4R2 cell line (CMAC(+) column) and HEK 293 cell line (CMAC(−) column) were prepared as previously described [27]. The columns were prepared using 50 × 106 cells of each cell line and 400 mg of an immobilized artificial membrane stationary phase (Rexchrom S12-300-IAM-PC) purchased from Regis Technologies (Morton Grove, IL, USA), and packed into a 20×5 mm Tricorn 5/20 column (Amersham Biosciences, Pittsburgh, PA, USA).
2.5. Specific approach for tobacco smoke condensate screening for α3β4 nAChR ligands
This approach is outlined in Table 1 and was as follows.
Table 1.
successive steps of the missing peak chromatography applied to the screening of nicotinic ligands in the tobacco smoke condensate.
| Process step | Operation |
|---|---|
| 1 | Extraction and fractionation to yield Fraction 1 |
| 2 | (S)-nicotine depletion to yield Fraction 1A |
| 3 | Chromatographic fingerprint of Fraction 1A |
| 4 | Chromatography of Fraction 1A on nAChR(+)-CMAC and nAChR(−)-CMAC |
| 5 | Determination of chromatographic fingerprints of collected fractions |
| 6 | Comparison of chromatographic fingerprints |
2.5.1. Extraction and fractionation to produce Fraction 1
Tobacco smoke condensate in acetone was purchased from Arista Laboratories (Richmond, VA, USA). The condensate (2g) was dissolved in 20 ml of methylene chloride:methanol (50:50, v/v), the resulting solution was centrifuged for 5 min at 200 × g and the supernatant was collected. 20 ml of acetic acid [1 M, pH 2.1] was added to the supernatant and the resulting solution was shaken, centrifuged for 5 min at 1600 × g and the aqueous fraction collected. This process was repeated 5 times, and the aqueous fractions were pooled and lyophilized to give 890 mg of brown viscous residue. The residue was dissolved in methanol to yield a 200 mg/ml solution. A 3 ml aliquot of the solution was mixed with 12 ml of water, the pH was adjusted to 1.5 using TFA, the solution was centrifuged at 2500 × g for 10 min and the supernatant (Solution A) was collected.
Solution A was fractionated using a semi-preparative C18 column (Zorbax SB-C18, 150×9.4 mm, 5 µm particle size, 80 Å pores), purchased from Agilent (Santa Clara, CA, USA) running on a Shimadzu LC10ADVp system (Shimadzu, Columbia, MD, USA) equipped with a SPD-M20A DAD-UV detector. Gradient elution was performed using mobile phases composed of (A) aqueous TFA [0.01% w/v] and (B) acetonitrile modified with TFA [0.01% w/v] using the following profile: 100% A to 95(A):5 (B) from 0–10 min; 95(A):5(B) to 30(A):70 (B) 10–30 min; 30(A):70 (B) to 100% B from 30–32 min; 100% (B) to 100% (A) from 32–48 min. The mobile phases were delivered at 4 ml/min at room temperature.
Multiple aliquots of Solution A were injected onto the system (30 × 500µl) and the eluents from 0 – 13 min (Fraction 1) and from 13 – 25 min (Fraction 2) were collected. The fractions from each run were combined and evaporated to dryness using a Rotavapor evaporator (Büchi Labortechnik, Flawil, Switzerland) at 40°C. Fraction 1 yielded 480 mg of dry residue and Fraction 2 yielded 185 mg of dry residue. Fraction 1 was dissolved in methanol to produce a 300 mg/ml solution.
2.5.2. Chromatographic fingerprint of Fraction 1
Fraction 1 was analyzed using a C18 column (Zorbax SB-C18, 250×4.6 mm, 3.5 µm particle size, 80 Å pores) running on an Agilent HP 1100 LC-UV-MS system using a MSD quadrupole mass spectrometer and an API-ES source. Gradient elution was performed using mobile phases composed of (A) aqueous TFA [0.01% w/v] and (B) acetonitrile modified with TFA [0.01% w/v] using the following profile: 100% A from 0–10 min; 100% (A) to 90(A):10 (B) 10–20 min; 90(A):10 (B) to 100% B from 20–40 min; 100% (B) from 40–55 min; from 100% (B) to 100% (A) from 55–60 min; 100% (A) from 60–75 min. The mobile phases were delivered at 1 ml/min at room temperature. After DAD-UV detection (range from λ = 200 to 350 nm), eluate was split 80:20 with the 20% fraction directed to a mixing tee (Upchurch, Oak Harbor, WA, USA) and combined with a make-up flow of isopropanol delivered at a flow rate of 0.2 ml/min to increase sensitivity and reduce the ion suppression caused by the TFA [28]. Detection was accomplished using SIM mode with the following settings: drying gas flow at 10 l/min and 350°C, nebulizer pressure at 20 psig, capillary voltage at 4000V, fragmentor at 50, gain at 2 and peak width at 0.10 s. Quantification of nicotine in the extract used a calibration curve (R2 = 0.9929) based on the area of peaks of the UV absorbance at λ = 265 nm, for triplicate injections of (S)-nicotine at 10, 25, 100, 500, 1000 and 2500 µM (Data not shown). Other standard compounds were injected (1 µl of a 500 µM solution) using the same chromatographic conditions.
2.5.3. Removal of (S)-nicotine from Fraction 1 to produce Fraction 1A
Forty-five 4 µl aliquots of Fraction 1 were chromatographed on a (S)-nicotine molecularly imprinted stationary phase (MIP) previously reported by Sambe, et al. [29], packed into a 100×6 mm column. The chromatography was carried out using a gradient elution employing mobile phase A: acetonitrile:ammonium acetate [10 mM, pH 5.0] (80:20, v/v) and mobile phase B: aqueous TFA (0.1% w/v). The elution program was: 100% A from 0–10 min; 100% A to 60(A):40(B) from 10–15 min; 60(A):40(B) to 100% (A) from 15–20 min; 100% (A) from 20–30 min. The mobile phase was delivered at a flow rate of 1.2 ml/min between 0–10 min and 1.0 ml/min between 10–30 min in order to remain below the 80 bar pressure limit of the MIP column. The eluents from 0–7 min (Fraction 1A) and 7–30 min were collected, pooled and evaporated with Rotavapor at 40°C under vacuum. The residue from Fraction 1A was dissolved in 1 ml of aqueous TFA [0.1% w/v] and fingerprinted as described above for Fraction 1.
2.5.4. Chromatography of Fraction 1A on the CMAC(+) and CMAC(−) columns
Fraction 1A was diluted 10-fold with ammonium acetate [1mM, pH 7.4] and 20 µl aliquots were chromatographed on the CMAC(+) or CMAC(−) column. The chromatography was carried out using a Shimadzu LC-10ADVP pump and a mobile phase composed of ammonium acetate [1mM, pH 7.4] delivered at a flow rate of 0.2 ml/min. Fractions were collected as follows: 0 to 20 min, 20 to 40 min, 40 min to 60 min, then every 60 min up to 300 min. The fractions were frozen, lyophilized in a ModulyoD (Thermo Fisher Scientific, Waltham, MA, USA) and the residues dissolved in 50 µl of aqueous TFA [0.01% w/v].
2.5.5. Fingerprinting of Fraction 1A and the fractions from the CMAC(+) and CMAC(−) columns
Fraction 1A and the fractions from the CMAC (+) and CMAC(−) columns were analyzed using the same column and condition as used for the fingerprinting of Fraction 1 described above. The monitored masses were based on masses present in the extract, and providing a significant peak area in fractions: m/z 112, 113, 115, 127, 134, 137, 138, 139, 148, 149, 150, 153, 154, 160, 161, 163, 164, 166, 169, 171, 172, 177, 178, 179, 185, 192, 193, 196, 199, 202, 204, 208, 210, 219, 221, 224, 236, 238, 243, 259, 263.
2.6. Relative affinity of identified compounds using frontal chromatography
Relative affinities of (S)-nicotine, (±)-anabasine, (±)-anatabine, N-nitrosonornicotine, (±)-nornicotine, N-methyl-γ-oxo-3-pyridinebutanamide and (1’S,2′S)-nicotine 1’-oxide were assessed displacement frontal chromatography on the CMAC(+) column. These experiments were performed on a β-RAM Model 3 scintillation flow detector (IN/US Systems, Tampa, FL, USA), using Ecoscint A as a scintillation liquid at a flow rate of 0.8 ml/min. Samples (20 to 40 ml) containing one standard at a time (10 µM) along with [3H]-epibatidine [60 pM] were loaded in a 50 ml Superloop by a Dynamax Model RP-1 peristaltic pump (Rainin, Oakland, CA, USA) and injected through a V7 valve (GE Healthcare, Chicago, IL, USA). The mobile phase was ammonium acetate [10 mM, pH 7.4] delivered at 0.2 ml/min using a Shimadzu LC-10ADVP pump.
3. Results and Discussion
3.1. Preparation and dereplication of Fraction 1A
Studies of the binding of agonists and competitive antagonists to the nAChR have demonstrated that these compounds usually contain a cationic moiety [30]. Thus, the cationic aqueous fraction of the tobacco smoke condensate (Fraction 1) was prepared using the classical extraction scheme developed for the isolation of alkaloids. Fraction 1 represented 44.5 % (w/w) of the condensate. The chromatographic fingerprint of Fraction 1 was obtained using diode array UV detection and contained a predominant peak that was identified as (S)-nicotine, which was present in a 45 mM concentration, Figure 2A.
Figure 2.
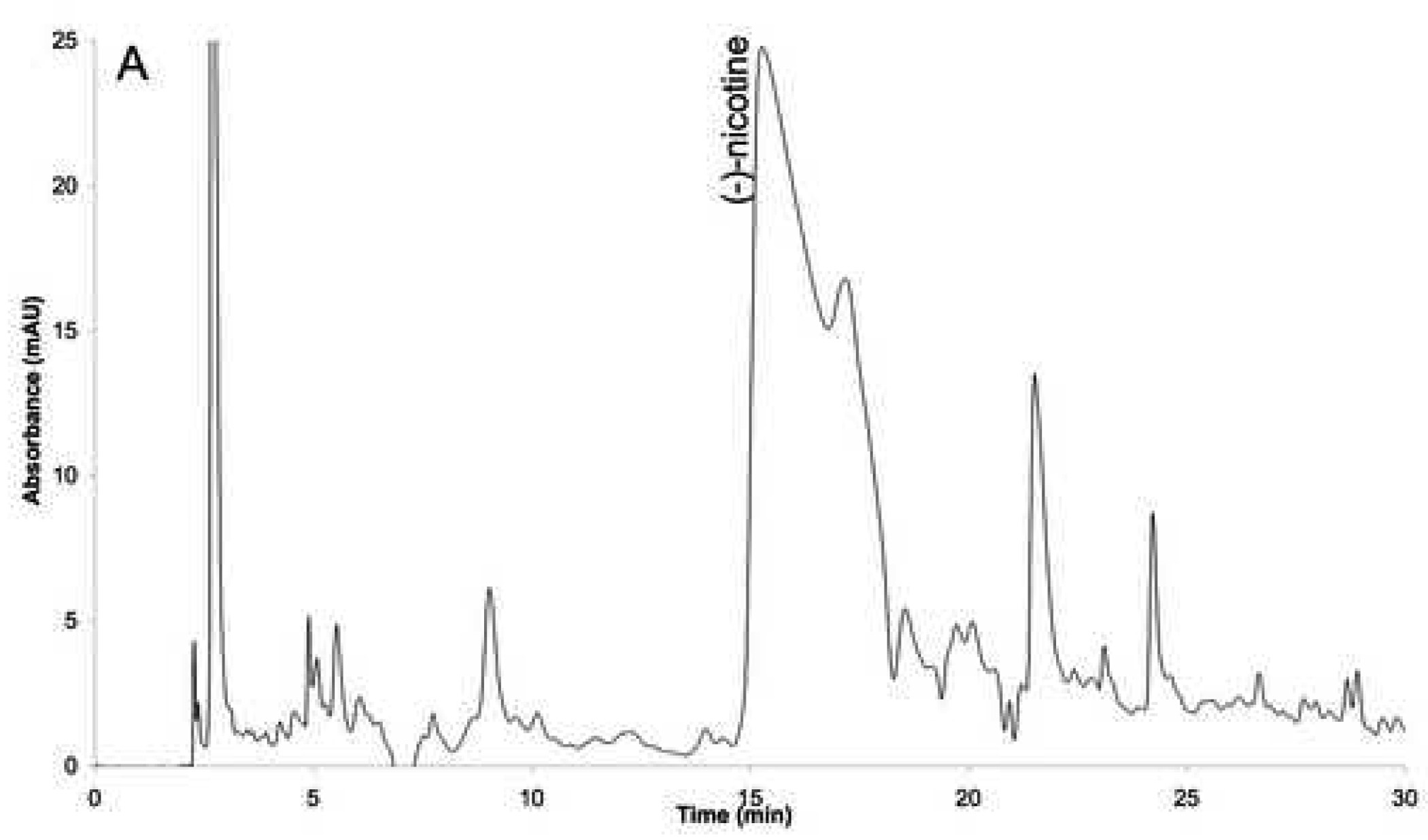
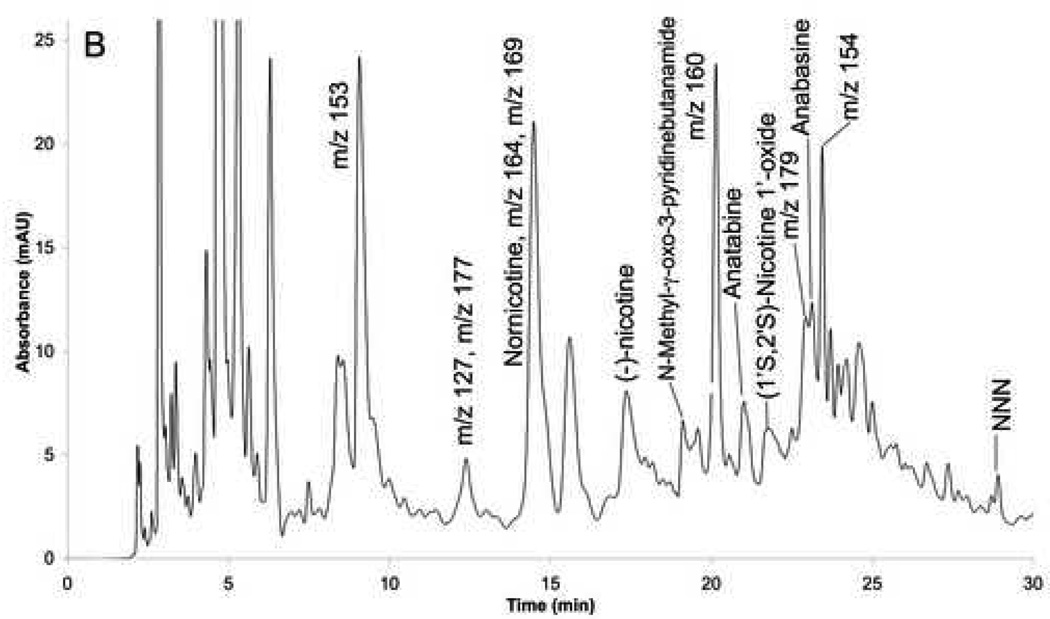
Fingerprints of the fractions: (A) before MIP (fraction 1), and (B) after MIP (fraction 1A).
The presence of a large concentration of (S)-nicotine raised the possibility that it would saturate the immobilized nAChRs making it impossible to identify other ligands. In addition, (S)-nicotine hindered the detection of other compounds within Fraction 1 making it impossible to obtain an accurate chromatographic fingerprint. Therefore, it was necessary to remove the majority of the (S)-nicotine from the fraction. This was accomplished using a previously reported molecular imprinted polymer (MIP) based on a (S)-nicotine template [29]. Multiple aliquots of Fraction 1, totaling 180 µl, were processed on the MIP, evaporated and reconstituted in 1 ml of water. The resulting solution, Fraction 1A, had a (S)-nicotine concentration of 50 µM, representing a 99.4 % reduction of the (S)-nicotine content. Fraction 1A was fingerprinted using UV diode array detection and over 50 peaks were observed in the chromatogram, Figure 2B.
The chromatographic fingerprint of Fraction 1A was obtained using UV detection since this approach is not destructive and can be used if the chromatography is scaled up for preparative studies. However, UV diode array detection was only able to definitively identify nicotine in the chromatographic fingerprint. Therefore, the detection format was changed to mass spectrometry, and the chromatographic retentions of 49 commercially available nicotine derivatives standards were determined using single ion monitoring (SIM). Based on retention time and mass, 6 additional components were unambiguously identified in the extract in addition to nicotine, Table 2.
Table 2.
The compounds identified in Fraction 1A of the tobacco smoke condensate using retention times (Rt) and molecular ions ([M+H]+) and their relative affinities for the immobilized α3β4 nAChR determined by the reduction in the retention volume of [3H]-epibatidine (Δml) produced by the addition of a 10 µM concentration of the compound to the mobile phase.
| Compounds | Rt (min) | [M+H]+ | Δml |
|---|---|---|---|
| (S)-Nicotine | 17.5 | 163.2 | 40 |
| (R,S)-Anatabine | 20.6 | 161.2 | 42 |
| N’-Nitrosonornicotine | 29.0 | 178.2 | 42 |
| (R,S)-Nornicotine | 14.5 | 149.2 | 36 |
| Anabasine | 23.1 | 163.2 | 32 |
| N-Methyl-γ-oxo-3-pyridinebutanamide | 19.0 | 193.4 | 18 |
| (1’S,2′S)-Nicotine 1’-oxide | 21.8 | 179.2 | 4 |
Under the chromatographic conditions used in this study, a majority of the peaks in the fingerprint were found to contain several compounds. For instance, using SIM at m/z of 177, the region on the chromatographic trace corresponding to a retention of 22.5 to 23.5 minutes contains a single peak which can be assigned to five different standards: (R,S)-N-ethylnornicotine, cotinine, N-formylnornicotine, N-methylanabasine and 5-methylnicotine. Since the MSD used in this study was only a single quadrupole instrument, we were unable to definitively identify the components. Therefore, only the seven identified components, Table 2, were followed through the affinity chromatography experiments.
3.2. Relative affinity of the identified components
Previous studies with the CMAC(nAChR) columns have demonstrated that frontal displacement chromatography can be used to determine the relative affinities of compounds for the immobilized receptor [31]. In these studies, [3H]-EB was used as the marker ligand and equimolar concentrations of the control ligands were added to the mobile phase. The ability of a given ligand to displace the marker, i.e. reduce the observed breakthrough volume of [3H]-EB, was used to qualitatively rank the affinities of the ligands. It is expressed in table 2 as Δml, the difference of [3H]-EB breakthrough time alone or when added ligand, multiplied by the flow rate.
In this study, the relative affinities of the 7 identified compounds were established using the same experimental approach, Table 2. Under the experimental conditions used in this study, the breakthrough volume of [3H]-EB was 46 ml. The addition of (S)-nicotine, N-nitrosonornicotine and anatabine reduced the breakthrough volume of [3H]-EB to less than 6 ml (Δml values ≥ 40) indicating that these compounds should have equivalent affinity for the immobilized α3β4 nAChR. This is consistent with previous data that N-nitrosonornicotine has a subnanomolar affinity for the nAChR [32]. The Δml values produced by nornicotine and anabasine were 36 and 32, respectively, indicating that these compounds should have a lower affinity for the receptor, relative to nicotine, while N-methyl-γ-oxo-3-pyridinebutanamide and (1’S,2’S)-nicotine-1’-oxide had little effect on the retention of the marker, Δml 18 and 4 ml respectively, and should have relatively little affinity for the receptor.
3.3. Chromatography of Fraction 1A on the CMAC(+) and CMAC(−) columns
In this study, the chromatographic peaks produced by components within Fraction1A produced broad, asymmetric peaks with extensive tailing when chromatographed on the CMAC(+) and CMAC(−) columns, Fig. 3A. This is consistent with the peak shapes predicted by the theory of non-linear chromatography, which describes the interactions between a ligand and a protein within an affinity chromatography system and which attributes the observed dissymmetry to the relatively slow dissociation of the ligand-protein complex [17]. Although these properties are useful in some experimental setting, they also hinder the direct application CMAC(+) columns in affinity screens.
Figure 3.
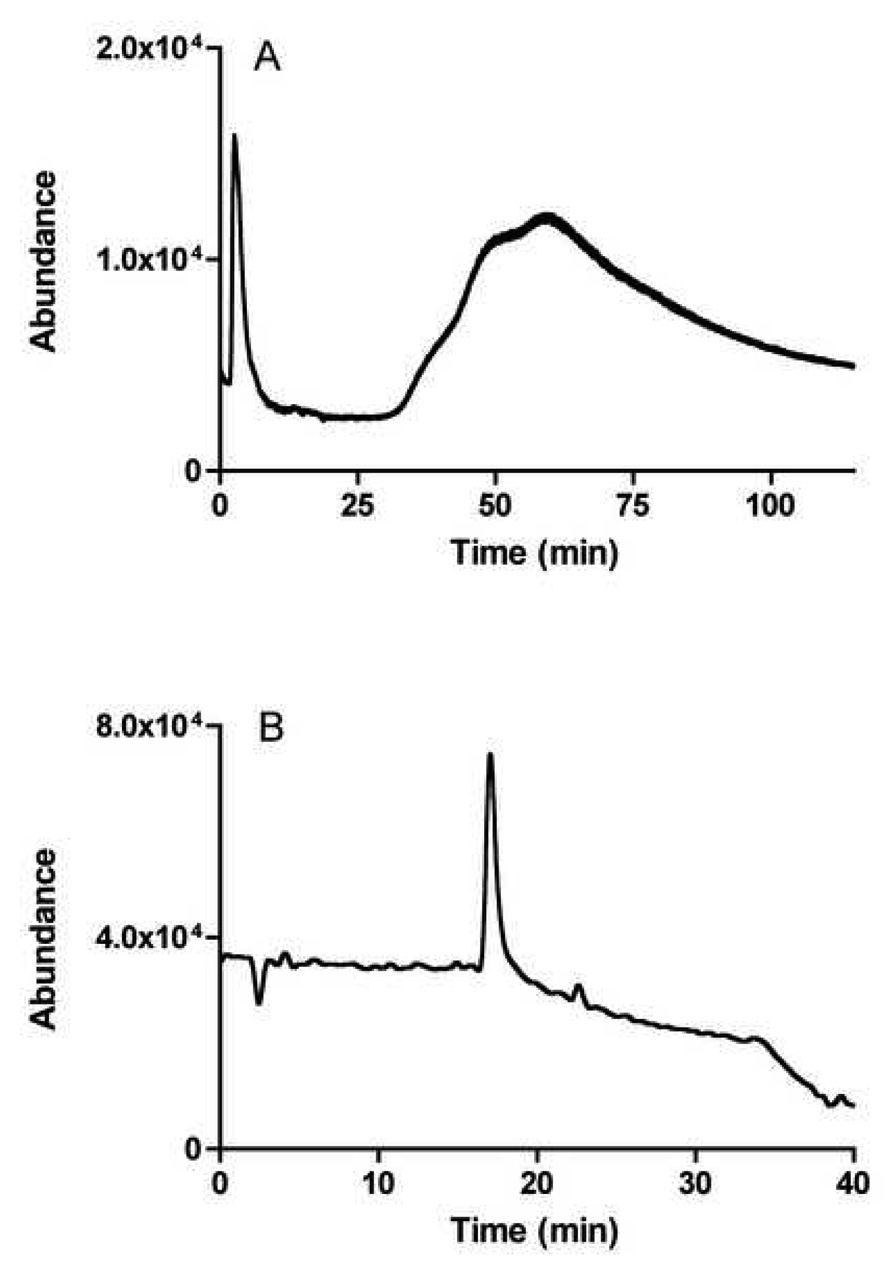
Chromatogram monitored at m/z 163 (nicotine mass) of the extract on the CMAC(+) (A) and of the fraction 2 collected on CMAC+ and processed by the missing peak chromatography method (B).
The direct application of CMAC columns to affinity screens is also hindered by the relative low capacity of the columns. Using the frontal displacement chromatography of EB [17]. The total specific binding capacity of the columns was shown to average ~0.5 pmoles/mg of stationary phase. The amount of ligand that can be injected in these columns without saturating the binding sites was in the same order of magnitude of the limit of detection of the MS instrument used in the study.
In our previous screening of a synthetic mixture of nAChR ligands and non-ligands [20], the coupling of the CMAC to the analytical column was accomplished online, while this study used off-line coupling as the timed fractions were collected, concentrated by 500-fold, and then injected onto a C18 column. The chromatographic trace on the C18 column of the second fraction obtained in this study from the CMAC(+) column and monitored at m/z 163 (nicotine) is presented in Fig. 3B.
While the non-linear chromatographic properties and low capacity of the CMAC columns lead to some problems for the use of these columns in the affinity screen of a complex mixture, the main challenge is the presence of cellular membrane fragments within the column. These fragments are a necessary part of the CMAC system since the preservation of membrane structure is essential for function and stability [33]. However, cellular membranes contain multiple sites for specific ligand interactions such as receptors, ion channels and transporters as well as sites for non-specific ionic and hydrophobic interactions. To solve this problem, an approach based on differential chromatography was used, in which Fraction 1A was chromatographed on CMAC(+) and CMAC(−) columns, as described in the Methodology section.
In this study, Fraction 1A was chromatographed on the CMAC(+) and CMAC(−) columns and 7 fractions were collected, concentrated and reanalyzed on the C18 column. As a consequence of the extensive tailing of the non-linear peaks, most compounds are present in several fractions. Thus, in order for the screen to work, it is necessary to establish the elution patterns of the compounds and to determine the distribution of the compound between the fractions. Using the “relative percent area” approach, two elution patterns can be observed for the seven compounds followed in this study, Fig. 4. In one type of pattern, the highest percentage of the total area is eluted later in CMAC(+) than in CMAC(−) columns. In the second elution pattern, the highest percentage of the total eluted area is found in the same fractions from both the CMAC(−) and CMAC(+) columns. The identification of ligands and non-ligands using relative percent area contained in different fractions has been previously established in “ligand fishing” studies using magnetic beads containing immobilized human serum albumin [34].
Figure 4.
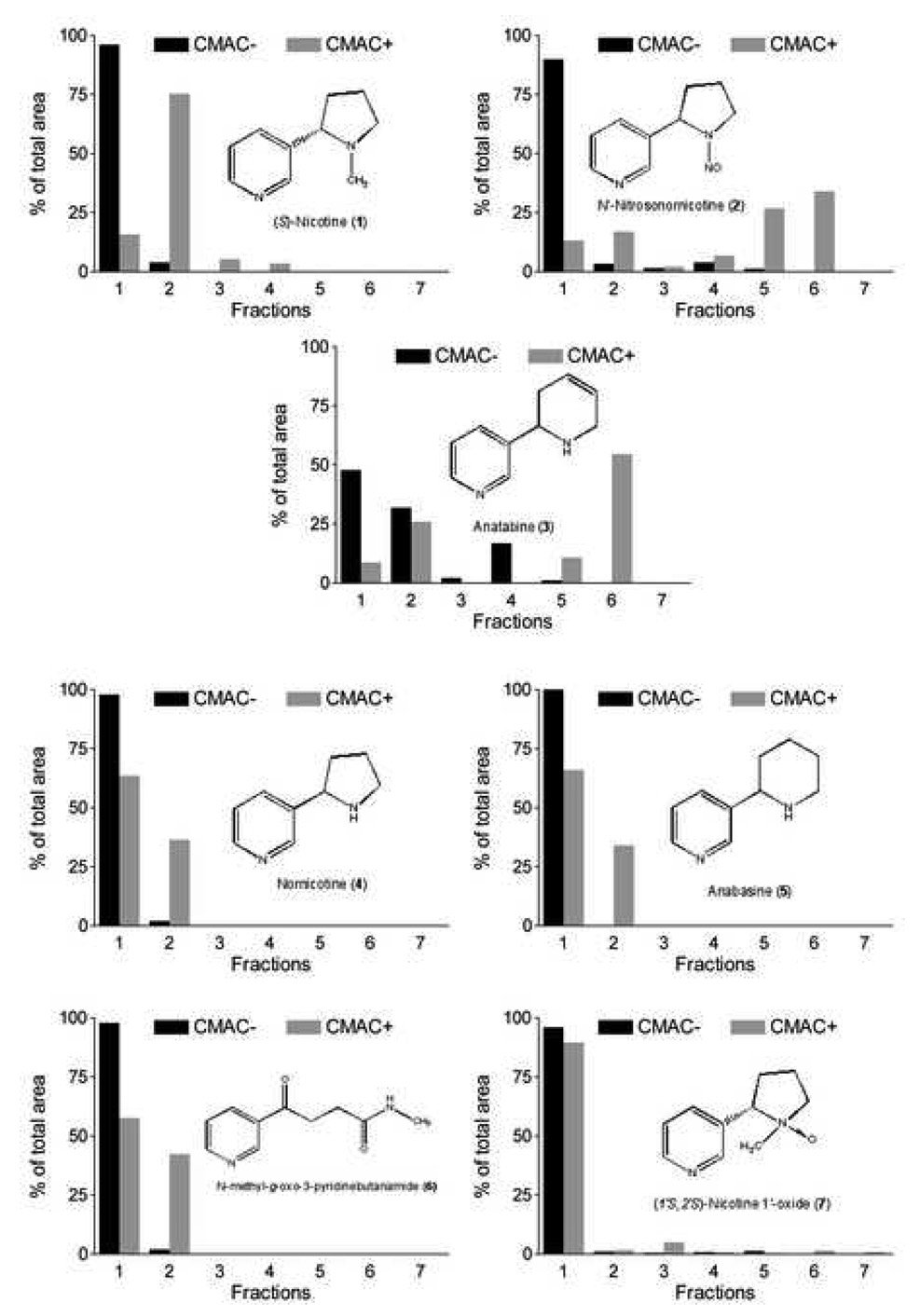
Differential elution profiles of identified compounds on CMAC− and CMAC+.
For nicotine, ~100% of the compound elutes from CMAC(−) column in the first fraction, while with the CMAC(+) column only 20% is found in the first fraction and 75% in the second fraction, Fig. 4. For N-nitrosonornicotine, the highest area percentage was also found in the first fraction eluted from the CMAC(−) vs fraction 5 and 6 for CMAC(+). The pattern also suggests that there are actually two compounds in the peak of N-nitrosonornicotine which elutes at 29.0 min, and produces significant ions at m/z 178. One eluted in the majority in fraction 2 and another eluted in the majority in fraction 6. Both compounds bind to the α3β4 nAChR with affinities ≥ nicotine. When the fractions were monitored at m/z 161 (anatabine) the majority of the compound eluted in the first fraction obtained from the CMAC(−) column, but there were significant concentrations also observed in fractions 2–4, suggesting that the compound(s) also interact with membrane components other than the nAChR. In the fractions from the CMAC(+), significant ion abundancies of m/z 161 were observed in the C18 fingerprint of fractions 2 and 6. This suggests that more than one compound is contained within the chromatographic peak and that only one of them has a significant affinity for the α3β4 nAChR.
For nornicotine, anabasine, N-methyl-γ-oxo-3-pyridinebutanamide and (1’S,2’S)- nicotine-1’-oxide, ~100% of the eluted area were found in the first fraction obtained from the CMAC(−) column and >50% in the first fraction obtained from the CMAC(+) column, with the remaining compound primarily found in the second fraction. In the frontal displacement studies, these compounds were identified as having a lower affinity for the nAChR relative to nicotine, Table 2, and therefore, compounds displaying this pattern can be considered weak or non-α3β4 nAChR ligands.
3.4. Detection of unidentified ligands by missing peak analysis
The two patterns observed with the identified compounds were also produced by a number of unidentified compounds, Fig. 2. For example, when the fractions were chromatographed on the C18 column and the eluate monitored at m/z 177, a peak at the retention time of 12 min was observed in the chromatograms with an elution profile similar to the one produced by weak and non-ligands, Fig. 5. This suggests that the identification and purification of this compound should not be pursued in a screen for nAChR ligands. Monitoring at m/z 169 and at a retention time of 13 min resulted in an elution pattern similar to the one produced by anatabine, which indicates that this peak would be of interest, Fig 5. When the fractions were analyzed at m/z 154, a peak at the retention time of 23 min had a retention pattern which had not been observed with the identified compounds, i.e. no retention on the CMAC(−) column and late elution from the CMAC(+) column, Fig. 5. This elution profile suggests a significant affinity for the α3β4 nAChR subtype and is consistent with the long elution times observed with allosteric inhibitors of this receptor [17].
Figure 5.
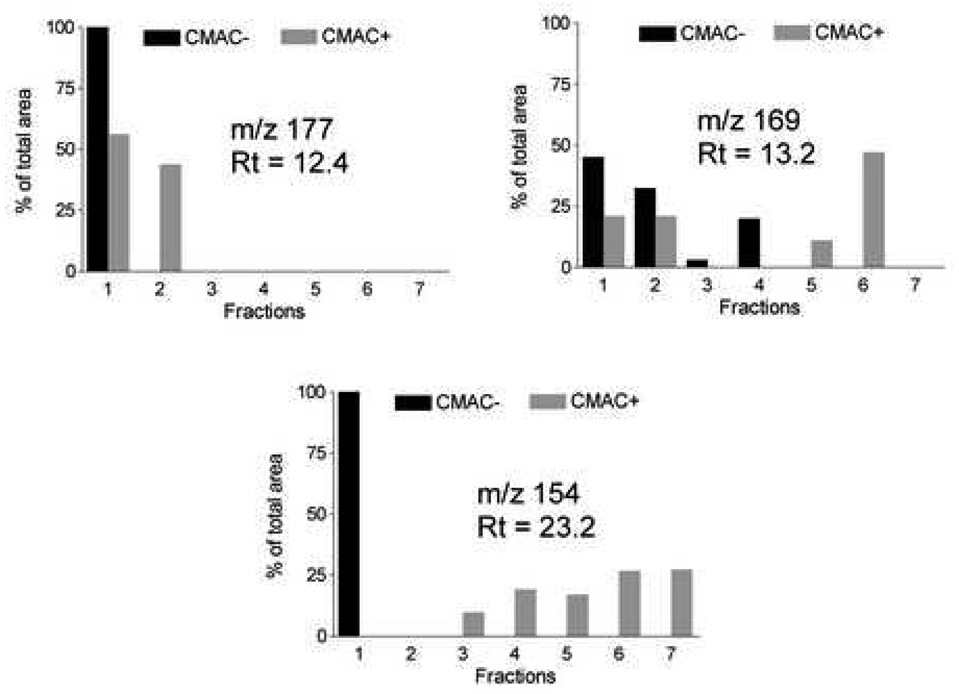
Differential elution profiles of unidentified compounds on CMAC− and CMAC+.
The next step in this screening approach would be the isolation of the components contained within the peaks which eluted at 13 and 23 min by the chromatography of Fraction 1A on a semi-preparative C18 column. The isolated compounds would then be identified and their pharmacological activity established. The results of this validation will be published upon completion.
4. Conclusions
The data from this study demonstrate that CMAC columns and the missing peak chromatography approach can be used in the online screening of complex chemical mixtures. The results also indicate that the application of this approach is not a straightforward process and requires the use of CMAC control columns in order to assess non-target interactions with the immobilized membranes as well as coupled column techniques to overcome the inefficiencies of the CMAC columns. However, the problems associated with CMAC screening are easily remedied by current state-of-the-art technology and by optimization of the specific CMAC columns and chromatographic techniques used in the study. The difficulties are also outweighed by the possibility to directly screen for compounds that bind to membrane receptors, ion channels and drug transporters. In addition, multiple-receptor CMAC columns created from cell lines constitutively expressing cannabinoid (CB 1 and 2), histaminic (H1 and H2), nicotinic (α7) (manuscript in preparation) and from human tissues present the possibility of simultaneous screening for compounds that interact with more than one receptor or with more than one isoform of the same receptor. The results from our ongoing studies with multi-isoform receptor CMAC columns and extracts from other natural sources will be reported elsewhere.
Acknowledgements
This work was supported by funds from the Intramural Research Program of the National Institutes of Health, National Institute on Aging.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Newman DJ, Cragg GM. J Nat Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 2.Chem Eng News. 2003;81 [Google Scholar]
- 3.Lambert M, Wolfender JL, Stærk D, Christensen SB, Hostettmann K, Jaroszewski JW. Anal Chem. 2007;79:727–735. doi: 10.1021/ac0616963. [DOI] [PubMed] [Google Scholar]
- 4.Rhee IK, Van Rijn RM, Verpoorte R. Phytochem Anal. 2003;14:127–131. doi: 10.1002/pca.675. [DOI] [PubMed] [Google Scholar]
- 5.Rhee IK, Appels N, Luijendijk T, Irth H, Verpoorte R. Phytochem Anal. 2003;14:145–149. doi: 10.1002/pca.695. [DOI] [PubMed] [Google Scholar]
- 6.De Jong CF, Derks RJE, Bruyneel B, Niessen W, Irth H. J Chromatogr A. 2006;1112:303–310. doi: 10.1016/j.chroma.2006.01.059. [DOI] [PubMed] [Google Scholar]
- 7.Kool J, Van Liempd SM, Van Rossum H, Van Elswijk DA, Irth H, Commandeur JNM, Vermeulen NPE. Drug Metab Dispos. 2007;35:640–648. doi: 10.1124/dmd.106.012245. [DOI] [PubMed] [Google Scholar]
- 8.Schenk T, Breel GJ, Koevoets P, Van Den Berg S, Hogenboom AC, Irth H, Tjaden UR, Van Der Greef J. J Biomol Screen. 2003;8:421–429. doi: 10.1177/1087057103255973. [DOI] [PubMed] [Google Scholar]
- 9.Van Elswijk DA, Diefenbach O, Van Der Berg S, Irth H, Tjaden UR, Van Der Greef J. J Chromatogr A. 2003;1020:45–58. doi: 10.1016/j.chroma.2003.08.055. [DOI] [PubMed] [Google Scholar]
- 10.Kool J, Eggink M, Van Rossum H, Van Liempd SM, Van Elswijk DA, Irth H, Commandeur JNM, Meerman JHN, Vermeulen NPE. J Biomol Screen. 2007;12:396–405. doi: 10.1177/1087057107299527. [DOI] [PubMed] [Google Scholar]
- 11.De Boer AR, Letzel T, Van Elswijk DA, Lingeman H, Niessen WMA, Irth H. Anal Chem. 2004;76:3155–3161. doi: 10.1021/ac035380w. [DOI] [PubMed] [Google Scholar]
- 12.Van Elswijk DA, Schobel UP, Lansky EP, Irth H, Van Der Greef J. Phytochemistry. 2004;65:233–241. doi: 10.1016/j.phytochem.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 13.Schobel U, Frenay M, Van Elswijk DA, Mcandrews JM, Long KR, Olson LM, Bobzin SC, Irth H. J Biomol Screen. 2001;6:291–303. doi: 10.1177/108705710100600503. [DOI] [PubMed] [Google Scholar]
- 14.Kool J, Ramautar R, Van Liempd SM, Beckman J, De Kanter FJJ, Meerman JHN, Schenk T, Irth H, Commandeur JNM, Vermeulen NPE. J Med Chem. 2006;49:3287–3292. doi: 10.1021/jm0507936. [DOI] [PubMed] [Google Scholar]
- 15.Zheng CJ, Han LY, Yap CW, Xie B, Chen YZ. Drug News Perspect. 2005;18:109–127. doi: 10.1358/dnp.2005.18.2.886480. [DOI] [PubMed] [Google Scholar]
- 16.Comess KM, Schurdak ME, Voorbach MJ, Coen M, Trumbull JD, Yang H, Gao L, Tang H, Cheng X, Lerner CG, Mccall JO, Burns DJ, Beutel BA. J Biomol Screen. 2006;11:743–754. doi: 10.1177/1087057106289971. [DOI] [PubMed] [Google Scholar]
- 17.Moaddel R, Jozwiak K, Wainer IW. Med Res Rev. 2007;27:723–753. doi: 10.1002/med.20091. [DOI] [PubMed] [Google Scholar]
- 18.Moaddel R, Wainer IW. J Pharm Biomed Anal. 2003;30:1715–1724. doi: 10.1016/s0731-7085(02)00513-7. [DOI] [PubMed] [Google Scholar]
- 19.Moaddel R, Bullock PL, Wainer IW. J Chromatogr B. 2004;799:255–263. doi: 10.1016/j.jchromb.2003.10.054. [DOI] [PubMed] [Google Scholar]
- 20.Baynham MT, Patel S, Moaddel R, Wainer IW. J Chromatogr B. 2002;772:155–161. doi: 10.1016/s1570-0232(02)00070-3. [DOI] [PubMed] [Google Scholar]
- 21.Moaddel R, Hamid R, Patel S, Bullock PL, Wainer IW. Anal Chim Acta. 2006;578:25–30. doi: 10.1016/j.aca.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 22.Schmeltz I, Hoffmann D. Chem Rev. 1977;77:295–311. [Google Scholar]
- 23.Daly JW. Cellular and Molecular Neurobiology. 2005;25:513–552. doi: 10.1007/s10571-005-3968-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holladay MW, Dart MJ, Lynch JK. J Med Chem. 1997;40:4169–4194. doi: 10.1021/jm970377o. [DOI] [PubMed] [Google Scholar]
- 25.Sharples CGV, Wonnacott S. Tocris Review. 2001 [Google Scholar]
- 26.Xiao Y, Meyer EL, Thompson JM, Surin A, Wroblewski J, Kellar KJ. Mol Pharmacol. 1998;54:322–333. doi: 10.1124/mol.54.2.322. [DOI] [PubMed] [Google Scholar]
- 27.Wainer IW, Zhang Y, Xiao Y, Kellar KJ. J Chromatogr B. 1999;724:65–72. doi: 10.1016/s0378-4347(98)00579-9. [DOI] [PubMed] [Google Scholar]
- 28.Apffel A, Fischer S, Goldberg G, Goodley PC, Kuhlmann FE. J Chromatogr A. 1995;712:177–190. doi: 10.1016/0021-9673(95)00175-m. [DOI] [PubMed] [Google Scholar]
- 29.Sambe H, Hoshina K, Moaddel R, Wainer IW, Haginaka J. J Chromatogr A. 2006;1134:88–94. doi: 10.1016/j.chroma.2006.08.073. [DOI] [PubMed] [Google Scholar]
- 30.Glennon RA, Dukat M. Pharmaceutica Acta Helvetiae. 2000;74:103–114. doi: 10.1016/s0031-6865(99)00022-9. [DOI] [PubMed] [Google Scholar]
- 31.Moaddel R, Jozwiak K, Yamaguchi R, Cobello C, Whittington K, Sarkar TK, Basak S, Wainer IW. J Chromatogr B. 2004;813:235–240. doi: 10.1016/j.jchromb.2004.09.042. [DOI] [PubMed] [Google Scholar]
- 32.Schuller HM. Life Sci. 2007;80:2274–2280. doi: 10.1016/j.lfs.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seddon AM, Curnow P, Booth PJ. BBA-Biomembranes. 2004;1666:105–117. doi: 10.1016/j.bbamem.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 34.Moaddel R, Marszałł M, Bighi F, Yang Q, Duan X, Wainer I. Anal Chem. 2007;79:5414–5417. doi: 10.1021/ac070268+. [DOI] [PubMed] [Google Scholar]