

Abstract
It is well-known that insoluble nickel compounds possess much more potent carcinogenic activities as compared with soluble nickel compounds. Although it is assumed that the different entry and clearance rate are responsible for the difference, the mechanisms underlying the different carcinogenic activities are still not well understood yet. In the present study, we found that exposure to soluble, but not insoluble nickel compounds, caused a significant inhibition of cell growth and G1/G0 cell cycle arrest, which was concomitant with a marked down-regulation of cylin D1, an essential nuclear protein for controlling G1/S transition, while both soluble and insoluble nickel compounds showed similar effects on NFκB activation, HIF-1α protein accumulation and TNF-α transcription and CAP43 protein expression at same doses range. The down-regulation of cyclin D1 is due to protein degradation rather than inhibition of transcription, because the nickel compounds treatment did not change cyclin D1 mRNA level, while MG132, the proteasome inhibitor, can rescue the degradation of cyclin D1 caused by soluble nickel compound. Moreover, the soluble nickel-induced cyclin D1 degradation is dependent on its Thr286 residue and requires IKKα, but not HIF-1α, which are both reported to be involved in cyclin D1 down-regulation. Taken together, we demonstrate that soluble, but not insoluble nickel compound, is able to cause cyclin D1 degradation and a cell growth arrest in an IKKα-dependent manner. Given the role of cyclin D1 and cell proliferation in carcinogenesis, we anticipate that the different effects of soluble and insoluble nickel compounds on cyclin D1 degradation and cell growth arrest may at least partially account for their different carcinogenic activities.
Keywords: nickel compounds, cell growth arrest, cyclin D1, HIF-1α, IKKα
Introduction
Nickel, a widely distributed natural metal, is industrially applied in alloys and industrial processes such as electroplating, foundries, catalysts, battery manufacturing, and coinage (Oller et al., 1997; Sunderman, 1989). Based on their water solubility, nickel compounds are categorized as soluble and insoluble forms. Epidemiological studies have shown that long-term inhalation exposures of workers employed in nickel refining operations to mixtures of soluble and insoluble nickel compounds in nickel refineries have been associated with excess lung cancer and nasal sinus cancer (1990; IARC, 1990). The carcinogenic activity of nickel compounds has been shown to inversely correlate to its solubility in water (National-Toxicology-Program, 1996a; National-Toxicology-Program, 1996b; National-Toxicology-Program, 1996c; Oller, 2002; Oller et al., 1997). The ability of insoluble nickel compounds to induce cancer formation has been verified by a series of animal carcinogenicity studies with different exposure routes in different research groups. Ottolenghi et al. has reported that the inhalation of insoluble nickel compounds induces adenomas and adenocarcinomas in the lungs of male and female F344 rats (Ottolenghi et al., 1975). Intramuscular (i.m.) injection of nickel compounds into male F344 rats induced sarcomas at the injection site (Sunderman and Maenza, 1976). Kasprzak et al. has showed that insoluble nickel compound is carcinogenic when injected into Wistar rats (Kasprzak et al., 1983). The U.S. National Toxicology Program (NTP) has conducted nickel animal carcinogenic studies by the inhalation route, which is most relevant to human carcinogenesis, and showed that insoluble nickel compounds induce lung tumors in rats and mice, but soluble nickel compounds do not (National-Toxicology-Program, 1996a; National-Toxicology-Program, 1996b; National-Toxicology-Program, 1996c). Several oral exposure studies in rats, mice and dogs with soluble nickel compounds also failed to induce tumors (Oller, 2002; Schroeder and Mitchener, 1975; Schroeder et al., 1974). These lines of evidence clearly indicate that insoluble nickel compounds are potent carcinogens, while soluble nickel compounds are less potent. Accordingly, the insoluble nickel compounds are classified as a human carcinogen, while soluble nickel compounds are considered as “not classifiable as a human carcinogen” (ACGIH, 1998) or “the carcinogenicity of soluble nickel compounds cannot be determined” (Haber et al., 2000a; Haber et al., 2000b).
After entry to cells, nickel compounds have been demonstrated to promote the generation of ROS (Landolph, 1999); interact directly or indirectly with nucleic acids and cause genotoxic damage (Sunderman, 1993); induce epigenetic alterations including DNA methylation and loss of histone acetylation (Sutherland and Costa, 2003); and activate signaling pathways such as PI-3K/Akt (Li et al., 2004), HIF-1α (Salnikow et al., 2000), NF-κB (Chen et al., 2001; Cruz et al., 2004; Huang et al., 2002), AP-1 (Cruz et al., 2004; Huang et al., 2002) and NFAT (Huang et al., 2001; Lu et al., 2005). All these events are believed to relate to their carcinogenic activities. Although the different entry and clearance rate as well as the persistence of water-soluble and water-insoluble nickel compounds in cells are presumably thought to be responsible for their different carcinogenic activities (Oller et al., 1997; Seilkop and Oller, 2003), so far, the mechanisms under the difference remains to be fully defined.
Carcinogenesis results from a synergism between genotoxic and nongenotoxic factors (Hecker, 1987; Zoumpourlis et al., 2003). The former induce irreversible genetic alterations (tumor initiation), whereas the latter promote tumor development by favoring the clonally outgrowth of the genetically altered cells. It is believed that the growth of genetic mutated cells is indispensable during the process of carcinogenesis (Charnley and Wilson, 1991; Cohen and Ellwein, 1988). Thus, it is of interest to investigate whether soluble and insoluble nickel compounds can differently affect cell growth. In the present study, we demonstrated that soluble, but not insoluble nickel, is able to cause significant cell growth arrest and cyclin D1 degradation in an IKKα-dependent manner. These results provide the first evidence that the different effect of soluble and insoluble nickel compounds on cyclin D1 down-regulation and cell growth arrest may be associated with their different carcinogenic activities.
Materials and Methods
Cell Culture and Reagents
Human lung carcinoma A549 cells were cultured in Ham’s F-12K medium supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 ig/ml streptomycin. Wild-type, IKKα−/− (Devin et al., 2001), HIF1α+/+ and HIF1α−/− (Ryan et al., 1998; Salnikow et al., 2000; Salnikow et al., 2003b) mouse embyo fibroblasts (MEFs) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Calbiochem, San Diego, CA). Human bronchial epithelial cell line HCCBE-3 (Ramirez et al., 2004) was cultured in defined keratinocyte serum-free medium (Invitrogene). The cultures were dissociated with trypsin and transferred to new 75-cm2 culture flasks (Fisher, Pittsburgh, PA) after centrifuge from one to three twice a week. FBS was purchased from Life Technologies, Inc.; DMEM was from Calbiochem (San Diego, CA); Nickel chloride (NiCl2) was purchased from Aldrich (Milwaukee, WI); nickel sulfide (NiS) was purchased from Alfa (Ward Hill, MA); Nickel subsulfide (Ni3S2) was obtained from INCO (Toronto, Canada). Antibodies against cyclin D1, p21, caspase-3 and CDK4 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA); antibodies against Phosphor-Cyclin D1 T286, ATF2 and IKKα were from Cell signaling Technology (Danvers, MA); RB antibody was from BD Biosciences (San Jose, CA); HIF-1α antibody was from Novus Biologicals (Littleton, CO); FLAG and β-Actin antibodies were purchased from Sigma (St. Louis, MO).
Cell growth inhibition analysis
1×103 of viable cells suspended in 100 μl complete medium supplemented with 10% FBS were added to each well of 96-well plates. The plates were incubated at 37°C in a humidified atmosphere of 5% CO2. 12 h later, the cells were exposed to nickel compounds for the times as indicated in figure legends. The exposed cells were taken pictures under microscopy, and then extracted with 50 μl lysis buffer. The cell growth was measured by using a CellTiter-Glo® Luminescent Cell Viability Assay kit (Promega, Madison WI) with a luminometer (Wallac 1420 Victor2 multipliable counter system) as described previously (Ouyang et al., 2006b). The results are expressed as inhibition of cell growth, which was calculated as following:
Cyclin D1 expression assay
2 ×105 of cells were cultured in each well of 6-well plates to 85–90% confluence. After exposure to nickel compounds together with or without MG132, a chemical inhibitor of proteasome-dependent protein degradation, for different times as indicated in the figure legends. The treated cells were washed once with ice-cold PBS and then extracted with SDS-sample buffer. The cell extracts were separated on polyacrylamide-SDS gels, transferred, and probed with specific antibodies against cyclin D1 and β-Actin which was used as a protein loading control. The protein bands specifically bound to primary antibodies were detected as indicated above.
Transfection and Western blot
293T cells were cultured in a 6-well plate until they reached 85–90% confluence. Five (5) μg of FLAG-cyclin D1 T286A/pCMV5 expression vector, which was described in the previous study (Kwak et al., 2005) were mixed with 10 μl of Lipofectamine 2000 reagent, and used to transfect each well in the absence of serum. After 6 h, the medium was replaced with 10% FBS DMEM. Approximately 48 h after the beginning of the transfection, the cells were exposed to 0.5mM of NiCl2 for 24 hrs, and then extracted for Western blot assay to detect cylin D1 level as describe above with antibodies against FLAG, cyclin D1 and β-actin. β-actin was used as a protein loading control.
RT-PCR
2×105 of cells were cultured in each well of 6-well plates to about 85% confluence. After exposure to nickel compounds for 12h, the cells were extracted with Trizol reagent (Invitrogen, CA), and the total RNA was isolated following the manufacturer’s instructions. 1 μg of total RNA was reverse-transcribed (Superscript II, Invitrogen), and the cDNA was subjected to PCR amplifications using primer pairs specific for murine cyclin D1 (forward, 5′-gtg cca tcc atg cgg aa-3′; backward, 5′-gga tgg tct gct tgt tct ca-3′), for murine β-actin (forward: 5′-cat ccg taa aga cct cta tgc c-3′; backword: 5′-acg cag ctc agt aac agt cc-3′ ), for human cyclin D1 (forward: 5′-gag caa aga tgg cgc ata at-3′; backward: 5′-aag ttt gcc gtg gtg ttt ct-3′) and human β-actin (forward: 5′-gcg aga aga tga ccc aga tca t-3′; backward: 5′-gct cag gag gag caa tga tct t-3′). The β-actins were used as the internal control. The PCR cycling conditions were: 25 cycles of 94 °C for 45 sec, 60 °C for 45 sec and 72 °C for 45 sec.
Cell cycle analysis
2 ×105 of cells were cultured in each well of 6-well plates to 70–80% confluence with normal culture medium. The cell culture medium was replaced with 0.1% FBS DMEM with 2 mM L-glutamine and 25 ig gentamicin and cultured for 24 h and then exposed to nickel compounds. The cells were harvested and fixed with 3 ml of ice-cold 80% ethanol overnight. The fixed cells were washed twice with PBS, and then suspended in propidium iodide staining solution (propidium iodide 50 μg/ml, RNAse A 10 mg/ml and 0.1% Triton X-100) (Sigma Chemical, St. Louis, MO) for at least 1 h at 4 °C. The DNA content was determined by flow cytometry using the Epics XL FACS (Beckman Coulter) and EXPO 32 software as described in our previous studies (Ouyang et al., 2006b; Ouyang et al., 2005).
Nuclear extraction
The cells were cultured in 100-mm dishes and treated with 0.5 mM nickel chloride for 6 h and 12 h. The nuclear proteins were extracted according to the protocol of Nuclear/Cytosol Fractionation Kit (BioVision, Mountain View, CA). In brief, cells were collected by centrifuge for 5 minutes at 600 × g, and lysized in 200 μl of Cytosol Extraction Buffer A including DTT and protease inhibitors. Then 11 μl of Cytosol Extraction Buffer B was added, and mixed by vortex vigorously for 5 seconds and incubated on ice for 1 min. The cell lysis was subjected to centrifuge immediately for 5 min at 16,000 × g. The supernatant was the cytoplasmic fraction. The crude nuclei pellet was resuspended in 100 μl ice-cold Nucelar Extraction Buffer and agitated at high speed for 40 minutes by vortex, and subjected to centrifuge for 10 minutes at 20,000× g. The supernatant was the nuclear extraction proteins.
Statistical Analysis
The significance of the difference between the treated and untreated groups was determined with the Student’s t test. The results are expressed as mean ± S.D.
Results
Exposure of A549 cells to soluble nickel compound caused cell growth arrest and cyclin D1 degradation
Soluble and insoluble nickel compounds have been demonstrated to possess different carcinogenic activities (National-Toxicology-Program, 1996a; National-Toxicology-Program, 1996b; National-Toxicology-Program, 1996c; Oller, 2002; Oller et al., 1997). However, previous findings by us and others have also demonstrated that the soluble and insoluble nickel compounds at certain dosages have similar effects on various cellular events, including generation of intracellular ROS and activation of cell signaling pathways, which include HIF-1α, NFκB and PI-3K/Akt (Costa et al., 2005; Huang et al., 2002; Li et al., 2004). To compare the differential effects of soluble and insoluble nickel compounds on cell cycle and proliferation, we first determined equivalent doses of both types of nickel compounds by evaluation of their activation of cell signaling pathways in different doses. As shown in Figs. 1a–1c, NiCl2 at doses ranged from 0.25 mM to 1 mM are equivalent to NiS at doses ranged from 0.25 μg/cm2 to 2 μg/cm2 in the activation of transcription factor NFκB and HIF-1α, and induction of TNF-α and CAP43 gene expression. In view of the role of cell growth in carcinogenesis, the lung being the primary target organ of nickel compounds, we examined the growth rate of human lung carcinoma A549 cells exposed to soluble and insoluble nickel compounds. As shown in Figs. 1d and 1e, the growth of A549 cells were significantly inhibited by the exposure of NiCl2, a soluble nickel compound, but not NiS, an insoluble nickel compound. The growth inhibition caused by soluble nickel but no insoluble nickel was also found in other type of cells, namely human bronchial epithelial cell line HCCBE-3 (Ramirez et al., 2004) and mouse skin epidermal cell line JB6 Cl41 (Li et al., 2004; Ouyang et al., 2006a) (Figs 1f–1i), indicating that the differential growth inhibition effects of soluble but not insoluble nickel compounds is a general phenomena, not cell type specific.
Fig. 1. Inhibition of cell growth of human lung A549 cells by NiCl2, but not NiS.
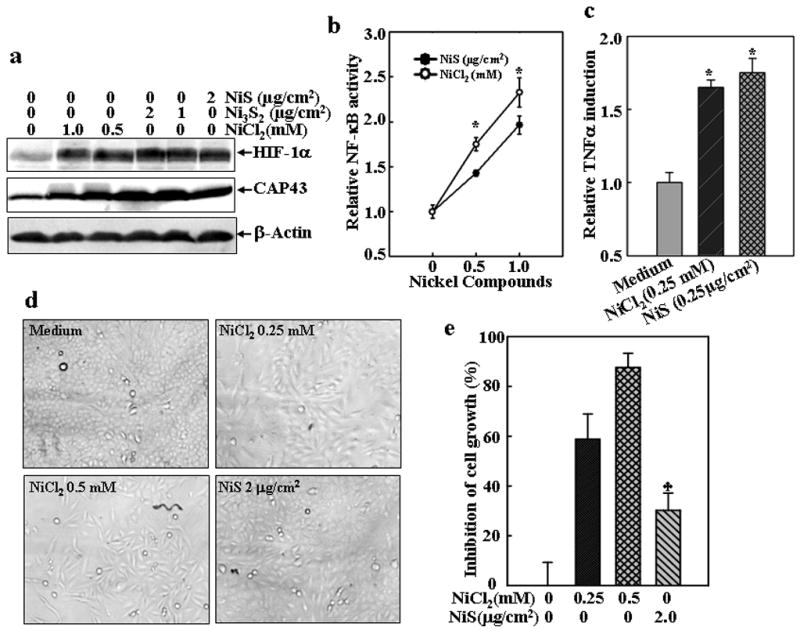
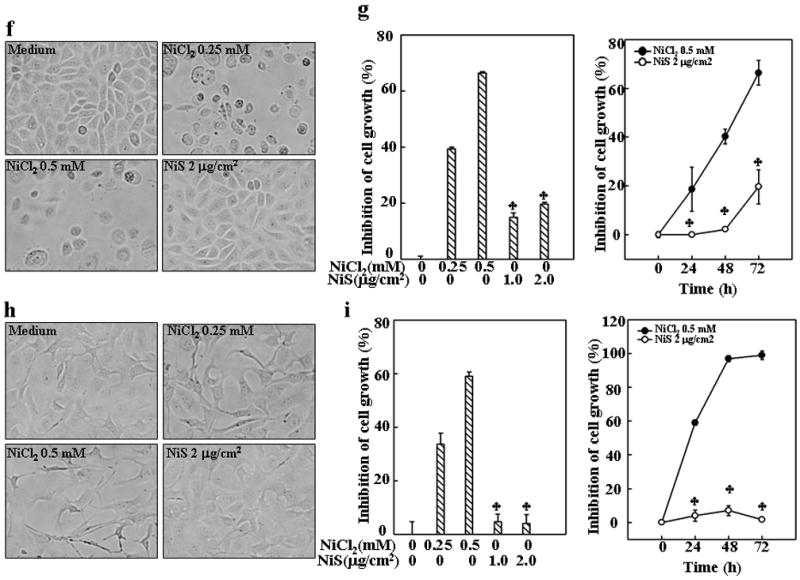
(a), The cells were treated with various concentrations of nickel compounds as indicated for 24 h and then extracted with SDS-sample buffer. Western blot was carried out with specific antibodies again HIF-1α, CAP43 or β-Actin as indicated. (b and c), TNF-α-Luc (b) or NF-κB-Luc reporter (c) stable transfectants were seeded into each well of 96-well plates, cultured in 10% FBS DMEM medium overnight, and then exposed to NiCl2 or NiS for 12 h. The cells were then extracted with lysis buffer, and the luciferase activity was measured using the Promega luciferase assay kit. The results were then presented as TNF luciferase activity relative to medium control (relative TNF induction) (b) or NF-κB transcription activity relative to the control (relative NF-κB activity) Each bar indicates the mean and standard deviation of triplicate assay wells. The symbol (*) indicates no significant changes between NiCl2 and NiS (p> 0.05). (d–i), 1×103 of lung carcinoma A549 cells (d and e), or human bronchial epithelial cell line HCCBE-3 (f and g), or mouse skin epidermal cell line Cl41 (h and i) were seeded into each well of 96-well plates, and then exposed to NiCl2 or NiS. The cells were photographed under microscopy 24 h (h) or 72 h later (d and f), and then extracted by lysis buffer for growth measurement using CellTiter-Glo® Luminescent Cell Viability Assay kit with a luminometer (e, g and i). The results are expressed as inhibition of cell growth which was calculated as described in Material and Methods. Each bar indicates the mean and standard deviation of triplicate assay wells. The symbol (♣) indicates a significant decrease from NiCl2 group (p< 0.05).
The growth inhibition was concomitant with an arrest of cell cycle progression at G1/G0 phase (Fig. 2a). The cells exposed to NiCl2 exhibited a significant increase in the proportion of G1/G0 phase, while with a marked decrease in the proportion of S phase (Fig. 2a). Since both NiCl2 and NiS showed similar biological effects on HIF-1α protein accumulation, NFκB activation, TNF-α and CAP43 expression at the same doses used in the studies of cell cycle and growth, this rules out the possibility that differential effects of NiCl2 and NiS on the inhibition of cell growth and cell cycle progression are due to either inactivation of insoluble nickel compounds or discrepancy of cytotoxicity to exposed cells.
Fig. 2. Cell cycle arrest and cyclin D1 degradation induced by NiCl2, but not NiS, in lung A549 cells.
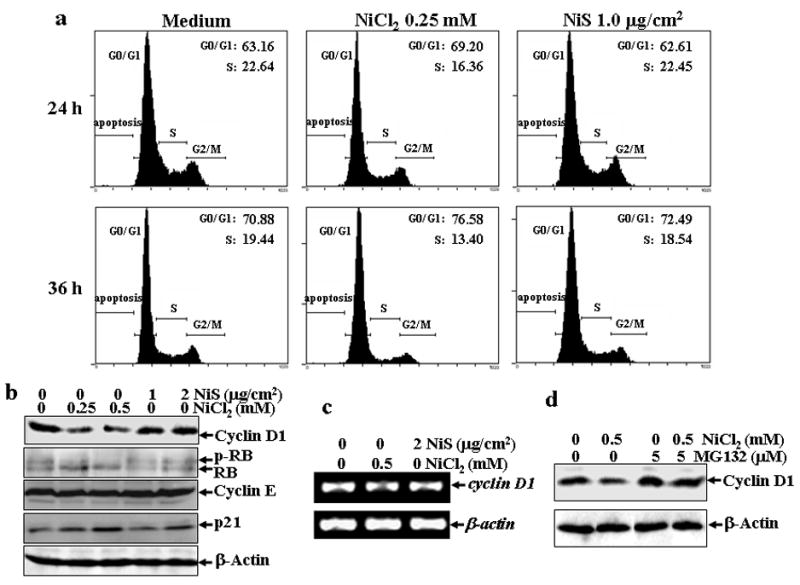
(a), A549 cells were seeded into each well of 6-well plates and cultured in Ham’s F-12K containing 10% FBS. After the cell density reached 70–80%, the cells were exposed to 0.25 mM NiCl2 or 1 μg/cm2 NiS for 24 or 36 hrs, and then were fixed and stained with propidium iodide as described in Materials and Methods. Cell cycle distribution was determined by flow cytometry. The data presented in this figure indicating the percentage of G0/G1 and S phases was one representative of three independent experiments. (b and d), The cells were treated with various concentrations of nickel compounds as indicated for 24 h (b), or 0.5 mM of NiCl2 in the presence or absence of MG132 for 12 h (d), and then extracted with SDS-sample buffer and Western blot was carried out. β-actin was used as a control for protein loading control. (c), The cells were exposed to 0.5 mM NiCl2 or 2 μg/cm2 NiS for 12 h, and then extracted with Trizol reagent for the total RNA isolation. Cyclin D1 was amplified with its specific primers by RT-PCR for 25 cycles. β-actin was used as an internal control.
Eukaryotic cell cycle progression is stringently regulated by a series of proteins. Cyclin D1 is one of the key regulators of G1/S transition (Serrano et al., 1993). To test whether the G1/G0 arrest in NiCl2-exposed A549 cells is due to change of cyclin D1 protein level, we analyzed the cyclin D1 protein levels in the A549 cells exposed to nickel compounds. As shown in Fig. 2b, the treatment of A549 cells with NiCl2 did cause a marked decrease in the cyclin D1 protein level, while the treatment of NiS only had a marginal effect. Consistently, the hypophosphorylation status of retinoblastoma (RB), the cyclin D1 downstream target which mediates G1/S phase transition by binding transcription factors such as E2F (Feliers et al., 2002), was observed following soluble nickel treatment, whereas insoluble exposure did not shown any inhibitory effects (Fig. 2b). Moreover, the other cell cycle regulating proteins, cyclin E and p21, were also detected. Cyclin E did not show any observed changes after nickel exposure, while p21 protein levels were increased by both nickel compounds even though insoluble nickel had much less effect (Fig. 2b). The decrease in the cyclin D1 protein levels seems to be caused by the proteasome-dependent proteolysis rather than the inhibition of the transcription, because the treatment of NiCl2 did not affect cyclin D1 mRNA level (Fig. 2c), while the co-incubation of cells with MG132, a chemical proteasome inhibitor, could reverse the down-regulation of cyclin D1 protein level caused by NiCl2 (Fig. 2d). These results indicate that soluble nickel and insoluble nickel compounds have differential effects on the down-regulation of cyclin D1 expression mediated by a proteasome-dependent cyclin D1 degradation.
Exposure of MEFs to soluble nickel compound also caused cell growth arrest and cyclin D1 degradation
The Cyclin D1 protein degradation has been shown to be regulated by selective kinases and transcription factors. Since MEFs with specific targeted gene knockouts are optimal cell models for the investigation of the kinase and transcription factor involved in the down-regulation of cyclin D1 by NiCl2, we examined the cell growth, cell cycle progression and cyclin D1 expression level in the MEFs exposed to NiCl2. The results indicated that the treatment of MEFs with NiCl2, but not NiS, also caused a cell growth arrest (Figs. 3a and 3b), a G1/G0 arrest (Fig. 4a) and a marked decrease in cyclin D1 protein level (Fig. 4b), but did not affect its mRNA level (Fig. 4c). These results are consistent with the observations in A549 cells, further confirming the effect of soluble nickel compounds on cell growth and cyclin D1 degradation and suggesting that the effect may have no cell-type specificity again.
Fig. 3. Inhibition of MEFs growth by NiCl2.
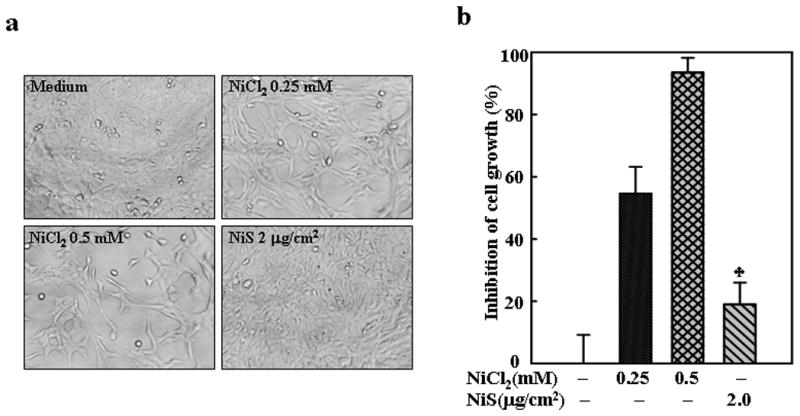
1 × 103 of MEFs were seeded into each well of 96-well plates, cultured in 10% FBS DMEM overnight, and then exposed to NiCl2 or NiS for 72 h. The cells were photographed under microscopy (a), and then extracted by lysis buffer for growth measurement using CellTiter-Glo® Luminescent Cell Viability Assay kit with a luminometer (b). The results are expressed as inhibition of cell growth which was calculated as described in Material and Methods. The symbol (♣) indicates a significant different from NiCl2 group (p< 0.05).
Fig. 4. Cell cycle arrest and cyclin D1 degradation induced by NiCl2 in MEFs.
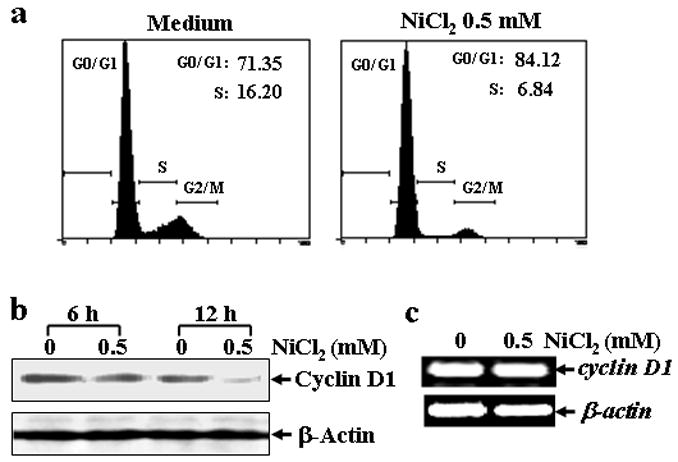
(a), MEFs were seeded into each well of a 6-well plates and cultured in DMEM containing 10% FBS at 37°C overnight. After the cell density reached 70–80%, the cells were exposed to 0.5 mM NiCl2 for 36 hrs, and then fixed and stained with propidium iodide. The cell cycle distribution was determined by flow cytometry. The data presented in this figure indicating the percentage of G0/G1 and S phases was one representative of three independent experiments. (b and c), MEFs were seeded into each well of 6-well plates, and cultured at 37°C overnight. The cells were treated with 0.5 mM NiCl2 for 6 or 12 hrs, washed once with ice-cold PBS and then extracted with SDS-sample buffer. The cyclin D1 protein levels in the extracts were analyzed by Western blot (b). The cells were treated with 0.5 mM NiCl2 for 12 hrs, and then extracted with Trizol reagent for the total RNA isolation. Cyclin D1 was amplified (c).
HIF-1α is not involved in cyclin D1 degradation caused by soluble nickel compounds
Since NiCl2 has a similar effect on cell growth and cyclin D1 down-regulation in MEFs and A549 cells, we employed the MEFs with specific gene deficiency to study the molecular mechanisms of the cyclin D1 degradation by NiCl2. Since HIF-1α is a key transcription factor mediating the biological functions of nickel compounds (Li et al., 2004; Salnikow et al., 2000), which is also reported to be involved in the regulation of cyclin D1 stability, we first examined the potential involvement of HIF-1α in the degradation of cyclin D1 upon NiCl2 exposure. Consistent with previous reports, exposure of MEFs to both NiCl2 and NiS caused a significant accumulation of HIF-1α to a similar extent (Fig. 5a). Nonetheless, HIF-1α was not required for the down-regulation of cyclin D1 in NiCl2-exposed cells because treatment of NiCl2 caused a significant decrease in cyclin D1 in both HIF-1α+/+ and HIF-1α−/− MEFs to a similar extent (Figs. 5b and 5c).
Fig. 5. HIF-1α is not involved in cyclin D1 degradation induced by NiCl2.
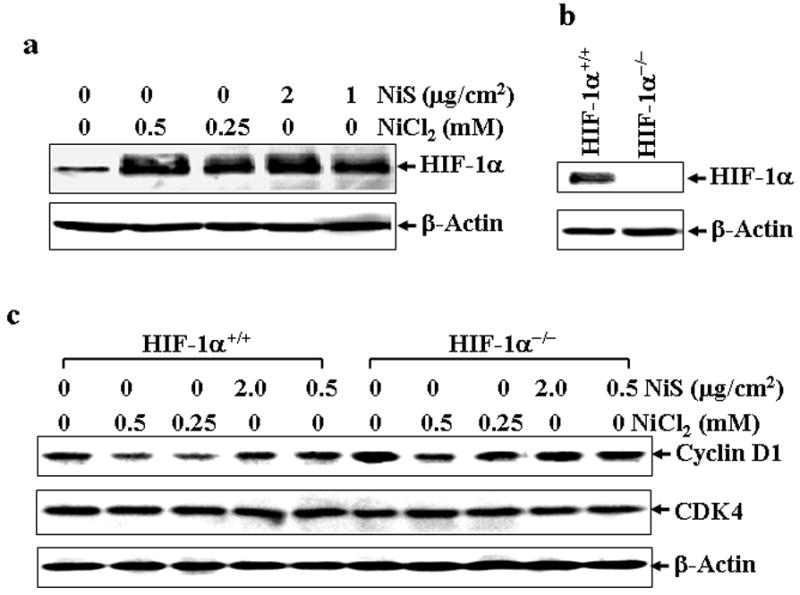
HIF-1α+/+ MEFs were seeded into each well of 6-well plates, and cultured in 10% FBS DMEM overnight and exposed to various concentrations of nickel compounds as indicated for 12 hrs, and then subjected to Western blot assay (a). HIF-1α−/− MEFs were identified using HIF-1α antibody (b). The effect of nickel compounds on cyclin D1 protein expression were compared between HIF-1α+/+ MEFs and HIF-1α−/− MEFs by Western blot assay. CDK4 and β-Actin were used as controls (c).
IKKα is required for cyclin D1 degradation and growth arrest caused by soluble nickel components
Phosphorylation at Thr286 and proteasomal degradation of cyclin D1 is considered as a prerequisite for normal progression of the S-phase (Diehl et al., 1997; Mukherji et al., 2008). Therefore, we detected cyclin D1 phosphorylation status following NiCl2 treatment. As shown in Fig. 6a, NiCl2 treatment in combination with MG132 could increase cyclin D1 phosphorylation at Thr286, while NiCl2 alone down-regulated phosphorylation at Thr286, which might be due to the decreased total cyclin D1 expression. Most recently, Kwak et al. has reported that IKKα plays a role in cyclin D1 turnover through the phosphorylation at Thr286 (Kwak et al., 2005). In view of our result that nickel compounds can activate NF-κB that is the downstream molecule of IKK complex (Chen et al., 2001; Cruz et al., 2004; Huang et al., 2002; Zandi et al., 1997), we examined the cyclin D1 protein levels in wild type (WT) and IKKα−/− MEFs (Fig. 6b) after exposure to various concentrations of nickel compounds. The results showed that treatment of NiCl2 caused a significant cyclin D1 decrease in WT MEFs, but did not cause any observed down-regulation of cyclin D1 in IKKα−/− MEFs (Fig. 6c). Again, the treatment of NiS has only a marginal effect on the cyclin D1 levels in both WT and IKKα−/− MEFs (Fig. 6c). Then we compared the subcellular distribution of cyclin D1 protein in WT and IKKα−/− MEFs following nickel treatment, since it was reported that IKKα-dependent cyclin D1 phosphorylation is responsible for cyclin D1 protein translocation from nuclear to cytosol where it is subjected to degradation (Cao et al., 2001; Kwak et al., 2005). As shown in Fig. 6d, cyclin D1 located mainly in cytosol rather than the nuclear part in both type of cells in absence of nickel treatment. Moreover, in WT cells nickel exposure triggered cyclin D1 protein decline in both cytosol and nuclear parts, which was blunt in IKKα−/− cells. These results strongly indicate that IKKα is required for the degradation of cyclin D1 by NiCl2.
Fig. 6. IKKα is a mediator for cyclin D1 degradation and cell cycle arrest induced by NiCl2.
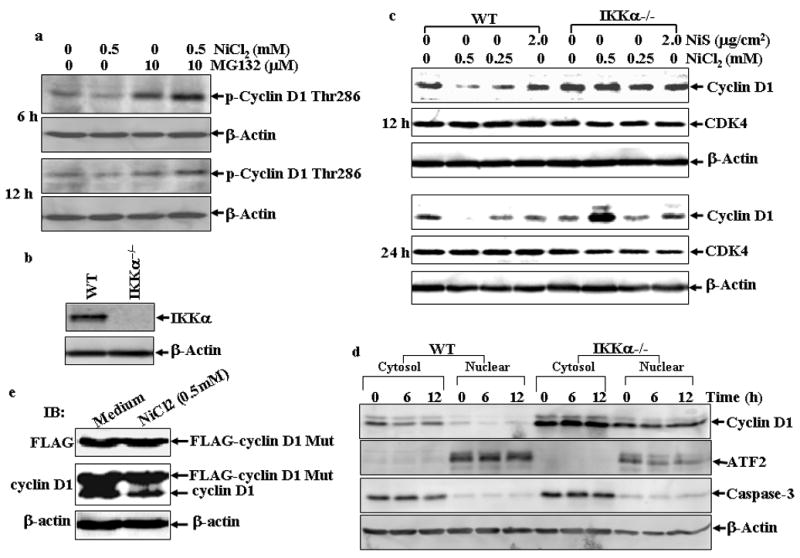
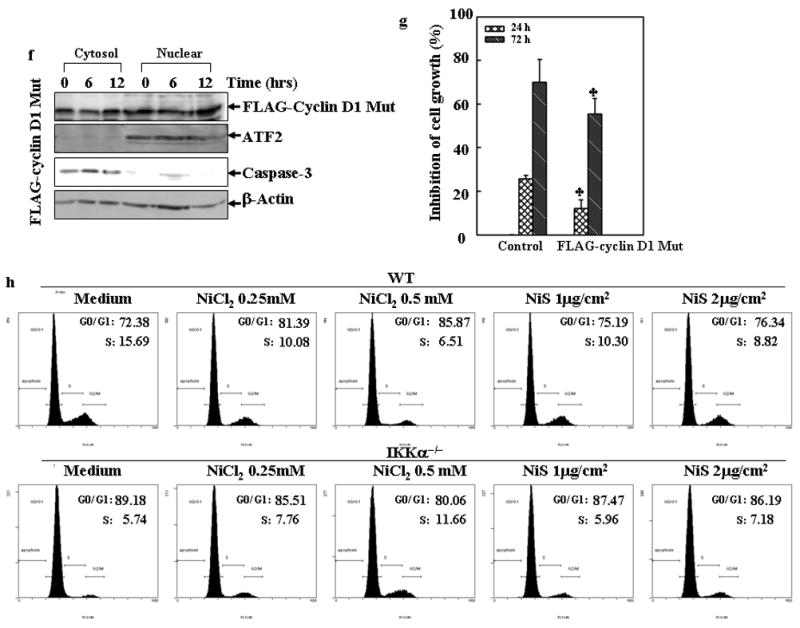
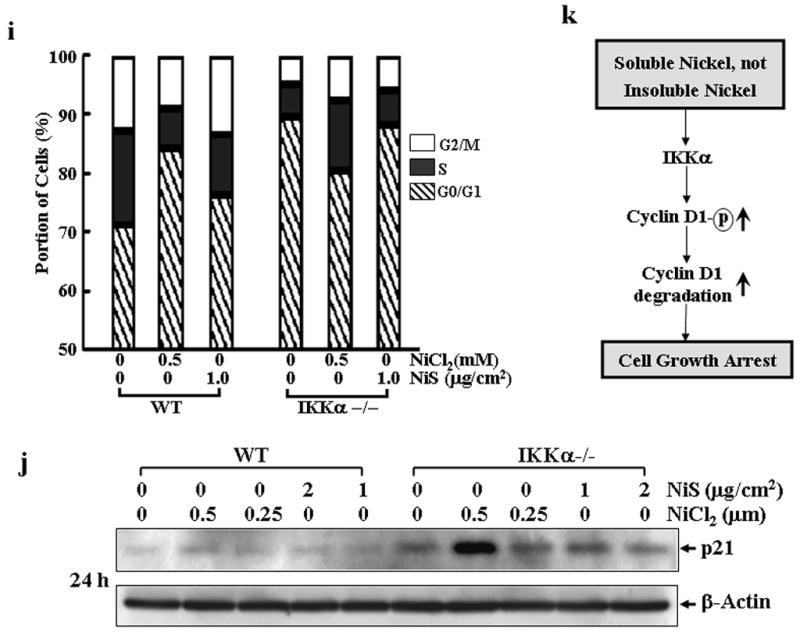
(a), A549 cells were treated with 0.5 mM of NiCl2 in the presence or absence of MG132 for 6 h and 12 h, and then extracted with SDS-sample buffer and Western blot was carried out. (b), IKKα−/− MEFs were identified by Western blot. (c), WT MEFs and IKKα−/− MEFs were seeded into each well of 6-well plates and cultured in DMEM containing 10% FBS at 37°C overnight. The cells were exposed to various concentrations of nickel compounds for 12 or 24 hrs, and then extracted with SDS-sample buffer. The cyclin D1 protein levels in the extracts were analyzed by Western blot. CDK4 and β-Actin were used as controls. (d), WT MEFs and IKKα−/− MEFs were seeded into 10-cm dishes and exposed to various concentrations of nickel compounds for 6 or 12 h, and then the nuclear and cytosol parts were extracted as described in “Materials and Methods”. The cyclin D1 protein levels in the extracts were analyzed by Western blot. ATF2 and caspase-3 were used as nuclear and cytosol marks individually. (e–g), 293T cells were transiently transfected with FLAG-cyclin D1 T286A expression vector as described in “Material and Methods”. 24 hrs after the transfection, the cells were treated with 0.5 mM NiCl2 for 12 hrs and then the FLAG-cyclin D1 T286A mutant and endogenous cyclin D1 protein in the extracts were analyzed by Western blot assay with antibodies against FLAG and cyclin D1 (e). Then the subcellular location of the FLAG-cyclin D1 T286 mutant was detected (f). The growth inhibitory effect of NiCl2, was compared between the vector control and FLAG-cyclin D1 T286 mutant cells. Each bar indicates the mean and standard deviation of triplicate assay wells. The symbol (♣) indicates a significant compared with vector control cells (p< 0.05) (g). (h–j), WT MEFs and IKKα−/− MEFs were seeded into each well of a 6-well plates and cultured in DMEM containing 10% FBS at 37°C overnight. The cells were exposed to various concentrations of nickel compounds as indicated for 24 hrs, and then fixed and stained with propidium iodide. The cell cycle distribution was determined by flow cytometry (h & i). The data presented in this figure indicating the percentage of G0/G1 and S phases was one representative of three independent experiments, or extracted with SDS sample buffer, and the cell extracts were analyzed by Western blot for p21 protein expression (j). β-Actin was used as protein loading control. (k), The overall scheme of the inhibitory effect on cell growth by soluble, but not insoluble nickel compounds.
Given that the regulation of cyclin D1 turnover by IKKα is through the phosphorylation of Thr 286 residue, we examined whether point mutation at T286A of cyclin D1 can prevent the degradation of cyclin D1 by NiCl2. As shown in Fig. 6e, the treatment of NiCl2 failed to cause the degradation of cyclin D1 with the point mutation though it caused marked endogenous cyclin D1 degradation. Consistently, the cyclin D1 protein degradation in both cytosol and nuclear parts was compromised by expression of cyclin D1 T286A mutant (Figs. 6d and 6f). Moreover, expression of cyclin D1 T286A mutant partially attenuated the growth inhibitory effect of NiCl2 (Fig. 6g). It may be noted that overexpression of cyclin D1 T286A mutant did not reverse the growth inhibitory effects of NiCl2 completely. The explanation for this might be due to either compromising effect of the endogenous cyclin D1 or other possibility that the growth inhibition by NiCl2 is largely, but not entirely, mediated by cyclin D1 downregulation.
Consistent with the failure to induce cyclin D1 degradation, NiCl2 did not induce an evident cell cycle arrest at G1/G0 phase in IKKα −/− MEFs (Figs. 6h and 6i). It may be notable that untreated IKKα−/− MEFs showed a much higher proportion of cells in the G1/G0 phase as compared with untreated WT MEFs (Figs. 6h and 6i), which seems contradictory to the higher stability of cyclin D1 protein level in IKKα−/− MEFs. This may be explained by the findings that cyclin D1 level oscillates during the cell cycle progression (Stacey, 2003). Cyclin D1 is induced specifically during the G2 phase, remains high through mitosis and into the G1 phase, and then declines as the cells enter the S phase. Although cyclin D1 is known to be critical for the transition of cell cycle from the G0/G1 to S phase, cyclin D1 levels must be low during the S phase, and the decline is a fundamental requirement for normal cell cycle progression (Stacey, 2003). Another explanation for the high cell proportion of IKKα−/− MEFs in the G0/G1 phase is that cyclin D1 can positively regulate the stability of p21 (Coleman et al., 2003), a key cyclin-dependent kinase inhibitor playing a role in the negative regulation of cell cycle progression from the G0/G1 to S phase, by forming complex with p21, which in turn blocks the binding of p21 with C8α subunit of the 20S proteasome complex. As shown in Fig. 6j, p21 protein level is much higher in IKKα−/− MEFs as compared with that in WT MEFs, which is consistent with the high level of cyclin D1 in IKKα−/− MEFs.
Discussion
Nickel compounds have been well documented as carcinogens in both humans and animals (IARC, 1990). Epidemiological studies have correlated occupational exposure to mixtures of insoluble and soluble nickel compounds with the increased incidence of respiratory, larynx, and nasal cancers (1990; IARC, 1990). Animal studies have shown that insoluble nickel compounds could cause significant tumor formations at the site of administration, whereas the soluble nickel compounds exhibited poor tumor induction (1990; IARC, 1990). Therefore, it is generally thought that insoluble nickel compounds are more potent carcinogens than the soluble. Although many studies have been carried out to explain the different carcinogenic activities of soluble and insoluble nickel compounds, including the studies on the entry and clearance of nickel (Benson et al., 1995), the genotoxicity (Danadevi et al., 2004), epigenetic alterations (Costa et al., 2005; Ke et al., 2006; Salnikow and Costa, 2000), the mechanisms are still not fully understood. In the present study, we demonstrated that soluble nickel compound, NiCl2, can induce a significant cell growth arrest and cyclin D1 down-regulation, while the insoluble nickel particle, NiS has only a marginal effect.
Cyclin D1 is regulated at both transcription and protein levels. The reduction of cyclin D1 induced by NiCl2 seems to be caused by proteolysis rather than inhibition of its transcription because the mRNA level of cyclin D1 did not show evident change after the treatment of NiCl2. Moreover, the pretreatment of MG132, a proteosome inhibitor can reverse the reduction of cyclin D1. Further, we demonstrated that the degradation of cyclin D1 by NiCl2 requires the IKKα rather than HIF-1α which are both reported to regulate cyclin D1 (Ghafar et al., 2003; Kwak et al., 2005).
Hypoxia-like response is one major consequence of nickel exposure, which is characterized by the accumulation of HIF-1α (Costa et al., 2005). HIF-1α is a transcription factor that was originally found to be activated in cells in response to hypoxia, which forms a heterodimer with HIF-1β, a constitutively expressed subunit in cells. Previous studies indicated that HIF-1α may be involved in deregulation of cyclin D1 level (Ghafar et al., 2003). However, in this study, HIF-1α was not required for the reduction of cyclin D1 by NiCl2 because exposure of HIF-1α−/− MEFs to NiCl2 still caused similar cyclin D1 reduction.
IKKα, one of the catalytic subunits of IKK complex, primarily mediates the NF-κB activation. Recently, Kwak et al demonstrated that IKKα can interact with and phosphorylate cyclin D1 at Thr286 residue within the PEST domain (Kwak et al., 2005). IKKα−/− cells showed exclusive nuclear localization and increased stability of cyclin D1 protein compared with parental MEF cells (Kwak et al., 2005). In this study, we demonstrated that IKKα was also required for the degradation of cyclin D1 by NiCl2 because the level of cyclin D1 in IKKα −/− cells did not show any reduction after the exposure to NiCl2, while the level of cyclin D1 in the parental cells showed a significant reduction. Consistently, the cyclin D1 with T286A point mutation is also resistant to the degradation caused by NiCl2. However, the grwoth inhibitory effect of NiCl2 was only partially rescued by cyclin D1 mutant, which could be explained by either compromising effect of the endogenous cyclin D1 or other possibility that the growth inhibition by NiCl2 is largely, but not entirely, mediated by cyclin D1 downregulation.
Interestingly, GSK-3β is also reported to regulate subcellular localization, ubiquitination, and proteolysis of cyclin D1 (Alt et al., 2000; Benzeno and Diehl, 2004; Diehl and Sherr, 1997). The previous studies suggest that GSK-3β is the physiological kinase mediating cyclin D1 phosphorylation at Thr286 (Alt et al., 2000). In this study, however, we failed to get convincing evidence to show the requirement of GSK3β for cyclin D1 degradation by NiCl2, because exposure of cells to NiCl2 did not induce the de-phosphorylation of GSK3β, which is required for its activation (Data not shown). In contrast, Akt, the upstream kinase of GSK3β, has been shown to be activated by the exposure of nickel compounds in our previous study in mouse epidermal Cl41 cells (Li et al., 2004), which may in turn phosphorylate and inactivate GSK3β (Cross et al., 1995). In addition, the finding that transfection of dominant negative mutant of GSK3β can not inhibit the reduction of cyclin D1 by NiCl2 (Data not shown) also suggests that GSK3β is not required for the degradation of cyclin D1 in the cell exposed to NiCl2.
Eukaryotic cell growth is stringently controlled by the well-defined cell cycle consisting of four distinct stages, G1, S, G2, and M. The cell cycle progression is mainly controlled by several key checkpoints, including the G1/S checkpoint, S-phase DNA damage checkpoint and G2/M spindle integrity checkpoint. Previous studies have demonstrated that faulty G1/S control plays a critical role in tumorigenesis. Cyclin D1 is one of the key regulators of G1/S transition (Johnson and Walker, 1999; Kastan and Bartek, 2004; Massague, 2004). Inducible cyclin D1 forms a complex with CDK4/6 which phosphorylate the retinoblastoma (Rb) tumor suppressor protein, helping to sequestrate its growth-inhibitory effects and enabling E2F transcription factors to activate genes required for entry into the DNA synthetic phase (S) of the cell division cycle. Accordingly, cyclin D1 is considered as a oncoprotein, and it has been demonstrated that the antisense to cyclin D1 is able to inhibit the growth and tumorigenicity of human colon cancer cells and induce apoptosis in human squamous carcinomas (Arber et al., 1997; Sauter et al., 1999). In this study, we demonstrated that exposure of cells to soluble nickel compound, but not insoluble nickel compound, caused a marked reduction of cyclin D1, which is consistent with their effects on cell cycle alternation. Given the indispensability of cell growth during carcinogenesis, these differences may be at least partially associated with their differential carcinogenic activities.
A critical issue with regard to this study is that the events mediate the different effects of soluble and insoluble nickel compounds on cell growth and cyclin D1 downregulation. Because we treated the cells without any wash, it seems that the persistence of nickel ion in the cells can not be used to explain the difference. Although it is well established that soluble and insoluble nickel compounds exhibit different entry and clearance rate in lungs of animal model, there is no clearance in the cell culture model. Moreover, constant exposure of cells to soluble and insoluble nickel compounds have showed similar profiles of consequences, including signal pathway activations (Huang et al., 2002; Li et al., 2004), target gene expressions (Salnikow et al., 2000; Salnikow et al., 2003a), epigenetic alterations (Ke et al., 2006) and cell transformations (Biedermann and Landolph, 1987; Trott et al., 1995). In our studies, we have also showed that exposure of cells to NiCl2 and NiS can induce NF-κB activation and TNFα expression to similar extents (Supplementary Fig. 1), so the difference can not attribute to the possibility of defective function of NiS. In addition, Ni3S2, another insoluble nickel compound, exhibited a similar defective effect on cyclin D1 downregulation, but an intact function in HIF-1α and CAP43 induction as compared with soluble nickel compound. Given that the degradation of cyclin D1 by NiCl2 requires IKKα, it is possible that soluble and insoluble nickel compounds may have different potency to activate IKKα. However, we failed to see any convincing difference by testing IKKα phosphorylation between the cells exposed to NiCl2 and NiS. In view of that the degradation of cyclin D1 is an ubiquitination- and proteosome-dependent proteolysis processes, it is possible that soluble and insoluble nickel compounds can differentially regulate this process. This possibility is being tested in our laboratory.
In summary, we demonstrate that soluble, but not insoluble nickel compounds are able to cause a significant cell growth arrest, which results from cyclin D1 degradation in an IKKα-dependent manner as shown in Fig. 6k. These differences may at least in part accounts for their different carcinogenic activities.
Acknowledgments
This work was supported in part by grants from NIH/NCI CA112557, CA094964, and CA103180, and from NIH/NIEHS ES012451. We appreciated Dr. Zheng-gang Liu (Cell and Cancer Biology Branch, Center for Cancer Research, National Cancer Institute, NIH) for providing us the IKKα−/−mouse embryo fibroblast.
References
- Report of the International Committee on Nickel Carcinogenesis in Man. Scand J Work Environ Health. 1990;16(1 Spec No):1–82. doi: 10.5271/sjweh.1813. [DOI] [PubMed] [Google Scholar]
- ACGIH. Threshold Limit Values and Biological Exposure Indices for Chemical Substances and Physical Agents. American Conference of Governmental Industrial Hygienists; Cincinnati, OH. 1998. [Google Scholar]
- Alt JR, Cleveland JL, Hannink M, Diehl JA. Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes Dev. 2000;14(24):3102–3114. doi: 10.1101/gad.854900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arber N, Doki Y, Han EK, Sgambato A, Zhou P, Kim NH, Delohery T, Klein MG, Holt PR, Weinstein IB. Antisense to cyclin D1 inhibits the growth and tumorigenicity of human colon cancer cells. Cancer Res. 1997;57(8):1569–1574. [PubMed] [Google Scholar]
- Benson JM, Chang IY, Cheng YS, Hahn FF, Kennedy CH, Barr EB, Maples KR, Snipes MB. Particle clearance and histopathology in lungs of F344/N rats and B6C3F1 mice inhaling nickel oxide or nickel sulfate. Fundam Appl Toxicol. 1995;28(2):232–244. doi: 10.1006/faat.1995.1164. [DOI] [PubMed] [Google Scholar]
- Benzeno S, Diehl JA. C-terminal sequences direct cyclin D1-CRM1 binding. J Biol Chem. 2004;279(53):56061–56066. doi: 10.1074/jbc.M411910200. [DOI] [PubMed] [Google Scholar]
- Biedermann KA, Landolph JR. Induction of anchorage independence in human diploid foreskin fibroblasts by carcinogenic metal salts. Cancer Res. 1987;47(14):3815–3823. [PubMed] [Google Scholar]
- Cao Y, Bonizzi G, Seagroves T, Greten F, Johnson R, Schmidt E, Karin M. IKKalpha provides an essential link between RANK signaling and cyclin D1 expression during mammary gland development. Cell. 2001;107(6):763–775. doi: 10.1016/s0092-8674(01)00599-2. [DOI] [PubMed] [Google Scholar]
- Charnley G, Wilson JD. Evaluation of the form of the cell growth rate function of the two-stage model for carcinogenesis. Prog Clin Biol Res. 1991;369:291–301. [PubMed] [Google Scholar]
- Chen F, Ding M, Castranova V, Shi X. Carcinogenic metals and NF-kappaB activation. Mol Cell Biochem. 2001;222(1–2):159–171. [PubMed] [Google Scholar]
- Cohen SM, Ellwein LB. Cell growth dynamics in long-term bladder carcinogenesis. Toxicol Lett. 1988;43(1–3):151–173. doi: 10.1016/0378-4274(88)90026-4. [DOI] [PubMed] [Google Scholar]
- Coleman ML, Marshall CJ, Olson MF. Ras promotes p21(Waf1/Cip1) protein stability via a cyclin D1-imposed block in proteasome-mediated degradation. Embo J. 2003;22(9):2036–2046. doi: 10.1093/emboj/cdg189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa M, Davidson TL, Chen H, Ke Q, Zhang P, Yan Y, Huang C, Kluz T. Nickel carcinogenesis: epigenetics and hypoxia signaling. Mutat Res. 2005;592(1–2):79–88. doi: 10.1016/j.mrfmmm.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378(6559):785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Cruz MT, Goncalo M, Figueiredo A, Carvalho AP, Duarte CB, Lopes MC. Contact sensitizer nickel sulfate activates the transcription factors NF-kB and AP-1 and increases the expression of nitric oxide synthase in a skin dendritic cell line. Exp Dermatol. 2004;13(1):18–26. doi: 10.1111/j.0906-6705.2004.00105.x. [DOI] [PubMed] [Google Scholar]
- Danadevi K, Rozati R, Banu BS, Grover P. Genotoxic evaluation of welders occupationally exposed to chromium and nickel using the Comet and micronucleus assays. Mutagenesis. 2004;19(1):35–41. doi: 10.1093/mutage/geh001. [DOI] [PubMed] [Google Scholar]
- Devin A, Lin Y, Yamaoka S, Li Z, Karin M, Liu Z. The alpha and beta subunits of IkappaB kinase (IKK) mediate TRAF2-dependent IKK recruitment to tumor necrosis factor (TNF) receptor 1 in response to TNF. Mol Cell Biol. 2001;21(12):3986–3994. doi: 10.1128/MCB.21.12.3986-3994.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl JA, Sherr CJ. A dominant-negative cyclin D1 mutant prevents nuclear import of cyclin-dependent kinase 4 (CDK4) and its phosphorylation by CDK-activating kinase. Mol Cell Biol. 1997;17(12):7362–7374. doi: 10.1128/mcb.17.12.7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 1997;11(8):957–972. doi: 10.1101/gad.11.8.957. [DOI] [PubMed] [Google Scholar]
- Feliers D, Frank MA, Riley DJ. Activation of Cyclin D1-Cdk4 and Cdk4-Directed Phosphorylation of RB Protein in Diabetic Mesangial Hypertrophy. Diabetes. 2002;51(11):3290–3299. doi: 10.2337/diabetes.51.11.3290. [DOI] [PubMed] [Google Scholar]
- Ghafar MA, Anastasiadis AG, Chen MW, Burchardt M, Olsson LE, Xie H, Benson MC, Buttyan R. Acute hypoxia increases the aggressive characteristics and survival properties of prostate cancer cells. Prostate. 2003;54(1):58–67. doi: 10.1002/pros.10162. [DOI] [PubMed] [Google Scholar]
- Haber LT, Diamond GL, Zhao Q, Erdreich L, Dourson ML. Hazard identification and dose response of ingested nickel-soluble salts. Regul Toxicol Pharmacol. 2000a;31(2 Pt 1):231–241. doi: 10.1006/rtph.2000.1378. [DOI] [PubMed] [Google Scholar]
- Haber LT, Erdreicht L, Diamond GL, Maier AM, Ratney R, Zhao Q, Dourson ML. Hazard identification and dose response of inhaled nickel-soluble salts. Regul Toxicol Pharmacol. 2000b;31(2 Pt 1):210–230. doi: 10.1006/rtph.2000.1377. [DOI] [PubMed] [Google Scholar]
- Hecker E. Three stage carcinogenesis in mouse skin--recent results and present status of an advanced model system of chemical carcinogenesis. Toxicol Pathol. 1987;15(2):245–258. doi: 10.1177/019262338701500221. [DOI] [PubMed] [Google Scholar]
- Huang C, Li J, Costa M, Zhang Z, Leonard SS, Castranova V, Vallyathan V, Ju G, Shi X. Hydrogen peroxide mediates activation of nuclear factor of activated T cells (NFAT) by nickel subsulfide. Cancer Res. 2001;61(22):8051–8057. [PubMed] [Google Scholar]
- Huang Y, Davidson G, Li J, Yan Y, Chen F, Costa M, Chen LC, Huang C. Activation of nuclear factor-kappaB and not activator protein-1 in cellular response to nickel compounds. Environ Health Perspect. 2002;110(Suppl 5):835–839. doi: 10.1289/ehp.02110s5835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IARC. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans Chromium, Nickel and Welding. WHO International Agency for Research on Cancer; Lyon, France. 1990. [PMC free article] [PubMed] [Google Scholar]
- Johnson DG, Walker CL. Cyclins and cell cycle checkpoints. Annu Rev Pharmacol Toxicol. 1999;39:295–312. doi: 10.1146/annurev.pharmtox.39.1.295. [DOI] [PubMed] [Google Scholar]
- Kasprzak KS, Gabryel P, Jarczewska K. Carcinogenicity of nickel(II)hydroxides and nickel(II)sulfate in Wistar rats and its relation to the in vitro dissolution rates. Carcinogenesis. 1983;4(3):275–279. doi: 10.1093/carcin/4.3.275. [DOI] [PubMed] [Google Scholar]
- Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432(7015):316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- Ke Q, Davidson T, Chen H, Kluz T, Costa M. Alterations of histone modifications and transgene silencing by nickel chloride. Carcinogenesis. 2006;27(7):1481–1488. doi: 10.1093/carcin/bgl004. [DOI] [PubMed] [Google Scholar]
- Kwak YT, Li R, Becerra CR, Tripathy D, Frenkel EP, Verma UN. IkappaB kinase alpha regulates subcellular distribution and turnover of cyclin D1 by phosphorylation. J Biol Chem. 2005;280(40):33945–33952. doi: 10.1074/jbc.M506206200. [DOI] [PubMed] [Google Scholar]
- Landolph JR. Role of free radicals in metal-induced carcinogenesis. Met Ions Biol Syst. 1999;36:445–483. [PubMed] [Google Scholar]
- Li J, Davidson G, Huang Y, Jiang BH, Shi X, Costa M, Huang C. Nickel compounds act through phosphatidylinositol-3-kinase/Akt-dependent, p70(S6k)-independent pathway to induce hypoxia inducible factor transactivation and Cap43 expression in mouse epidermal Cl41 cells. Cancer Res. 2004;64(1):94–101. doi: 10.1158/0008-5472.can-03-0737. [DOI] [PubMed] [Google Scholar]
- Lu H, Shi X, Costa M, Huang C. Carcinogenic effect of nickel compounds. Mol Cell Biochem. 2005;279(1–2):45–67. doi: 10.1007/s11010-005-8215-2. [DOI] [PubMed] [Google Scholar]
- Massague J. G1 cell-cycle control and cancer. Nature. 2004;432(7015):298–306. doi: 10.1038/nature03094. [DOI] [PubMed] [Google Scholar]
- Mukherji A, Janbandhu VC, Kumar V. GSK-3[beta]-dependent destabilization of cyclin D1 mediates replicational stress-induced arrest of cell cycle. FEBS Letters. 2008;582(7):1111–1116. doi: 10.1016/j.febslet.2008.02.068. [DOI] [PubMed] [Google Scholar]
- National-Toxicology-Program. NTP Toxicology and Carcinogenesis Studies of Nickel Oxide (CAS No. 1313–99–1) in F344 Rats and B6C3F1 Mice (Inhalation Studies) Natl Toxicol Program Tech Rep Ser. 1996a;451:1–381. [PubMed] [Google Scholar]
- National-Toxicology-Program. NTP Toxicology and Carcinogenesis Studies of Nickel Subsulfide (CAS No. 12035–72–2) in F344 Rats and B6C3F1 Mice (Inhalation Studies) Natl Toxicol Program Tech Rep Ser. 1996b;453:1–365. [PubMed] [Google Scholar]
- National-Toxicology-Program. NTP Toxicology and Carcinogenesis Studies of Nickel Sulfate Hexahydrate (CAS No. 10101–97–0) in F344 Rats and B6C3F1 Mice (Inhalation Studies) Natl Toxicol Program Tech Rep Ser. 1996c;454:1–380. [PubMed] [Google Scholar]
- Oller AR. Respiratory carcinogenicity assessment of soluble nickel compounds. Environ Health Perspect. 2002;110(Suppl 5):841–844. doi: 10.1289/ehp.02110s5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oller AR, Costa M, Oberdorster G. Carcinogenicity assessment of selected nickel compounds. Toxicol Appl Pharmacol. 1997;143(1):152–166. doi: 10.1006/taap.1996.8075. [DOI] [PubMed] [Google Scholar]
- Ottolenghi AD, Haseman JK, Payne WW, Falk HL, MacFarland HN. Inhalation studies of nickel sulfide in pulmonary carcinogenesis of rats. J Natl Cancer Inst. 1975;54(5):1165–1172. doi: 10.1093/jnci/54.5.1165. [DOI] [PubMed] [Google Scholar]
- Ouyang W, L J, Ma Q, Huang C. Essential roles of PI-3K/Akt/IKK{beta}/NF{kappa}B pathway in cyclin D1 induction by arsenite in JB6 Cl41 cells. Carcinogenesis. 2006a;27(4):864–873. doi: 10.1093/carcin/bgi321. [DOI] [PubMed] [Google Scholar]
- Ouyang W, Li J, Ma Q, Huang C. Essential roles of PI-3K/Akt/IKKbeta/NFkappaB pathway in cyclin D1 induction by arsenite in JB6 Cl41 cells. Carcinogenesis. 2006b;27(4):864–873. doi: 10.1093/carcin/bgi321. [DOI] [PubMed] [Google Scholar]
- Ouyang W, Ma Q, Li J, Zhang D, Liu ZG, Rustgi AK, Huang C. Cyclin D1 induction through IkappaB kinase beta/nuclear factor-kappaB pathway is responsible for arsenite-induced increased cell cycle G1-S phase transition in human keratinocytes. Cancer Res. 2005;65(20):9287–9293. doi: 10.1158/0008-5472.CAN-05-0469. [DOI] [PubMed] [Google Scholar]
- Ramirez RD, Sheridan S, Girard L, Sato M, Kim Y, Pollack J, Peyton M, Zou Y, Kurie JM, DiMaio JM, Milchgrub S, Smith AL, Souza RF, Gilbey L, Zhang X, Gandia K, Vaughan MB, Wright WE, Gazdar AF, Shay JW, Minna JD. Immortalization of Human Bronchial Epithelial Cells in the Absence of Viral Oncoproteins. Cancer Res. 2004;64(24):9027–9034. doi: 10.1158/0008-5472.CAN-04-3703. [DOI] [PubMed] [Google Scholar]
- Ryan HE, Lo J, Johnson RS. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. Embo J. 1998;17(11):3005–3015. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salnikow K, Blagosklonny MV, Ryan H, Johnson R, Costa M. Carcinogenic nickel induces genes involved with hypoxic stress. Cancer Res. 2000;60(1):38–41. [PubMed] [Google Scholar]
- Salnikow K, Costa M. Epigenetic mechanisms of nickel carcinogenesis. J Environ Pathol Toxicol Oncol. 2000;19(3):307–318. [PubMed] [Google Scholar]
- Salnikow K, Davidson T, Kluz T, Chen H, Zhou D, Costa M. GeneChip analysis of signaling pathways effected by nickel. J Environ Monit. 2003a;5(2):206–209. doi: 10.1039/b210262p. [DOI] [PubMed] [Google Scholar]
- Salnikow K, Davidson T, Zhang Q, Chen LC, Su W, Costa M. The involvement of hypoxia-inducible transcription factor-1-dependent pathway in nickel carcinogenesis. Cancer Res. 2003b;63(13):3524–3530. [PubMed] [Google Scholar]
- Sauter ER, Nesbit M, Litwin S, Klein-Szanto AJ, Cheffetz S, Herlyn M. Antisense cyclin D1 induces apoptosis and tumor shrinkage in human squamous carcinomas. Cancer Res. 1999;59(19):4876–4881. [PubMed] [Google Scholar]
- Schroeder HA, Mitchener M. Life-term effects of mercury, methyl mercury, and nine other trace metals on mice. J Nutr. 1975;105(4):452–458. doi: 10.1093/jn/105.4.452. [DOI] [PubMed] [Google Scholar]
- Schroeder HA, Mitchener M, Nason AP. Life-term effects of nickel in rats: survival, tumors, interactions with trace elements and tissue levels. J Nutr. 1974;104(2):239–243. doi: 10.1093/jn/104.2.239. [DOI] [PubMed] [Google Scholar]
- Seilkop SK, Oller AR. Respiratory cancer risks associated with low-level nickel exposure: an integrated assessment based on animal, epidemiological, and mechanistic data. Regul Toxicol Pharmacol. 2003;37(2):173–190. doi: 10.1016/s0273-2300(02)00029-6. [DOI] [PubMed] [Google Scholar]
- Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366(6456):704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- Stacey DW. Cyclin D1 serves as a cell cycle regulatory switch in actively proliferating cells. Curr Opin Cell Biol. 2003;15(2):158–163. doi: 10.1016/s0955-0674(03)00008-5. [DOI] [PubMed] [Google Scholar]
- Sunderman FW., Jr Mechanisms of nickel carcinogenesis. Scand J Work Environ Health. 1989;15(1):1–12. doi: 10.5271/sjweh.1888. [DOI] [PubMed] [Google Scholar]
- Sunderman FW., Jr Search for molecular mechanisms in the genotoxicity of nickel. Scand J Work Environ Health. 1993;19(Suppl 1):75–80. [PubMed] [Google Scholar]
- Sunderman FW, Jr, Maenza RM. Comparisons of carcinogenicities of nickel compounds in rats. Res Commun Chem Pathol Pharmacol. 1976;14(2):319–330. [PubMed] [Google Scholar]
- Sutherland JE, Costa M. Epigenetics and the environment. Ann N Y Acad Sci. 2003;983:151–160. doi: 10.1111/j.1749-6632.2003.tb05970.x. [DOI] [PubMed] [Google Scholar]
- Trott DA, Cuthbert AP, Overell RW, Russo I, Newbold RF. Mechanisms involved in the immortalization of mammalian cells by ionizing radiation and chemical carcinogens. Carcinogenesis. 1995;16(2):193–204. doi: 10.1093/carcin/16.2.193. [DOI] [PubMed] [Google Scholar]
- Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell. 1997;91(2):243–252. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- Zoumpourlis V, Solakidi S, Papathoma A, Papaevangeliou D. Alterations in signal transduction pathways implicated in tumour progression during multistage mouse skin carcinogenesis. Carcinogenesis. 2003;24(7):1159–1165. doi: 10.1093/carcin/bgg067. [DOI] [PubMed] [Google Scholar]