

Abstract
Macroautophagy (hereafter referred to as autophagy) can increase or decrease the amount of cell death in response to various stimuli. To test if autophagy also controls the characteristics associated with dying cells, we studied tumor cell killing by Epidermal Growth Factor Receptor (EGFR)-targeted diphtheria toxin (DT-EGF). DT-EGF kills epithelial and glioblastoma tumor cells with similar efficiency but by different mechanisms that depend on whether the cells activate autophagy when treated with the drug. Dying cells in which autophagy is induced selectively release the immune modulator HMGB1 without causing lysis of the cell membrane and classical necrosis. Conversely, cells that are killed by DT-EGF where autophagy is blocked, activate caspases but retain HMGB1. These data suggest that it may be feasible to manipulate the immunogenicity of dying cells by increasing or decreasing autophagy.
INTRODUCTION
Autophagy (1) regulates the amount of cell death that occurs in response to specific stimuli (2, 3) e.g. by blocking apoptosis after growth factor depletion (4, 5) or exposure of cells to various other apoptotic stimuli (6), protecting against caspase-independent death (7) or preventing necrosis in apoptosis-incompetent cells that are subjected to severe metabolic stress (8). Although it is still unclear under which conditions autophagy leads to cell death (9), autophagic cell death (3) has been reported in response to external insults including DNA damaging agents (10), caspase inhibitors (11) (although a more recent study raised questions about this conclusion (12)), and a large variety of anti-cancer drugs (13). Autophagy can also contribute to cell death during normal development (14). Thus autophagy can both positively and negatively regulate the amount of cell death that occurs in response to a stimulus.
The biological response to cell death stimuli such as treatment with anti-cancer drugs is determined both by the absolute amount of cell death and the characteristics of the dying cells. For example, apoptosis is usually non-inflammatory while necrosis usually causes an immune response. This difference is determined by the release of immunogenic stimulators especially the DNA-binding protein high-mobility group B1 (HMGB1), which is usually (but not always (15)) retained in apoptotic nuclei but released during necrosis (16). While it is well established that autophagy can regulate the amount of cell death in response to a death stimulus, it is less clear how autophagy regulates characteristics of dying cells such as the release of HMGB1. To address this question, we asked how autophagy affects cells that are destined to die in different ways by treating tumor cells with a targeted toxin (17), DT-EGF, which blocks protein synthesis and kills tumor cells in an EGFR-dependent manner as demonstrated by loss of metabolic activity (18), clonogenicity, and the regression of established tumors (19).
RESULTS
We evaluated the cytotoxicity of DT-EGF (18) on EGFR-positive glioblastoma (U87MG and U373MG) and epithelial (HeLa and HN12) cancer cells using clonogenic survival (Fig. 1A) assays by treating with increasing doses of the targeted toxin for 24 hours, then washing away the drug and allowing surviving cells to recover and undergo clonogenic growth for up to two weeks. In all cases we obtained similar, dose-dependent cell death as demonstrated by loss of clonogenic capacity. Previous studies (19) demonstrated that in addition to loss of metabolic activity as measured in cell viability assays, treatment with DT-EGF causes the regression of established glioblastoma tumors indicating that the drug causes bona fide glioblastoma cell death rather than growth arrest. Like many other anti cancer agents, targeted toxins usually kill tumor cells by activating caspase-dependent apoptosis (reviewed in (20)). However, time-lapse microscopy (Fig. 1B) showed that the morphology of the dying cells was markedly different with only the epithelial cells displaying typical characteristics of caspase-dependent apoptosis such as membrane blebbing and cellular fragmentation, while dying glioma cells rounded up but showed no signs of apoptosis or membrane rupture. Western blotting of the caspase substrate PARP (Fig. 1C) and measurements of the activity of effector caspases (Supp Fig 1A) showed that DT-EGF activated caspases in the epithelial cells but not in the glioblastoma cells. However both U87MG and HeLa cells displayed robust caspase activation after treatment with lexatumumab, an antibody that activates the TNF-related apoptosis inducing ligand (TRAIL) receptor DR5 (Supp Fig. 1B) indicating that both cell types contain functional caspases and could undergo apoptosis. Furthermore caspase inhibition protected against DT-EGF in HeLa cells but not in U87MG cells (Supp Fig. 1C).
Figure 1. DT-EGF kills glioblastoma and epithelial tumor cells by different mechanisms.

Panel A, quantification of clonogenic growth assay after treatment of HeLa, HN12, U87MG and U373MG cells for 24 hours with increasing doses of DT-EGF. All the cell lines die with similar dose responses (mean +/- SD from three experiments). Panel B shows frames from time-lapse movies of DT-EGF-treated HeLa cells and U87MG cells, showing that the morphological characteristics of the dying cells differs with only the epithelial cells displaying characteristics of apoptosis. Panel C, western analysis of PARP cleavage after 24 hours of DT-EGF treatment. Only the epithelial cells contain caspase-cleaved PARP.
We analyzed the proteome of dying U87MG cells treated with lexatumumab or DT-EGF. 2D-DIGE allows simultaneous analysis of different protein samples by labeling each sample with a different CyDye then combining the samples and separating them on the same gel. Lexatumumab caused numerous changes to the whole cell proteome, shown by many red and green spots that indicate differences between the samples (Fig. 2A). Extensive changes in the proteome are expected in apoptotic cells where caspases cleave hundreds of substrates (21). In contrast, U87MG cells dying as a result of DTEGF treatment displayed very few changes to the whole cell proteome as shown by the lack of green and red spots and the abundance of yellow spots (Fig. 2A). When the 2D gels were quantitated using a 2 fold threshold as the cut-off for counting differences; 241 out of 2,464 (9.8%) of spots were different between the control and lexatumumab-treated samples whereas only 2.4% of proteins were different between the control and DT-EGF-treated cells. These data indicate that the U87MG cells treated with DT-EGF cells are not undergoing apoptosis or apoptosis-like death caused by other proteases that cleave many substrates. Moreover, there was no apparent loss of soluble proteins as would be expected if there was abrupt lysis of the cell membrane as in typical necrosis.
Figure 2. DT-EGF induces autophagy in glioblastoma cells.
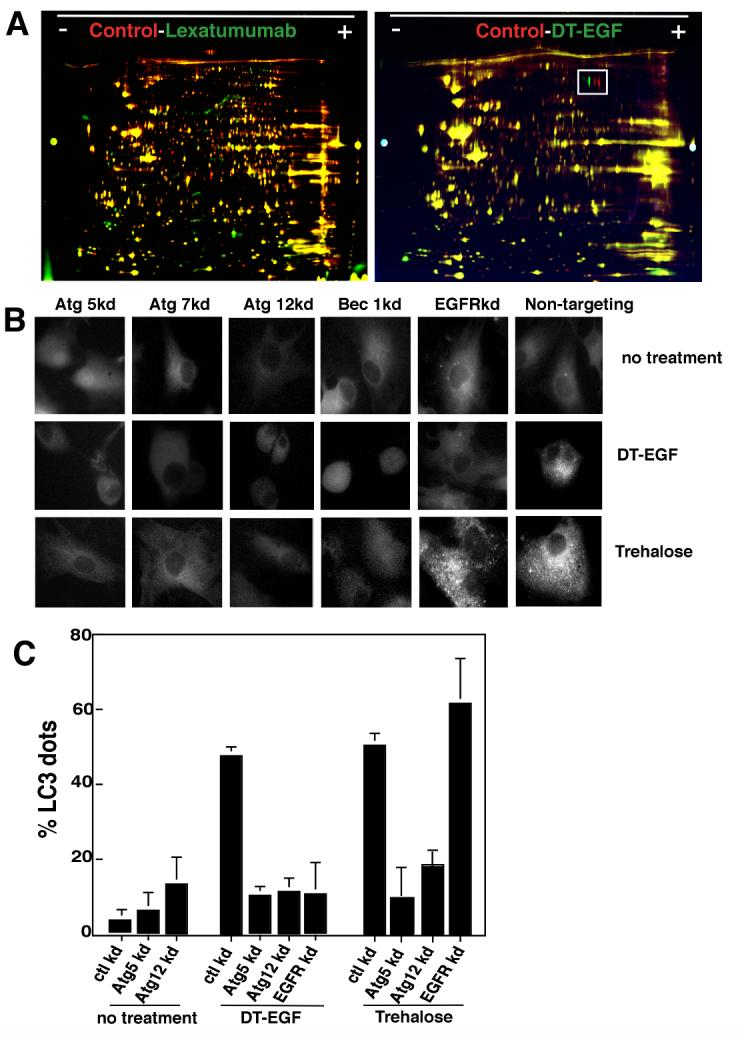
Panel A, 2D-DIGE of lexatumumab or DT-EGF-treated U87MG cells compared with untreated controls. Lexatumumab treatment causes many changes to the proteome whereas DT-EGF treatment shows few changes. The boxed area identifies a post-translational change involving phosphorylation of eEF2 that is induced by DT-EGF treatment. Panel B, U87MG cells stably expressing GFP-LC3 were treated with DT-EGF or trehalose after transfection of siRNAs as indicated. Aggregation of LC3 was monitored by fluorescence microscopy after 24 hours and quantitated (Panel C) by counting the percentage of cells with > 10 GFP-labeled dots, data shown is the mean +/- SD from two experiments. Both DT-EGF and trehalose induce LC3 aggregation that is abolished by knockdown of autophagy regulators; only DT-EGF-induced aggregation is blocked by EGFR knockdown.
One notable proteomic change induced by DT-EGF involved an apparent pI shift in a protein (Fig. 2A boxed region) that was identified by mass spectrometry as isoforms of the translation elongation factor eEF2alpha (eEF2). This protein is the direct catalytic target of diphtheria toxin, which ADP ribosylates eEF2 on a unique dipthamide residue. However, the pI shift and western analysis (not shown) indicated that the modification caused by DT-EGF also involved phosphorylation. Because eEF2 kinase regulates autophagy in glioblastoma cells (22), we asked if DT-EGF induces autophagy in U87MG cells. Upon induction of autophagy, the microtubule-associated light-chain 3 (LC3) is conjugated to phosphatidylethanolamine and incorporated into autophagosomes (1). This can be visualized by looking for the formation of foci of a green fluorescent protein tagged version of LC3 (GFP-LC3) (23). DT-EGF induced GFP-LC3 foci in glioblastoma cells (Fig. 2B, C) similar to that seen with the strong autophagy inducer trehalose (24). siRNA knockdown of the autophagy regulators Beclin1, Atg 5, Atg 12 or Atg 7 (knockdown was confirmed by western blotting, Supp Fig. 2) prevented both trehalose and DT-EGF-induced GFP-LC3 aggregation in the glioblastoma cells. However, only DT-EGF-induced autophagy was inhibited by EGFR knockdown. Autophagic flux was induced by DT-EGF in the U87MG cells as demonstrated (Fig. 3A) by increased LC3-II formation relative to the actin control when lysosomal protease inhibitors were used together with DT-EGF (25) and processing of the autophagy cargo substrate betaine homocysteine methyltransferase (BHMT) using a GST-tagged BHMT construct (26) (Fig. 3B). As has been noted in other systems, detection of LC3-I was variable. At later times after DT-EGF or trehalose treatment, we detected increased numbers of mCherry but not GFP labeled structures from a tandem labeled GFP-mCherry-LC3 expression construct. This protein displays green and red fluorescence in autophagosomes but only shows red fluorescence after the autophagosomes fuse with lysosomes (27, 28) indicating that DT-EGF causes the maturation of autophagosomes and fusion with lysosomes (Supp Fig. 3). DT-EGF also induced autophagy in U373MG cells (Fig. 3B) as shown using the GST-BHMT assay. DT-EGF treatment of the epithelial cell lines that we tested did not increase autophagy as indicated by LC3-II formation (Fig 3A), BHMT processing (Fig. 3B) or increased GFP-LC3 foci (Supp Fig. 4). Two other DT-EGF-sensitive glioblastoma cell lines (8MGBA and SNB19) also activated processing the the autophagy cargo BHMT in response to DT-EGF treatment (Supp Fig. 5). These data indicate that DT-EGF induces autophagosome formation and autophagic flux in some but not all tumor cells suggesting that the different characteristics of the dying cells could be related to the drug’s ability to activate autophagy in the different cell types.
Figure 3.
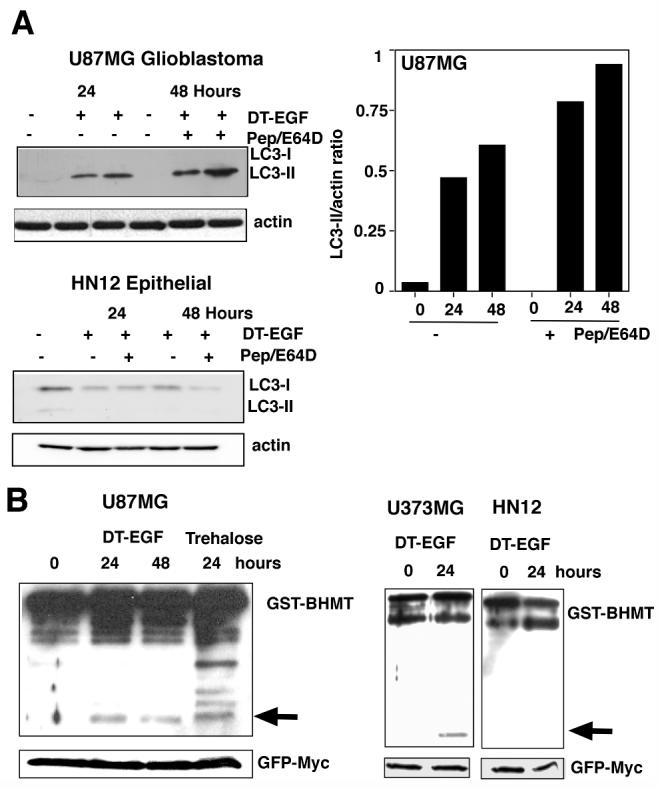
Panel A, western analysis of LC3 processing in the presence or absence of lysosomal protease inhibitors pepstatin A and E64D. DT-EGF causes increased LC3-II, which was further stimulated by treatment with pepstatin and E64D in the glioblastoma cells but not in the HN12 epithelial cells. The histogram shows quantitation of the ratio of LC3-II compared to actin for U87MG cells. Panel B, processing of the autophagy cargo protein betaine homocysteine methyltransferase (BHMT). The arrow indicates the processed form of the protein that is produced in autophagolysosomes. U87MG and U373MG glioblastoma cells display DT-EGF-induced BHMT processing, HN12 epithelial cells do not. GFP-Myc indicates the loading control, which was expressed from the GST-BHMT by an IRES sequence (26).
To test if autophagy controls the amount of DT-EGF-induced death, we performed siRNA knockdown of autophagy regulators, treated with increasing doses of DT-EGF for 24 hours, then removed the drug and cultured cells for two weeks to allow colony growth of surviving cells. Alternatively, autophagy was increased by pre-treatment with trehalose and cell death after treatment with DTEGF was monitored by time-lapse microscopy or clonogenic survival. Inhibition of autophagy increased the efficiency of DT-EGF killing in the clonogenic growth assay (Fig. 4A). Conversely, increasing autophagy with trehalose delayed (Fig 4B) and slightly inhibited (Fig. 4C) DT-EGF-induced glioblastoma cell death. In the U87MG cells, inhibition of autophagy by siRNA knockdown of Atg5 allowed induction of caspase 3/7 activity in response to DT-EGF (Fig. 4D). These data indicate that the glioblastoma cells do not undergo autophagic cell death but rather are protected by the DT-EGF-induced autophagy and suggest that this occurs because autophagy prevents more efficient caspase-dependent death. Increasing autophagy prior to DT-EGF treatment also protected HN12 epithelial cells by clonogenic assay (Supp. Fig. 6A) and time-lapse microscopy (Supp. Fig. 6B). The protection conferred by trehalose was greater in the epithelial cells than the glioblastoma cells consistent with the idea that in glioblastoma cells, trehalose-induced autophagy was merely added to that produced by DT-EGF on its own.
Figure 4. Autophagy protects against DT-EGF-induced death.

Panel A, clonogenic survival assay in U87MG cells after siRNA knockdown as indicated and treatment with increasing doses of DT-EGF, data represents the mean +/- SD of four experiments each performed in triplicate. Knockdown of autophagy regulators increases DT-EGF-induced cell death. Panel B, frames from a time-lapse microscopy experiment of U87MG cells treated with DT-EGF with or without 24 hours pre-treatment with trehalose to further increase autophagy; increased autophagy delays DT-EGF-induced death. Panel C, clonogenic survival assays in U87MG cells treated with DT-EGF in the presence or absence of trehalose to increase autophagy (mean +/- SD from three experiments). Increased autophagy inhibits DT-EGF-induced death. Panel D, caspase 3/7 activity in DT-EGF-treated U87MG cells after siRNA treatment (mean +/- SD from three replicates). Control and EGFR siRNA, DT-EGF-treated cells show no caspase activity, DT-EGF causes caspase activity in Atg5 siRNA-treated cells that is similar to the level achieved by treatment with Fas ligand plus cycloheximide.
We next asked if the cells that activate autophagy after DT-EGF treatment released HMGB1. DT-EGF-treated U87MG cells released HMGB1 into the extracellular media as shown by time-lapse microscopy of GFP-HMGB1-transfected cells (Fig. 5A) and western blotting (Fig. 5B). Moreover, this release was controlled by autophagy as shown by knockdown of Atg5, Atg7 or Atg12, which prevented HMGB1 release in DT-EGF-treated U87MG cells as detected by western blotting or time-lapse microscopy of GFP-HMGB1 (Fig. 5 A,B, Supp Fig. 8A). These data indicate that the formation of autophagosomes is required for the release of HMGB1. Release occurred after the GFP-HMGB1 protein became localized in a punctate pattern as shown in the magnified image in Fig. 5A. These HMGB1-positive structures, co-localized with a few of the LC3-positive dots (see arrows in Supp Fig. 7) consistent with the idea that a subset of autophagosomal structures contains HMGB1 prior to its release from the cell.
Figure 5. Autophagy regulates selective release of HMGB1 from DT-EGF-treated cells.
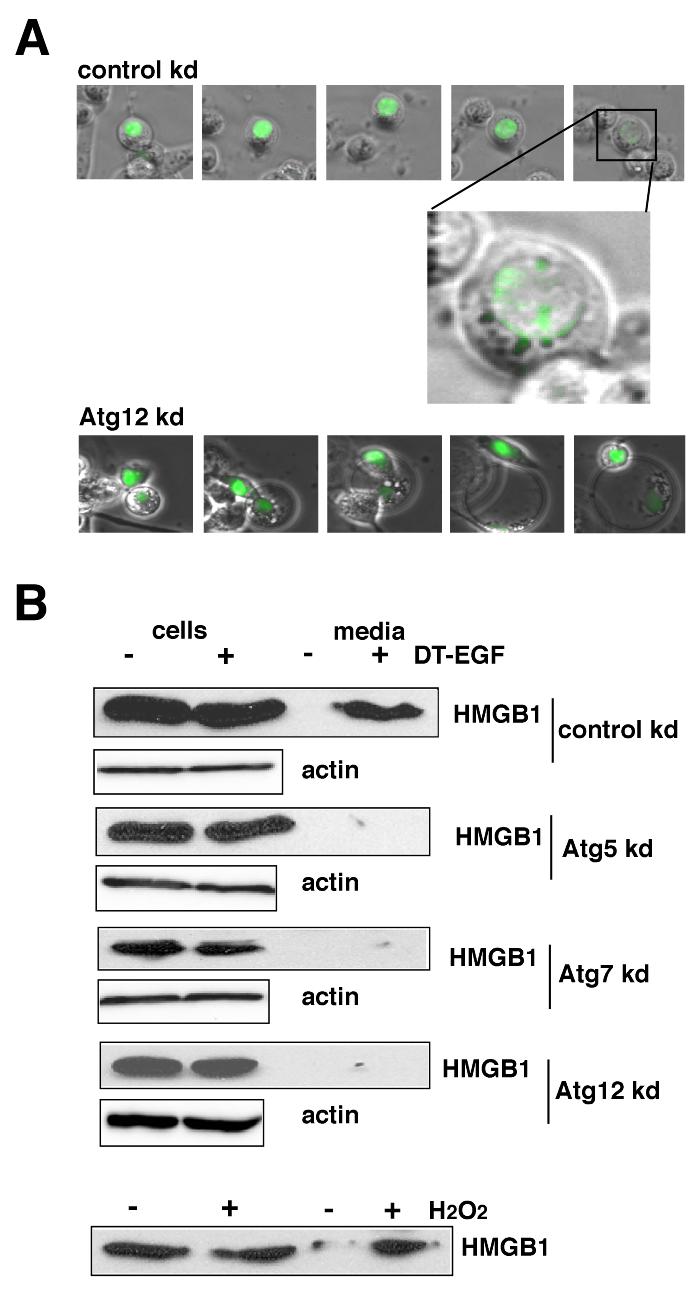
Panel A, U87MG cells transfected with GFP-HMGB1 and control (non-specific) or Atg12 siRNAs were monitored by time-lapse microscopy after treatment with DT-EGF. DT-EGF causes release of HMGB1 in control cells, Atg12 knockdown causes the retention of HMGB1 in fragmented apoptotic nuclei. Panel B, western blot analysis of cell extract and media for HMGB1 protein after DT-EGF treatment for 48 hours. HMGB1 is not released into the media in DT-EGF-treated cells when autophagy regulators are knocked down. Control shows cells treated for 3.5 hours with 10mM H2O2.
HMGB1 release usually occurs in cells that are undergoing classical necrotic cell death marked by abrupt membrane lysis and the release of soluble proteins (16). However, unlike the necrotic, H2O2-treated U87MG cells, which also released lactate dehydrogenase (LDH) (Fig. 6C) and were stained with propidium iodide (Fig. 6A,B) indicating that their cytoplasmic membranes were lysed, the DT-EGF-treated cells did not display other necrotic characteristics. Indeed there was greater propidium iodide staining in DT-EGF-treated cells where autophagy had been inhibited by Atg5 knockdown, due to cells in late stages of apoptosis. Together with the limited changes in the whole cell proteome in the dying DT-EGF-treated U87MG cells (Fig 2A), these data indicate that the HMGB1 release from these dying cells is not associated with membrane lysis and necrosis but instead represents selective release of the HMGB1 protein from the cell. Conversely, DT-EGF-treated epithelial cells (which activate caspases and undergo apoptosis) did not release significant amounts of HMGB1 (Fig. 6D and Supp Fig. 8). However, when we stimulated autophagy with trehalose, then treated HN12 epithelial cells with DT-EGF, this caused HMGB1 release from the dying cells (Fig. 6D) without significant release of LDH (Fig. 6E). This release is due to the autophagy itself because similar levels of HMGB1 release occur with treatment by trehalose alone (Fig. 6D).
Figure 6. DT-EGF treatment causes HMGB1 release without causing membrane lysis and necrosis.

Panel A, propidium iodide staining of U87MG cells after treatment with DT-EGF, 10mM H2O2, or DT-EGF after knockdown of Atg5. DT-EGF alone does not cause increased propidium iodide staining compared with living, untreated cells and displays less propidium iodide staining than in DT-EGF cells with Atg5 knockdown. Panel B, Quantitation of percentage of propidium iodide positive nuclei after treatment with DT-EGF, H2O2, or DT-EGF after knockdown of Atg5 (mean +/- SD from 3 replicates). Panel C, lactate dehydrogenase (LDH) release in U87 MG cells treated with DT-EGF or 10mM H2O2, no LDH is released in DT-EGF-treated cells (mean +/- SD from three replicates). Panel D, western analysis of HMGB1 in cells and media from HN12 epithelial cells treated with DT-EGF in the presence or absence of trehalose to stimulate autophagy; trehalose treatment promotes HMGB1 release into the media. Panel E, LDH release in the HN12 cells after treatment with DT-EGF with or without trehalose cells; trehalose treatment does not increase LDH release (mean +/- SD from three replicates).
DISCUSSION
Manipulation of autophagy and necrosis may be useful in cancer therapy (29, 30). For example, it may be possible to increase the efficiency of tumor cell killing in response to chemotherapeutic drugs by simultaneously inhibiting autophagy (31). Our data support these ideas but add a new level of complexity by showing that in addition to regulating the amount of cell death, autophagy can control the characteristics of the dying cells by regulating the selective release of HMGB1 and thus selectively controlling an important characteristic of dying cells that is usually associated with necrosis. DT-EGF induces autophagy more effectively in the glioblastoma cell lines that we tested compared with the epithelial tumor cell lines tested, however further studies will be needed to determine if this is a generally consistent difference between cell types, and, if it is to uncover the basis for the difference. Our data show that even in cells such as U87MG cells, which die through a mechanism that displays characteristics of neither apoptosis or necrosis, the autophagy that occurs in the dying cells protects the tumor cells from the drug (i.e. causes a leftward shift in the dose response curve); i.e. although the drug induces autophagy and then the cells die, this is not a case of autophagic cell death. In fact, the autophagy that is induced by DT-EGF in U87MG cells inhibits caspase activation and thus explains why they don’t activate apoptosis. Tumor cells that are dying with increased autophagy (either caused by DT-EGF alone in the case of the glioblastoma cells or when autophagy is further increased with trehalose in epithelial tumor cells) selectively release the nuclear HMGB1 protein but do so without displaying other characteristics of necrosis such as LDH release or loss of membrane integrity such that propoidium iodide can stain DNA. Although HMGB1 release is usually considered to be a marker of necrosis, activated monocytes and macrophages can secrete HMGB1 without undergoing necrosis (32) and some apoptotic stimuli can result in the release of HMGB1 under some conditions (15, 33). We speculate that these instances of HMGB1 release may occur when the apoptotic cells also have high levels of autophagy.
HMGB1 enhances the immune response to poorly immunogenic apoptotic cells (34) and, when dying tumor cells release HMGB1, they activate a Toll-like Receptor 4-dependent, tumor-specific, immune response (33). This has led to the idea that by manipulating the death mechanisms to promote immunogenic tumor cell killing, it may be feasible to increase the long term effectiveness of anti-cancer treatment by inducing an immune response that subsequently seeks out and destroys tumor cells that have avoided being killed by the drug (35-37). To achieve such a response it is necessary that tumor cells die (so that tumor antigens can be generated and recognized) and that the immune system is stimulated by the HMGB1. Our data suggest a way to achieve this manipulation by increasing autophagy in cells that are destined to die and thus cause the dying cells to release HMGB1. In situations where the drug not only kills the cells but also induces autophagy (as with DT-EGF treatment of the U87MG cells), the same agent may achieve both objectives. Alternatively, one could imagine adopting a strategy where one might inhibit autophagy to switch the tumor cell death mechanism towards caspase-dependent apoptosis without release of HMGB1. Further studies will be necessary to determine which scenario may be more effective to maximize anti tumor treatment in the long term and to determine whether the ability of autophagy to reduce the amount of tumor cell killing is a benefit or a handicap compared with the ability of autophagy to cause HMGB1 release and potentially increase the immunogenicity of the dying cells.
There are numerous mechanistic connections between apoptosis, necrosis and autophagy (38, 39) suggesting that different death mechanisms are intimately related. We suggest that a spectrum of characteristics can be displayed by a dying cell and that this is regulated in part by the amount of autophagy that occurs as they die. When autophagy is not activated, it is easier to activate caspases and dying cells undergo apoptosis; conversely, when autophagy is high, caspase activation is blocked but cells that are destined to die can release HMGB1 without membrane lysis and true necrosis. Thus, we propose that autophagy can regulate the amount of death in response to a particular stimulus and the characteristics associated with the dying cells and that the overall biological effect of autophagy manipulation during treatment with targeted anti-cancer drugs like DT-EGF that kill tumor cells may depend on both aspects.
MATERIALS & METHODS
Cells and reagents
HN12 cells were provided by Andrew Yeudall, other cells were obtained from ATCC. Lexatumumab, was provided by Human Genome Sciences and used as previously described (40). Trehalose, a potent autophagy inducer (24), was obtained from Sigma (St. Louis, MO) and used at 100 mM. zVAD.fmk was used at 50μM.
Cell death and autophagy assays
For clonogenic growth assays, cells in triplicate treated with 3 fold dilutions up to 10 ng/ml DT-EGF for 24 hours, drug was removed, cells were washed and maintained in growth media for 10-14 days, fixed and stained with 0.4% crystal violet. Because the glioma cells do not form tight colonies, stained cells were solubilized in 33% acetic acid and absorbance was read at 540nm. Values were normalized to non-treated sample. Clonogenic assays and HMGB1 release experiments with siRNA knockdown were performed 3 days after siRNA knockdown using siRNAs and transfection reagents from Dharmacon (Lafayette, CO). For time-lapse microscopy and other experiments with a single dose of DT-EGF, the toxin was used at 50 or 100ng/ml. Caspase assays were performed using Promega Caspase-Glo 3/7 reagent. LDH release was measured using LDH Cytotoxicity Detection Kit from Takara Bio Inc. (Seta 3-4-1, Otsu, Shiga, Japan). Time-lapse microscopy was performed on a Zeiss Axiovert 200 microscope equipped with an environmental chamber to maintain cells at 37°C/5%CO2. Images were captured with a Hamamatsu Orca CCD camera every 15-30 minutes using the Improvision Volocity program. Formation of autophagic vesicles was monitored by GFP-LC3 aggregation in stably expressing polyclonal cell lines, for quantitation the percentage of cells with >10 GFP-LC3 dots were scored as positive after counting ∼100 randomly chosen cells from two separate experiments. Autophagic flux assays were performed by western blotting for LC3-II formation after treatment with DT-EGF in the presence or absence of lysosomal protease inhibitors as recommended by Mizushima and Yoshimori (25), by monitoring the processing of a GST-tagged version of the autophagy cargo protein betaine homocysteine methyltransferase as described by Mercer et.al. (26) and by stably expressing a tandem GFP, mCherry-labeled LC3 protein that retains both green and red fluorescence in autophagosomes but retains only red fluorescence after fusion with lysosomes as described by Kimura et.al. (27).
Immunoblotting
PVDF membranes were blotted with antibodies that recognize PARP, eEF2, peEF2 (Thr 56), Beclin 1, Atg 5, which recognizes the Atg5-12 conjugate protein (Cell Signaling Technologies, Danvers, MA), EGFR, HMGB1 (Abcam, Cambridge, MA), Atg7 (Santa Cruz Biotechnology, Santa Cruz, CA), βActin (Sigma, St. Louis, MO) or LC3 (MBL, Woburn, MA), Myc (9B11) (Cell Signaling), GST (Z5) (Santa Cruz Biotechnology).
2D-DIGE
Supplies and protocols were from Amersham Biosciences/GE Healthcare (Piscataway, NJ). 50 μg of protein were minimally labeled with Cy3 or Cy5 Dye on ice for 30 minutes, combined and brought to a final volume of 450μl in 7 M urea, 2 M thiourea, 4% CHAPS, 40mM DTT, 0.002% bromophenol blue and 0.5% pharmalytes, rehydrated overnight onto a 24 cm immobilized pH gradient strip (IPG 3-10 NL) and isoelectric focused at 500 V for 100 Vhr, 1,000 V for 1,000 Vhr, and 8,000 V for 62,500 Vhr on a IPGphor apparatus. IPG strips were equilibrated for 15 minutes in a DTT solution, followed by reaction with iodoacetamide solution and applied to a precast 8-16% SDS-PAGE gel (Jule Inc., Milford, CT) for electrophoresis at a constant 20 W/gel at 25°C in an Ettan DALT 12 electrophoresis unit. Labeled proteins were visualized by scanning the gels on a Typhoon 9400 scanner. DeCyder software (v. 5.0) was used for spot detection and relative quantification of proteins based on the fluorescence images. Peak volumes and ratios were measured for each protein spot in the two fluorescent channels (Cy3 and Cy5). Proteins to be excised and identified were selected using the DeCyder Differential In-gel Analysis (DIA) Module. Individual DIGE gels were matched using the Biological Variation Analysis (BVA) Module. Protein spot excision and in-gel enzymatic digestion were performed automatically. Digests were analyzed by MALDI-TOF MS (Voyager DE-PRO, Applied Biosystems, Foster City, CA) and spectra were collected over the range m/z 500-5000. Spectra were processed using ProTS Data (Efeckta Technologies, Steamboat Springs, CO) to generate centroided peak lists that were submitted to Mascot (v 2.1, Matrix Science, Boston, MA) and Protein Prospector (v 4.21.1, UCSF, CA) for database searching.
Supplementary Material
Acknowledgements
We thank Drs. A. Yeudall, M. Bianchi, T. Yoshimori, N. Mizushima, J. Debnath and P. Dennis for providing critical reagents and Patrick O’Toole and Corinne Lowe for help with some experiments. Supported by NCI grant CA111421.
REFERENCES
- 1.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21(Nov 15)(22):2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 2.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115(Oct)(10):2679–2688. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsujimoto Y, Shimizu S. Another way to die: autophagic programmed cell death. Cell Death Differ. 2005;12(Nov)(Suppl 2):1528–1534. doi: 10.1038/sj.cdd.4401777. [DOI] [PubMed] [Google Scholar]
- 4.Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25(Feb)(3):1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120(Jan 28)(2):237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 6.Ravikumar B, Berger Z, Vacher C, O’Kane CJ, Rubinsztein DC. Rapamycin pre-treatment protects against apoptosis. Hum Mol Genet. 2006;15(Apr 1)(7):1209–1216. doi: 10.1093/hmg/ddl036. [DOI] [PubMed] [Google Scholar]
- 7.Colell A, Ricci JE, Tait S, Milasta S, Maurer U, Bouchier-Hayes L, et al. GAPDH and Autophagy Preserve Survival after Apoptotic Cytochrome c Release in the Absence of Caspase Activation. Cell. 2007;129(Jun 1)(5):983–997. doi: 10.1016/j.cell.2007.03.045. [DOI] [PubMed] [Google Scholar]
- 8.Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(Jul)(1):51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scarlatti F, Granata R, Meijer AJ, Codogno P. Does autophagy have a license to kill mammalian cells? Cell Death Differ. 2008;(Jul 4) doi: 10.1038/cdd.2008.101. [DOI] [PubMed] [Google Scholar]
- 10.Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6(Dec)(12):1221–1228. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 11.Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, et al. Regulation of an ATG7-beclin 1 Program of Autophagic Cell Death by Caspase-8. Science. 2004;304(May 6):1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 12.Wu YT, Tan HL, Huang Q, Kim YS, Pan N, Ong WY, et al. Autophagy Plays a Protective Role during zVAD-Induced Necrotic Cell Death. Autophagy. 2008;4(Feb 1)(4) doi: 10.4161/auto.5662. [DOI] [PubMed] [Google Scholar]
- 13.Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5(Sep)(9):726–734. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- 14.Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell. 2007;131(Dec 14)(6):1137–1148. doi: 10.1016/j.cell.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bell CW, Jiang W, Reich CF, Pisetsky DS. The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell Physiol. (3rd) 2006;291(Dec)(6):C1318–1325. doi: 10.1152/ajpcell.00616.2005. [DOI] [PubMed] [Google Scholar]
- 16.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418(Jul 11)(6894):191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 17.Pastan I, Hassan R, Fitzgerald DJ, Kreitman RJ. Immunotoxin therapy of cancer. Nat Rev Cancer. 2006;6(Jul)(7):559–565. doi: 10.1038/nrc1891. [DOI] [PubMed] [Google Scholar]
- 18.Liu TF, Cohen KA, Ramage JG, Willingham MC, Thorburn AM, Frankel AE. A diphtheria toxin-epidermal growth factor fusion protein is cytotoxic to human glioblastoma multiforme cells. Cancer Res. 2003;63(Apr 15)(8):1834–1837. [PubMed] [Google Scholar]
- 19.Liu TF, Hall PD, Cohen KA, Willingham MC, Cai J, Thorburn A, et al. Interstitial diphtheria toxin-epidermal growth factor fusion protein therapy produces regressions of subcutaneous human glioblastoma multiforme tumors in athymic nude mice. Clin Cancer Res. 2005;11(Jan 1)(1):329–334. [PubMed] [Google Scholar]
- 20.Thorburn A, Thorburn J, Frankel AE. Induction of apoptosis by tumor cell-targeted toxins. Apoptosis. 2004;9:19–25. doi: 10.1023/B:APPT.0000012118.95548.88. [DOI] [PubMed] [Google Scholar]
- 21.Luthi AU, Martin SJ. The CASBAH: a searchable database of caspase substrates. Cell Death Differ. 2007;14(Apr)(4):641–650. doi: 10.1038/sj.cdd.4402103. [DOI] [PubMed] [Google Scholar]
- 22.Wu H, Yang JM, Jin S, Zhang H, Hait WN. Elongation factor-2 kinase regulates autophagy in human glioblastoma cells. Cancer Res. 2006;66(Mar 15)(6):3015–3023. doi: 10.1158/0008-5472.CAN-05-1554. [DOI] [PubMed] [Google Scholar]
- 23.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000;19(Nov 1)(21):5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282(Feb 23)(8):5641–5652. doi: 10.1074/jbc.M609532200. [DOI] [PubMed] [Google Scholar]
- 25.Mizushima N, Yoshimori T. How to Interpret LC3 Immunoblotting. Autophagy. 2007;3(Jun 19)(6):542–545. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 26.Mercer CA, Kaliappan A, Dennis PB. Macroautophagy-dependent, intralysosomal cleavage of a betaine homocysteine methyltransferase fusion protein requires stable multimerization. Autophagy. 2008;4(Jan 1)(2):185–194. doi: 10.4161/auto.5275. [DOI] [PubMed] [Google Scholar]
- 27.Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3(Jan 1)(5):452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 28.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(Aug 17)(33):24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 29.Amaravadi RK, Thompson CB. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin Cancer Res. 2007;13(Dec 15)(24):7271–7279. doi: 10.1158/1078-0432.CCR-07-1595. [DOI] [PubMed] [Google Scholar]
- 30.Hippert MM, O’Toole PS, Thorburn A. Autophagy and cancer: good bad or both? Cancer Research. 2006;66(19):9349–9351. doi: 10.1158/0008-5472.CAN-06-1597. [DOI] [PubMed] [Google Scholar]
- 31.Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117(Jan 18):326–336. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. Embo J. 2003;22(Oct 15)(20):5551–5560. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13(Aug 19):1050–1059. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 34.Rovere-Querini P, Capobianco A, Scaffidi P, Valentinis B, Catalanotti F, Giazzon M, et al. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep. 2004;5(Aug)(8):825–830. doi: 10.1038/sj.embor.7400205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Apetoh L, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Lidereau R, et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev. 2007;220(Dec):47–59. doi: 10.1111/j.1600-065X.2007.00573.x. [DOI] [PubMed] [Google Scholar]
- 36.Zitvogel L, Kroemer G. The immune response against dying tumor cells: avoid disaster, achieve cure. Cell Death Differ. 2008;15(Jan)(1):1–2. doi: 10.1038/sj.cdd.4402267. [DOI] [PubMed] [Google Scholar]
- 37.Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. 2008;8(Jan 1)(1):59–73. doi: 10.1038/nri2216. [DOI] [PubMed] [Google Scholar]
- 38.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8(Sep)(9):741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 39.Thorburn A. Apoptosis and autophagy: regulatory connections between two supposedly different processes. Apoptosis. 2007;13(Nov 8)(1):1–9. doi: 10.1007/s10495-007-0154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomas LR, Henson A, Reed JC, Salsbury FR, Thorburn A. Direct binding of FADD to the TRAIL receptor DR5 is regulated by the death effector domain of FADD. J Biol Chem. 2004;279(Jun 1):32780–32785. doi: 10.1074/jbc.M401680200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.