

Abstract
This paper introduces a flexible assay for characterizing the activities of the histone deacetylase enzymes. The approach combines mass spectrometry with self-assembled monolayers that present acetylated peptides and enables a label-free and one-step assay of this biochemical activity. The assay was used to characterize the activity of HDAC8 toward peptides taken from the N-terminal tail of the H4 histone and reveals that a distal region of the peptide substrate interacts with the deacetylase at an exosite and contributes to the activity of the substrate. Specifically, a peptide corresponding to residues 8−19 of H4 and having lysine 12 acetylated is an active substrate, but removal of the KRHR (residues 16−19) sequence abolishes activity. Mutation of glycine 11 to arginine in the peptide lacking the KRHR sequence restores activity, demonstrating that both local and distal sequences act synergistically to regulate the activity of the HDAC. Assays with peptides bearing multiply acetylated residues, but in which each acetyl group is isotopically labeled, permit studies of the processive deacetylation of peptides. Peptide substrates having an extended sequence that includes K20 were used to demonstrate that methylation of this residue directly affects HDAC8 activity at K12. This work provides a mechanistic basis for the regulation of HDAC activities by distal sequences and may contribute to studies aimed at evaluating the role of the histone code in regulating gene expression.
The discovery that gene activity is highly correlated with the acetylation state of histones, and that this modification is reversible, has spurred intense efforts to understand the role of this modification in regulating gene expression (1). The histones have multiple lysine residues that are substrates for histone acetyltransferases (HATs) and histone deacetylases (HDACs). These opposing activities determine the position and extent of acetylation and their associated effects on gene transcription (2). Numerous studies have identified several patterns of post-translational modifications that are correlated with specific gene expression activities, but an understanding of the biochemical mechanisms that give rise to these preferred states is lacking (3). Some reports suggest that specific HAT and HDAC activities are recruited by large protein complexes that bind to particular post-translationally modified forms of the histones, such as the bromodomain that binds to acetylated lysines (4). Other studies note that adapter proteins that bind to particular DNA sequences can recruit enzymatic activity to specific histone residues to bring about the various cellular events (5). Additionally, these enzymes have intrinsic biochemical properties that are characterized by distinct substrate specificities. While the specificity of many HATs is beginning to be determined (6,7), the current knowledge of HDAC biochemistry is limited by a lack of activity assays that are convenient and compatible with peptide substrates that suitably mimic the endogenous histone substrates.
Many assays use peptide substrates containing isotopically labeled acetylated lysyl residues to monitor deacetylation (8–11). In one example, Lopez-Rodas and co-workers used the SPOT technique (12) to synthesize an array of 12 acetylated peptides on cellulose membranes (13). The arrays were treated with two pea HDAC complexes, HD1 and HD2, and then treated with [14C]acetic anhydride to attach a radiolabel to those peptides that were active substrates for the enzyme. Apart from the labor and safety concerns associated with radioisotopes, this method does not permit the use of peptide substrates having nonacetylated lysine residues, and it is not able to distinguish the specific sites of HDAC activity for those peptides that contain more than one acetylated lysine. In an alternative to radiolabels, Schwienhorst and co-workers developed a fluorogenic assay that uses a tripeptide coupled to a coumarin molecule. Treatment of the peptide with the protease trypsin results in release of the coumarin molecule, producing a fluorescent signal, only for peptides that have been deacetylated (14). A limitation with these formats is that the fluorescent residues can alter the activity of the substrate (15). In another fluorescence-based assay, Denu and co-workers used a split pool synthesis to generate a bead library of 105000 acetylated lysine pentapeptides. Deacetylated peptides were labeled with biotin, labeled with quantum dots, and finally sorted by fluorescence (16). This strategy was limited by the use of short peptide substrates and the fact that any hit had to be sequenced to obtain substrate specificity information.
Several recent reports suggest that the activities of histone-modifiying enzymes, including methylases, kinases, and the HDACs, depend on both the immediate sequence surrounding the modified residue and sequences that are several amino acids removed from this site: we refer to these regions as local and distal sequences of the substrate, respectively. The current assays used in studies of HDAC activity make it difficult to understand the role that distal sequence has in regulating activity and the ways in which multiple acetylated residues combine to influence activity. For example, Guarante and co-workers found that the histone deacetylase SIRT1 displayed no substrate specificity on 10-mer peptide substrates; however, they note that the substrate specificity may involve protein−protein interactions mediated by additional sites on SIRT1 (17). Zhu and co-workers showed that the histone methylase SET8 requires the full sequence RHRK20VLRDN for methylation at K20 on the N-terminal tail of histone 4 (18). In another methylation study, Pradhan and co-workers demonstrated that G9a methylates K9 on the H3 tail and requires the full sequence TARK9STG. This finding is important because there is a similar motif on the H3 tail, ARK27ST, yet no methylase activity by G9a has been observed at this site, consistent with their results showing that a longer sequence is essential for G9a activity (19). This dependence of substrate activity on the length of the peptide was also observed for demethylase LSD1, which acts on mono- and dimethylated H3K4. Mattevi and co-workers showed that modifications or mutations to both local and distal residues within the 21mer H3 peptide substrate can drastically affect activity at H3K4 (20).
To gain an understanding of the relationship between structure and activity, Marmorstein and co-workers have crystallized both the acetyltransferase, GCN5 (21), and the deacetylase, Hst2 (22), in complex with peptide substrates. These structures show that long-range contacts are mediated between the substrate and enzyme, though some of these contacts appear to depend only on the peptide backbone. The first crystal structure of HDAC8 with a peptide substrate was recently reported by Di Marco and co-workers (23). This complex of a catalytically inactive HDAC8 enzyme bound to the p53 tetramer peptide RHKAcKAc containing a coumarin molecule showed that Asp101 of HDAC8 forces the second acetylated lysine into a conformation that allows it to enter the active site. This crystal structure, however, does not clearly explain why the enzyme is specific for the second acetylated lysine, and it does not address whether the neighboring coumarin molecule is involved in any interactions with the enzyme, which could affect substrate binding.
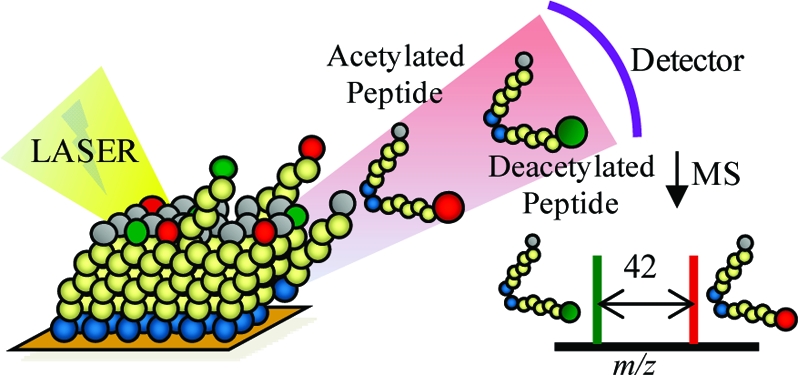
In this work, we report a straightforward assay of HDAC activity that uses mass spectrometry to analyze peptide substrates that are immobilized to a self-assembled monolayer of alkanethiolates on gold. The SAMDI assay provides a label-free route for analyzing deacetylation reactions, permits the simultaneous characterization of peptide substrates having multiple acetylated lysine residues, and is readily applicable to assays of other histone-modifying enzymes. We use this assay to show that the activity of HDAC8 for the N-terminal peptide of histone H4 depends on both local and distal sequences of the substrate and that the native sequence of this histone is not optimized for activity toward this deacetylase.
Materials and Methods
Reagents
All reagents were used as received without further purification. HDAC8 was purchased from Millipore and stored as recommended. Trifluoroacetic acid (TFA) was purchased from VWR. All amino acids and peptide synthesis reagents were purchased from Anaspec unless noted. FMOC-Lys(Me3)-OH was purchased from Bachem. The HDAC8 assay buffer for enzymatic reactions was 25 mM Tris-HCl (pH 8.0) containing 125 mM NaCl and 10% glycerol.
Synthesis of Acetylated Lysine Peptides
Peptides were synthesized manually using standard protocols on FMOC-Rink amide 4-methylbenzhydrylamine resin. Briefly, residues were coupled for 30 min with 4 equiv of PyBop (ChemPep Inc., Miami, FL) and N-methylmorpholine (Sigma), and coupling was monitored using ninhydrin (Sigma). FMOC was deprotected with 20% piperidine (Sigma) in dimethylformamide. The FMOC-Lys(Ac)-OH residue was used to incorporate acetylated lysine residues. To incorporate deuterated acetyl groups, we used the FMOC-Lys(Alloc)-OH reagent, and after the synthesis was complete, we deprotected this side chain using palladium and phenylsilane as previously reported (24) and then treated the peptide with deuterated acetic anhydride. The N-terminal FMOC was deprotected and acetylated, and the peptide was cleaved from the resin using TFA, triisopropylsilane (Sigma), and ethanedithiol (Sigma). All peptides were purified by reverse-phase HPLC on a C18 column (Waters).
Preparation of Gold Substrate
Glass coverslips were sonicated for 30 min first in deionized ultrafiltered (DIUF) water and then in ethanol and dried under a stream of nitrogen. Titanium (4 nm) and gold (29 nm) were evaporated onto the glass coverslips using an electron beam evaporator (Boc Edwards) at a rate of 0.05−0.10 nm/s and at a pressure of 1.0 × 10−6 Torr.
Preparation of SAMs Presenting Maleimide Groups
The maleimide-presenting SAMs were prepared as previously reported (25). Briefly, gold-coated coverslips were immersed in an ethanolic solution of maleimide-terminated disulfide and tri(ethylene glycol)-terminated disulfide in a ratio of 4:25 for 48 h at 4 °C (total concentration of disulfide of 1 mM). The substrates were washed with DIUF water and ethanol and dried under nitrogen.
HDAC8 Assay
Peptides were immobilized by applying 1−2 µL of 0.1 mM cysteine-terminated peptides (pH 6.5−7.0) to maleimide-presenting SAM biochips (∼5 mm2) and incubated at 30 °C for 1 h. Following immobilization, monolayers were rinsed with DIUF water and ethanol and dried under a stream of nitrogen. HDAC8 was diluted to 500 nM in HDAC8 buffer, and 1−2 µL was applied to each monolayer and incubated at 37 °C for 1 h. The monolayers were rinsed with DIUF water, a 10 µM hydrochloric acid solution, and ethanol and dried under a stream of nitrogen. Monolayers were then treated with matrix (2,4,6-trihydroxyacetophenone, 0.3 µL, 5 mg/mL in acetone), dried, and analyzed by SAMDI to produce a mass spectrum for each chip. In these studies, we compare the relative activities of a panel of peptides under conditions where the parent peptide gave an approximately 40% conversion to the deacetylated product. In this way, the assay has a dynamic range that can identify mutations in the substrates that lead to higher or lower activity.
Mass Spectrometry
Mass analysis was performed using a Voyager DE-PRO biospectrometry mass spectrometer (Applied Biosystems, Framingham, MA). A 337 nm nitrogen laser was used as a desorption/ionization source, and all spectra were acquired with a 20 kV accelerating voltage using positive reflector mode. The extraction delay was 50 ns; 600 laser shots were applied for each spectrum, and for each spectrum, the entire surface of the chip was sampled. All spectra were Gaussian-smoothed and baseline-corrected. Each spectrum was calibrated using the EG3 background disulfide as an internal standard.
Ionization Efficiency of Peptides and Quantitative Analysis
Equal concentrations of different sets of peptides, where each set differed by only an acetyl group, were immobilized onto the monolayers as previously described. SAMDI spectra showed that each set of differing peptides had similar relative intensities, suggesting that peak intensities for differently acetylated peptides can be used to semiquantitatively compare reaction yields. For quantitation, the relative amount of each component was calculated by measuring the relative intensities of each molecular ion peak (M+) [relative amount of x = Ix/(Ix + Iy), where x refers to the deacetylated peak and y refers to the parent ion]. The assay was performed in quadruplicate and averaged. Error bars indicate the standard deviation.
Results
Label-Free Assay of HDAC8 Activity
The approach we use to measure HDAC activities is based on a general strategy we have developed for solid-phase assays of biochemical activities (26). We use self-assembled monolayers of alkanethiolates on gold that present a peptide substrate for an enzyme. Because the monolayers are compatible with matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI TOF MS), spectra acquired before and after the monolayer has been treated with the enzyme give mass-to-charge peaks that correspond to the mass of the peptide−alkanethiolate conjugates and reveal any reactions that have modified the structure of the peptide. We have applied this technology to a broad range of enzymatic assays, including protease (27), kinase (28), glycosyltransferase (29), and methyl transferase activities (30). The monolayers are well-suited to biochip applications because they provide rigorous control over the density and orientation of the attached peptides (25,31), and monolayers that present the oligo(ethylene glycol) group are very effective at preventing nonspecific interactions between proteins and the surface (32).
For assays of HDAC activity, we synthesized cysteine-terminated peptide substrates containing acetylated lysine residues and immobilized the peptides to monolayers presenting maleimide groups at a density of 8% against a background of triethylene glycol (EG3) groups. In one example, we prepared a monolayer presenting the peptide Ac-GAKRHRKAcVC-NH2 (KAc denotes acetyllysine) and treated the monolayer with HDAC8 (500 nM in HDAC8 buffer at 37 °C for 1 h) (Figure 1). A SAMDI spectrum of the initial monolayer shows a peak at m/z 1992.9 corresponding to the proton adduct of the peptide−alkanethiolate conjugate. After treatment with HDAC8, the spectrum reveals a new peak at m/z 1950.8 that is consistent with the product of a deacetylation reaction. In this work, we use HDAC8 to demonstrate the assay because this enzyme is active in the absence of protein complexes (33) and the H4 N-terminal tail, which protrudes from the nucleosome core and can interact with DNA, neighboring nucleosomes, and other proteins, has been suggested to have implications for chromatin conformation and gene expression (34).
Figure 1.
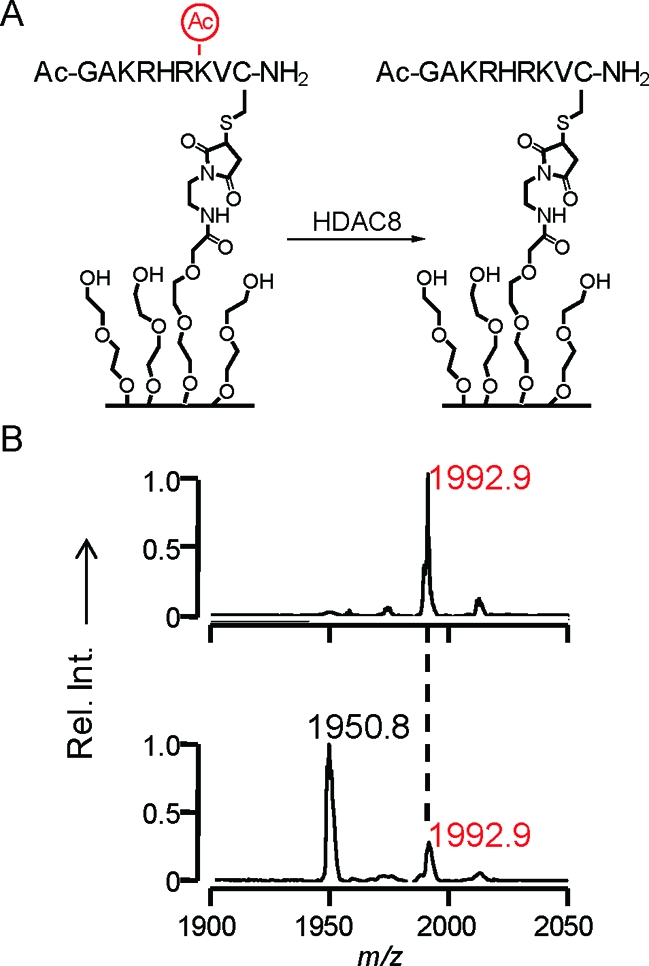
This work used the SAMDI mass spectrometry method to assay the activities of histone deacetylase enzymes. (A) The monoacetylated peptide, Ac-GAKRHRKAcVC-NH2, is immobilized to a monolayer presenting a maleimide group and then treated with HDAC8 to give the deacetylated peptide. (B) The SAMDI spectrum shows a shift of m/z 42, indicating the loss of an acetyl group.
HDAC8 Activity on the H4 N-Terminal Tail
The first 19 residues of the H4 N-terminal tail (S1GRGK5GGK8GLGK12GGAK16RHR...) include four lysines (K5, K8, K12, and K16) that have been observed in their acetylated forms in in vivo studies (6). Three of these residues (K5, K8, and K12) are found within a GKG motif, and the fourth (K16) is found within an AKR motif. To assess the effect that the immediate neighboring residues have on the activity at these lysines, we synthesized four short cysteine-terminated pentapeptides (GGKAcGGC, GRKAcGGC, GGKAcRGC, and GRKAcRGC) and treated the immobilized peptides with HDAC8. Figure 2A shows that the peptide having two neighboring glycine residues is a poor substrate, with a yield of approximately 5% for the deacetylated peptide. The mutation of glycine to an arginine in the P1′ position leads to an approximately 8-fold higher activity, whereas mutation of glycine to arginine at the P1 position yields a poor substrate, as does substitution of arginine for both glycine residues. Importantly, these data show that short peptides can serve as active substrates for HDAC8 and that the residues positioned adjacent to the acetylated lysine have a significant effect on activity. Furthermore, since the local sequence of the active peptide is not found among K5, K8, K12, and K16, these data could be intuitively interpreted to mean that the H4 tail is not a substrate for HDAC8, a conclusion that we show below to be incorrect.
Figure 2.

Activity profile of HDAC8. (A) Relative activity of HDAC8 on pentapeptides where arginine is substituted at the P1, P1′, and both P1 and P1′ positions to the acetylated lysine. This profile shows that an arginine in the P1′ position is ~8-fold more active toward HDAC8 deacetylation. (B) The proximity of the KRHR domain does not significantly change the extent of reaction at K12. (C) Mutations to the KRHR distal sequence show that R17 and R19 are essential for activity at K12. (D) Both the local and distance sequences can affect HDAC8 activity.
With recent studies reporting that long-range contacts between the peptide and the enzyme are important for activity, we prepared a series of longer peptides that derives from the native H4 N-terminal tail (Table 1). We found that the peptide Ac-K8GLGKAcGGAC-NH2 (ΔKRHR, 1) was inactive. We then evaluated two longer peptides, which had an extended sequence either in the N-terminal or in the C-terminal direction, Ac-S1GRGKAcGGKAcGLGKAcGGAC-NH2 (not in Table 1) and Ac-K8GLGKAcGGAKRHRC-NH2 (K8−R19, 2). The first of these peptides was not active, suggesting that distal sequence positioned at the N-terminal side of the local sequence is not important, whereas the second peptide (K8−R19, 2, hereafter termed the parent peptide) was converted to the deacetylated product in ~40% yield. Despite having a local sequence very similar to that of the short GGKAcGGC peptide, the parent peptide was ~8-fold more active (Figure 2B). This result suggests that the KRHR distal sequence plays a role in the activity of HDAC8. We then synthesized a series of peptides, shown in Table 1, to more clearly understand the sequence determinants that give rise to substrate activity with the longer peptide substrate.
Table 1. Peptide Sequences Derived from the H4 N-Terminal Tail and Their Relative Activities When Treated with HDAC8a.

X indicates the absence of an amino acid, and acetylated lysines are shown in bold.
Spatial Recognition of the KRHR Domain
On the basis of the experiments described above, we term the tripeptide sequence containing the acetylated lysine the local sequence and the KRHR tetrapeptide the distal sequence. To determine whether there is a strict requirement for a fixed distance between these two regions, we assayed four peptides having one (A Ext., 4) or two (AA Ext., 3) alanine residues inserted between the local and distal sequences, or having A15 (A Del., 5) or G14A15 (GA Del., 6) removed from the intervening sequence. We found that none of these variants had a significant impact on the activity of the peptide substrate (Figure 2B). In fact, only the peptide in which the KRHR sequence was extended by one alanine (A Ext., 4) varied by more than 20% from the parent, while the activities of the other derivatives where unchanged within error. This finding provides evidence that it is not the precise location of the KRHR domain that is crucial for HDAC8 substrate specificity, but rather its presence within the sequence. These data can be accommodated by a model in which the distal sequence interacts with an “exosite” on the enzyme surface to increase the effective concentration of the local sequence. This model would also suggest that the K5 and K8 residues might also be substrates for HDAC8, despite the greater distance from the KRHR domain, and we address this question further below.
Role of the KRHR Domain
The presence of the distal tetrapeptide sequence was able to rescue activity of a local sequence that on its own is a poor substrate. We evaluated several peptides having mutations in the distal sequence to identify the determinants that give rise to enhanced activity. Acetylation of the lysine residue (K16KAc, 7) or substitution with arginine (K16R, 8) resulted in a conversion to the deacetylated product similar to that of the parent peptide, suggesting that K16 is not mediating specific or electrostatic interactions with HDAC8 (Figure 2C). We then evaluated four peptides in which each of the four residues was substituted with alanine and found that substitution of the lysine (K16A, 9) or histidine (H18A, 10) gave a small enhancement in activity and substitution of either arginine (R17A, 11; R19A, 12) gave a slight decrease in activity. A double mutant having both arginine residues substituted with alanine (R17A/R19A, 13), however, was inactive toward the enzyme (Figure 2C). These data suggest that R17 and R19 contact HDAC8 in a manner that favorably allows deacetylation at K12 and may be consistent with a general electrostatic effect that requires both basic residues in the sequence. More importantly, these experiments establish that a sequence distal to the active residue can drastically affect the activity of the substrate and again support the proposal that the distal peptide interacts with an exosite on the surface of the enzyme.
Synergy of Local and Distal Sites
The N-terminal tail of histone 4 contains the KRHR distal sequence that we find is important for substrate activity, but it does not contain the RKG local sequence that we find exhibits activity even in the absence of the distal sequence. To determine whether the effects of these sequence determinants are additive, we assayed peptides that had various combinations of local and distal sequence. We first introduced a G11R mutation into a peptide lacking the distal sequence (ΔKRHR G11R, 14), which we previously found to be inactive, and as anticipated found that the mutation to the local sequence rescued activity and relaxed the need for a distal sequence (column 3, Figure 2D). When we assayed a peptide that contained the optimal local sequence and the native distal sequence (Full G11R, 15), we observed essentially complete conversion to the deacetylated product (column 4, Figure 2D). These data show that the local and distal sequences work synergistically in their effect on HDAC8 specificity, both making essential contacts with the enzyme that facilitate deacetylation at K12. Interestingly, the sequence motif with an arginine in the P1′ position is found only on the H4 histone at K20, which to date has only been observed to be a substrate for methylation (35). We also assayed peptides 14 and 15 under varying ionic conditions to assess the role of electrostatic interactions in the enzyme−substrate interface. We used the same buffers supplemented with NaCl, MgCl2, or MnCl2 (500 mM). While peptide 14 exhibited hardly any conversion to the deacetylated product under all three conditions, we observed a 33% decrease in activity for peptide 15 with added NaCl and essentially no activity after addition of MgCl2 or MnCl2 (data not shown). These results point to the importance of electrostatic interactions between the substrate and enzyme.
HDAC8 Discrete Pattern Behavior
In an experiment aimed at determining the effect of acetylation of K16 on deacetylation at K12, we synthesized a peptide that was acetylated at both sites (K16KAc, 7). This experiment could not distinguish whether the reaction resulted in deacetylation of only K12 or a mixture of two products derived from the loss of the acetyl group at K12 and K16. To resolve the activities at different residues, we synthesized a peptide that was modified at K12 with a deuterated acetyl group, therefore marking this acetyl group with a mass 3 Da larger than that of the acetyl group at K16 (16). SAMDI spectra of this peptide after treatment with HDAC8 reveal that the product peak differs from the original peak by m/z 45, corresponding to activity solely at K12 (Figure 3A). Furthermore, no dideacetylated peptide was observed.
Figure 3.
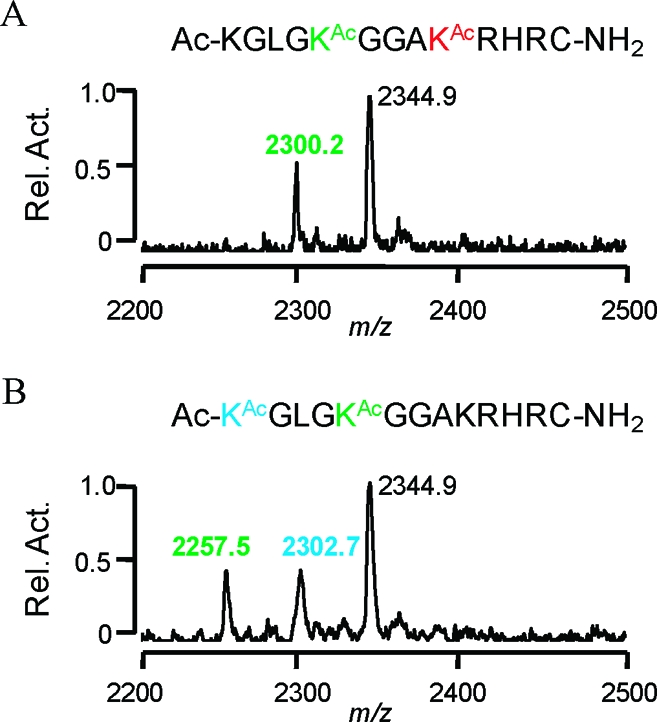
Mass resolvable deacetylation by HDAC8 on peptides derived from the H4 N-terminal tail. (A) SAMDI spectra of a diacetylated peptide at K16 and K12, where the acetyl group at K12 is deuterated (colored green), show a shift of m/z ~45 corresponding to deacetylation at the K12 position. (B) HDAC8 reaction on a diacetylated peptide at K8 (colored blue) and K12, where K12 contains a deuterated acetyl group (colored green), produces a SAMDI spectrum where the monodeacetylation product is represented by a shift of m/z 42, and di-deacetylation is shown by another shift of m/z 45, indicating that K8 is deacetylated prior to K12.
To determine if acetylation of K8 affected deacetylation at K12, we also synthesized a peptide in which both K8 and K12 were acetylated (17). Treatment with HDAC8 gave three peaks in the SAMDI spectra, one corresponding to the parent peptide, one for a monodeacetylated peptide, and one for the dideacetylated peptide. The first deacetylation product could either be the result of random deacetylation by HDAC8 at both sites or reflect a specificity requirement that renders one of the two residues more active for the first reaction. To distinguish among these possibilities, we again synthesized the diacetylated peptide having a deuterated acetyl group at K12 (18). The SAMDI spectra showed that the first product peak occurred at a difference of m/z 42 while the second peak corresponded to a loss of m/z 45 (Figure 3B). This result shows specific pattern behavior in which HDAC8 deacetylates K8 prior to activity at K12 and confirms the earlier suggestion that the proximity of the KRHR sequence to an active lysine is not as important as its mere presence.
Effect of Methylation of H4K20 on H4K12 Deacetylation
Finally, we demonstrate that the assay reported here can be applied to the investigation of the effects of lysine methylation on HDAC activity. We synthesized four additional peptides based on the H4 N-terminal tail (Ac-K8GLGKAcGGAKRHRKC-NH2) that each had K12 in its acetylated form and K20 present in either the unmodified, monomethylated, dimethylated, or trimethylated state (Figure 4A). We found that the addition of unmodified K20 enhanced activity nearly 20% relative to that of the parent peptide (peptide 2). The peptides having the monomethylated K20 and dimethylated K20 residues exhibited an increase in activity over the parent peptide of nearly 40 and 50%, respectively. However, the peptide that was trimethylated at K20 was less active than the unmethylated peptide for deacetylation at K12 (Figure 4B). These data support earlier findings of a negative correlation between trimethylation of K20 and acetylation of the H4 tail and are discussed further below.
Figure 4.

Methylation at K20 influences deacetylation at K12. (A) SAMDI-MS spectra of the Ac-K8GLGKAcGGAKRHRK20C-NH2 peptides, where K20 is unmodified or mono-, di-, or trimethylated. (B) The presence of unmodified K20 increases the activity by approximately 20% relative to that of the parent peptide, while the mono- and dimethylated derivatives have approximately 40 and 50% increased activity, respectively. The trimethylated derivative has a decreased activity relative to that of the unmethylated peptide.
Discussion
Local and Distal Sequence Determinants
The most significant result of this work is the finding that the activity of HDAC8 depends on both the local and distal sequences of the substrate. By local sequence we refer to the tripeptide sequence that contains the acetylated lysine residue at the central position, and by distal sequence we refer to sequences that are more than three residues from the acetylated lysine residue and, in this specific instance, to the KRHR tetrapeptide sequence. In this way, the local sequence interacts with the active site of the deacetylase enzyme and the distal sequence is proposed to interact with a different region of the enzyme. Our first evidence for the mutual roles of local and distal sequence in determining the activity of the substrate came from finding that a peptide corresponding to residues 8−15 of histone H4 (1, with the acetylated lysine residue at position 12) was an inactive substrate but could be converted to an active substrate by either mutating the local sequence (G11R, 14) or lengthening the peptide sequence with four additional residues (8−19, 2) (Figure 5). A peptide that combined this local sequence with the KRHR distal sequence (15) exhibited enhanced activity. Taken together, these results establish that the activity of a substrate is influenced by both the local and distal sequences of the substrate, although the latter is not strictly required for activity, and that mutation of either can synergistically contribute to activity. Hence, these experiments point to an exosite model for HDAC8 activity, wherein the substrate makes separate interactions with a surface region of the enzyme and may serve to increase the Michaelis constant. Our finding that insertions or deletions of residues between the local and distal sequences did not significantly influence activity suggests that these two sites interact independently with the enzyme and are indeed separate.
Figure 5.
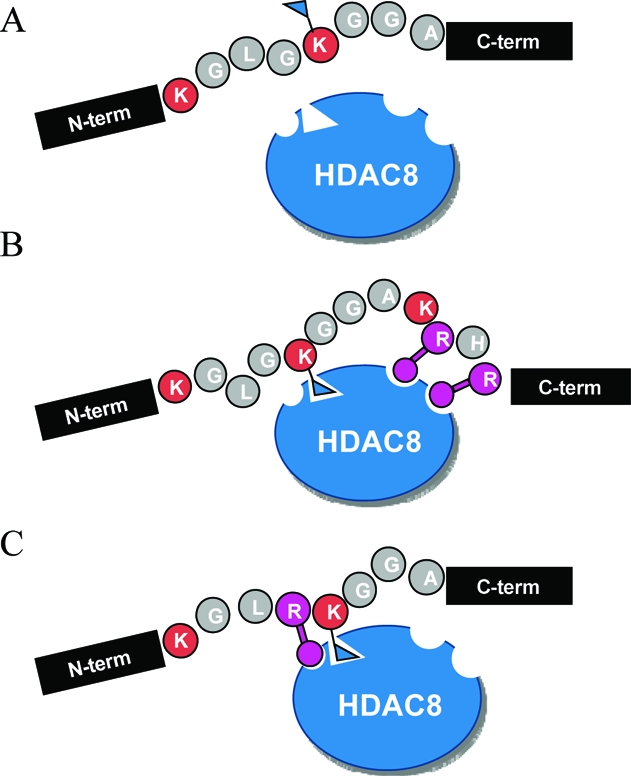
Proposed model of substrate recognition by HDAC8 at both the local and distal residues to the active lysine. (A) With a poor local sequence and in the absence of the favorable distal sequence, HDAC8 is not active. (B) With a poor local sequence but in the presence of the distal sequence, HDAC8 is active. (C) Despite the lack of the distal sequence, if a favorable local sequence is presented, HDAC8 activity is rescued.
Other Examples of Exosites
Several other classes of enzymes have been reported to utilize exosites in the recognition of their polypeptide substrates, including matrix metalloproteinases (MMPs) (36), kinases (37), and the thrombin protease (38). The exosite is usually a surface region of the enzyme that is distinct from the active site and that binds to a region of the peptide substrate to increase the affinity of the enzyme for its substrate. In this way, the exosite increases the activity of a substrate and can also contribute to the specificity with which enzymes discriminate among many possible substrates. Recent work has shown that the sirtuin enzymes, a class of deacetylases that use β-nicotinamide adenine dinucleotide (NAD+) to mediate deacetylation of both histone and non-histone proteins, employ an exosite in recognizing their substrates (39). Recent work has found that the small molecule resveratrol interacts with SIRT1 at an exosite and can allosterically increase the activity of the enzyme for its substrates (40).
We do not yet know whether the role of an exosite in HDAC8 is shared by the other class I (HDACs 1−3 and 8) and class II (HDACs 4−7 and 9−11) deacetylases. It is known that HDACs 1−11 have a conserved catalytic core, even though the enzymes differ in size, domain structure, and expression pattern (41), and it is therefore tempting to speculate that the specificities of the members of this family derive from interactions of the substrate or separate adaptor proteins with the exosites. Current work in our laboratory is characterizing the substrate specificities and the exosite binding specificities of the HDAC members to address this mechanistic possibility. We also suggest that the role of an exosite may be relevant to understanding the roles for and maintenance of a “histone code” which we address next.
Relevance to the Histone Code
The histone code hypothesis claims that various post-translational modifications are integrated into a code that operates as a global control mechanism for regulating gene expression (42). Many studies have revealed that the modification states of the histones are not evenly distributed among all possible states but rather reflect correlations of the modified forms of specific residues, yet there is an absence of biochemical studies that provide mechanisms that clearly account for the generation of only a small subset of the possible states. Our finding that the activity of a local sequence can be influenced by distal sequence provides a clear manner in which modifications at one part of the histone tail can affect subsequent modifications in a distal region. Further, our results show that the local sequences at K5, K8, and K12 are not optimized for deacetylation by HDAC8, thereby stressing the importance of the distal sequence in activity and suggesting that the modification state of the distal sequence can be a regulator of the activity at the local site.
Finally, the assays of peptides that are methylated at K20 provide an avenue for investigating the proposal by Allis and colleagues of “binary switches” on histone proteins, where modified residues can regulate substrate activity at adjacent or nearby residues (43). Recent studies have found a correlation between the methylation states of H4K20 and acetylation of lysine residues 5, 8, 12, and 16 on the H4 tail (44,45). In one example, Nishioka et al. used site specific antibodies to determine that methylation of K20 reduced the level of acetylation at K16 by the HAT p300 but had little effect on acetylation at K12, though this study did not characterize the activities of peptides having distinct methylation states at K20 (44). Lindner and co-workers found that histones that were trimethylated at K20 were present largely in the non- and monoacetylated states, whereas the di-, tri-, and tetraacetylated forms of the histone lacked the trimethylation mark at K20. This work and related work suggest that trimethylation at K20 and acetylation at K5, K8, K12, and K16 are mutually exclusive, but these studies do not address the mechanisms by which the correlations are established and maintained (46). In the work presented here, we show that the activity of HDAC toward K12 increases with mono- and dimethylation of K20 but that trimethylation results in decreased activity of the deacetylase. Hence, the intrinsic enzyme activity of HDAC8 is consistent with the correlations observed in the post-translational modification states of endogenous histone proteins. Our current efforts are in part directed at identifying the binary switches that operate at the level of enzyme−substrate specificity, as opposed to those that require adaptor proteins and higher-order complexes, to provide a biochemical basis for understanding the regulation of histone modification states.
Importance of Flexible Assays
The still-limited understanding of HDAC biochemistry largely stems from a lack of convenient and flexible assays available for this class of enzymes. The SAMDI assay offers many advantages in that it does not require labels, does not put any restrictions on the size of the peptide substrates, and can distinguish activities on peptides containing unmodified lysines, methylated lysines, and peptides with multiple acetylated lysine residues. The label-free format is important because it avoids the incorporation of large fluorophores at sites proximal to the active residue, which can alter the activity of the substrate. Further, the SAMDI method minimizes the number of steps that are required in the assay and avoids, for example, the sequences of reactions that are used to selectively modify deacetylated residues.
The most significant limitation of current HDAC assays is the use of short peptide substrates and, therefore, the difficulty in investigating the complementary roles of local and distal sequence on activity. The SAMDI assay was critical to our identification of the importance of distal sites in recognition of the substrate by HDAC8 and studies of the effect of methylation state at K20. This limitation on the length of the peptide is a lesser concern with proteases, where the substrate specificity is often determined mainly by scanning the prime and nonprime sequence near the scissile bond (47–50), but recent reports of the activities of enzymes that modify histone substrates, including HDACs, suggest that the local sequence may not be the lone factor in how the enzymes entertain their substrates. For example, Marmorstein and co-workers observed that residues as far as six positions from the acetylated lysine made contacts with the HAT GCN5 and the yeast HDAC Hst2. They interpret their results to suggest that specificity may be derived from regions of the substrate distal from the acetyllysine site (21,22).
A final benefit of the SAMDI assay is its ability to distinguish specific activity on multiply acetylated peptide substrates. Whereas other assays rely on site specific antibodies to determine specific sites for activity (51), with the attendant challenges of obtaining specific and high-affinity antibodies, the incorporation of isotopes into the acetyl position renders each acetyl group distinct in the mass spectrum and allows SAMDI to determine not only the site of activity but also the order in which multiple deacetylation reactions take place. These biochemical data complement the ongoing proteomic studies of histone modification patterns. Burlingame and co-workers used a bottom-up approach with a combination of MALDI-TOF MS and nanoelectrospray tandem MS to idenfity the specific acetylation sites on histone H4. The authors suggest a “zipper”-type behavior in which HATs acetylate lysine residues toward the N-terminus of the peptide and therefore HDACs must function in the reverse direction (52). Our results show that while the order of deacetylation proceeds from K8 to K12, HDAC8 is not active on K16. Furthermore, the observation of intermediate forms of the modified peptide demonstrates that HDAC8 does not remain associated with the peptide substrate during the course of multiple reactions but instead dissociates from the substrate after each deacetylation reaction.
Top-down proteomic studies from the laboratories of Freitas and Kelleher have also provided extensive information concerning the complexity and large number of distinct modification patterns that can occur on histones, but these methods do not address the substrate specificities inherent to individual enzymes and do not explicitly define the stepwise progression that connects two states (53,54). In an analogous problem, Sakmar and co-workers determined the order of tyrosine sulfonation by tyrosylprotein sulfotransferases and used a combination of reverse-phase HPLC, proteolytic digestion, and MALDI (55). The SAMDI method, in combination with isotopically labeled residues, provides a straightforward method for identifying the sequence of modifications that underlie histone modification. Further, these studies complement those that analyze histone modification patterns in the nucleus and will contribute to the mechanistic understanding of the roles that histone modification plays in gene expression.
This work reports two significant advances. First, the application of the SAMDI method to biochemical assays of HDAC activities—and, by extension, to other activities directed toward the modification of histones—provides a straightforward and flexible route to characterizing and comparing the activities of members of this important family of enzymes. The label-free method permits studies of the role of local and distal sequence determinants and of substrates having multiple modifications. Second, this work has made clear that the activity of HDAC8 for substrates that derive from histone H4 depends on interactions of the enzyme and substrate that are distinct from those at the active site and covalent modification of that distal sequence can direct enzyme activity. This finding will motivate studies of HDAC regulation in the other members of the family and will frame hypotheses for further study of histone modification in the nucleus.
Funding Statement
National Institutes of Health, United States
References
- Allfrey V. G.; Faulkner R.; Mirsky A. E. (1964) Acetylation + Methylation of Histones + Their Possible Role in Regulation of RNA Synthesis. Proc. Natl. Acad. Sci. U.S.A. 51, 786–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T.; Allis C. D. (2001) Translating the histone code. Science 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- Shahbazian M. D.; Grunstein M. (2007) Functions of site-specific histone acerylation and deacetylation. Annu. Rev. Biochem. 76, 75–100. [DOI] [PubMed] [Google Scholar]
- Dhalluin C.; Carlson J. E.; Zeng L.; He C.; Aggarwal A. K.; Zhou M. M. (1999) Structure and ligand of a histone acetyltransferase bromodomain. Nature 399, 491–496. [DOI] [PubMed] [Google Scholar]
- Pazin M. J.; Kadonaga J. T. (1997) What’s up and down with histone deacetylation and transcription?. Cell 89, 325–328. [DOI] [PubMed] [Google Scholar]
- Peterson C. L.; Laniel M. A. (2004) Histones and histone modifications. Curr. Biol. 14, R546−R551. [DOI] [PubMed] [Google Scholar]
- Sterner D. E.; Berger S. L. (2000) Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 64, 435–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolle D.; Brosch G.; Lechner T.; Lusser A.; Loidl P. (1998) Biochemical methods for analysis of histone deacetylases. Methods 15, 323–331. [DOI] [PubMed] [Google Scholar]
- DarkinRattray S. J.; Gurnett A. M.; Myers R. W.; Dulski P. M.; Crumley T. M.; Allocco J. J.; Cannova C.; Meinke P. T.; Colletti S. L.; Bednarek M. A.; Singh S. B.; Goetz M. A.; Dombrowski A. W.; Polishook J. D.; Schmatz D. M. (1996) Apicidin: A novel antiprotozoal agent that inhibits parasite histone deacetylase. Proc. Natl. Acad. Sci. U.S.A. 93, 13143–13147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heltweg B.; Trapp J.; Jung M. (2005) In vitro assays for the determination of histone deacetylase activity. Methods 36, 332–337. [DOI] [PubMed] [Google Scholar]
- Wegener D.; Hildmann C.; Schwienhorst A. (2003) Recent progress in the development of assays suited for histone deacetylase inhibitor screening. Mol. Genet. Metab. 80, 138–147. [DOI] [PubMed] [Google Scholar]
- Frank R. (1992) Spot-Synthesis: An Easy Technique for the Positionally Addressable, Parallel Chemical Synthesis on a Membrane Support. Tetrahedron 48, 9217–9232. [Google Scholar]
- Clemente S.; Franco L.; Lopez-Rodas G. (2001) Distinct site specificity of two pea histone deacetylase complexes. Biochemistry 40, 10671–10676. [DOI] [PubMed] [Google Scholar]
- Riester D.; Hildmann C.; Schwienhorst A.; Meyer-Almes F. J. (2007) Histone deacetylase inhibitor assay based on fluorescence resonance energy transfer. Anal. Biochem. 362, 136–141. [DOI] [PubMed] [Google Scholar]
- Su J.; Rajapaksha T. W.; Peter M. E.; Mrksich M. (2006) Assays of endogenous caspase activities: A comparison of mass spectrometry and fluorescence formats. Anal. Chem. 78, 4945–4951. [DOI] [PubMed] [Google Scholar]
- Garske A. L.; Denu J. M. (2006) SIRT1 top 40 hits: Use of one-bead, one-compound acetyl-peptide libraries and quantum dots to probe deacetylase specificity. Biochemistry 45, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blander G.; Olejnik J.; Olejnik E. K.; Mcdonagh T.; Haigis M.; Yaffe M. B.; Guarente L. (2005) SIRT1 shows no substrate specificity in vitro. J. Biol. Chem. 280, 9780–9785. [DOI] [PubMed] [Google Scholar]
- Yin Y. L.; Liu C. D.; Tsai S. N.; Zhou B.; Ngai S. M.; Zhu G. (2005) SET8 recognizes the sequence (RHRKVLRDN)-V-20 within the N terminus of histone H4 and mono-methylates lysine 20. J. Biol. Chem. 280, 30025–30031. [DOI] [PubMed] [Google Scholar]
- Chin H. G.; Pradhan M.; Esteve P. O.; Patnaik D.; Evans T. C.; Pradhan S. (2005) Sequence specificity and role of proximal amino acids of the histone H3 tail on catalysis of murine G9a lysine 9 histone H3 methyltransferase. Biochemistry 44, 12998–13006. [DOI] [PubMed] [Google Scholar]
- Forneris F.; Binda C.; Dall’Aglio A.; Fraaije M. W.; Battaglioli E.; Mattevi A. (2006) A highly specific mechanism of histone H3-K4 recognition by histone demethylase LSD1. J. Biol. Chem. 281, 35289–35295. [DOI] [PubMed] [Google Scholar]
- Clements A.; Poux A. N.; Lo W. S.; Pillus L.; Berger S. L.; Marmorstein R. (2003) Structural basis for histone and phosphohistone binding by the GCN5 histone acetyltransferase. Mol. Cell 12, 461–473. [DOI] [PubMed] [Google Scholar]
- Zhao K. H.; Chai X. M.; Marmorstein R. (2003) Structure of the yeast Hst2 protein deacetylase in ternary complex with 2 ′-O-acetyl ADP ribose and histone peptide. Structure 11, 1403–1411. [DOI] [PubMed] [Google Scholar]
- Vannini A.; Volpari C.; Gallinari P.; Jones P.; Mattu M.; Carfi A.; De Francesco R.; Steinkuhler C.; Di Marco S. (2007) Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8-substrate complex. EMBO Rep. 8, 879–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thieriet N.; Alsina J.; Giralt E.; Guibe F.; Albericio F. (1997) Use of Alloc-amino acids in solid-phase peptide synthesis. Tandem deprotection-coupling reactions using neutral conditions. Tetrahedron Lett. 38, 7275–7278. [Google Scholar]
- Houseman B. T.; Gawalt E. S.; Mrksich M. (2003) Maleimide-functionalized self-assembled monolayers for the preparation of peptide and carbohydrate biochips. Langmuir 19, 1522–1531. [Google Scholar]
- Gurard-Levin Z. A.; Mrksich, M. (2008) Combining Self-Assembled Monolayers and Mass Spectrometry for Applications in Biochips. Annu. Rev. Anal. Chem. 1, XXX–XXX(in press). [DOI] [PubMed] [Google Scholar]
- Min D. H.; Tang W. J.; Mrksich M. (2004) Chemical screening by mass spectrometry to identify inhibitors of anthrax lethal factor. Nat. Biotechnol. 22, 717–723. [DOI] [PubMed] [Google Scholar]
- Min D. H.; Su J.; Mrksich M. (2004) Profiling kinase activities by using a peptide chip and mass spectrometry. Angew. Chem., Int. Ed. 43, 5973–5977. [DOI] [PubMed] [Google Scholar]
- Houseman B. T.; Mrksich M. (1999) The role of ligand density in the enzymatic glycosylation of carbohydrates presented on self-assembled monolayers of alkanethiolates on gold. Angew. Chem., Int. Ed. 38, 782–785. [DOI] [PubMed] [Google Scholar]
- Min D. H.; Yeo W. S.; Mrksich M. (2004) A method for connecting solution-phase enzyme activity assays with immobilized format analysis by mass spectrometry. Anal. Chem. 76, 3923–3929. [DOI] [PubMed] [Google Scholar]
- Houseman B. T.; Mrksich M. (2002) Towards quantitative assays with peptide chips: A surface engineering approach. Trends Biotechnol. 20, 279–281. [DOI] [PubMed] [Google Scholar]
- Mrksich M.; Whitesides G. M. (1997) Using self-assembled monolayers that present oligo(ethylene glycol) groups to control the interactions of proteins with surfaces. ACS Symp. Ser. 680, 361–373. [Google Scholar]
- Somoza J. R.; Skene R. J.; Katz B. A.; Mol C.; Ho J. D.; Jennings A. J.; Luong C.; Arvai A.; Buggy J. J.; Chi E.; Tang J.; Sang B. C.; Verner E.; Wynands R.; Leahy E. M.; Dougan D. R.; Snell G.; Navre M.; Knuth M. W.; Swanson R. V.; McRee D. E.; Tari L. W. (2004) Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 12, 1325–1334. [DOI] [PubMed] [Google Scholar]
- Dion M. F.; Altschuler S. J.; Wu L. F.; Rando O. J. (2005) Genomic characterization reveals a simple histone H4 acetylation code. Proc. Natl. Acad. Sci. U.S.A. 102, 5501–5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Reinberg D. (2001) Transcription regulation by histone methylation: Interplay between different covalent modifications of the core histone tails. Genes Dev. 15, 2343–2360. [DOI] [PubMed] [Google Scholar]
- Overall C. M. (2001) Matrix metalloproteinase substrate binding domains, modules and exosites: Overview and experimental strategies. Methods Mol. Biol. 151, 79–120. [PubMed] [Google Scholar]
- Biondi R. M.; Nebreda A. R. (2003) Signalling specificity of Ser/Thr protein kinases through docking-site-mediated interactions. Biochem. J. 372, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnaswamy S. (2005) Exosite-driven substrate specificity and function in coagulation. J. Thromb. Haemostasis 3, 54–67. [DOI] [PubMed] [Google Scholar]
- Sauve A. A.; Wolberger C.; Schramm V. L.; Boeke J. D. (2006) The biochemistry of sirtuins. Annu. Rev. Biochem. 75, 435–465. [DOI] [PubMed] [Google Scholar]
- Howitz K. T.; Bitterman K. J.; Cohen H. Y.; Lamming D. W.; Lavu S.; Wood J. G.; Zipkin R. E.; Chung P.; Kisielewski A.; Zhang L. L.; Scherer B.; Sinclair D. A. (2003) Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425, 191–196. [DOI] [PubMed] [Google Scholar]
- Holbert M. A.; Marmorstein R. (2005) Structure and activity of enzymes that remove histone modifications. Curr. Opin. Struct. Biol. 15, 673–680. [DOI] [PubMed] [Google Scholar]
- Strahl B. D.; Allis C. D. (2000) The language of covalent histone modifications. Nature 403, 41–45. [DOI] [PubMed] [Google Scholar]
- Fischle W.; Wang Y. M.; Allis C. D. (2003) Binary switches and modification cassettes in histone biology and beyond. Nature 425, 475–479. [DOI] [PubMed] [Google Scholar]
- Nishioka K.; Rice J. C.; Sarma K.; Erdjument-Bromage H.; Werner J.; Wang Y. M.; Chuikov S.; Valenzuela P.; Tempst P.; Steward R.; Lis J. T.; Allis C. D.; Reinberg D. (2002) PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol. Cell 9, 1201–1213. [DOI] [PubMed] [Google Scholar]
- Latham J. A.; Dent S. Y. R. (2007) Cross-regulation of histone modifications. Nat. Struct. Mol. Biol. 14, 1017–1024. [DOI] [PubMed] [Google Scholar]
- Sarg B.; Helliger W.; Talasz H.; Koutzamani E.; Lindner H. H. (2004) Histone H4 hyperacetylation precludes histone H4 lysine 20 trimethylation. J. Biol. Chem. 279, 53458–53464. [DOI] [PubMed] [Google Scholar]
- Deng S. J.; Bickett D. M.; Mitchell J. L.; Lambert M. H.; Blackburn R. K.; Carter H. L.; Neugebauer J.; Pahel G.; Weiner M. P.; Moss M. L. (2000) Substrate specificity of human collagenase 3 assessed using a phage-displayed peptide library. J. Biol. Chem. 275, 31422–31427. [DOI] [PubMed] [Google Scholar]
- Harris J. L.; Backes B. J.; Leonetti F.; Mahrus S.; Ellman J. A.; Craik C. S. (2000) Rapid and general profiling of protease specificity by using combinatorial fluorogenic substrate libraries. Proc. Natl. Acad. Sci. U.S.A. 97, 7754–7759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazif T.; Bogyo M. (2001) Global analysis of proteasomal substrate specificity using positional-scanning libraries of covalent inhibitors. Proc. Natl. Acad. Sci. U.S.A. 98, 2967–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk B. E.; Huang L. L.; Piro E. T.; Cantley L. C. (2001) Determination of protease cleavage site motifs using mixture-based oriented peptide libraries. Nat. Biotechnol. 19, 661–667. [DOI] [PubMed] [Google Scholar]
- Liu C. L.; Kaplan T.; Kim M.; Buratowski S.; Schreiber S. L.; Friedman N.; Rando O. J. (2005) Single-nucleosome mapping of histone modifications in S-cerevisiae. PLoS Biol. 3, 1753–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K. L.; Williams K. E.; Huang L.; Yau P.; Siino J. S.; Bradbury E. M.; Jones P. R.; Minch M. J.; Burlingame A. L. (2002) Histone acetylation and deacetylation: Identification of acetylation and methylation sites of HeLa histone H4 by mass spectrometry. Mol. Cell. Proteomics 1, 500–508. [DOI] [PubMed] [Google Scholar]
- Su X. D.; Zhang L. W.; Lucas D. M.; Davis M. E.; Knapp A. R.; Green-Church K. B.; Marcucci G.; Parthun M. R.; Byrd J. C.; Freitas M. A. (2007) Histone H4 acetylation dynamics determined by stable isotope labeling with amino acids in cell culture and mass spectrometry. Anal. Biochem. 363, 22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C. E.; Kelleher N. L.; Mizzen C. A. (2006) Mass spectrometric characterization of human histone H3: A bird’s eye view. J. Proteome Res. 5, 240–247. [DOI] [PubMed] [Google Scholar]
- Seibert C.; Cadene M.; Sanfiz A.; Chait B. T.; Sakmar T. P. (2002) Tyrosine sulfation of CCR5 N-terminal peptide by tyrosylprotein sulfotransferases 1 and 2 follows a discrete pattern and temporal sequence. Proc. Natl. Acad. Sci. U.S.A. 99, 11031–11036. [DOI] [PMC free article] [PubMed] [Google Scholar]