

Abstract
One of the earliest documented molecular events after sciatic nerve injury in adult rats is the rapid, long-term upregulation of the immediate early gene transcription factor c-Jun mRNA and protein in lumbar dorsal root ganglion (DRG) neurons, suggesting that c-Jun may regulate genes that are important both in the early post-injury period and during later peripheral axonal regeneration. However, neither the mechanism through which c-Jun protein is increased nor the level of its post-injury transcriptional activity in axotomized DRGs has been characterized. To determine whether transcriptional activation of c-Jun occurs in response to nerve injury in vivo and is associated with axonal regeneration, we have assayed axotomized adult rat DRGs for evidence of jun kinase activation, c-Jun phosphorylation, and activator protein-1 (AP-1) binding. We report that sciatic nerve transection resulted in chronic activation of c-Jun amino-terminal kinase-1 (JNK) in L4/L5 DRGs concomitant with c-Jun amino-terminal phosphorylation in neurons, and lasting AP-1 binding activity, with both c-Jun and JunD participating in DNA binding complexes. The timing of JNK activation was dependent on the distance of the axotomy site from the DRGs, suggesting the requirement for a retrograde transport-mediated signal. AP-1 binding and c-Jun protein returned to basal levels in DRGs as peripheral regeneration was completed but remained elevated in the case of chronic sprouting, indicating that c-Jun may regulate target genes that are involved in axonal outgrowth.
Keywords: AP-1, axotomy, c-Jun, dorsal root ganglion, JNK, phosphorylation, regeneration, sciatic nerve
Peripheral nerve transection in adult rats induces a highly complex morphological response in affected lumbar dorsal root ganglion (DRG) neuronal cell bodies. In the initial stages of the post-injury period, neurons undergo disintegration of nuclear chromatin, a shift in nuclear position, and cell body swelling, changes cumulatively referred to as the axon reaction (Lieberman, 1971). Surviving neurons establish a cellular and molecular program directed toward axonal elongation and reinnervation of the denervated target tissue (Lieberman, 1971). In regenerating sensory neurons, anterograde transport of structural proteins increases (Jacob and McQuarrie, 1991), synthesis of some neurotransmitters, such as substance P (Nielsch and Keen, 1989), declines, and there is increased transcription and synthesis of tubulin isoforms (Miller et al., 1989;Moskowitz and Oblinger, 1995) and GAP43 (VanderZee et al., 1989; Woolf et al., 1990; Somervaille et al., 1991; Wiese et al., 1992), proteins that are used during axonal outgrowth.
The activation of heretofore quiescent genes suggests that increased transcription factor activity is required in injured neurons. Peripheral axotomy induces a rapid, prolonged increase in levels of the immediate early gene transcription factor c-Jun mRNA (Jenkins and Hunt, 1991; De Leon et al., 1995) and protein (Jenkins and Hunt, 1991;Herdegen et al., 1992, Kenney and Kocsis, 1997) in DRG neurons via a retrograde transport-mediated signal (Leah et al., 1991; Kenney and Kocsis, 1997). However, specific axotomy-induced genes regulated by c-Jun in vivo in DRG neurons have not yet been identified, and functional activation of c-Jun in DRGs after axotomy and during regeneration has not been demonstrated explicitly.
In cultured cells, the transcriptional activation of c-Jun is stimulated by phosphorylation at two amino-terminal serine residues (Binetruy et al., 1991; Smeal et al., 1991, 1992) by c-Jun amino-terminal kinases (JNKs) (Hibi et al., 1993; Derijard et al., 1994), also known as stress-activated protein kinases (SAPKs) (Kyriakis et al., 1994). In vitro, these kinases are rapidly activated in response to environmentally stressful stimuli or specific membrane receptor interactions (for review, see Davis, 1994; Kyriakis and Avruch, 1996). However, the behavior of JNK/SAPKs remains uncharacterized in the intact nervous system, where stimulation or environmental perturbation may occur at a remote site from the soma, i.e., along the axon, rather than directly at the cell body as in the case of cultured cells.
To test the hypothesis that peripheral axotomy in vivo leads to c-Jun activation in DRGs in a manner analogous to that of cultured cells subjected to environmentally stressful stimuli, we assayed adult rat DRGs for JNK activity, c-Jun amino-terminal phosphorylation, and DNA binding by c-Jun and jun family members after peripheral axotomy. We compared the timing of JNK activation after injury at two distances from the DRG cell bodies to determine whether nerve injury sends an instantaneous signal or one requiring retrograde transport to activate the JNK pathway. To assess the correlation between c-Jun activity and axonal regeneration, we compared c-Jun protein levels and c-Jun-mediated activator protein-1 (AP-1) binding behavior in DRGs after sciatic nerve transection under regeneration-permissive (crush) or -nonpermissive (ligation with cuff) conditions.
MATERIALS AND METHODS
Surgical procedures. All surgeries were performed using adult female Wistar rats (160–180 gm, 8–10 weeks of age), anesthetized with ketamine (40 mg/kg, i.p.) and xylazine (2.5 mg/kg, i.p.), with the right side being the experimental side. For proximal peripheral nerve transections, the sciatic nerve was exposed at the convergence of the L4 and L5 spinal nerves, after a dorsal midline incision, retraction of the paraspinal muscles, and removal of a small sacral bone fragment. The spinal nerves were ligated with silk 4.0 suture (Ethicon, Somerville, NJ) and transected with iridectomy scissors. A 2.0 mm segment of the distal nerve stump was removed. For distal nerve transections, the sciatic nerve was exposed at the level of the pyriform tendon. The nerve was ligated with silk 4.0 suture and sutured into a silicone tubing cuff (Baxter Scientific, Boston, MA) sealed with silicone rubber (Dow-Corning). Lack of regeneration after proximal or distal nerve transections was verified at the time the rats were killed by visual inspection of the transection site. Nerve crushes were performed by exposing the nerve at the pyriform tendon level and squeezing with Dumont number 5 forceps (Roboz, Rockville, MD) for 20 sec. Regeneration after nerve crush was assessed in two ways at the time rats were killed: (1) by obtaining hind footprints to determine the extent of functional recovery of the denervated limb (Dellon and Dellon, 1991; Walker et al., 1994) and (2) by dissecting free the injured nerve and squeezing gently with forceps distal to proximal toward the crush site until a reflex response was shown, as described by Rich et al. (1984). The distance between the crush site and the point of response was the distance the regenerating axons had progressed. For all surgical procedures, the contralateral (left) side served as a sham-operated control, wherein the same site was exposed but not transected or crushed. After surgery, the overlying skin and muscles were sutured closed with silk 4.0 suture, and the wound was treated with Betadine (Purdue Frederick, Norwalk, CT) to prevent infection. Recovery was uneventful in all cases, and animals showed no signs of autotomy. At various times after surgery, the animals were anesthetized briefly with C02 and exsanguinated by carotid section or perfused intracardially with ice-cold 4.0% paraformaldehyde (PFA) (Sigma, St. Louis, MO)/0.14 m Sorensen’s phosphate buffer. Transected and contralateral sham-operated L4 and L5 dorsal root ganglia were rapidly dissected for use in the assays described below. For each analysis, three animals were used for every time point assayed for each surgery.
Immune-complex kinase assays. Unless indicated otherwise, all chemicals were purchased from Sigma. In a protocol based onKyriakis et al. (1994), L4 and L5 DRG pairs were mechanically homogenized on ice in 1.5 ml lysis buffer containing 20 mmHEPES, pH 7.4, 2 mm EGTA, 100 mm sodium orthovanadate, 50 mm β-glycerophosphate, 10% glycerol, 1% Triton X-100, 5 mg/ml leupeptin, 400 mm PMSF, 10 mg/ml aprotinin, 400 mm diisopropylfluorophosphate, and 1 mm DTT. Samples were sonicated for 20 sec and kept on ice for 15 min, and cellular debris was collected by centrifugation at 14,000 rpm, 4°C, for 5 min. Protein concentration of the supernatants was determined by a modified Bradford protein assay (Bio-Rad, Hercules, CA). Fifty micrograms of each sample were diluted to 300 ml with lysis buffer and added to 40 ml of swollen protein A-Sepharose CL-4B beads (Sigma) diluted 1:1 with lysis buffer. Anti-JNK (C-17, Santa Cruz Biotechnology, Santa Cruz, CA), 1:1000, was used in immunoprecipitation for 6 hr at 4°C with rotation. Beads were collected by microcentrifugation and washed once with lysis buffer, twice with high ionic strength buffer (500 mm LiCl, 100 mmTris, pH 7.4, 0.1% Triton X-100, 1 mm DTT), and three times in kinase buffer (20 mm MOPS, pH 7.2, 10 mm MgCl2, 2 mm EGTA, 0.1% Triton X-100, 1 mm DTT). The final wash was aspirated thoroughly, and to the 20 ml of beads was added 20 ml of kinase buffer and 20 ml of 0.3 mg/ml GST-c-Jun (1–135) substrate, diluted in kinase buffer. Fifteen microliters of 50 mmMgCl2/125 mm ATP containing 10 mCi g-32P-ATP (Amersham, Arlington Heights, IL) were added to start the reaction. Samples were incubated at 30°C for 20 min, with frequent mixing, before the reaction was terminated by the addition of 20 ml of 6× SDS-PAGE loading buffer. Samples were vortexed and boiled for 10 min, before separation on a 12.5% SDS-polyacrylamide gel. Dried gels were exposed to Kodak XOMAT-LS film. Phosphorylated GST-c-Jun was visible as a 46 kDa doublet. The intensity of the GST-c-Jun signal was analyzed by densitometry using a Bioimage Whole Band Analysis system (Millipore, Bedford, MA), and phosphorylation of c-Jun substrate in the dried gels was also quantified by electronic autoradiography with an Instantimager (Packard).
Immunoblot analysis. To prepare samples for JNK Western blotting, L4/L5 DRGs were dissected onto dry ice and then boiled for 10 min in 150 ml 3.0% SDS. An equal volume of 0.3 m sucrose was added, and samples were homogenized in the presence of 500 mm PMSF. Insoluble matter was pelleted by centrifugation at 14,000 rpm, 4°C, for 10 min. Protein concentration was determined by a modified Lowry assay (Peterson, 1977) and performed in duplicate, and 20 mg of protein was separated by SDS-PAGE through a 12.5% gel. Proteins were transferred to an Immobilon membrane (Millipore) for immunoblotting with anti-JNK (C-17, Santa Cruz Biotechnology) (1:2000). For c-Jun (antibody PC06S, 2 mg/ml; Calbiochem, La Jolla, CA) Western blots, 50 mg of each sample prepared for electrophoretic mobility shift assays (see below) was run through a 12.5% SDS-polyacrylamide gel and transferred to Immobilon. Primary antibody incubations were performed in 5% dry milk, 0.1% Triton X-100, and 1× Tris-buffered saline (TBS) overnight at 4°C. Blots were washed three times in the milk solution, three times in 1× TBS, 0.1% Triton X-100, and then incubated for 1 hr in HRP-conjugated goat anti-rabbit secondary antibody (Pierce, Rockford, IL) (1:8000). Washes were repeated as above. Immunoreactivity was visualized after development with Amersham ECL reagents and exposure to Kodak XOMAT-AR film. ECL films were analyzed by densitometry using a Bioimage Whole Band Analysis System (Millipore).
Immunohistochemistry. After intracardial perfusion with ice-cold 4% PFA, 24 hr axotomized L4 and L5 DRGs were immediately removed into 10% sucrose/4% PFA, where they were stored for 3 hr at 4°C before being transferred to 30% sucrose/4% PFA for overnight storage at 4°C. DRGs were frozen in OTC in liquid nitrogen, and 35 mm sections were cut on a cryostat. Sections were washed twice in 1× TBS and then incubated for 1 hr at room temperature in blocking solution containing 5% normal goat serum/0.3% Triton X-100/0.02% sodium azide in TBS. After overnight incubation at 4°C with phospho-specific c-Jun (Ser63) II antibody (New England Biolabs, Beverly, MA) (1:200) in 1% normal goat serum/0.3% Triton X-100/0.02% sodium azide in TBS, sections were washed four times with TBS, then incubated for 3 hr with goat anti-rabbit secondary antibody (Sigma; 1:100), washed, and treated with rabbit peroxidase anti-peroxidase (Sigma; 1:400) for 1 hr. Immunoreactivity was visualized using 0.4% diaminobenzidine/0.003% hydrogen peroxide in TBS, with all sections developed for 8 min before the reaction was quenched with 0.02% sodium azide in TBS. Sections were applied to poly-l-lysine coated slides, air-dried overnight, and then dehydrated and cleared through a series of graded ethanols before they were coverslipped with Cytoseal 60 (Stephens Scientific).
Electrophoretic mobility shift assays (EMSAs). Unless stated otherwise, all chemical reagents are from Sigma. L4/L5 DRGs were homogenized on ice in 500 ml of EMSA lysis buffer, pH 7.9, containing 20 mm HEPES, 0.4 m NaCl, 20% glycerol, 5 mm MgCl2, 500 mm EDTA, 500 mm EGTA, 1% Nonidet P-40, 0.1 m benzamidine, 10 mg/ml leupeptin, 1 mg/ml pepstatin A, 1 mm okadaic acid (Life Technologies, Gaithersburg, MD), 1 mm PMSF, and 5 mm DTT. After sonication for 20 sec, samples were placed on ice for 20 min and then centrifuged for 25 min at 14,000 rpm, 4°C. Protein concentrations were determined by a modified Bradford assay (Bio-Rad), and 6 mg of protein was used in each binding reaction. Human metallothionein IIA AP-1 consensus and mutant oligonucleotides derived from Lee et al. (1987) were purchased from Santa Cruz Biotechnology. Probes, labeled to high specific activity (50,000–100,000 cpm/ng) using T4 polynucleotide kinase (New England Biolabs) and γ-32P-ATP (Amersham), were purified twice over G-25 spin columns (Boehringer Mannheim, Indianapolis, IN), and 1 ng was used per reaction. Binding reactions were performed for 20 min at room temperature in binding buffer containing 10 mmTris, pH 7.5, 25 mm NaCl, 1 mm EDTA, 5% glycerol, 1 mg polydI-dC (Pharmacia, Piscataway, NJ), and 5 mm DTT. For competition studies, 1, 10, or 20 ng of unlabeled consensus or mutant oligonucleotides were incubated with protein samples in binding buffer for 15 min at room temperature, before addition of 1 ng of labeled probe as above. For supershift assays, 1–2 mg of antibody (c-Jun Ab-1, Calbiochem; junB (N-17) X and JunD (329) X, Santa Cruz Biotechnology) was incubated with the protein sample in binding buffer for 30 min at room temperature before addition of the labeled probe. Reactions were separated through a 6% nondenaturing polyacrylamide gel and then dried and exposed for 24 hr to Kodak XOMAT-LS film at −80 C. Autoradiographs were quantified by densitometry and electronic autoradiography as described above.
RESULTS
Axotomy induces prolonged activation of JNK in DRGs
To determine whether peripheral nerve axotomy is a stimulus for the in vivo activation of jun kinases, we assayed the level of JNK activity in axotomized DRGs in comparison with the contralateral, untransected DRGs, using an immune complex kinase assay. We also compared the timing of JNK activation after nerve transections either close to or distant from the DRG cell bodies to ascertain whether JNK activation results from an instantaneously communicated signal or is dependent on one that is axonally transported from the injury site. At times from 15 min to 30 d after sciatic nerve transection, JNK was immunoprecipitated from DRGs and tested for its activity toward a c-Jun substrate. For each time point, three animals were used, and JNK activity was determined by densitometry or direct quantitation of substrate phosphorylation (see Materials and Methods) (Fig. 1). Within 30 min of proximal transection (∼1 cm from the DRG cell bodies), there was a 60% increase in the level of JNK activity in axotomized DRGs, which continued to increase to threefold over control levels (Fig.1a). By 30 d after axotomy, JNK activity was still at least double that of the contralateral, uninjured side (Fig.1a). Sciatic nerve transection at the mid-thigh, which is ∼4 cm from the DRGs, did not induce significant increases in JNK activity until 3 hr after axotomy (Fig. 1b). After mid-thigh transection, JNK activity rose to threefold over control levels by 7 d, where it remained until at least 30 d post-injury, the latest time point evaluated (Fig. 1b). We also observed that the level of JNK activity in axotomized DRGs appeared to vary more after proximal than after distal nerve transection (Fig.1a,b). Although JNK activity at each time point from 30 min to 30 d after proximal axotomy was significantly higher than that of the contralateral, sham-operated DRGs (Fig. 1a), when comparisons were made between injured DRGs, values for JNK activity from 6 hr to 7 d after proximal axotomy did not differ significantly from each other.
Fig. 1.

JNK is activated in vivo in dorsal root ganglia (DRG) after peripheral nerve transection, with timing dependent on distance of the lesion from the DRGs. JNK activity in L4/L5 DRGs was assessed using an immune complex kinase assay at times from 15 min to 30 d after proximal (a) or distal (b) nerve transection. Levels of JNK activation were determined as a ratio of the activity in the injured versus contralateral, uninjured DRGs. The graph shows the mean levels of activation in injured DRGs, with error bars indicating SEM. Thedotted line represents the average level of JNK activity in right versus left DRGs of three unoperated rats. For each time point, n = 3 animals. p values were calculated using the Student’s t test; *p < 0.05 or **p < 0.01 in comparison of injured versus control DRGs. Below each graph are representative autoradiographs from selected time points, showing GST-c-Jun fusion protein phosphorylated by immunoprecipitated JNK isolated from injured (I) and contralateral uninjured (C) DRGs. EachI/C represents DRGs from an individual rat.
The timing of JNK activation after either proximal or distal transection preceded that reported for c-Jun protein induction after injury at each site, respectively: c-Jun protein increases have been observed 3 hr after proximal axotomy and 6 hr after distal axotomy (Kenney and Kocsis, 1997). c-Jun upregulation occurs after activation of c-Jun-containing complexes that bind an AP-1 site in the c-Jun promotor (Angel et al., 1988, Van Dam et al., 1993). The axotomy-induced prolonged activation of JNK that we report here is likely to contribute to the long-lasting upregulation of c-Jun mRNA and protein in DRGs after axotomy. The delay in protein upregulation may reflect the time required for complex activation and subsequent mRNA and protein synthesis to reach levels within the limits of detection by the immunoblot method used previously.
The surgical procedure for proximal transections is more traumatic than that for mid-thigh transection, raising the possibility that earlier JNK activation could reflect the invasiveness of the procedure. However, kinase activity in each L4/L5 DRG sample was determined as a ratio of that in the contralateral, sham-operated side, which underwent the same surgery, with the exception of nerve transection. Basal JNK activity was observed in contralateral sham-operated DRGs after both proximal and distal transection, which did not reflect a systemic response to surgery, because similar levels of JNK activity were observed in DRGs removed from unoperated animals (data not shown). It is possible that this activation occurred when the animals were killed, although the speed with which anesthesia, dissection, and homogenization were accomplished makes this unlikely. Thus, the rapidity of JNK induction after proximal as compared with distal peripheral axotomy indicates that the timing of JNK activation is likely to be dependent on the length of axon between the transection and the DRGs.
JNK protein levels do not change after axotomy
In vitro, JNK is activated by phosphorylation at Thr183 and Tyr185 (Derijard et al., 1994). JNK activity in cultured cells is frequently transient, lasting minutes to several hours after stimulation (Derijard et al., 1994; Chen et al., 1996; Zhang et al., 1996). However, peripheral nerve transection invoked JNK activation in DRGs lasting at least 30 d. The high level of JNK activity that we observed in uninjured DRGs (Fig. 1) suggested that there may be a pool of constitutively active JNK in vivo in DRGs, raising the possibility that the long-term maintenance of increased JNK activity could be attributed to greater levels of JNK protein in axotomized DRGs. We measured JNK protein levels in DRGs by Western blotting at various times after proximal or distal peripheral nerve transection. Axotomy did not induce increased JNK protein at times up to 21 d after axotomy (Fig. 2). Therefore, JNK activity in injured DRGs is likely to be maintained by persistent phosphorylation of the constitutively expressed protein at Thr183 and Tyr185
Fig. 2.

JNK protein levels do not change after axotomy. Protein extracts prepared from injured and contralateral uninjured DRGs were immunoblotted for JNK at progressive times after proximal (•) or distal (▪) axotomy. The intensity of the JNK signal was measured by densitometric analysis of ECL autoradiographs, and a ratio between injured and contralateral DRGs was determined. Each data point is representative of the injured/contralateral JNK ratio in three different animals. Error bars represent SEM. There was no statistically significant difference in JNK levels between injured and contralateral, uninjured DRGs at any time, for either surgical procedure.
Phosphorylated c-Jun is present in axotomized DRG neurons
Substrates of JNK include c-Jun (Hibi et al., 1993; Derijard et al., 1994), ATF2, and Elk1 (Gupta et al., 1995, 1996; Whitmarsh et al., 1995), and to a lesser extent JunD (Kallunki et al., 1996). Amino-terminal phosphorylation of c-Jun by JNK does not affect its ability to bind to target DNA sequences, but rather allows it to interact with CREB-binding protein (Arias et al., 1994), which in turn can bind to components of the basal transcriptional machinery (Kwok et al., 1994). Using immunohistochemistry of free-floating DRG sections with an antibody specific for amino-terminal (Ser63) phosphorylated c-Jun, we detected intense, punctate nuclear staining in DRG neurons, but not satellite cells, examined 24 hr after axotomy (Fig.3a). Some faint nuclear immunoreactivity was seen in the contralateral, uninjured DRGs (Fig.3b). However, we have commented above on the apparent constitutive activity of JNK, and immunoblots for c-Jun protein show strong expression in unoperated DRGs (Fig. 6b,d). These immunostaining results confirm that increased c-Jun protein levels are accompanied by increased amounts of the phosphorylated protein in DRG neurons. Ser63-phosphorylated c-Jun immunoreactivity was observed in both small and large DRG neurons (Fig. 3), indicating that like c-Jun protein upregulation, c-Jun phosphorylation is a general response to axotomy shared by all classes of axotomized DRG neurons.
Fig. 3.
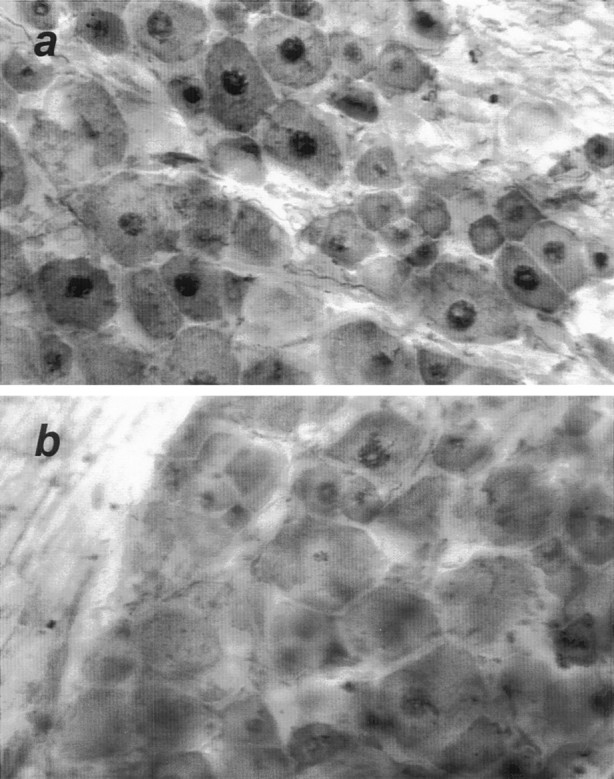
Phosphorylated c-Jun is localized to axotomized DRG neurons. Twenty-four hours after proximal nerve transection, axotomized (a) and contralateral untransected (b) DRGs were removed and processed as 35 mm free-floating sections for immunostaining with an antibody specific for serine-63 phosphorylated c-Jun. Intense nuclear staining was restricted to the nuclei of DRG neurons and was strongly evident in axotomized DRGs. Sections shown are representative of immunostaining in DRGs from three separately analyzed animals.
Fig. 6.

AP-1 binding and c-Jun protein levels in affected DRGs after distal ligation with cuff or sciatic nerve crush. At the indicated times after mid-thigh sciatic nerve transection with cuff (a, b) or crush (c, d), injured (I) or contralateral uninjured (C) DRGs were analyzed by EMSA with Hmt IIA AP-1 oligonucleotides (a, c) or enhanced chemiluminescent Western blotting for c-Jun protein (b, d). Shown are autoradiographs representative of samples analyzed from three rats at each time point, for each surgery. All animals were included in quantitative densitometric analysis described in Results. The level of injury-induced AP-1 binding activity in each sample was determined as a ratio of AP-1 binding activity in injured versus contralateral, uninjured samples. The participation of c-Jun in AP-1 binding activity was determined by the ability of an antibody against the c-Jun DNA binding domain to inhibit formation of AP-1 binding complexes. Samples treated with the c-Jun antibody are indicated above the lane (*). The specific AP-1 binding complex signal is shown by <. Samples analyzed by EMSA were also immunoblotted for c-Jun, under previously optimized experimental conditions that have been established to quantify changes in c-jun protein levels in axotomized DRGs using chemiluminescent detection (Kenney and Kocsis, 1997, 1998), also described in Materials and Methods. The c-Jun antibody used recognizes four major species: 80, 60, 46, and 30 kDa. Only the doublet appearing at 46 kDa is regulated by axonal transection. When extracts from UV-irradiated NIH 3T3 cells were immunoblotted for c-Jun using this antibody, the same hybridization pattern was observed, and the 46 kDa doublet was the only species induced by UV light. Therefore, this species was understood to represent c-Jun and was quantified by densitometric analysis of ECL autoradiographs. A ratio of c-jun immunoreactivity in injured versus the contralateral uninjured DRGs was then established to determine axotomy-induced changes in c-jun protein levels.
AP-1 binding activity is induced in axotomized DRGs
c-Jun belongs to the basic, leucine zipper group of transcription factors, which initiate gene transcription after binding in dimeric form to a recognition sequence, known as 12-O-tetradecanoylphorbol-13-acetate response element (TRE) or AP-1 binding site, (Angel and Karin, 1991), located in the promotor region of target genes. c-Jun may form homodimers or may heterodimerize with other basic leucine zipper transcription factors, including members of the fos, jun, and CRE-binding protein families (Angel and Karin, 1991). The selection of target genes of c-Jun as well as the levels of transcriptional activation of c-Jun therefore may be controlled by the identity of its DNA-binding partners. To determine whether axotomy in vivo induces DNA binding by c-Jun, and to identify possible partners for c-Jun in DNA binding, we investigated the effect of axotomy on the formation and composition of AP-1 binding complexes in DRGs at various times after injury, using an AP-1 consensus sequence based on the human metallothionein IIA(Hmt IIA) promotor in electrophoretic mobility shift assays with antibodies to various jun family members. We did not examine DRGs for AP-1 binding by c-fos, a highly stable partner for c-Jun (Angel and Karin, 1991), because studies using various techniques have not detected this transcription factor in DRGs (Herdegen et al., 1992; Plantinga et al., 1994). ATF2, a member of the CRE-binding protein family, can dimerize with c-Jun to initiate c-Jun transcription from a specialized TRE in the c-Jun promotor (van Dam et al., 1993) but has been shown not to bind to the consensus AP-1 sequence used in our study (Hai and Curran, 1991).
We observed specific, axotomy-induced binding to the Hmt IIA AP-1 sequence; incubation of 72 hr axotomized DRG extracts with unlabeled Hmt IIA AP-1 oligonucleotides prevented binding to the labeled oligonucleotide, whereas treatment with a mutant oligonucleotide differing by one base pair in the consensus AP-1 sequence did not affect activity of the induced complexes (Fig. 4). There was no specific binding activity associated with the labeled mutant oligonucleotide (Fig. 4). Similar results were obtained with DRGs assayed 24 hr after proximal transection (data not shown).
Fig. 4.

Specific AP-1 binding activity in axotomized DRGs. Protein extracts from injured (lanes 1, 3–9) and contralateral uninjured (lanes 2,10) DRGs were assayed for their ability to bind radiolabeled Hmt IIA AP-1 (lanes 1–8) or mutant (lanes 9, 10) sequences, at 72 hr after proximal axotomy. Incubation of injured DRG extracts with the indicated amounts of unlabeled AP-1 (lanes 3–5) reduced the amount of axotomy-induced binding to radiolabeled AP-1, whereas there was no reduction in specific AP-1 binding in the presence of unlabeled mutant AP-1 oligonucleotides (lanes 6–8). A band of more rapid mobility appearing in both injured and uninjured DRGs was competed by both AP-1 and mutant oligonucleotides, indicating that this protein complex does not specifically recognize the Hmt IIA AP-1 sequence, nor does it have high affinity for the mutant AP-1 (lanes 9, 10). We focused our studies on the more slowly migrating, injury-induced complex, indicated by <.
De Leon et al. (1995) reported that AP-1 binding activity is present in DRGs examined at 5 d after mid-thigh sciatic nerve transection. Supporting and expanding on these results, we observed elevated AP-1 binding in axotomized L4/L5 DRGs assayed from 12 hr to 30 d after proximal nerve transection (Fig. 5). At 12 hr post-axotomy, AP-1 binding activity in injured DRGs was eightfold (±0.8 SEM; n = 3) that of control levels. At 3, 7, and 14 d, increased AP-1 binding activity persisted in axotomized DRGs (ninefold that of control; ±0.9 SEM; n = 3 animals per time point). However, by 30 d after proximal axotomy, AP-1 binding activity dropped to 3.5-fold (±0.4 SEM; n = 3) that of control levels. This drop may represent a reduction in c-Jun activity, because it coincides with a decrease in JNK activity at this time (Fig.1a). It is also possible that the diminished JNK activity and AP-1 binding in 30 d proximally axotomized DRGs is indicative of some neuronal cell death, which is more prevalent in proximally axotomized DRGs during the weeks after nerve transection (Ygge, 1989).
Fig. 5.

AP-1 binding activity by c-Jun and JunD in L4/L5 DRGs after proximal sciatic nerve transection. At the indicated times after proximal peripheral nerve transection, DRG protein extracts from injured (I) and contralateral uninjured (C) DRGs were assayed for activity toward an Hmt IIA AP-1 sequence, in the presence of antibodies against various jun family members. Shown are autoradiographs representative of DRGs from three separately analyzed rats at each time point. All animals were included in quantitative densitometric analysis discussed in the text (see Results). The level of AP-1 binding activity in each sample was determined as a ratio of AP-1 binding activity in injured versus contralateral, uninjured samples. In the case of antibody supershift experiments, a ratio was drawn between AP-1 binding activity in injured DRG samples with antibody versus without antibody. Antibodies used are indicated above each lane. A diffuse signal strongly present in extracts from injured DRGs (←AP-1) was not affected by incubation with antibodies to junB. Antibodies to c-Jun blocked formation of this complex, because these antibodies interfere with the DNA-binding domain of c-Jun. JunD antibodies diminished formation of the injury-induced complex and caused a portion to be retained near the top of the gel. The extent to which junD antibodies diminished formation of the specific AP-1 complex was quantified by densitometric analysis of autoradiographs, as described above and in Results. ← NS indicates a nonspecific, noninduced complex.
Treatment of proximally transected DRG extracts with an antibody against the DNA-binding domain of c-Jun before incubation with the AP-1 oligonucleotide interfered with formation of the induced complex at all times after injury (Fig. 5), suggesting that AP-1 binding complexes in axotomized DRGs contain c-Jun, as a homodimer or in complex with other AP-1 binding proteins. c-Jun antibodies caused a 60–80% reduction in AP-1 complex formation at each time point, with three animals analyzed per time point. The increased AP-1 binding activity of c-Jun after axotomy indicates that in addition to positive regulation of c-Jun through activation of JNK, nerve injury may influence signaling pathways, leading to dephosphorylation of negative regulatory sites adjacent to the DNA binding domain of c-Jun (Boyle et al., 1991), one mechanism that can lead to increased DNA binding affinity of c-Jun.
An increase of junB mRNA (De Leon et al., 1995) and transient increases in junB protein (Kenney and Kocsis, 1998) have been reported in DRGs several days after axotomy. JunB may downregulate c-Jun activity by forming heterodimers that are less efficient at binding to DNA (Deng and Karin, 1993) and are not efficiently phosphorylated by jun kinases (Kallunki et al., 1996). Treatment of axotomized DRG extracts with an antibody to junB altered neither the formation nor the mobility of the Hmt IIA AP-1 binding complexes (Fig. 5). Increasing the amount of antibody used in the binding reactions or varying the time and temperature at which reactions took place did not have any effect. When tested by ECL immunoblotting on 72 hr and 7 d axotomized DRGs, the junB supershift antibody recognized a 46 kDa species (data not shown), in agreement with previously reported junB immunoblotting in DRGs (Kenney and Kocsis, 1998), indicating that the JunB supershift antibody that we used can bind to junB, albeit in the denatured form for SDS-PAGE and electrophoretic transfer. However, the lack of well established positive controls for in vivo junB AP-1 supershifts prevents us from completely ruling out the participation of junB in Hmt IIA AP-1 binding complexes in axotomized DRGs.
In contrast, antibodies to JunD caused a reduction in the specific AP-1 binding complex signal, accompanied by retention of a portion of the AP-1 binding complex near the origin of the gel (Fig. 5). The signal shifted toward the top of the gel by the junD antibody represents specific activity of this antibody toward components of the AP-1 binding complex; formation and mobility of complexes binding to CRE (cAMP/Ca2+ response element) oligonucleotides, for which jun family members have less affinity (Angel and Karin, 1991), were not affected by addition of the junD antibody, whereas the addition of a CREB (CRE-binding protein) antibody prevented CRE-binding complex formation (data not shown). When autoradiographs were analyzed by densitometry, the amount of axotomy-induced AP-1 binding complex formed in the presence of the JunD antibody was reduced by 30–50% at all times after proximal axotomy, with three animal analyzed at each time point. JunD protein, normally strongly present in both neurons and satellite cells of DRGs (Herdegen et al., 1992; De Leon et al., 1995), has been reported to increase modestly in DRGs after axotomy (Herdegen et al., 1992; Kenney and Kocsis, 1998). Because c-Jun antibodies abolished more of the AP-1 binding complexes, we suggest that JunD may be heterodimerizing with c-Jun, in which context JunD can be phosphorylated by jun kinases (Kallunki et al., 1996) to activate AP-1 responsive transcription, although the transactivation capacity of these heterodimers may be less than that of c-Jun/c-Jun homodimers (Angel and Karin, 1991).
c-Jun-containing AP-1 complexes are present during axonal outgrowth
We have observed that nerve transection induces persistent activation of JNK and AP-1 binding with c-Jun and JunD for at least 30 d post-injury in adult rat DRGs, suggesting that c-Jun is activated and mediates gene transcription both early and later after nerve injury. Using immunohistochemistry with an antibody recognizing all jun family members, Leah et al. (1991) reported that jun protein levels diminish in adult rat DRGs because process regeneration is completed after sciatic nerve crush. To learn whether c-Jun activity is correlated with axonal regeneration, we compared AP-1 binding activity between axotomized DRGs under regeneration-permissive or -nonpermissive conditions, and we used quantitative immunoblotting (Kenney and Kocsis, 1997, 1998) to quantify c-Jun protein levels in the same samples. One day after both nerve crush, which permits target reconnection, and transection with cuff, which results in chronic neurite outgrowth and neuroma formation, AP-1 binding activity and c-Jun protein levels were increased (Fig. 6). Incubation of injured DRG extracts with c-Jun antibodies reduced formation of AP-1 binding complexes (Fig. 6a,c) by 60–80% (n = 3 animals per surgery, per time point), indicating that both distal sciatic nerve transection and crush increase the DNA binding behavior of c-Jun.
Two methods were used to assess the progression of axonal regeneration after nerve crush. First, hind footprints of the nerve-crushed and contralateral uninjured foot were obtained. Second, the sciatic nerve was dissected free and pinch-tested to determine the advance of the axonal regeneration front (see Materials and Methods) before DRG removal. Twenty-four hours after sciatic nerve crush, the right (axotomized) foot was curled (Fig. 7), showing foot drop, and was little used by the animals, coincident with increased AP-1 binding. A 1.5-fold (±0.15 SEM; n = 3 animals per surgery, per time point) increase in c-Jun protein levels in affected DRGs (Fig. 6c,d) was observed after either crush or transection with cuff. At 72 hr and 7 d post-crush, c-Jun protein levels in injured DRGs were 2.4-fold (±0.25 SEM at 72 hr, ±0.3 SEM at 7 d; n = 3 animals per surgery, per time point) that of control levels (Fig. 6d). Eight- to 10-fold increased AP-1 binding activity was present in the same animals at 24 and 72 hr after crush. By 7 d after nerve crush, AP-1 activity was reduced, although c-jun protein levels remained elevated (Fig. 6d). The earlier reduction in AP-1 binding activity in comparison with c-Jun protein levels may reflect activity of pathways leading to phosphorylation of c-Jun at sites near its C terminal, which can interfere with its DNA binding activity (Boyle et al., 1991). By comparison, AP-1 binding activity continued to be present at similarly high levels at all time points, with three animals analyzed per time point, after transection with cuff. As target reinnervation after crush progressed, the amount of foot drop was reduced (Fig. 7), until at 30 d post-crush the rats showed comparable use of both hindfeet. The length of nerve between the original crush site and the site at which nerve pinching elicited local muscle reflex activity, indicating the leading edge of regenerating axons, increased from 1–1.5 cm at 7 d to 2.5 cm at 14 d post-injury. At 21 and 30 d it was no longer possible to access any portion of nerve that did not induce a muscle reflex in response to pinching.
Fig. 7.

Hind footprints of animals at progressive times after right-side sciatic nerve crush. Regeneration of the sciatic nerve axons after crush was assessed just before animals were killed by obtaining hind footprints after the animals were anesthetized, and then during the dissection procedure by measuring the anatomical site of the axonal regeneration front (see Results). Footprints are shown for selected times after right-side nerve crush. n = 3 animals for each time point. DRGs removed from these animals were analyzed by EMSA and c-Jun immunoblotting (see Results and Fig.6).
As regeneration became complete, AP-1 binding activity declined (Fig.6c), and by 30 d post-crush c-Jun protein levels in injured DRGs were no longer significantly different from contralateral uninjured DRGs (Fig. 6d), as measured by densitometric analysis of ECL immunoblot autoradiographs. In contrast, chronically axotomized DRGs showed elevated AP-1 binding activity and c-Jun protein throughout the 30 d of study (Fig. 6a,b). The protein analysis results agree with previously reported kinetics of c-Jun protein regulation after distal axotomy (Kenney and Kocsis, 1997). A pattern of increased AP-1 binding activity closely followed c-Jun protein upregulation in distally transected DRGs. AP-1 binding activity can be affected by complex composition and post-translational modification of components. This is reflected in the larger fold-induction of AP-1 binding activity in comparison with c-Jun protein level increases. c-Jun protein elevation can be both a cause and an effect of AP-1 binding activity.
DISCUSSION
Axotomy-induced c-Jun activation
We have found that in the intact peripheral nervous system, axotomy is a stimulus that causes rapid, long-term activation of JNK and is associated with c-Jun amino-terminal phosphorylation and DNA binding by c-Jun and JunD in DRG neurons. We have also shown that persistent c-Jun-mediated DNA binding occurs during chronic target disconnection, whereas peripheral nerve regeneration is accompanied by decreased AP-1 binding and c-Jun protein levels over time. Other physiologically relevant JNK activators in the nervous system in vivo have not been identified. In neuronal tissue in vitro, glutamate has recently been shown to induce JNK activation and c-Jun transcriptional activity in primary neuronal cultures from striatum (Schwarzschild et al., 1997). Zhang et al. (1996) recently reported that in primary glial cell cultures, JNK activation is induced by the same types of environmental insults shown to activate JNK in other cultured cell systems.
The mechanisms underlying the axotomy-induced in vivo JNK activity and long-term c-Jun upregulation and activity in DRGs remain unknown. We found that JNK protein levels are not altered after axotomy, suggesting that long-term post-translational modification of constitutively expressed JNK takes place in axotomized DRGs. The 3 hr delay before JNK activation in DRGs after mid-thigh sciatic nerve transection in comparison with the rapid (30 min) activation after proximal nerve transection that we report here fits well with the fast component axonal transport rate of 50–400 mm/d (Jacob and McQuarrie, 1991), suggesting that a retrogradely transported signal mediates axonal injury-induced activation of JNK.
Wu et al. (1993) showed that blockage of normally transported molecules can alter neuronal gene expression. In a similar manner, nerve transection could disinhibit JNK activity by interfering with the somatopedal flow of a constitutively transported modulatory molecule. However, the present study cannot differentiate between deprivation of a negative control signal and generation of a positive signal at the injury site that travels in a retrograde manner to the DRG cell body to stimulate the JNK cascade. An example of intra-axonal post-translational modification and retrograde transport of signaling molecules can be found in Aplysia after nerve injury (Povelones et al., 1997), where axotomy results in rapid inactivation of an NF-κB homolog in axons. In rat sciatic nerve, retrograde transport of molecules in the ERK pathway has been observed (Johanson et al., 1995). Thus, sciatic nerve transection could cause JNK or other JNK/SAPK pathway components to be activated at the site of nerve injury, then translocated to the DRG soma, leading to c-Jun phosphorylation, which we observed in axotomized DRG neurons.
Long-term maintenance of the activated JNK pathway after peripheral nerve transection may be supported by many signals that are generated within the axotomized DRG neurons and throughout the injured nerve. For example, the cellular swelling experienced by axotomized neurons (Lieberman, 1971) could cause changes in osmotic balance, which can activate JNK in Chinese hamster ovary cells (Galcheva-Gargova, 1994). Axotomy leads to proliferation of resident and invading macrophages (Lu and Richardson, 1993), and TNFα, which activates JNKs in vitro (Kyriakis, 1994; Sluss, 1994; Su, 1994), has also been detected in axotomized DRGs (Murphy et al., 1995). The association of an inflammatory response in DRGs with axonal regeneration (Lu and Richardson, 1991) and increased c-Jun mRNA in DRG neurons (Lu and Richardson, 1995) provides further evidence of a relationship between pathways that can activate JNK in DRGs, c-Jun upregulation, and axonal regeneration.
c-Jun activity in regenerating DRG neurons
In many systems, JNK activation and c-Jun activity are often associated with either proliferation (Derijard et al., 1994; Su et al., 1994) or cell death (Estus et al., 1994, Ham et al., 1995; Verheij et al., 1996). However, peripherally axotomized adult rat DRG neurons are nonmitotic cells, the majority of which do not die as a result of injury (Arvidsson et al., 1986; Himes and Tessler,1989; Swett et al., 1995) but rather initiate axonal extension. We report here that in peripherally axotomized DRG neurons, the activation domain of c-Jun is phosphorylated and there is an increase in AP-1 binding activity in DRGs, with c-Jun and JunD participating in binding complexes. Although downstream target genes are not known, we found that increased AP-1 binding by c-Jun, and elevated c-Jun protein levels, persist for at least 30 d after axotomy if successful regeneration is blocked, but diminish over time if axons are permitted to proceed to target reinnervation after nerve crush, suggesting that c-Jun is active in DRGs while they are making regenerative attempts and that c-Jun activity persists as long as DRG neurons have not reestablished contact with peripheral target tissue.
UV-induced c-Jun phosphorylation in vitro (Devary et al., 1992), and in vivo JNK activity in perfused heart (Bogoyevitch et al., 1996) or exercised muscle (Goodyear et al., 1996) have been proposed to have a protective or growth-promoting function. JNK activation and c-Jun activity in DRGs could serve in a similar capacity, initially promoting survival after injury and later operating as part of a cellular growth program. To support the increased energy requirements imposed by a growth rate of 2 mm/d (Jacob and McQuarrie, 1991), regenerating neurons show an increase in the activity of enzymes involved in oxidative metabolism (Harkonen, 1964). c-jun could play a protective role in metabolic control in regenerating DRG neurons. A similar model has been found in rat vascular smooth muscle cells, where AP-1 activity and growth have been linked to oxidative stress (Rao et al., 1996). In yeast, the c-Jun homolog PAR1/YAP1 protects cells against oxidative damage by regulating genes involved in oxygen detoxification (Schnell et al., 1992; Wu and Moye-Rowley, 1994), and in some mammalian cells under oxidative stress, AP-1 regulates proteins involved in detoxification of free radicals (Pinkus et al., 1996;Rahman et al., 1996a,b).
c-Jun may regulate genes specifically pertinent to axonal elongation, such as GAP43, which has AP-1 binding sites in its promotor (Eggen et al., 1994). Like c-Jun, GAP43 expression increases in DRGs within hours after sciatic nerve crush, and GAP43 mRNA levels drop as regeneration is completed (VanderZee et al., 1989). Vasoactive intestinal peptide, a neuropeptide the expression in vivo of which is also upregulated after peripheral axotomy (Shehab and Atkinson, 1986; Nielsh and Keen, 1989) and which can have neuroprotective effects (Gozes and Brenneman, 1996), is a target for c-Jun transcriptional regulation in cultured DRG neurons (Mulderry and Dobson, 1996).
Concluding remarks
We have found that axotomy elicits chronic activation of the JNK pathway in adult rat DRGs in vivo. We have shown that this pathway may contribute to increased levels and trans-activating capacity of c-Jun in DRG neurons by demonstrating that JNK activation is concurrent with amino-terminal phosphorylation of c-Jun in DRG neurons, and increased AP-1 binding activity by c-Jun, as well as JunD. Although it is difficult to parse out the signals specifically responsible for activation and maintenance of the JNK signal transduction pathway, or the effector genes downstream of c-Jun transcriptional activation, using in vivo models, our observations support the hypothesis that peripheral nerve transection induces prolonged transcriptional activation of c-Jun in vivo, through which genes important for neuroprotection as well as axonal regrowth may be regulated.
It is known that the kinetics of c-Jun protein regulation in axotomized peripheral neurons differs from that in axotomized CNS neurons (Leah et al., 1993), which do not normally reconnect with their targets after transection. This may indicate that different signal transduction pathways are activated by CNS injury, leading to a coordination of c-Jun expression with other transcription factors that fail to foster a regenerative response. It has also been suggested that the CNS contains factors that inhibit c-jun expression (Vaudano et al., 1996). Interestingly, although the cell bodies of sciatic nerve motor neurons reside in the CNS, after peripheral axotomy they manifest a c-Jun response resembling that of transected peripheral ganglion sensory neurons (Herdegen et al., 1992), and they regenerate their axons after peripheral transection, with jun family protein levels declining after completion of target tissue reinnervation (Leah et al., 1991). This suggests that the peripheral environment provides unique signals that regulate activity of transcription factors like c-Jun in a manner that promotes a growth response in neurons, and that these signals may be absent or inhibited in the CNS.
An increased clarification of activated intracellular signaling pathways and transcription factor functions in injured peripheral neurons in vivo will aid in the understanding of molecular events that regulate both adaptive and maladaptive responses to nerve injury. For example, the regenerative role of c-jun may have implications for the phenomenon of conditioned collateral sprouting, wherein nerve injury and peripheral axon regrowth is associated with the simultaneous vigorous sprouting (McMahon and Kett-White, 1991) and formation of inappropriate synaptic connections in the spinal cord by central processes of regenerating peripheral axons (Woolf et al., 1995). This may contribute to the pathophysiology of allodynia, in which light touch sensations are perceived as painful stimuli. In addition, characterization of the signals that permit or encourage peripheral nerve regeneration may help identify factors the absence or inhibition of which prevents successful regeneration and recovery of function after injury to CNS neurons.
Footnotes
This work was supported in part by National Institutes of Health (NS 10174) and the Medical Research Service of the Department of Veterans’ Affairs. We thank John Kyriakis (Massachusetts General Hospital/Harvard University, Boston, MA) for his permission to use c-jun (1–135) GST fusion protein. We are sincerely grateful to Michael A. Schwarzschild for his generous advice and thoughtful review of this manuscript. We also thank Philippe Male for his assistance with quantitation.
REFERENCES
- 1.Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta. 1991;1072:129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- 2.Angel P, Hattori K, Smeal T, Karin M. The jun proto-oncogene is positively autoregulated by its product, Jun/AP-1. Cell. 1988;55:875–885. doi: 10.1016/0092-8674(88)90143-2. [DOI] [PubMed] [Google Scholar]
- 3.Arias J, Alberts AS, Brindle P, Claret FX, Smeal T, Karin M, Feramisco J, Montminy M. Activation of cAMP and mitogen responsive genes relies on a common nuclear factor. Nature. 1994;370:226–229. doi: 10.1038/370226a0. [DOI] [PubMed] [Google Scholar]
- 4.Arvidsson J, Ygge J, Grant G. Cell loss in lumbar dorsal root ganglia and transganglionic degeneration after sciatic nerve resection in the rat. Brain Res. 1986;373:15–21. doi: 10.1016/0006-8993(86)90310-0. [DOI] [PubMed] [Google Scholar]
- 5.Binetruy B, Smeal T, Karin M. Ha-Ras augments c-Jun activity and stimulates phosphorylation of its activation domain. Nature. 1991;351:122–127. doi: 10.1038/351122a0. [DOI] [PubMed] [Google Scholar]
- 6.Bogoyevitch MA, Gillespie-Brown J, Ketterman AJ, Fuller SJ, Ben-Levy R, Ashworth A, Marshall CJ, Sugden PH. Stimulation of the stress-activated mitogen activated protein kinase subfamilies in perfused heart. Circ Res. 1996;79:162–173. doi: 10.1161/01.res.79.2.162. [DOI] [PubMed] [Google Scholar]
- 7.Boyle WJ, Smeal T, Defize LHK, Angel P, Woodgett JR, Karin M, Hunter T. Activation of protein kinase C decreases phosphorylation of c-Jun at sites that negatively regulate its DNA-binding activity. Cell. 1991;64:573–584. doi: 10.1016/0092-8674(91)90241-p. [DOI] [PubMed] [Google Scholar]
- 8.Chen YR, Wang X, Templeton D, Davis RJ, Tan TH. The role of c-Jun N-terminal kinase (JNK) in apoptosis induced by ultraviolet C and γ radiation. J Biol Chem. 1996;271:31929–31936. doi: 10.1074/jbc.271.50.31929. [DOI] [PubMed] [Google Scholar]
- 9.Davis RJ. MAPKs: new JNK expands the group. Trends Biochem Sci. 1994;19:170–173. doi: 10.1016/0968-0004(94)90132-5. [DOI] [PubMed] [Google Scholar]
- 10.DeLeon M, Nahin RL, Molina CA, DeLeon DD, Ruda MA. Comparison of c-Jun, junB, and JunD mRNA expression and protein in the rat dorsal root ganglia following sciatic nerve transection. J Neurosci Res. 1995;42:391–401. doi: 10.1002/jnr.490420314. [DOI] [PubMed] [Google Scholar]
- 11.Dellon ES, Dellon AL. Functional assessment of neurologic impairment: track analysis in diabetic and compression neuropathies. Plast Reconstr Surg. 1991;88:686–694. doi: 10.1097/00006534-199110000-00020. [DOI] [PubMed] [Google Scholar]
- 12.Deng T, Karin M. JunB differs from c-Jun in its DNA binding and dimerization domains, and represses c-Jun by formation of inactive heterodimers. Genes Dev. 1993;7:479–490. doi: 10.1101/gad.7.3.479. [DOI] [PubMed] [Google Scholar]
- 13.Derijard B, Hibi M, Wu IW, Barrett T, Su B, Deng T, Karin M, Davis RJ. JNK: a protein kinase that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 14.Devary Y, Gottlieb RA, Smeal T, Karin M. The mammalian ultraviolet response is triggered by activation of Src tyrosine kinases. Cell. 1992;71:1081–1091. doi: 10.1016/s0092-8674(05)80058-3. [DOI] [PubMed] [Google Scholar]
- 15.Eggen BJL, Nielander HB, Rensen-de Leeuw MGA, Schotman P, Gispen WH, Schrama LH. Identification of two promotor regions in the rat B-50/GAP-43 gene. Mol Brain Res. 1994;23:221–234. doi: 10.1016/0169-328x(94)90229-1. [DOI] [PubMed] [Google Scholar]
- 16.Estus S, Zaks WJ, Freeman RS, Gruda M, Bravo R, Johnson EM. Altered gene expression in neurons during programmed cell death: identification of c-Jun as necessary for neuronal apoptosis. J Cell Biol. 1994;127:1717–1727. doi: 10.1083/jcb.127.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galcheva-Gargova Z, Derijard B, Wu IW, Davis RJ. An osmosensing signal transduction pathway in mammalian cells. Science. 1994;265:806–808. doi: 10.1126/science.8047888. [DOI] [PubMed] [Google Scholar]
- 18.Goodyear LJ, Chang PY, Sherwood DJ, Dufresne SD, Moller DE. Effects of exercise and insulin on mitogen activated kinase signalling pathways in rat skeletal muscle. Am J Physiol. 1996;271:E403–408. doi: 10.1152/ajpendo.1996.271.2.E403. [DOI] [PubMed] [Google Scholar]
- 19.Gozes I, Brenneman DE. Activity-dependent neurotrophic factor (ADNF): an extracellular neuroprotective chaperonin? J Mol Neurosci. 1996;7:235–244. doi: 10.1007/BF02737061. [DOI] [PubMed] [Google Scholar]
- 20.Gupta S, Campbell D, Derjard B, Davis RJ. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science. 1995;267:389–393. doi: 10.1126/science.7824938. [DOI] [PubMed] [Google Scholar]
- 21.Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Derijard B, Davis RJ. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996;15:2760–2770. [PMC free article] [PubMed] [Google Scholar]
- 22.Hai T, Curran T. Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc Natl Acad Sci USA. 1991;88:3720–3724. doi: 10.1073/pnas.88.9.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, Rubin LL. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- 24.Harkonen M. Carboxylic esterases, oxidative enzymes, and catecholamines in the superior cervical ganglion of the rat and the effect of pre- and post-ganglionic nerve division. Acta Physiol Scand. 1964;[Suppl 63]237:1–94. [Google Scholar]
- 25.Herdegen T, Fiallos-Estrada CE, Schmid W, Bravo R, Zimmerman M. The transcription factors c-Jun, JunD and CREB, but not fos and KROX-24, are differentially regulated in axotomized neurons following transection of rat sciatic nerve. Mol Brain Res. 1992;14:155–165. doi: 10.1016/0169-328x(92)90170-g. [DOI] [PubMed] [Google Scholar]
- 26.Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 27.Himes BT, Tessler A. Death of some dorsal root ganglion neurons and plasticity of others following sciatic nerve section in adult and neonatal rats. J Comp Neurol. 1989;284:215–230. doi: 10.1002/cne.902840206. [DOI] [PubMed] [Google Scholar]
- 28.Jacob JM, McQuarrie IG. Axotomy accelerates slow component b of axonal transport. J Neurobiol. 1991;22:570–582. doi: 10.1002/neu.480220603. [DOI] [PubMed] [Google Scholar]
- 29.Jenkins R, Hunt SP. Long-term increases in the levels of c-Jun mRNA and Jun protein-like immunoreactivity in motor and sensory neurons following axon damage. Neurosci Lett. 1991;129:107–110. doi: 10.1016/0304-3940(91)90731-8. [DOI] [PubMed] [Google Scholar]
- 30.Johanson SO, Crouch MF, Hendry IA. Retrograde transport of signal transduction proteins in rat sciatic nerve. Brain Res. 1995;690:55–53. doi: 10.1016/0006-8993(95)00587-g. [DOI] [PubMed] [Google Scholar]
- 31.Kallunki T, Deng T, Hibi M, Karin M. C-Jun can recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell. 1996;87:929–939. doi: 10.1016/s0092-8674(00)81999-6. [DOI] [PubMed] [Google Scholar]
- 32.Kenney AM, Kocsis JD. Timing of c-Jun protein induction in lumbar dorsal root ganglia after sciatic nerve transection varies with lesion distance. Brain Res. 1997;751:90–95. doi: 10.1016/s0006-8993(96)01402-3. [DOI] [PubMed] [Google Scholar]
- 33.Kenney AM, Kocsis JD (1998) Temporal variability of jun family transcription factor levels in peripherally or centrally transected adult rat dorsal root ganglia. Mol Brain Res, in press. [DOI] [PubMed]
- 34.Kwok RP, Lundblad JR, Chrivia JC, Richards JP, Bachinger HP, Brennan RG, Roberts SG, Green MR, Goodman RH. Nuclear factor CBP is a coactivator for the transcription factor CREB. Nature. 1994;370:223–226. doi: 10.1038/370223a0. [DOI] [PubMed] [Google Scholar]
- 35.Kyriakis JM, Avruch J. Sounding the alarm: protein kinase cascades activated by stress and inflammation. J Biol Chem. 1996;271:24313–24316. doi: 10.1074/jbc.271.40.24313. [DOI] [PubMed] [Google Scholar]
- 36.Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie E, Ahmad MF, Avruch J, Woodgett JR. The stress-activated protein kinase family of c-Jun kinases. Nature. 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 37.Leah JD, Herdegen T, Bravo R. Selective expression of Jun proteins following axotomy and axonal transport block in peripheral nerves in the rat: evidence for a role in the regeneration process. Brain Res. 1991;566:198–207. doi: 10.1016/0006-8993(91)91699-2. [DOI] [PubMed] [Google Scholar]
- 38.Leah JD, Herdegen T, Murashov A, Dragunow M, Bravo R. Expression of immediate early gene proteins following axotomy and inhibition of axonal transport in the central nervous system. Neuroscience. 1993;57:53–66. doi: 10.1016/0306-4522(93)90111-r. [DOI] [PubMed] [Google Scholar]
- 39.Lee W, Mitchell P, Tjian R. Purified transcription factor AP-1 interacts with TPA-inducible enhancer elements. Cell. 1987;49:741–752. doi: 10.1016/0092-8674(87)90612-x. [DOI] [PubMed] [Google Scholar]
- 40.Lieberman AR. The axon reaction: a review of the principal features of perikaryal responses to axon injury. Int Rev Neurobiol. 1971;14:49–124. doi: 10.1016/s0074-7742(08)60183-x. [DOI] [PubMed] [Google Scholar]
- 41.Lu X, Richardson PM. Inflammation near the nerve cell body enhances axonal regeneration. J Neurosci. 1991;11:972–978. doi: 10.1523/JNEUROSCI.11-04-00972.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu X, Richardson PM. Responses of macrophages in rat dorsal root ganglia following peripheral nerve injury. J Neurocytol. 1993;22:334–341. doi: 10.1007/BF01195557. [DOI] [PubMed] [Google Scholar]
- 43.Lu X, Richardson PM. Changes in neuronal mRNAs induced by a local inflammatory reaction. J Neurosci Res. 1995;41:8–14. doi: 10.1002/jnr.490410103. [DOI] [PubMed] [Google Scholar]
- 44.McMahon SB, Kett-White R. Sprouting of peripherally regenerating primary sensory neurons in the adult central nervous system. J Comp Neurol. 1991;304:307–315. doi: 10.1002/cne.903040211. [DOI] [PubMed] [Google Scholar]
- 45.Mendelson KG, Contois LR, Tevosian SG, Davis RJ, Paulson KE. Independent regulation of JNK/p38 mitogen activated protein kinases by metabolic oxidative stress in the liver. Proc Natl Acad Sci USA. 1996;93:12908–12913. doi: 10.1073/pnas.93.23.12908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller FD, Tetzlaff W, Bisby MA, Fawcett JW, Milner RJ. Rapid induction of the major embryonic α-tubulin mRNA, Tα1, during nerve regeneration in adult rats. J Neurosci. 1989;9:1452–1463. doi: 10.1523/JNEUROSCI.09-04-01452.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moskowitz PF, Oblinger MM. Sensory neurons selectively upregulate synthesis and transport of the bIII-tubulin protein during axonal regeneration. J Neurosci. 1995;15:1545–1555. doi: 10.1523/JNEUROSCI.15-02-01545.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mulderry PK, Dobson SP. Regulation of VIP and other neuropeptides by c-Jun in sensory neurons: implications for the neuropeptide response to axotomy. Eur J Neurosci. 1996;8:2479–2491. doi: 10.1111/j.1460-9568.1996.tb01542.x. [DOI] [PubMed] [Google Scholar]
- 49.Murphy PG, Grondin J, Altares M, Richardson PM. Induction of interleukin-6 in axotomized sensory neurons. J Neurosci. 1995;15:5130–5138. doi: 10.1523/JNEUROSCI.15-07-05130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nielsch U, Keen P. Reciprocal regulation of tachykinin and vasoactive intestinal peptide gene expression in rat sensory neurones following cut and crush injury. Brain Res. 1989;481:25–30. doi: 10.1016/0006-8993(89)90481-2. [DOI] [PubMed] [Google Scholar]
- 51.Peterson GL. A simplification of the protein assay method of Lowry et al which is more generally applicable. Anal Biochem. 1977;83:346–356. doi: 10.1016/0003-2697(77)90043-4. [DOI] [PubMed] [Google Scholar]
- 52.Pinkus R, Weiner LM, Daniel V. Role of oxidants and antioxidants in the induction of AP-1, NF-κB, and glutathione S-transferase gene expression. J Biol Chem. 1996;271:13422–13429. doi: 10.1074/jbc.271.23.13422. [DOI] [PubMed] [Google Scholar]
- 53.Plantinga LC, Verhaagen J, Wong SL, Edwards PM, Bar PR, Gispen WH. The neurotrophic peptide Org2766 does not influence the expression of the immediate early gene c-fos following sciatic nerve crush in the rat. Int J Dev Neurosci. 1994;12:117–125. doi: 10.1016/0736-5748(94)90004-3. [DOI] [PubMed] [Google Scholar]
- 54.Povelones M, Tran K, Thanos D, Ambron RT. An NF-kB-like transcription factor in axoplasm is rapidly inactivated after nerve injury in Aplysia. J Neurosci. 1997;17:4915–4920. doi: 10.1523/JNEUROSCI.17-13-04915.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rahman I, Bel A, Mulier B, Lawson MF, Harrison DJ, MacNee W, Smith CAD. Transcriptional regulation of γ-glutamylcysteine synthetase-heavy subunit by oxidants in human alveolar epithelial cells. Biochem Biophys Res Commun. 1996a;229:832–837. doi: 10.1006/bbrc.1996.1888. [DOI] [PubMed] [Google Scholar]
- 56.Rahman I, Smith CAD, Lawson MF, Harrison DJ, MacNee W. Induction of γ-glutamylcysteine synthetase by cigarette smoke is associated with AP-1 in human alveolar epithelial cells. FEBS Lett. 1996b;396:21–25. doi: 10.1016/0014-5793(96)01027-7. [DOI] [PubMed] [Google Scholar]
- 57.Rao GN, Glasgow WC, Eling TE, Runge MS. Role of hydroperoxyeicosatetraenoic acids in oxidative stress induced activating protein 1 (AP-1) activity. J Biol Chem. 1996;271:27760–27764. doi: 10.1074/jbc.271.44.27760. [DOI] [PubMed] [Google Scholar]
- 58.Rich KM, Yip HK, Osborne PA, Schmidt RE, Johnson EM. Role of nerve growth factor in the adult dorsal root ganglion neuron and its response to injury. J Comp Neurol. 1984;230:110–118. doi: 10.1002/cne.902300110. [DOI] [PubMed] [Google Scholar]
- 59.Schnell N, Krems B, Entian KD. The PAR1 (YAP1/SNQ3) gene of Saccharomyces cerevisiae, a c-Jun homologue, is involved in oxygen metabolism. Curr Genet. 1992;21:269–273. doi: 10.1007/BF00351681. [DOI] [PubMed] [Google Scholar]
- 60.Schwarzschild MA, Cole RL, Hyman SE. Glutamate, but not dopamine, stimulates stress-activated protein kinase and AP-1-mediated transcription in striatal neurons. J Neurosci. 1997;17:3455–3466. doi: 10.1523/JNEUROSCI.17-10-03455.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shehab SAS, Atkinson ME. Vasoactive intestinal polypeptide (VIP) increases in the spinal cord after peripheral axotomy of the sciatic nerve originate from primary afferent neurons. Brain Res. 1986;372:37–44. doi: 10.1016/0006-8993(86)91456-3. [DOI] [PubMed] [Google Scholar]
- 62.Sluss HK, Barrett T, Derijard B, Davis RJ. Signal transduction by tumor necrosis factor mediated by JNK protein kinases. Mol Cell Biol. 1994;14:8376–8384. doi: 10.1128/mcb.14.12.8376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Smeal T, Binetruy B, Mercola D, Birrer M, Karin M. Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature. 1991;354:494–496. doi: 10.1038/354494a0. [DOI] [PubMed] [Google Scholar]
- 64.Smeal T, Binetruy B, Mercola D, Grover-Bardwick A, Heidecker G, Rapp UR, Karin M. Oncoprotein-mediated signalling cascade stimulates c-Jun activity by phosphorylation of serines 63 and 73. Mol Cell Biol. 1992;12:3507–3515. doi: 10.1128/mcb.12.8.3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Somervaille T, Reynolds ML, Woolf CJ. Time-dependent differences in the increase in GAP43 expression in dorsal root ganglion cells after peripheral axotomy. Neuroscience. 1991;45:213–220. doi: 10.1016/0306-4522(91)90117-7. [DOI] [PubMed] [Google Scholar]
- 66.Su B, Jacinto E, Hibi M, Kallunki T, Karin M, Ben-Neriah Y. JNK is involved in signal integration during costimulation of T lymphocytes. Cell. 1994;77:727–736. doi: 10.1016/0092-8674(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 67.Swett JE, Hong CZ, Miller PG. Most dorsal root ganglion neurons of the adult rat survive nerve crush injury. Somatosens Mot Res. 1995;12:177–189. doi: 10.3109/08990229509093656. [DOI] [PubMed] [Google Scholar]
- 68.Van Dam H, Duyndam M, Rottier R, Bosch A, deVries-Smits L, Herrlich P, Zantema A, Angel P, Van der Eb A. Heterodimer formation of cJun and ATF-2 is responsible for induction of c-Jun by the 243 amino acid adenovirus E1A protein. EMBO J. 1993;12:479–487. doi: 10.1002/j.1460-2075.1993.tb05680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Van der Zee CEEM, Nielander H, Vos JP, Lopes da Silva S, Verhaagen J, Oestreicher AB, Schrama LH, Schotman P, Gispen WH. Expression of growth-associated protein B-50 (GAP43) in dorsal root ganglia and sciatic nerve during regenerative sprouting. J Neurosci. 1989;9:3505–3512. doi: 10.1523/JNEUROSCI.09-10-03505.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vaudano E, Campbell G, Hunt SP. Change in the molecular phenotype of Schwann cells upon transplantation into the central nervous system: down-regulation of c-jun. Neuroscience. 1996;73:553–565. doi: 10.1016/0306-4522(96)00199-6. [DOI] [PubMed] [Google Scholar]
- 71.Verheij M, Bose R, Lin XH, Yao B, Jarvis WD, Grant S, Birrer MJ, Szabo E, Zon L, Kyriakis JM, Haimovitz-Friedman A, Fuks Z, Kolesnick RN. Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature. 1996;380:75–79. doi: 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- 72.Walker JL, Evans JM, Resig P, Guarnieri S, Meade P, Sisken BS. Enhancement of functional recovery following a crush lesion to the sciatic nerve by exposure to pulsed electromagnetic fields. Exp Neurol. 1994;125:302–305. doi: 10.1006/exnr.1994.1033. [DOI] [PubMed] [Google Scholar]
- 73.Whitmarsh AJ, Shore P, Sharrocks AD, Davis RJ. Integration of the MAP kinase signal transduction pathways at the serum response element. Science. 1995;269:403–407. doi: 10.1126/science.7618106. [DOI] [PubMed] [Google Scholar]
- 74.Wiese UH, Ruth JL, Emson PC. Differential expression of growth-associated protein (GAP43) mRNA in rat primary sensory neurons after peripheral nerve lesion: a non-radioactive in situ hybridisation study. Brain Res. 1992;592:141–156. doi: 10.1016/0006-8993(92)91669-6. [DOI] [PubMed] [Google Scholar]
- 75.Woolf CJ, Shortland P, Reynolds M, Ridings J, Doubell T, Coggeshall RE. Reorganization of central terminals of myelinated primary afferents in the rat dorsal horn following peripheral axotomy. J Comp Neurol. 1995;360:121–134. doi: 10.1002/cne.903600109. [DOI] [PubMed] [Google Scholar]
- 76.Wu AL, Moye-Rowley WS. GSH-1, which encodes γ-glutamylcysteine synthetase, is a target gene for yAP-1 transcriptional regulation. Mol Cell Biol. 1994;14:5832–5839. doi: 10.1128/mcb.14.9.5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu W, Mathew TC, Miller FD. Evidence that loss of homeostatic signals induces regeneration-associated alterations in neuronal gene expression. Dev Biol. 1993;158:456–466. doi: 10.1006/dbio.1993.1203. [DOI] [PubMed] [Google Scholar]
- 78.Ygge J. Neuronal loss in lumbar dorsal root ganglia after proximal compared to distal sciatic nerve resection: a quantitative study in the rat. Brain Res. 1989;478:193–195. doi: 10.1016/0006-8993(89)91497-2. [DOI] [PubMed] [Google Scholar]
- 79.Zhang P, Miller BS, Rosenzweig SA, Bhat NR. Activation of c-Jun N-terminal kinase/stress-activated protein kinase in primary glial cell cultures. J Neurosci Res. 1996;46:114–121. doi: 10.1002/(SICI)1097-4547(19961001)46:1<114::AID-JNR14>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]