

Abstract
Many diseases are easier to treat and control when detected at an early stage of disease progression. Often, disease-related antigens or biomarkers are shed from the primary site and present in the blood. Unfortunately, there are very few tests capable of detecting these rare biomarkers in the blood. A blood test would be very useful to diagnose the disease earlier, monitor effectiveness of treatments, predict recurrence, and monitor recurrence. There is certainly a need to develop assays that are ultra-sensitive, non-invasive and high-throughput. Here we describe several highly sensitive immunological assays we have developed to detect rare serum antigens. Initially we created an assay named immunodetection amplified by T7 RNA polymerase (IDAT). To enhance the effectiveness and streamline the procedure, this assay was amended to the facile amplification system termed fluorescent amplification catalyzed by T7 polymerase technique (FACTT). These assays have been used to analyze the tumor antigen HER2 and the prion protein PrPSc. They can also be applied to other tumor markers or antigens from a variety of diseases such as cardiovascular disease, rheumatoid arthritis, Alzheimer’s disease, Parkinson’s disease, and hepatitis. These tests are not limited to testing only serum, but may also be applicable to detecting biomarkers in tissue, saliva, urine, cerebrospinal fluid, etc. Clearly, the FACTT-based technology represents an important step in the detection of rare molecules in fluids or tissues for a variety of diseases.
Keywords: T7 RNA polymerase, RiboGreen, ELISA, IDAT, FACTT, biomarker, blood test, high-throughput, HER2, prion
Introduction
Effective therapies for many diseases, such as cancer and infective diseases, require early detection. Herceptin therapy for breast cancer, an example of targeted therapy for the HER2 receptor, highlights the growing dependence of therapy on an effective diagnosis at the molecular level [1]. Immunohistochemistry (IHC) and fluorescence in situ hybridization (FISH), which are performed on tissue slides from patients, are considered invasive techniques. A non-invasive immunological test to detect an identified biomarker only using serum or plasma would be extremely useful to monitor the effectiveness of on-going treatment or to predict disease recurrence after treatment.
Conventional methodologies for the determination and quantification of antigens have been limited to gel electrophoresis and antibody binding. The main problem with these methodologies is their low-throughput nature, which limits the usefulness of these methods in the clinic. Recent progress in proteomics has led to the development and broad use of mass spectrometry (MS), a technology that has the potential to identify unknown proteins in the analytes. However, it has been recognized that MS has a sensitivity of approximately 1 μg/ml, and, thus, is not ideal for low abundant proteins [2].
The enzyme-linked immunosorbent assay (ELISA) is high-throughput and suitable for diagnostic antigen detection; however, it only has a detection limit of 0.01-50 ng/ml [3]. That limit is greatly dependent on antibody affinities, in particular, the capture antibody. In general, the signal is amplified with horseradish peroxidase, alkaline phosphatase or β-galactosidase marker enzymes that are conjugated to the detection antibodies [4]. The amplification performed by these nonlinear enzymes improves the sensitivity of the ELISA assay; however, the capability of analyzing low abundant antigens using these enzymes is limited due to “substrate inhibition” [5]. This is a phenomenon referring to the competition between abundant substrates for limited enzymes in the system when antigens are at extremely low levels.
Immuno-PCR is a method in which PCR techniques are teamed up with immunodetection methods [6]. The method uses a linker molecule, with affinity for both DNA and antibody, which specifically binds a DNA template to an antibody-antigen complex. The template is then amplified by PCR using the proper primers and a set number of cycles. The presence of the PCR product, through gel electrophoresis or threshold cycle analysis by real-time PCR, reveals the existence of the complexes and thus is used to calculate the presence of the antigen [6]. One advantage of the immuno-PCR method is that it is approximately 105-fold more sensitive than alkaline phosphatase-conjugated ELISA antigen detection [6, 7]. Unfortunately, temperature changes during the PCR cycles are detrimental to the stability of proteins in the system, and denatured proteins can greatly interrupt the PCR reaction thus reducing the reliability of this approach.
Recently we have developed several immunoassays utilizing the T7 RNA polymerase to amplify signals. This technique overcomes many of the problems associated with the assays described above and provides a novel approach to quantify low-abundance antigens within limited amounts of serum or tissue.
The development of sensitive and high-throughput assays
IDAT
To address the limitations of other immunological assays, we first developed the immunodectection amplified by T7 RNA polymerase (IDAT) assay which has been described in detail [8]. Briefly, a 384-well plate is coated with a capture antibody, excess antibody is washed away, and a patient blood sample is added to each well. The antigen of interest binds to the capture antibody and the plate is washed to remove unwanted antigens. Next, a detection antibody that binds to a different epitope of the antigen and is conjugated to a double-stranded oligonucleotide (ds-oligo) containing the T7 promoter is added to each well. After unbound detection antibody is washed away, T7 RNA polymerase is used to amplify RNA in a linear manner from the ds-oligo [9-11], and the quantity of RNA is determined by gel electrophoresis. As proof of principle, this assay was used to detect and quantify the receptor tyrosine kinase HER2 [8]. HER2 was detected from the lysate of T6-17 cells at a 1013 dilution which is 109-fold more sensitive than the conventional ELISA method and 1011–fold more sensitive than Western blot analysis. Also, IDAT was sensitive enough to identify HER2 in the serum of mice bearing early stage HER2 overexpressing tumors.
There are many benefits of the IDAT assay. First, this assay has a greater sensitivity than the enhanced chemiluminescence-Western blot assay, conventional ELISA, RCA, and immunoPCR. IDAT is also a better alternative to HPLC, two-dimensional chromatography, and mass spectrometry-based methods of protein detection because it is more sensitive, less cumbersome, less time consuming and extremely versatile. For instance, the potent T7 amplification procedure facilitates the use of low affinity antibodies. The versatility of the system allows not only intact antibodies but also single-chain fragment variable (ScFv) fragments and complementarity determining region (CDR) peptides of the detection antibody to be used at antigen detectors [8]. In addition, the ds-oligo is coupled to the detection antibody at a common epitope to create a universal detector species so virtually any cellular antigen can be detected. Plus, RNA detection can be quantified using fluorescence or radioactivity. Lastly, this system is also capable of monitoring proteins, lipids, and metabolites and their modifications at the single-cell level [8]. For example, IDAT can detect the functional status of a protein by analyzing subtle post-translational modifications such as phosphorylation of specific residues.
Although IDAT combines the specificity of antigen–antibody interactions with the sensitivity powered by the liner amplification ability of T7 RNA polymerase, there are several limitations to this procedure. For instance, the specific binding affinity between any antigen and its antibody or antibody derivatives can be restrictive. In addition, it takes about five hours to couple the ds-oligo to the detection antibody before this complex is added to the well, and this procedure has to be done for each detection antibody used. Lastly, to quantify the RNA, this procedure uses electrophoresis, which is tedious and not a high-throughput method.
FACTT
To address the drawbacks of the IDAT assay, a second generation assay was created. A modular, facile amplification system termed fluorescent amplification catalyzed by T7 polymerase technique (FACTT) was described previously [12] (Figure 2). Similar to the IDAT procedure, capture antibodies on the bottom of the wells of a 384-well plate are used to bind an antigen of interest from a blood sample. Next, a biotinylated detection antibody, which binds to a different epitope of the antigen is added and the excess is washed away. FACTT varies from IDAT in that streptavidin tetramer is used to bridge the detection antibody to the double-stranded, biotinylated amplification module. The biotinylated amplification module is approximately 600 bps (approximately 10 times the length of the IDAT ds-oligo), contains a T7 promoter and terminator sequence, and is biotinylated at one end. The T7 RNA polymerase and then RiboGreen, a fluorescent RNA intercalating dye, are sequentially added to the well and a fluorimeter is used to quantify the yield of amplified RNA.
Figure 2.
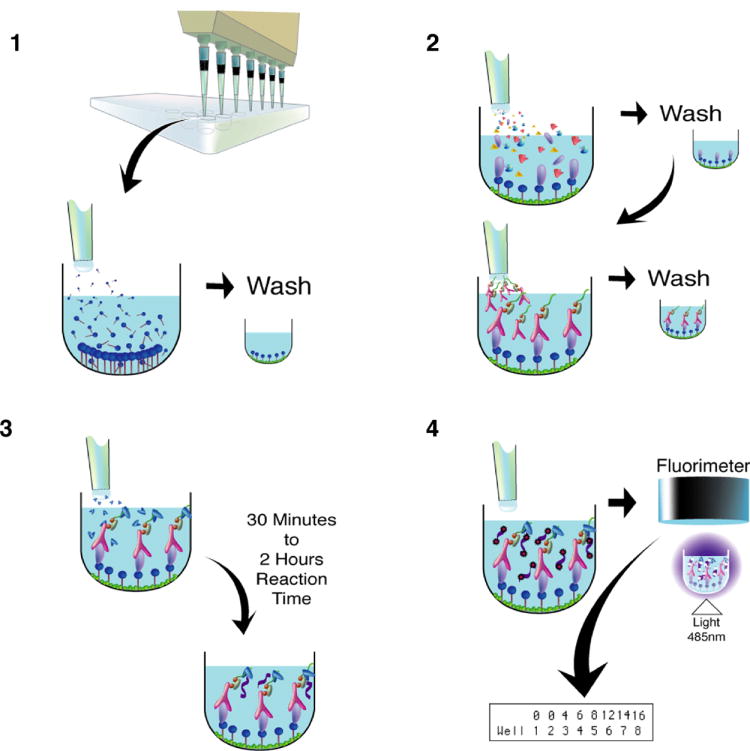
Schematic diagram depicting the fluorescent amplification catalyzed by T7 polymerase technique (FACTT) procedure. Step 1) Capture antibodies (dark blue) are added to each well of a 384-well plate. Excess unbound antibodies are washed away and the plate is blocked to prevent non-specific binding. Step 2) Patient or animal sample is administered to each well and the antigen of interest binds to the capture antibody. After washing, a biotinylated detection antibody (pink), which recognizes a different epitope or the same epitope if the target is a multimeric complex, is linked by streptavidin (gray) to the double-stranded, biotinylated amplification module (green). The detection antibody-Streptavidin-DNA complexes can be pre-assembled before they are added to the plate. Step 3) The T7 RNA polymerase (medium blue) is added and generates RNA (purple) during a 30 minute to 2 hour incubation period. Step 4) The fluorescent RNA intercalating dye RiboGreen (red) is dispensed into each well and a fluorimeter measures the yield of amplified RNA at wavelengths of 485 nm (ex) / 535 nm (em).
The FACTT assay has numerous benefits over other standard assays. First, it is a linear, quantitative assay that is isothermal. FACTT utilizes RiboGreen, which simplifies the assay for use in the 384-well plate format. With a detection limit of approximately 1 ng/ml RNA in solution, RiboGreen is 200-fold more sensitive than ethidium bromide–based assays [13]. Another advantage is that RiboGreen quantifies amplified RNA and thus eliminates the use of radioactivity as well as the tedious electrophoresis. In addition, the detection requires only 5–10 minutes to determine the amount of amplified RNA using a standard fluorimeter. The FACTT procedure is similar to that of ELISA and is compatible with current high-throughput liquid handling robots for reagent dispersal and repeated plate washing. The FACTT technique also has the potential to study multiple antigens because streptavidin is used to bridge any detection antibody to the double-stranded, biotinylated amplification module. This system is 103-to 105-fold more sensitive than the ELISA technology and can detect protein targets specifically at subfemtomolar levels (~0.08 fM). Thus, this assay is an efficient tool for analyzing low-abundance proteins in serum and can be adapted to high-throughput screening and automation. Indeed, this technology will enable monitoring of antigen levels at a much earlier disease stage, as well as, during treatment and remission.
However, there are still a few limitations of FACTT. The RNA amplification and detection is costly since the RiboGreen dye and a fluorimeter are required. The quality of the T7 RNA polymerase enzyme also is a limiting factor. The quality varies between vendors and the unit activity is not always consistent and reproducible when obtained from different enzyme sources. This assay is also dependent on RNA stability. Although FACTT shows better reproducibility than the original IDAT assay, the FACTT assay can be further optimized by using different antibody conjugates or detection reagents [14].
FACTT 2.0
The sensitivity of FACTT can be further improved by incorporating additional DNA template to the detection antibody. In the original protocol, the DNA template, with a biotin-label at one end, was added to bind to the streptavidin and was subsequently subjected to T7 RNA polymerase amplification [12]. In the modified FACTT 2.0 procedure the DNA template (BB) is biotin-labeled at both ends (Figure 3A). This is achieved by using biotinylated 5’ and 3’ primers to prepare the DNA template. With two biotin haptens, BB can bind to the streptavidin attached to the biotinylated antibody and still have one free biotin hapten to function as the docking site for subsequently added streptavidin. The newly captured streptavidin then becomes the host for more DNA templates. To compare the FACTT 2.0 with FACTT, we coated streptavidin directly to the plate. Using the standard FACTT procedure, the reading for 50 fg/ml streptavidin was not significantly different from the control. Using the FACTT 2.0 procedure, the reading for 50 fg/ml was enhanced over the background and the difference was significant (Figure 3B). Theoretically, more DNA templates could be attached with additional cycles of streptavidin and BB. However, under current conditions, we do not see any benefit for sensitivity beyond two cycles (data not shown).
Figure 3.
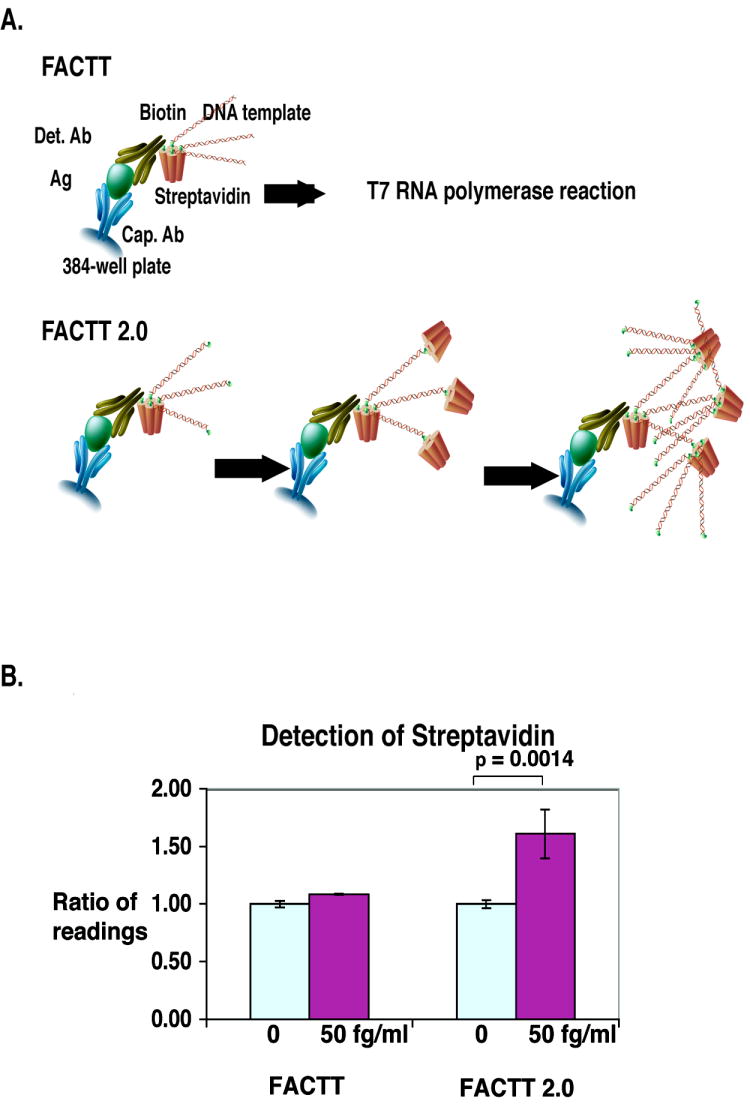
Further improvement of sensitivity of FACTT by streptavidin-based amplification of the signal. A) Schematic diagram illustrating the streptavidin-based amplification in FACTT 2.0. B) Comparison of FACTT and FACTT 2.0 in the detection of streptavidin. Streptavidin (50 fg/ml) was coated onto a 384-well high binding plate and the wells were blocked with 1% casein. For FACTT, RNA polymerase amplification was performed after the DNA template was incubated with streptavidin, and the unbound templates were removed by washing. For FACTT 2.0, before T7 RNA polymerase amplification, the streptavidin-coated wells were incubated sequentially with the double biotinylated DNA template (BB), streptavidin and BB again. Unbound streptavidin and BB were removed by a PBST wash. Experiments were performed in triplicate.
Clinical Significance
HER2
The receptor tyrosine kinase HER2 (Neu or p185her2/neu) is a therapeutic and diagnostic biomarker overexpressed in approximately 25-30% of primary breast, ovarian and pancreatic tumors [15-19]. The ectodomain of HER2 is shed from the cell surface and can be detected in the sera of breast cancer patients [20]. In addition, higher serum concentrations of HER2 correlate with a lower response rate to therapy and shorter survival time after recurrence [21, 22]. The most commonly used HER2 tests are immunohistochemistry (IHC) and fluorescence in situ hybridization (FISH) [23]. Individuals who are found to be HER2-positive by these tests have a response rate of approximately 34-35% to Herceptin therapy [24]; however, both of these clinical tests are invasive and low-throughput.
Monitoring HER2 serum levels with a FACTT-based assay is a sensitive, quantitative and non-invasive strategy. Studies have shown that serum HER2 levels, prior to treatment, positively correlate with tumor size, number of positive lymph nodes and histological scores [25]. In fact, serum HER2 levels were a better indicator for stage IV breast cancer than were IHC scores [26]. Serum HER2 levels were also an effective prognostic indicator for metastasis-free and disease-specific survival after treatment cessation [25]. FACTT was used to detect and quantify HER2 in both rodent and human blood [12]. FACTT was five orders of magnitude more sensitive than ELISA in the detection of recombinant HER2 in the serum. In addition, FACTT detected HER2 in the blood of mice with very early HER2 overexpressing tumor masses, whereas conventional detection methods were unable to detect HER2 in mice bearing tumors as large as 100 mm3. Lastly, when human serum samples were subjected to FACTT, individuals with HER2 positive breast cancer (determined by IHC and FISH) were shown to have elevated serum HER2 levels compared to HER2-negative individuals and healthy controls. However, the ELISA assay only found 20% of HER2-positive samples had elevated HER2 serum levels. Clearly, FACTT-based assays provide a sensitive and non-invasive alternative to conventional HER2 detection methods.
Other tumor markers
The FACTT-based assays are applicable to other tumor markers. Early detection for diagnosing and treating cancer in its pre-invasive state, before metastasis, is extremely important for controlling cancer. Unfortunately, ovarian cancer is generally diagnosed at a late stage. A widely used ovarian cancer serum biomarker, CA 125, is used to monitor treatment and recurrence of ovarian disease [27]. If FACTT-based assays were used to detect CA 125 at very low levels in the blood, diagnosis might be possible at a much earlier stage of tumor progression. Similarly, effective early detection and screening are currently not available for pancreatic ductal adenocarcinoma (PDAC), which is the fourth leading cause of cancer-related death in the United States [28]. These tumors are also typically diagnosed at a late stage, frequently after metastasis. Due to the low sensitivity and specificity of most detection assays that are currently used in the clinic, existing pancreatic serum biomarkers, such as CA19-9, are not adequate as early detection markers [28]. Thus, a great need exists for ultra-sensitive and specific FACTT-based assays to detect CA19-9 and the recently discovered serum protein biomarkers. These assays would greatly aid in the early detection of cancer and allow for more effective treatments.
Prion disease
Transmissible spongiform encephalopathy (TSE), or prion disease, is a group of fatal neurodegenerative diseases [29, 30]. The exhibition of spongiform lesions in the brains of infected animals or humans by histopathology was the only diagnostic test for decades [29, 30]. There was no reliable in vitro diagnostic test available until the discovery that TSEs are caused by the conversion of a normal cellular prion protein, PrPC, into the pathogenic, protease resistant scrapie PrP isoform, PrPSc [31-34]. In addition, a study in 2004 reported that two patients who received blood from donors who had died from variant Creutzfeldt-Jakob disease [35, 36], also developed the disease, indicating that the infectious prion in the blood is a public health concern. Unfortunately, current prion tests are mostly postmortem, invasive, insensitive and nonquantitative. In fact, all in vitro diagnostic tests for prion diseases require either the demonstration of protease-resistant PrPSc in brain or lymphatic tissue [37, 38] or the uncovering of hidden epitopes after denaturation [38, 39]. However, an in vitro test that can detect PrPSc in blood would greatly enhance our ability to monitor the occurrence and spread of prion diseases.
Recently, FACTT has been integrated with aggregate-specific ELISA to detect prion proteins in the blood of infected animals [40]. In this method (Am-A-FACTT), an in vitro PrPSc amplification step (Am) is included to further improve the detection limit. This methodology is based on the Protein Misfolding Cyclic Amplification (PMCA) which allows an undetectable amount of PrPSc to convert exogenously provided PrPC to PrPSc with [41-43] or without [44] multiple cycles of sonication. Similiarly, in the Am-A-FACTT, plasma or tissue homogenate from normal animals is incubated with infected samples to amplify PrPSc in the sample. In addition, only one antibody is used as both the capture antibody and the detecting antibody since the protein of interest, PrPSc, is known to form aggregates. The ultra-sensitive Am-A-FACTT assay had 100% sensitivity in detecting disease-associated prion aggregates in blood of infected mice at late but asymptomatic stages of disease (70 days postinoculation) [40]. At a very early stage (35 days postinoculation), Am-A-FACTT had a sensitivity of 50% and a specificity of 100%. This difference reflects the lower levels of PrPSc in the circulation early in the disease progression. Am-A-FACTT was also able to detect, with a high degree of specificity and sensitivity, disease-associated prion aggregates in presymptomatic mule deer that were naturally or experimentally infected with the chronic wasting disease agent. Overall, the Am-A-FACTT assay is minimally invasive, more quantitative and less time-consuming than other assays, such as PMCA, and can detect naturally infected pathogenic prion aggregates in the blood. Clearly, this FACTT-based assay is an incredibly sensitive tool that can detect prion diseases in asymptomatic carriers.
Versatility of FACTT for diseases
FACTT-based assays have the potential to detect low-abundance proteins present in patients with a wide variety of diseases. For instance, we have applied FACTT to the detection of TNF-alpha, G-CSF and other proteins (to be reported separately). All proteins have been detected with a sensitivity about 103 to 104-fold higher than by ELISA [45], confirming that FACTT-based assays have broad applications. Blood tests are currently being used to detect biomarkers for cardiovascular disease [46-48], rheumatoid arthritis [49-51], Alzheimer’s disease [52, 53], and hepatitis [54, 55]. Detecting and quantitatively monitoring the serum concentrations of cytokine inhibitors or soluble cytokine receptors, as well as the clinical response of patients to treatment with cytokine antagonists, might generate important information for monitoring autoinflammatory diseases [56]. The enhanced sensitivity of the FACTT-based assays would enable detection of antigens at levels beyond the existing ELISA detection limit. This would allow currently used antigens or novel low-abundance antigens to be detected and analyzed earlier during disease progression. The FACTT-based assays are not limited only to the detection of antigens found in the blood, but can also be applied to antigens found in the saliva, urine, cerebrospinal fluid, etc [12]. Finally, the principle of the FACTT-based assays for prion may be applicable to other diseases caused by abnormal protein aggregation, such as Alzheimer’s disease or Parkinson’s disease [40].
Conclusion
The sensitivity of the ELISA and other immunological assays does not satisfy the current need for the clinical detection of marker antigens in extremely low concentrations in patients with disease. The FACTT-based technology represents a significant step in the detection of low abundance molecules in fluids or tissues. With far greater sensitivity than the ELISA assay and with adaptability to high-throughput techniques, FACTT-based assays will facilitate the detection of these antigens as well as more abundant antigens from much smaller sample volumes. In addition, this assay can be modified to accommodate molecules of interest derived from cells, tissues and body fluids under different physiological conditions. Through the application of FACTT-based assays, we were able to detect the appearance of the tumor marker HER2 and the prion marker PrPSc at far earlier time points during disease progression than with any other assays. With carefully chosen antibodies, FACTT-based assays can be a practical tool for quantitatively profiling changes to crucial proteins, in terms of amount and post-translational modifications. These assays are clearly useful methods to enhance current ELISAs in the clinic for a wide variety of diseases.
Figure 1.

The potential therapeutic uses of an ultra-sensitive serum biomarker assay at various stages of cancer progression.
Acknowledgments
This work was supported by grants from the Abramson Family Cancer Research Institute and the National Institutes of Health to M.I.G. This work was also supported by R21 CA116103 from NIH and the start-up funds from the Department of Pathology and Lab Medicine of University of Pennsylvania to H.Z.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bilous M, Dowsett M, Hanna W, Isola J, Lebeau A, Moreno A, Penault-Llorca F, Ruschoff J, Tomasic G, van de Vijver M. Modern Pathology. 2003;16:173–182. doi: 10.1097/01.MP.0000052102.90815.82. [DOI] [PubMed] [Google Scholar]
- 2.Gygi SP, Rist B, Griffin TJ, Eng J, Aebersold R. Journal of Proteome Research. 2002;1:47–54. doi: 10.1021/pr015509n. [DOI] [PubMed] [Google Scholar]
- 3.Porstmann T, Kiessig ST. J Immunol Methods. 1992;150:5. doi: 10.1016/0022-1759(92)90061-w. [DOI] [PubMed] [Google Scholar]
- 4.Porstmann B, Porstmann T, Nugel E, Evers U. J Immunol Methods. 1985;79:27. doi: 10.1016/0022-1759(85)90388-6. [DOI] [PubMed] [Google Scholar]
- 5.Degn H. Nature. 1968;217:1047–1050. doi: 10.1038/2171047b0. [DOI] [PubMed] [Google Scholar]
- 6.Sano T, Smith CL, Cantor CR. Science. 1992;258:120–122. doi: 10.1126/science.1439758. [DOI] [PubMed] [Google Scholar]
- 7.Sawada T, Nishihara T, Yamamoto A, Teraoka H, Yamashita Y, Okamura T, Ochi H, Ho JJ, Kim YS, Hirakawa K. Japanese Journal of Cancer Research. 1999;90:1179–1186. doi: 10.1111/j.1349-7006.1999.tb00693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang HT, Kacharmina JE, Miyashiro K, Greene MI, Eberwine J. Proc Natl Acad Sci U S A. 2001;98:5497–5502. doi: 10.1073/pnas.101124598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Gelder RN, von Zastrow ME, Yool A, Dement WC, Barchas JD, Eberwine JH. Proc Natl Acad Sci U S A. 1990;87:1663–1667. doi: 10.1073/pnas.87.5.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo L, Salunga RC, Guo H, Bittner A, Joy KC, Galindo JE, Xiao H, Rogers KE, Wan JS, Jackson MR, Erlander MG. Nat Med. 1999;5:117–122. doi: 10.1038/4806. [DOI] [PubMed] [Google Scholar]
- 11.Chee M, Yang R, Hubbell E, Berno A, Huang XC, Stern D, Winkler J, Lockhart DJ, Morris MS, Fodor SP. Science. 1996;274:610–614. doi: 10.1126/science.274.5287.610. [DOI] [PubMed] [Google Scholar]
- 12.Zhang H, Cheng X, Richter M, Greene MI. Nature Medicine. 2006;12:473–477. doi: 10.1038/nm1378. [DOI] [PubMed] [Google Scholar]
- 13.Jones LJ, Yue ST, Cheung CY, Singer VL. Anal Biochem. 1998;265:368. doi: 10.1006/abio.1998.2914. [DOI] [PubMed] [Google Scholar]
- 14.Nam JM, Thaxton CS, Mirkin CA. Science. 2003;301:1884. doi: 10.1126/science.1088755. [DOI] [PubMed] [Google Scholar]
- 15.Slamon DJ. Science. 1989;244:707. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- 16.Cohen JA. Oncogene. 1989;4:81. [PubMed] [Google Scholar]
- 17.Lodato RF, Maguire HC, Greene MI, Weiner DB, LiVolsi VA. Mod Pathol. 1990;3:449. [PubMed] [Google Scholar]
- 18.Maguire HC, Greene MI. Pathobiology. 1990;58:297. doi: 10.1159/000163601. [DOI] [PubMed] [Google Scholar]
- 19.Williams TM, Weiner DB, Greene MI, Maguire HC. Pathobiology. 1991;59:46. doi: 10.1159/000163614. [DOI] [PubMed] [Google Scholar]
- 20.Kandl H, Seymour L, Bezwoda WR. Br J Cancer. 1994;70:739. doi: 10.1038/bjc.1994.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fehm T, Maimonis P, Katalinic A, Jager WH. Oncology. 1998;55:33–38. doi: 10.1159/000011832. [DOI] [PubMed] [Google Scholar]
- 22.Yamauchi H, O’Neill A, Gelman R, Carney W, Tenney DY, Hosch S, Hayes DF. J Clin Oncol. 1997;15:2518–2525. doi: 10.1200/JCO.1997.15.7.2518. [DOI] [PubMed] [Google Scholar]
- 23.Di Leo A, Dowsett M, Horten B, Penault-Llorca F. Oncology. 2002;63(Suppl 1):25–32. doi: 10.1159/000066204. [DOI] [PubMed] [Google Scholar]
- 24.Vogel CL. J Clin Oncol. 2002;20:719. doi: 10.1200/JCO.2002.20.3.719. [DOI] [PubMed] [Google Scholar]
- 25.Saghatchian M, Guepratte S, Hacene K, Neumann R, Floiras J-L, Pichon M-F. Int J Biol Markers. 2004;19:14–22. doi: 10.1177/172460080401900102. [DOI] [PubMed] [Google Scholar]
- 26.Luftner D, Henschke P, Kafka A, Anagnostopoulos I, Wiechen K, Geppert R, Stein H, Wernecke KD, Kreienberg R, Possinger K. International Journal of Biological Markers. 2004;19:1–13. doi: 10.1177/172460080401900101. [DOI] [PubMed] [Google Scholar]
- 27.Visintin I, Feng Z, Longton G, Ward DC, Alvero AB, Lai Y, Tenthorey J, Leiser A, Flores-Saaib R, Yu H, Azori M, Rutherford T, Schwartz PE, Mor G. Clin Cancer Res. 2008;14:1065–1072. doi: 10.1158/1078-0432.CCR-07-1569. [DOI] [PubMed] [Google Scholar]
- 28.Misek DE, Patwa TH, Lubman DM, Simeone DM. J Natl Compr Canc Netw. 2007;5:1034–1041. doi: 10.6004/jnccn.2007.0086. [DOI] [PubMed] [Google Scholar]
- 29.Gajdusek DC, Zigas V. N Engl J Med. 1957;257:974–978. doi: 10.1056/NEJM195711142572005. [DOI] [PubMed] [Google Scholar]
- 30.Johnson RT, Gibbs CJ., Jr N Engl J Med. 1998;339:1994–2004. doi: 10.1056/NEJM199812313392707. [DOI] [PubMed] [Google Scholar]
- 31.Bolton DC, McKinley MP, Prusiner SB. Science. 1982;218:1309–1311. doi: 10.1126/science.6815801. [DOI] [PubMed] [Google Scholar]
- 32.Prusiner SB. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 33.Cohen FE, Prusiner SB. Annu Rev Biochem. 1998;67:793–819. doi: 10.1146/annurev.biochem.67.1.793. [DOI] [PubMed] [Google Scholar]
- 34.Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE, et al. Proc Natl Acad Sci U S A. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Llewelyn CA, Hewitt PE, Knight RS, Amar K, Cousens S, Mackenzie J, Will RG. Lancet. 2004;363:417–421. doi: 10.1016/S0140-6736(04)15486-X. [DOI] [PubMed] [Google Scholar]
- 36.Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW. Lancet. 2004;364:527–529. doi: 10.1016/S0140-6736(04)16811-6. [DOI] [PubMed] [Google Scholar]
- 37.Kretzschmar HA. Clin Lab Med. 2003;23:109–128. viii. doi: 10.1016/s0272-2712(02)00043-4. [DOI] [PubMed] [Google Scholar]
- 38.Safar JG, Geschwind MD, Deering C, Didorenko S, Sattavat M, Sanchez H, Serban A, Vey M, Baron H, Giles K, Miller BL, Dearmond SJ, Prusiner SB. Proc Natl Acad Sci U S A. 2005;102:3501–3506. doi: 10.1073/pnas.0409651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB. Nat Med. 1998;4:1157–1165. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 40.Chang B, Cheng X, Yin S, Pan T, Zhang H, Wong P, Kang SC, Xiao F, Yan H, Li C, Wolfe LL, Miller MW, Wisniewski T, Greene MI, Sy MS. Clin Vaccine Immunol. 2007;14:36–43. doi: 10.1128/CVI.00341-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Castilla J, Saa P, Soto C. Nat Med. 2005;11:982–985. doi: 10.1038/nm1286. [DOI] [PubMed] [Google Scholar]
- 42.Saborio GP, Permanne B, Soto C. Nature. 2001;411:810–813. doi: 10.1038/35081095. [DOI] [PubMed] [Google Scholar]
- 43.Soto C, Anderes L, Suardi S, Cardone F, Castilla J, Frossard MJ, Peano S, Saa P, Limido L, Carbonatto M, Ironside J, Torres JM, Pocchiari M, Tagliavini F. FEBS Lett. 2005;579:638–642. doi: 10.1016/j.febslet.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 44.Supattapone S. J Mol Med. 2004;82:348–356. doi: 10.1007/s00109-004-0534-3. [DOI] [PubMed] [Google Scholar]
- 45.Arnold S. Biotechnol Bioeng. 2001;72:548. [PubMed] [Google Scholar]
- 46.Kucur M, Isman FK, Karadag B, Vural VA, Tavsanoglu S. Coron Artery Dis. 2007;18:391–396. doi: 10.1097/MCA.0b013e328241d991. [DOI] [PubMed] [Google Scholar]
- 47.Inoue T, Kotooka N, Morooka T, Komoda H, Uchida T, Aso Y, Inukai T, Okuno T, Node K. Am J Cardiol. 2007;100:569–574. doi: 10.1016/j.amjcard.2007.03.062. [DOI] [PubMed] [Google Scholar]
- 48.Whitaker DC, Green AJ, Stygall J, Harrison MJ, Newman SP. Perfusion. 2007;22:267–272. doi: 10.1177/0267659107083243. [DOI] [PubMed] [Google Scholar]
- 49.Sahin M, Aydintug O, Tunc SE, Tutkak H, Naziroglu M. Clin Biochem. 2007;40:6–10. doi: 10.1016/j.clinbiochem.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 50.Klimiuk PA, Fiedorczyk M, Sierakowski S, Chwiecko J. Scand J Rheumatol. 2007;36:345–350. doi: 10.1080/03009740701406460. [DOI] [PubMed] [Google Scholar]
- 51.Knudsen LS, Christensen IJ, Lottenburger T, Svendsen MN, Nielsen HJ, Nielsen L, Horslev-Petersen K, Jensen JE, Kollerup G, Johansen JS. Biomarkers. 2007:1–20. doi: 10.1080/13547500701615017. [DOI] [PubMed] [Google Scholar]
- 52.Tan ZS, Beiser AS, Vasan RS, Roubenoff R, Dinarello CA, Harris TB, Benjamin EJ, Au R, Kiel DP, Wolf PA, Seshadri S. Neurology. 2007;68:1902–1908. doi: 10.1212/01.wnl.0000263217.36439.da. [DOI] [PubMed] [Google Scholar]
- 53.Rentzos M, Michalopoulou M, Nikolaou C, Cambouri C, Rombos A, Dimitrakopoulos A, Vassilopoulos D. J Neurol Sci. 2005;228:129–135. doi: 10.1016/j.jns.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 54.El Awady MK, El Abd YS, Shoeb HA, Tabll AA, Hosny Ael D, El Shenawy RM, Atef K, Bader El Din NG, Bahgat MM. Virol J. 2006;3:67. doi: 10.1186/1743-422X-3-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clemente M, Nunez O, Lorente R, Rincon D, Matilla A, Salcedo M, Catalina MV, Ripoll C, Iacono OL, Banares R, Clemente G, Garcia-Monzon C. J Viral Hepat. 2006;13:625–632. doi: 10.1111/j.1365-2893.2006.00733.x. [DOI] [PubMed] [Google Scholar]
- 56.Dayer E, Dayer JM, Roux-Lombard P. Nat Clin Pract Rheumatol. 2007;3:512–520. doi: 10.1038/ncprheum0572. [DOI] [PubMed] [Google Scholar]