

Abstract
Braak and colleagues have proposed that, within the central nervous system, Parkinson’s disease (PD) begins as a synucleinopathy in non-dopaminergic structures of the lower brainstem or in the olfactory bulb. The brainstem synucleinopathy is postulated to progress rostrally to affect the substantia nigra (SN) and cause parkinsonism at a later stage of the disease. In the context of a diagnosis of PD, made on the basis of current clinical criteria, the pattern of lower brainstem involvement accompanying mesencephalic synucleinopathy is often observed. However, outside of that context, the patterns of synucleinopathy described by Braak are often not observed, particularly in dementia with Lewy bodies and when synucleinopathy occurs in the absence of neurological manifestations. The concept that lower brainstem synucleinopathy represents “early PD” rests on the supposition that it has a substantial likelihood of progressing within the human lifetime to involve the mesencephalon and thereby cause the SN pathology and clinical parkinsonism that have heretofore defined the disease. However, the predictive validity of this concept is doubtful, based on numerous observations made in populations of aged individuals who, in spite of the absence of neurologic signs, have brain synucleinopathy ranging up to Braak Stages 4 to 6 at postmortem. Furthermore, there is no relationship between Braak stage and the clinical severity of PD. We conclude that the relationship between patterns of abnormal synuclein immunostaining in the human brain and the disease entity now recognized as PD remains to be determined.
The neurobiology of α-synuclein has been central to the study of Parkinson’s disease (PD) since the discovery that missense mutations in this protein cause familial PD1. The discovery that α-synuclein is a major component of Lewy bodies in idiopathic PD heightened interest in the protein2, as did the observation that duplication or triplication of the α-synuclein gene also causes PD3. Synuclein had yet another important impact on thinking about PD when Braak and colleagues published elegant descriptions of abnormal patterns of α-synuclein immunostaining in human brains, and proposed a neuropathologic staging scheme4. They proposed that the earliest stage of PD (Stage 1) is characterized by abnormal α-synuclein immunostaining confined to the medulla oblongata or the olfactory bulb, and that clinical parkinsonism and neuron loss in the substantia nigra pars compacta (SNpc), both of which are now required for a definitive diagnosis of PD, occur late in the disease course.
This proposed staging scheme has had a profound impact on many aspects of current thinking about PD. The concept that the disease may begin in non-dopaminergic structures of the brainstem, or perhaps even in the peripheral autonomic nervous system4,5 has influenced how we think about early diagnosis of PD, how we might develop biomarkers6 and how we think about the “best” animal models for the disease7. The Braak staging scheme for PD has already begun to receive wide acceptance8,9. Given the significant influence of the Braak staging scheme and its likely impact on future PD research directions, it seems critical to assess whether its scientific validity has been affirmed, and to consider issues that will require further investigation.
Braak Staging and the Clinical Manifestations of PD
Braak staging of the pathology of PD
Braak and colleagues have outlined their concept of staging PD with great clarity in many publications4,5,10,11, so it will suffice here to summarize their principal findings. Their observations are depicted schematically in Figure 1 (based on Braak, 200611). Some brains showed abnormal synuclein immunostaining only in the medulla oblongata (in the dorsal motor nucleus (DMN) of the vagus) or the olfactory bulb; these regions are identified as region “1”. Other brains showed not only involvement of these structures, but also rostrally adjacent structures, including the caudal raphe nuclei and the locus ceruleus (LC) (region “2” in Figure 1). Since each group of brains with more widespread changes always included the involvement of the medulla oblongata and pons, as observed in the mildly involved brains, Braak and colleagues proposed that the varying degrees of synuclein pathology depict a temporal sequence of events: involvement limited to region “1” is the earliest phase (“Stage 1”), involvement of regions “1” and “2” is the next phase (“Stage 2”), and so on. Braak and colleagues proposed that it is only when the synuclein pathology reaches Stage 3 or 4 that SN neuron loss occurs, clinical signs of parkinsonism appear, and it becomes possible to diagnose “PD” by current criteria. Thus the Braak hypothesis proposes that synuclein pathology in the lower brainstem is necessary for the later appearance of PD, and, further, that it is sufficient; i.e., this synuclein pathology so predictably evolves to PD that it can be considered to represent “early PD”.
Fig. 1.
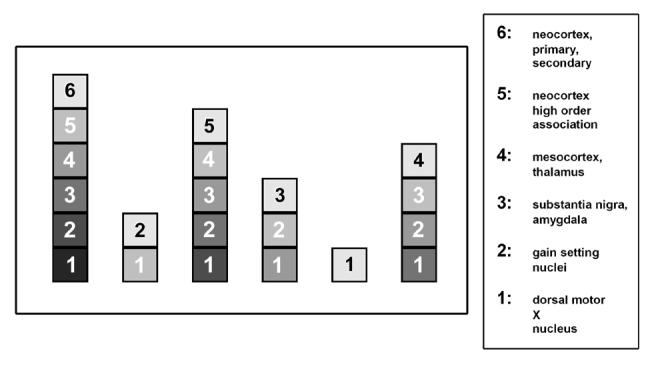
Patterns of abnormal immunostaining for α-synuclein identified by Braak and colleagues (adapted from Figures 1c and d, Braak et al, 2006). Six patterns of immunostaining were observed. In the pattern with the least extent of abnormal staining, involvement was observed only in region #1, which contains the dorsal motor nucleus of the vagus. In the pattern with the next most limited distribution, staining was observed, in addition to region #1, in region #2, which included the locus ceruleus and other “gain setting nuclei”. In the pattern with the next most involvement, abnormal staining was observed not only in regions #1 and #2, but also region #3, which included the SN and the amygdala. Since each succeeding pattern of increased rostral involvement included pathology in the adjacent more caudal regions, Braak and colleagues proposed that PD begins in region #1 (Stage 1), then proceeds rostrally to region #2 (Stage 2), and so on. Clinical signs of parkinsonism and Lewy pathology in the mesencephalon, both of which are now required for a definitive diagnosis of PD, are hypothesized to occur late in the disease, at Stage 3.
Patterns of synuclein pathology in human brain
We shall first consider the evidence that lower brainstem synuclein pathology is necessary for the eventual occurrence of clinical PD, as it is now recognized. In Braak’s original data4, of the patients who had been clinically diagnosed to have PD, all had synuclein pathology not only in the SNpc, but also in either the LC, the nucleus raphe, or the DMN. This observation, that in the context of a clinical diagnosis of PD, synuclein pathology is always observed caudal to the mesencephalon, has been largely borne out by other investigators12,13. However, this observation is not enough to be certain that in these instances, the clinical diagnosis of PD was universally preceded by lower brain stem synucleinopathy. To consider whether such may be the case, it is useful to consider patterns of synuclein deposition in cases of “incidental Lewy body disease”, individuals without dementia or parkinsonism who harbor synuclein pathology, including in the SN. There is substantial evidence that such cases may represent “pre-clinical” PD; they have reduced numbers of SN dopamine neurons to a slightly less degree than that in PD patients 14. Contrary to what would be predicted by the Braak hypothesis, many of these brains with synuclein pathology in the mesencephalon, likely to represent “pre-clinical” PD, do not show a lower brainstem synucleinopathy. Parkkinen and colleagues identified 79 brains with synuclein pathology in the mesencephalon. Among these, 13 (16%) did not have synuclein pathology in the DMN or LC12. Most of these patients were neurologically normal; some had had strokes; one had a diagnosis of dementia with Lewy bodies (DLB).
DLB is a particular problem for the Braak scheme, because such patients were excluded4. The exclusion of cases of DLB from the Braak series cannot be justified, because there is a growing consensus that the extensive overlap in the clinical and pathological features of PD, PD with dementia and DLB do not permit meaningful distinctions among them 15,16. These disorders are more appropriately considered as manifestations of a single underlying pathogenic mechanism encompassed by the terms “Lewy body disorders” 15 or “Lewy body disease” 16. Among 226 brains with synuclein pathology more recently analyzed by Parkkinen and colleagues, there was a clinical diagnosis of dementia in 74. Among these 28% had synuclein pathology which did not fit the Braak scheme. Given the presence of synuclein pathology, many of these cases were likely to have DLB17.
The Braak scheme also does not address the occurrence of “amygdala predominant” synuclein pathology18. In their series, Zaccai and colleagues identified synuclein pathology in 39% of aged individuals followed prospectively in two population-based cohorts. Among the 76 brains with synuclein pathology, 35 (46%) did not conform to the Braak staging scheme. Among these 22 (29%) had amygdala predominant synuclein pathology. They identified no relationship between amygdala synuclein pathology and neurofibrillary Alzheimer changes18.
Thus, while the patterns described by Braak and colleagues usually appear to be confirmed when a clinical diagnosis of PD has been made by current clinical criteria, they often are not confirmed outside of that context, particularly in clinically asymptomatic individuals and in individuals with DLB. Given the evidence that individuals with incidental Lewy body disease may represent preclinical PD, and the frequent observation that individuals with an initial diagnosis of DLB evolve to a condition that is indistinguishable from PD, it is not tenable to propose that lower brainstem synuclein pathology is a necessary occurrence prior to the onset of PD, as it is now recognized.
Does Braak Stage 1 (or 2) represent “early PD”?
The Braak hypothesis not only proposes that lower brainstem pathology is a necessary pre-condition for the occurrence of PD, but also that it is sufficient. In other words, there is such a compelling liklihood that Stage 1 or Stage 2 synuclein pathology will evolve to Stages 3 or 4, and, more importantly, evolve to manifest clinical parkinsonism that this pathology can be considered to represent “early PD”, as has been claimed9. A number of observations do not support the claim that Stage 1 and Stage 2 Braak synuclein pathology can be meaningfully considered to represent “early PD”. Data from elderly individuals without detectable neurologic impairment (Fig. 2) reveal that many harbor early Braak pathology. Given the advanced age of many of these individuals, it is not meaningful to speculate that they would have developed PD, had they lived longer. We do not know how long these individuals harbored this pathology nor do we know how long it would take for it to evolve, if ever.
Fig. 2.

Relationship between subject age at time of death and Braak PD Stage in 116 individuals without dementia or parkinsonism. The data presented are from Braak4, Parkkinen12, and Bloch44. There is no relationship between age at death and Braak Stage (r = 0.09, NS). Braak Stage 1 to 6 synuclein pathology is observed in neurologically unimpaired individuals who live decades beyond the average life expectancy at birth for industrialized nations.
A more worrisome concern is whether the evolution of synuclein pathology to Braak Stages 4, 5, or 6, even if it occurs, necessarily means that the individual will clinically manifest any of the motor or non-motor disabilities currently recognized as clinical manifestations of PD. In Figure 2, it is apparent that many individuals die harboring Braak synuclein pathology Stages 4-6 without ever having had neurological impairment, and many additional studies support this observation19-23. Therefore, we cannot be certain that early Braak Stages will evolve to higher stages in the human lifetime, and even when they do, they will not necessarily be associated with the clinical disabilities that we now identify as PD. Thus, the relationship between early Braak stages of abnormal synuclein staining and the disease entity that we now identify as PD is unknown.
Is there a relationship between Braak Stages and either the duration or the clinical severity of PD?
If the Braak staging scheme represents the actual temporal evolution of PD, there should be a relationship between Braak Stages and either the duration or the severity of PD. Braak and colleagues did not examine any possible relationship between stages and duration of disease. However, their data demonstrate that there is no relationship between Braak stage and Hoehn and Yahr score (Fig. 3). Many patients with Braak Stages 4 and higher did not have signs of parkinsonism. On the other hand, many individuals with Braak Stages 3 and 4 (when signs of parkinsonism are predicted to first appear) had severe PD (Hoehn and Yahr 5). In fact, Braak’s data appear to cluster into two distinct sets of patients: those with PD and those without.
Fig. 3.

Relationship between Braak PD Stage and Hoehn and Yahr PD score at the time of death in 96 individuals. The data presented are from Braak, 2003. The 68 individuals with a Hoehn and Yahr score of 0 are the same individuals as shown in Fig. 2, with the single exception that Braak Case 105, with a diagnosis of Alzheimer’s, is excluded in Fig. 2. Among 29 patients with PD (Hoehn and Yahr 1 or more), there is no relationship between Hoehn and Yahr score and Braak Stage (r = - 0.2, NS). The data appears to be clustered into two populations: those with PD and those without.
Is REM sleep behavior disorder (RBD) an early, non-dopaminergic stage of PD due to brainstem synucleinopathy?
In support of the Braak scheme, it has been proposed that RBD may be an early nondopaminergic manifestation of PD due to synuclein pathology identified in Stage 26,9,24. There is no question that RBD can precede signs of parkinsonism in a number of neurodegenerative diseases24,25. The question is whether such instances support the concept that PD universally begins with non-dopaminergic involvement of the brainstem. The proposal that RBD represents a clinical manifestation of Stage 2 synucleinopathy would depend foremost on compelling evidence that the pathology described provides a plausible neuropathologic substrate. The neural structures of the human brain that mediate muscle atonia during REM sleep are not precisely known24,25. It has been proposed that the subceruleus region in cat, and its analogue in rat, the sublateral dorsal nucleus, are crucial for atonia during REM sleep24. Although the human homologue of this structure is not defined24, we can consider whether Stage 2 Braak pathology is found ventromedial to the LC, the region thought important for RBD24. While Braak and colleagues have described Lewy pathology in the LC, they have not provided an explicit description of the region postulated to be the analogue of the rat sublateral dorsal nucleus. Even if such Lewy pathology were found, it would not provide a compelling basis for the postulated cellular dysfunction. Indeed, abnormal synuclein accumulation does not prove cellular dysfunction (discussed further below), and it is unknown whether Stage 2 Braak is accompanied by neuron loss or gliosis in the medulla or pons4. Thus, there is no compelling evidence that the synuclein pathology described in Braak Stage 2 provides a neural substrate for RBD.
If RBD cannot be specifically attributed to the abnormal synuclein staining observed in Braak Stages 1 and 2, then we must evaluate other evidence that RBD, in the setting of neurodegenerative synucleinopathies, is in fact due to involvement of brainstem nondopaminergic systems. One potential source of supportive data would be postmortem analysis in the context of such diseases that RBD can occur in the absence of SN involvement. To date there have been postmortem analyses of 27 RBD brains associated with synucleinopathies25. Among these, the SN was examined in 7 brains, and all of these brains exhibited SN neuron loss and synuclein pathology. Thus, we are unaware of a single case in which RBD was associated with a synucleinopathy without co-existing involvement of the SN.
An alternative source of data to evaluate the proposition that RBD is a non-dopaminergic manifestation of the synucleinopathies is to assess the relationship between RBD and nigrostriatal dopaminergic pathways assessed by neuroimaging techniques. Eisensehr and colleagues examined striatal dopamine transporter binding by [123I]IPT-SPECT in 5 patients with RBD and with normal neurological examinations. The RBD patients had reduced [123I]IPT-SPECT26. Similar observations were made by Albin et al using [11C]DTBZ -PET in 6 patients with RBD and no signs of parkinsonism27. Gilman et al examined the status of nigrostriatal dopaminergic projections by use of [11C]DTBZ -PET in 13 patients with RBD and a clinical diagnosis of MSA28. [11C]DTBZ striatal binding values were diminished among these patients, and there was a significant inverse correlation between [11C]DTBZ binding and severity of RBD28. Eisensehr and colleagues identified reduced [123I]IPT-SPECT values even in subclinical RBD patients29. Thus there is evidence of dopaminergic dysfunction in clinical and even subclinical RBD without other neurologic manifestations. These studies do not provide evidence to support the notion that RBD is due to non-dopaminergic involvement in the synucleinopathies; on the contrary, they indicate that mild and even subclinical RBD is associated with dopaminergic dysfunction.
It has often been pointed out that RBD may precede parkinsonism by many years. However, if the Braak scheme is a valid and general description of disease progression in PD, then it would be predicted that RBD would always precede parkinsonism. However, more often the converse is true. In a series of 195 patients referred to a PD clinic, assessed by Scaglione and coworkers, 64 (33%) had RBD. Of these, the signs of parkinsonism preceded RBD in 73%, and among these patients RBD appeared an average of 8 years after the onset of parkinsonism30. In a series of 60 patients with RBD reported by De Cock and colleagues, the manifestations of RBD preceded parkinsonism in only 22% of instances31. Thus, even if RBD in the context of synucleinopathies could be attributed to a specific degenerative change in the brainstem, and even if such change could be attributed to the synuclein pathology described by Braak, then the above clinical observations would suggest that involvement of mesencephalic dopaminergic systems in the majority of PD cases precedes pontine involvement, contrary to the Braak scheme.
While the occurrence of RBD prior to parkinsonism in PD has often been offered as evidence that specifically supports the Braak staging scheme of PD, it cannot be accepted as such. In many cases, RBD precedes parkinsonism in the multiple system atrophies (MSA)32,33. While MSA is also a synucleinopathy, characterized by synuclein-positive glial inclusions, it does not manifest intraneuronal synucleinopathy, and the Braak staging scheme cannot be proposed to occur.
What is the Pathophysiologic Significance of Lewy Pathology?
Another challenge for the Braak staging scheme is the uncertain pathophysiological significance of Lewy pathology. The Braak scheme assumes that Lewy pathology reliably detects neurons that are in a state of PD-related cellular dysfunction. However, synuclein pathology may not identify such neurons with sensitivity, and, conversely, neurons with abnormal synuclein immunostaining may not be in a state of PD-related dysfunction. With regard to the ability of synuclein immunostaining to detect early PD neuronal dysfunction, patients with PD-causing LRRK2 mutations are informative. While the majority of patients with the most common LRRK2 mutation (G2019S) show classic Lewy body pathology, some do not. And strikingly, many patients with other pathogenic mutations of LRRK2 do not show Lewy pathology. Indeed, the initial clinical report of LRRK2-related PD describes multiple cases of “pure nigral degeneration”34. Thus, in a genetically defined patient population it is clear that Lewy pathology is not a reliable indicator of PD cellular dysfunction. Therefore, the assumption that synuclein immunopathology will define where PD “begins”, as in the Braak scheme, is not tenable.
Conversely, cell biological studies of synuclein and related protein aggregation disorders raise questions about the pathophysiologic significance of protein aggregates. Increased expression of synuclein protein has been identified in neurons in a variety of injury models35-37, and in these contexts it has, in fact, been postulated to possibly play a protective role. In a Drosophila model in vivo, formation of synuclein inclusions correlates with reduced cellular toxicity38. In human brain, an inverse relationship has been noted between the presence of LB’s and neuron death39. Related work in Huntington’s disease demonstrates that protein inclusions mark surviving cells that are effectively sequestering misfolded protein40. In conclusion, the absence of abnormal immunostaining for synuclein in neurons cannot be interpreted as evidence that the cell is free of PD-related dysfunction, and, on the other hand, the presence of such staining cannot be interpreted as evidence that the cell is dysfunctional or that synuclein is responsible.
Are There Alternative Hypotheses for the Regional Patterns of Synuclein Pathology Observed by Braak and Colleagues?
If there is a consensus that the regional patterns of synuclein pathology described by Braak and colleagues are in most instances confirmed in the context of a clinical diagnosis of PD, and yet there is reason also to question the caudal-to-rostral evolution of the disease, as proposed, then we must consider whether there are alternative hypotheses to account for the patterns observed. One alternative hypothesis is that patterns are due to the relative likelihood of different brain regions to manifest Lewy pathology. Without invoking any assumptions about the timing of events, we might hypothesize that the DMN is a brain region very likely to reveal such pathology, the LC next most likely, and so on. Without any assumptions about which region, if any, was affected “first”, it would be proposed that observations made at postmortem reflect these probabilities. Such a concept would account for the observation that it is unlikely to encounter SN Lewy pathology in the absence of pathology in the DMN and/or LC. On the other hand, this concept would also account for the instances in which atypical patterns are observed. The likelihood of different brain regions to manifest Lewy pathology would be determined by many cellular characteristics of the neurons in these regions: their level of expression of synuclein, the likelihood that the cellular environment would be permissive for synuclein aggregation, the ability of these neurons to clear synuclein aggregates, and so on. The DMN, LC, and the SNpc are distinguished for their abundant levels of expression of α-synuclein mRNA41 (Fig. 4). There is, in fact, a striking correlation between the human brain structures identified in “early” Braak stages and structures with the highest levels of synuclein expression in the rodent brain: Stage 1: DMN and olfactory tubercle; Stage 2: LC; Stage 3: amygdala, SN, the pedunculopontine nucleus, and the nucleus of the diagonal band. In human brain high levels of synuclein expression have been observed in the LC and SNpc42.
Fig. 4.

Patterns of expression of α-synuclein mRNA in rat brain. The Figure is adapted from Maroteaux and Scheller, 1991, Figure 4. The images are darkfield micrographs of in situ hybridization for mRNA, so intense hybridization appears white on a black background. Different brain sections are arranged in ascending order according to the regions involved at different Braak Stages of synucleinopathy. In the bottom panels it is apparent that both the olfactory tubercle (LOT: nucleus of the lateral olfactory tract) and the DMN of the vagus (“10”) express high levels of α-synuclein mRNA (red arrows). In the middle panel, it can be seen that the locus ceruleus (LC) also expresses high levels. In the top panels, the amygdala (Am), and the SNpc express high levels.
Conclusions
The elegant and thorough morphologic investigations of Braak and colleagues have revealed intriguing patterns of synuclein pathology in the brains of patients with a clinical diagnosis of PD. For the most part, these patterns have been confirmed by other investigators. However, there has been little exploration of these patterns in patients with a clinical diagnosis of DLB, and the existing evidence suggests that non-conformance with the Braak patterns occurs. This question bears importantly on the meaning of the Braak patterns for the pathogenesis of PD, because PD and DLB are arguably variant presentations of a single entity16. Most importantly, this assessment of the Braak staging scheme suggests that there is insufficient evidence to claim that human PD begins in non-dopaminergic structures of the brainstem, and then evolves in a caudal-to-rostral progression over the course of the disease. A number of currently unresolved questions lead to this assessment. First, the prognostic implications of the presence of synuclein pathology in the brainstem are unknown. There are numerous instances of synuclein pathology in the brainstem, and, in fact, even more widely distributed in the brain, in neurologically intact elderly individuals. To propose that these individuals would have developed PD “had they lived long enough” is speculative and meaningless, given their advanced age. We do not know how long these individuals harbored this pathology, or if it would have progressed at all. Secondly, we do not understand the relationship between abnormal synuclein immunostaining in the brain and the selective loss of neurons that is the cardinal manifestation of PD. There are reported individuals with only Braak Stage 2 synucleinopathy and yet who have clinical parkinsonism17, but, on the other hand, there are many individuals with Braak Stage 6 who do not. Not surprisingly, in view of these observations, there is no relationship between Braak stages and the clinical severity of PD. In conclusion, the regional patterns of synuclein pathology in PD brains described by Braak and colleagues do not provide compelling evidence for the caudal-to-rostral temporal progression of the disease that they postulate, because these patterns may be accounted for alternatively by considerations related to the propensity of brain regions to display such pathology, based on regional differences in levels of synuclein expression and other factors.
The resolution of these questions will require much more study, including additional assessments of the patterns of synucleinopathy in the human brain, and, most importantly, how these patterns relate to validated biomarkers for progression of PD43. At a more basic level, we need a better understanding of the neurobiology of synuclein. While there is much evidence that overexpression of synuclein can be deleterious to neurons, it is unlikely to be just that simple. While time and again the neurobiology of synuclein has offered opportunities to understand the pathogenesis of PD, these are as yet only opportunities, and their fulfillment will require much additional work and critical analysis.
Acknowledgements
This work was supported by NS26836, NS38370, K02 NS045798, The Anne and Bernard Spitzer Center for Cell and Genetic Therapy For Parkinson’s Disease, and the Parkinson’s Disease Foundation. We are indebted to Dr. Irina Alafuzoff, Kuopio University, and Dr Kurt Jellinger, Institute of Clinical Neurobiology, for their review of the manuscript and helpful comments.
Footnotes
Statement of Conflict: The authors have no financial interests to disclose
References
- 1.Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the α-synuclein gene identified in families with Parkinson’s Disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 2.Spillantini MG, Schmidt ML, Lee VMY, et al. α-Synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 3.Singleton AB, Farrer M, Johnson J, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 4.Braak H, Del Tredici K, Rub U, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 5.Braak H, Sastre M, Bohl JR, et al. Parkinson’s disease: lesions in dorsal horn layer I, involvement of parasympathetic and sympathetic pre- and postganglionic neurons. Acta Neuropathol. 2007;113:421–429. doi: 10.1007/s00401-007-0193-x. [DOI] [PubMed] [Google Scholar]
- 6.Langston JW. The Parkinson’s complex: parkinsonism is just the tip of the iceberg. Ann Neurol. 2006;59:591–596. doi: 10.1002/ana.20834. [DOI] [PubMed] [Google Scholar]
- 7.Fernagut PO, Chesselet MF. Alpha-synuclein and transgenic mouse models. Neurobiol Dis. 2004;17:123–130. doi: 10.1016/j.nbd.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 8.Moore DJ, West AB, Dawson VL, et al. Molecular pathophysiology of Parkinson’s disease. Annu Rev Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- 9.Ahlskog JE. Beating a dead horse: dopamine and Parkinson disease. Neurology. 2007;69:1701–1711. doi: 10.1212/01.wnl.0000296942.14309.4a. [DOI] [PubMed] [Google Scholar]
- 10.Braak H, Del Tredici K, Bratzke H, et al. Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson’s disease (preclinical and clinical stages) J Neurol. 2002;249(Suppl 3):III/1–III/5. doi: 10.1007/s00415-002-1301-4. [DOI] [PubMed] [Google Scholar]
- 11.Braak H, Bohl JR, Muller CM, et al. Stanley Fahn Lecture 2005: The staging procedure for the inclusion body pathology associated with sporadic Parkinson’s disease reconsidered. Mov Disord. 2006;21:2042–2051. doi: 10.1002/mds.21065. [DOI] [PubMed] [Google Scholar]
- 12.Parkkinen L, Kauppinen T, Pirttila T, et al. Alpha-synuclein pathology does not predict extrapyramidal symptoms or dementia. Ann Neurol. 2005;57:82–91. doi: 10.1002/ana.20321. [DOI] [PubMed] [Google Scholar]
- 13.Jellinger KA. Lewy body-related alpha-synucleinopathy in the aged human brain. J Neural Transm. 2004;111:1219–1235. doi: 10.1007/s00702-004-0138-7. [DOI] [PubMed] [Google Scholar]
- 14.Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain. 1991;114:2283–2301. doi: 10.1093/brain/114.5.2283. [DOI] [PubMed] [Google Scholar]
- 15.Lippa CF, Duda JE, Grossman M, et al. DLB and PDD boundary issues: diagnosis, treatment, molecular pathology, and biomarkers. Neurology. 2007;68:812–819. doi: 10.1212/01.wnl.0000256715.13907.d3. [DOI] [PubMed] [Google Scholar]
- 16.McKeith I. Dementia with Lewy bodies and Parkinson’s disease with dementia: where two worlds collide. Pract Neurol. 2007;7:374–382. doi: 10.1136/jnnp.2007.134163. [DOI] [PubMed] [Google Scholar]
- 17.Parkkinen L, Pirttila T, Alafuzoff I. Applicability of current staging/categorization of alpha-synuclein pathology and their clinical relevance. Acta Neuropathol. 2008;115:399–407. doi: 10.1007/s00401-008-0346-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zaccai J, Brayne C, McKeith I, et al. Patterns and stages of alpha-synucleinopathy: Relevance in a population-based cohort. Neurology. 2008;70:1042–1048. doi: 10.1212/01.wnl.0000306697.48738.b6. [DOI] [PubMed] [Google Scholar]
- 19.Mikolaenko I, Pletnikova O, Kawas CH, et al. Alpha-synuclein lesions in normal aging, Parkinson disease, and Alzheimer disease: evidence from the Baltimore Longitudinal Study of Aging (BLSA) J Neuropathol Exp Neurol. 2005;64:156–162. doi: 10.1093/jnen/64.2.156. [DOI] [PubMed] [Google Scholar]
- 20.Saito Y, Ruberu NN, Sawabe M, et al. Lewy body-related alpha-synucleinopathy in aging. J Neuropathol Exp Neurol. 2004;63:742–749. doi: 10.1093/jnen/63.7.742. [DOI] [PubMed] [Google Scholar]
- 21.Wakisaka Y, Furuta A, Tanizaki Y, et al. Age-associated prevalence and risk factors of Lewy body pathology in a general population: the Hisayama study. Acta Neuropathol. 2003;106:374–382. doi: 10.1007/s00401-003-0750-x. [DOI] [PubMed] [Google Scholar]
- 22.Klos KJ, Ahlskog JE, Josephs KA, et al. Alpha-synuclein pathology in the spinal cords of neurologically asymptomatic aged individuals. Neurology. 2006;66:1100–1102. doi: 10.1212/01.wnl.0000204179.88955.fa. [DOI] [PubMed] [Google Scholar]
- 23.Ding ZT, Wang Y, Jiang YP, et al. Characteristics of alpha-synucleinopathy in centenarians. Acta Neuropathol. 2006;111:450–458. doi: 10.1007/s00401-005-0015-y. [DOI] [PubMed] [Google Scholar]
- 24.Boeve BF, Silber MH, Saper CB, et al. Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain. 2007;130:2770–2788. doi: 10.1093/brain/awm056. [DOI] [PubMed] [Google Scholar]
- 25.Gagnon JF, Postuma RB, Mazza S, et al. Rapid-eye-movement sleep behaviour disorder and neurodegenerative diseases. Lancet Neurol. 2006;5:424–432. doi: 10.1016/S1474-4422(06)70441-0. [DOI] [PubMed] [Google Scholar]
- 26.Eisensehr I, Linke R, Noachtar S, et al. Reduced striatal dopamine transporters in idiopathic rapid eye movement sleep behaviour disorder. Comparison with Parkinson’s disease and controls. Brain. 2000;123(Pt 6):1155–1160. doi: 10.1093/brain/123.6.1155. [DOI] [PubMed] [Google Scholar]
- 27.Albin RL, Koeppe RA, Chervin RD, et al. Decreased striatal dopaminergic innervation in REM sleep behavior disorder. Neurology. 2000;55:1410–1412. doi: 10.1212/wnl.55.9.1410. [DOI] [PubMed] [Google Scholar]
- 28.Gilman S, Koeppe RA, Chervin RD, et al. REM sleep behavior disorder is related to striatal monoaminergic deficit in MSA. Neurology. 2003;61:29–34. doi: 10.1212/01.wnl.0000073745.68744.94. [DOI] [PubMed] [Google Scholar]
- 29.Eisensehr I, Linke R, Tatsch K, et al. Increased muscle activity during rapid eye movement sleep correlates with decrease of striatal presynaptic dopamine transporters. IPT and IBZM SPECT imaging in subclinical and clinically manifest idiopathic REM sleep behavior disorder, Parkinson’s disease, and controls. Sleep. 2003;26:507–512. doi: 10.1093/sleep/26.5.507. [DOI] [PubMed] [Google Scholar]
- 30.Scaglione C, Vignatelli L, Plazzi G, et al. REM sleep behaviour disorder in Parkinson’s disease: a questionnaire-based study. Neurol Sci. 2005;25:316–321. doi: 10.1007/s10072-004-0364-7. [DOI] [PubMed] [Google Scholar]
- 31.De Cock VC, Vidailhet M, Leu S, et al. Restoration of normal motor control in Parkinson’s disease during REM sleep. Brain. 2007;130:450–456. doi: 10.1093/brain/awl363. [DOI] [PubMed] [Google Scholar]
- 32.Olson EJ, Boeve BF, Silber MH. Rapid eye movement sleep behaviour disorder: demographic, clinical and laboratory findings in 93 cases. Brain. 2000;123(Pt 2):331–339. doi: 10.1093/brain/123.2.331. [DOI] [PubMed] [Google Scholar]
- 33.Plazzi G, Corsini R, Provini F, et al. REM sleep behavior disorders in multiple system atrophy. Neurology. 1997;48:1094–1097. doi: 10.1212/wnl.48.4.1094. [DOI] [PubMed] [Google Scholar]
- 34.Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 35.Kholodilov NG, Neystat M, Oo TF, et al. Increased expression of rat synuclein1 in the substantia nigra pars compacta identified by differential display in a model of developmental target injury. J Neurochem. 1999;73:2586–2599. doi: 10.1046/j.1471-4159.1999.0732586.x. [DOI] [PubMed] [Google Scholar]
- 36.Purisai MG, McCormack AL, Langston WJ, et al. Alpha-synuclein expression in the substantia nigra of MPTP-lesioned non-human primates. Neurobiol Dis. 2005;20:898–906. doi: 10.1016/j.nbd.2005.05.028. [DOI] [PubMed] [Google Scholar]
- 37.Manning-Bog AB, McCormack AL, Purisai MG, et al. Alpha-synuclein overexpression protects against paraquat-induced neurodegeneration. J Neurosci. 2003;23:3095–3099. doi: 10.1523/JNEUROSCI.23-08-03095.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen L, Feany MB. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci. 2005;8:657–663. doi: 10.1038/nn1443. [DOI] [PubMed] [Google Scholar]
- 39.Tompkins MM, Basgall EJ, Zamrini E, et al. Apoptotic-like changes in Lewy-body-associated disorders and normal aging in substantia nigral neurons. Am J Path. 1997;150:119–131. [PMC free article] [PubMed] [Google Scholar]
- 40.Arrasate M, Mitra S, Schweitzer ES, et al. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 41.Maroteaux L, Scheller RH. The rat brain synucleins; family of proteins transiently associated with neuronal membrane. Mol Br Res. 1991;11:335–343. doi: 10.1016/0169-328x(91)90043-w. [DOI] [PubMed] [Google Scholar]
- 42.Solano SM, Miller DW, Augood SJ, et al. Expression of alpha-synuclein, parkin, and ubiquitin carboxy-terminal hydrolase L1 mRNA in human brain: genes associated with familial Parkinson’s disease. Ann Neurol. 2000;47:201–210. [PubMed] [Google Scholar]
- 43.Brooks D.Examining Braak’s hypothesis by imaging Parkinson’s disease Mov Disord 2008; in press [DOI] [PubMed] [Google Scholar]
- 44.Bloch A, Probst A, Bissig H, et al. Alpha-synuclein pathology of the spinal and peripheral autonomic nervous system in neurologically unimpaired elderly subjects. Neuropathol Appl Neurobiol. 2006;32:284–295. doi: 10.1111/j.1365-2990.2006.00727.x. [DOI] [PubMed] [Google Scholar]