

Abstract
Forkhead transcription factor FoxO3a, also known as DAF-16 in C. Elegans nematodes, is a key regulator of the insulin receptor (IR)/IGF-1 signaling pathway mediated extension of lifespan in worms and yeast. In this study, we report that calorie restriction (CR) - mediated activation of IR signaling pathway leads to hyperphosphorylation of FoxO3a transcription factor and, consequently, its exclusion from nucleus. This inactivation of FoxO3a activity is correlated with attenuation of Alzheimer’s disease (AD)-type amyloid neuropathology and with preservation of spatial reference memory in the Tg2576 mouse model of AD. Further in vitro studies reveal that exogenous expression of viral triple mutant constitutively active (CA) FoxO3a resulting in increased nuclear FoxO3a activity in primary neuron cultures derived from Tg2576 mouse embryos, causally promotes AD amyloid-β peptide (Aβ) levels by inhibiting non-amyloidogenic α-secretase activity, indicating the existence of an inverse correlation between FoxO3a activity and cerebral Aβ amyloidosis. Moreover, we report for the first time that the exclusion of the FoxO3a transcription factor from the nucleus in combination with inhibition of nuclear FoxO3a activity by SIRT1-mediated deacetylation in response to CR is a mechanism resulting in the repression of Rho-associated protein kinase-1 (ROCK1) gene expression, thereby activating non-amyloidogenic α-secretase processing of APP and lowering Aβ generation. This study provides a novel metabolic pathway for prevention and/or treatment of AD.
Keywords: Alzheimer’s disease, amyloid-β peptide, calorie restriction, Forkhead transcription factor FoxO3a
1. Introduction
One critical effect of CR on lifespan extension in several species is to increase insulin sensitivity1. The insulin/IGF-1 receptor pathway is perhaps the most prominent pathway that regulates lifespan in worms and flies. In the nematode C. Elegans, mutants of DAF-2, a homolog of the insulin/IGF-1 receptor, live longer than wild type animals2. DAF-2 is a tyrosine kinase receptor that binds to an insulin-like molecule and then activates a phosphatidylinositol-3-OH kinase (a homolog of mammalian PI3K), ultimately resulting in the phosphorylation and downregulation of the forkhead transcription factor DAF-16, a homolog of mammalian FoxO3a transcription factor2. In the presence of environmental hazards and starvation, there is decreased signaling from DAF-2, allowing DAF-16 to be translocated into the nucleus, promoting dauer formation (arrested juvenile form) and metabolic changes towards energy conservation (fat accumulation), protection from oxidative stress and longer lifespan2. Thus, cellular localization of the forkhhead transcription factor DAF-16 (FoxO3a) may play a critical role in the regulation of gene expression related to life span extension.
In mammals, there are four DAF-16 homologues, namely the members of the forkhead FoxO family FoxO1, −3, −4 and −6, that are known to be negatively regulated by the IR-PI3K-Akt signaling pathway3. Mammalian FoxO factors control various biological functions, including apoptosis, cell cycle, differentiation, or the expression of genes involved in DNA repair and oxidative stress resistance in a cell-type-specific manner1,4. These particular functions of FoxO transcription factors may be relevant to FoxO’s ability to control longevity1. Most importantly and highly relevant to the present study, recent evidence strongly suggests a functional link between mammalian SIRT1 (an ortholog of yeast Sir2) and FoxO where SIRT1 regulates the activity of the FoxO family members in the nucleus and influences FoxO-induced gene expression, ranging from activation to repression3–8.
Based on these considerations we sought to test the hypothesis that a mechanism through which CR may beneficially influence Alzheimer’s disease- type amyloid neuropathology9,10 and spatial reference memory dysfunction is through activation of IR signaling pathway and SIRT1 deacetylase activity eventually resulting in the inhibition of FoxO3a-mediated ROCK1 gene expression. Our study for the first time indicates that the IR-PI3k-Akt-FoxO3a signaling pathway is involved in CR-mediated prevention of Alzheimer’s disease-type amyloid neuropathology in vivo. The mechanism for modulation of AD pathology appears to be due to the regulation of the expression of Rho-activated protein kinase-1 (ROCK1) by FoxO3a. Moreover, we identify a new pathway of crosstalk involving FoxO3a, SIRT1 deacetylase activity and ROCK1, providing a secondary mechanism for enhancing non-amyloidogenic α-secretase activity and lowering Aβ generation.
2. Materials and methods
2.1. Tg2576 mice and diets
The quantification of AD-type neuropathology, assessments of cognitive deterioration and other biochemical studies reported in the present paper were assessed in the same cohort of mice presented in previous studies from the lab9,10. In this study 4 month-old female Tg2576 mice11 (Taconic, Inc. Germantown Inc.) were randomly assigned to CR or ad libitum (AL) dietary regimens. Female mice were monitored for reproductive cycle status by the evaluation of appearance of the vagina at various stages of the estrous cycle, which is reliable as the customary method of vaginal smearing. CR was achieved by feeding Tg2576 mice 70% of the calories consumed by the pair-controlled AL animals, as previously reported9,10. At 10 months of age, mice were sacrificed and brain specimens harvested as previously described9. All animal studies were conducted following protocols approved by the Mount Sinai School of Medicine Institutional Animal Care and Use Committee.
2.2. Analysis of Y1162/1163 -IRβ phosphorylation
The assessment of Y1162/1163 -IRβ phosphorylation (a direct index of IR tyrosine kinase activity) was conducted as previously described12. Briefly, IRβ in brain lysates was first immuno-precipitated with the IRβ C-19 antibody. Contents of the Y1162/1163-phosphorylated IRβ and total IRβ in the immunoprecipitated fraction were assessed by Western blot analysis using a polyclonal pY1162/1163 IRβ-phosphospecific antibody (Biosource, Camarillo, CA) and the IRβ C-19 antibody, respectively, following a procedure essentially as previously described13. β-actin immunoreactivity (identified with anti-actin antibody, 1:1,000 dilution, Sigma) controlled for selectivity of changes. Immunoreactivities in these experiments, as well as all immunoblot analyses described below were visualized autoradiographically by chemiluminescence (SuperSignal; Pierce Biotechnology, Rockford, IL).
2.3. Western blot analysis
An aliquot of frozen cerebral cortex homogenate was further extracted in phosphate-buffered saline containing final concentrations of 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 0.1 mM EDTA plus protease inhibitors. Tissue lysate was recovered following centrifugation (14,000 × g at 4°C for 10 min). Total IRβ-chain protein content in the cerebral cortex of insulin-resistant Tg2576 vs. control Tg2576 mice was quantified by Western blot analysis using an anti-IRβ antibody (IRβ polyclonal C-19 antibody, Santa Cruz Biotechnology, Inc., Santa Cruz, CA). For assessment of PI3K activity the content of the 85 kDa regulatory α-subunit (p85) was assessed by Western blot assay (1:200 dilution; Santa Cruz Biotechnology, Inc.). Changes in pS473-AKT/PKB (a direct index of AKT/PKB activity) were assessed in a parallel Western blot analysis with anti pS473 phosphospecific antibody (1:500 dilution; Upstate, Lake Placid, NY). The same protein gel blots were used to assess total cerebral cortical AKT/PKB content with anti-AKT/PKB antibody (1:5,000 dilution; Cell Signaling, MA). To detect FoxO3a activity, the content of phospho-Thr32-FoxO3a and total FoxO3a in the cerebral cortex was immunodetected using anti-phospho-FHRL1/FoxO3a (Thr32) (1:1,000 dilution; Upstate Biotechnology, Inc.) and anti-FKHRL-1 (FoxO3a) (1:1,000 dilution; Upstate Biotechnology, Inc.) antibodies, respectively. In these studies β-actin immunoreactivity (1:1,000 dilution; Sigma, St. Louis) controlled for selectivity of changes.
Other antibodies used in this study include: polyclonal anti-SIRT1 antibody (1:3,000 dilution; Upstate, Lake Placid, NY), polyclonal anti-amyloid precursor protein C-terminal (751–770) antibody (anti-O443, 1:5,000 dilution; Calbiochem, San Diego, CA), monoclonal 6E10 antibody (1:1,000 dilution; Senetek, St. Louis, MO), rabbit polyclonal antibody against ROCK1 (1:5,000 dilution; Chemicon International, Temecula, CA).
2.3. Cell culture
Embryonic (E14) neocortical primary neuronal cultures derived from Tg2576 transgenic mice (Tg2576 neurons) were prepared as previously described14. Chinese hamster ovary (CHO) cells expressing human APP carrying the K670N, M671L Swedish mutation (APPswe) (gift from Dr. Robakis) were grown in McCoy’s 5A medium supplemented with 10% fetal bovine serum, 1% streptomycin/penicillin (GIBCO), and 200 μg/ml G418 (Life Technologies, Carlsbad, CA). For adenoviral infection studies, Tg2576 neuronal (5-days old) or CHO-APPswe (18 hr after plating) cultures were infected with WT SIRT115, CA FoxO3a, Lac Z or GFP control adenoviruses at doses defined as multiplicities of infection (MOI). Conditioned medium was collected at indicated time points for Aβ detection. Cell viability was assessed by LDH assay.
2.5. FoxO3a immunocytochemical localization
For assessment of FoxO3a immuno-localization in brain, neocortical tissue sections (50 μm) adjacent to those used for assessment of amyloid plaque pathology in CR Tg2576 mice were washed with Tris-buffered saline (TBS; 20 mM, pH 7.4), followed by incubation with 0.2% TritonX-100 for 30 min for permabilization. Endogenous peroxidase activity was quenched by incubating the sections with 0.3% hydrogen peroxide for 5 min. For detection of FoxO3a immunolocalization in Tg2576 neurons, primary neuron cells were grown on tissue culture chamber slides and post-fixed with 4% paraformaldehyde for 30 min, then treated with 0.2% TritonX-100 for 20 minutes. Following a rinse with TBS, the sections were incubated with blocking buffer (10% goat serum in TBS) for 120 min. The sections were then incubated with primary anit-FKHRL-1 (FoxO3a) (1:100 dilution) antibody in blocking buffer at 4°C overnight. The sections were washed three times with TBS and then incubated with Texas-Red conjugated goat anti-rabbit antibody (1:1,000 dilution; Vector Laboratories, Burlingame, CA). Nuclei counterstaining was done with DAPI (Molecular Probe, Carlsbad, CA). Cell images were examined by a Zeiss fluorescence microscope and captured with Axiovision software.
2.6. FoxO3a acetylation assays
FoxO3a lysine acetylation was analyzed by immuno-precipitation of FoxO3a followed by western blot using acetyl-lysine antibodies. Brain and cell extracts were obtained as described above. FoxO3a was immunoprecipitated using anti-FKHRL-1 (FoxO3a) antibody. Total FoxO3a levels and acetylated FoxO3a were detected using specific antibodies for FoxO3a and monoclonal anti-acetyl-lysine (1:500 dilution; Upstate Biotechnology, Inc), respectively.
2.7. Aβ ELISA assay
The quantitative assessment of Aβ peptides in brain and cultured cells was performed as previously described16.
2.8. Reporter plasmid construction
ROCK1 (−2549 to −2017) containing 3 putative FOXO consensus binding sites was amplified from mouse genomic DNA (BAC clone RP24-132C7; ATCC, Manassas, VA) using the Genomic PCR amplification kit (Clontech). The fragment was purified and subcloned into SalI and XhoI sites of pGL3-SV40 vector (Promega).
2.9. Luciferase Activity Assay
CHO-APPswe cells were seeded at a density of 1×105 cells/well in 24-well-plates. After 18 hours, cells were transiently transfected with FHRE-luc or ROCK1-luc using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, California), according to the manufacture’s instruction. pRL-TK (Promega) was included in every transfection as an internal control. Medium was changed 5 hours later and cells were infected with Adeno-GFP, CA FoxO3a or SIRT1, respectively. Firefly and Renilla luciferase activities were measured 24h after transfection using the Dual-Luciferase Reporter Assay System (Promega).
2.10. Statistical analysis
All values are expressed as mean ± standard error of the mean (SEM). Differences between means were analyzed using a two-tailed Student t test. In all analyses, the null hypothesis was rejected at the 0.05 level. All statistical analyses were performed using the Prism Stat program (GraphPad Software, Inc., San Diego CA).
3. Results
3.1. CR-mediated activation of insulin signaling pathway reduces FoxO3a activity in the brains of Tg2576 mice
In this study, we first investigated if changes in the IR signaling pathway (IR-PI3k-Akt-FoxO3a) in response to CR regimen are correlated to CR-mediated prevention of Alzheimer’s disease-type amyloid neuropathology in Tg2576 mice. We found that ~ 6 months, 30% CR dietary regimen resulted in a significant ~ 1.5 fold (P < 0.05) elevation of both total IRβ expression and Y1162/1163-IRβ autophosphorylation, an index of increased IRβ activation (Fig. 1A, B), coincidental to elevated of PI3K p85 subunit expression, an index of increased PI3K signaling in the brain of ~10 months old CR Tg2576 mice (Fig. 1C, P<0.05), relative to ad libitum fed (AL)-fed Tg 2576 mice. More interestingly, we found an ~1.5 fold induction of cerebral cortical pS473- AKT phosphorylation (indicative of increased AKT activity) relative to total cerebral cortical AKT protein content (Fig. 1D, P<0.05). Based on the fact that FoxO3a is downstream factor in the IR signaling pathways and target of AKT phosphorylation we continued to explore the regulation of FoxO3a.
Fig. 1. CR activates IR signaling cascades in the brains of Tg2576 mice.
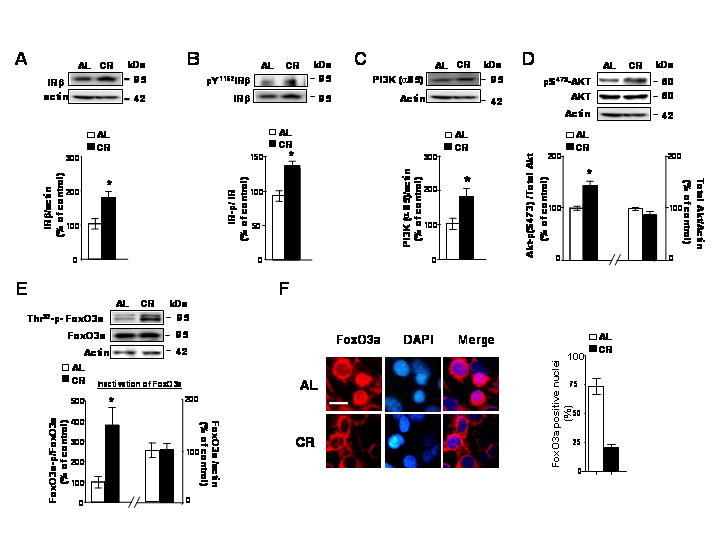
In this study ~10 month-old Tg2576 transgenic mice were assessed for indexes of IR signaling in the cerebral cortex in response to CR or AL dietary regimens. (A) Total IRβ and (B) Y1162/1163-phosphorylated IRβ protein contents in the cerebral cortex. Total IRβ expression is normalized to actin; IRβ Y1162/1163 phosphorylation is expressed as the ratio of IRβ Y1162/1163 phosphorylated protein relative to IRβ in the same cerebral cortex samples; (A and B) insets, representative western blot analysis of IRβ and Y1162/1163 phosphorylated IRβ proteins. (C) PI3K (p85α) protein content in the cerebral cortex normalized to actin; (c inset), representative PI3K (p85α) western blot analysis. (D) pS473AKT expressed as a ratio of actin; Total AKT expressed as a ratio of actin; (D) insets, representative western blot analysis of pS473AKT and total AKT. (E) Thr32-p-FoxO3a expressed as a ratio of total FoxO3a; (E) inset, representative western blot analysis of Thr32-p-FoxO3a and total FoxO3a protein. In (A–E), values represent means ± SEM, n = 4–5 per group; * P < 0.05 vs. control group (2-tailed Student’s t test). (F) Cellular distribution of neocortical FoxO3a immunoreactivity in the contralateral neocortex used for (A–E) Western blot analysis. The brain section were fixed and immunofluorescence staining for FoxO3a (a) using anit-FKHRL-1 antibody, which is visualized using Texas Red. Nuclei were identified using DAPI staining (blue). Scale bar: 20μM. Data are expressed as mean ± SEM for 3 animals per group (n = 100 cells per animal).
Interestingly, we found a commensurate increase in phosphorylation of FoxO3a (Fig. 1E), an index of increased levels of inactive Thr32-FoxO3a, eventually resulting in its sequestration away from the nucleus into the cytoplasm (Fig. 1F) in the brain of CR Tg2576 mice relative to AL-fed control mice. This event coincided with significant decreased Aβ1-40 and Aβ1-42 peptide contents10 and decreased Alzheimer’s disease-type amyloid plaque neuropathology in the contralateral hemisphere10 and a significant elevation of glucose utilization as assessed by Intra-peritoneal glucose tolerance test (IGTT) (Data not Shown).
Based on this evidence we proceeded to assess the potential beneficial role of CR on Alzheimer’s disease-type hippocampal spatial reference memory deterioration using Barnes maze17,18. Barnes maze was used in this study primarily because of the lack of buoyancy and swimming in lean CR Tg2576 mice preventing accurate assessment of spatial memory function in Morris water maze test. We found a significant improvement in overall escape latency in CR Tg2576 mice relative to the AL-fed control group. For learning trial: two-way ANOVA repeated measure, p<0.001, F12,156 = 3.929 for time; and p<0.05, F1,13 = 4.830 for dietary treatment. For reversal study: Two-way ANOVA repeated measure: p<0.01, F1,13 = 10.59 for dietary treatment. Collectively, this behavioral study suggests that CR not only is capable to promote learning of new tasks but also capable to promote retention of learned tasks.
CR in strain-, age-, and gender-matched wild-type (WT) mice had no significant influence on spatial reference memory performance in the Barnes test, compared to untreated control WT mice (Data not shown). This finding strongly supports the hypothesis that CR may attenuate memory attenuation through mechanism involving Aβ accumulation in the brain. Thus, CR-mediated phosphorylation and exclusion of FoxO3a from nucleus in response to activation of IR signaling appeared to play a major role in the prevention of Alzheimer’s disease-type condition resulting in the attenuation of spatial cognitive deterioration in Tg2576 mice.
3.2. Increased nuclear expression of FoxO3a causally inhibits α-secretase activity and promotes Aβ peptides generation in primary Tg2576 neuron cultures
Next we continued to explore mechanistically the role of FoxO3a in the regulation of non-amyloidogenic processing of APP and Aβ generation in vitro. To do this, we virally expressed triple mutant constitutively active (CA) FoxO3a which is not phosphorylatable and thus fully localized in the nuclear region19 in primary cortico-hippocampal neuron cultures derived from Tg2576 mouse embryos (Tg2576 neurons). As expected, we found that CA FoxO3a viral infection resulted in >2-fold increased levels of (total) FoxO3a (Fig. 2A), that resulted in ~ 30% decreased levels of phosphorylated Thr32-FoxO3a, indicative of primarily nuclear translocation of CA FoxO3a (see Fig. 2B showing CA FoxO3a immunoreactive signal primarily colocalized with DAPI nuclear staining in Tg2576 neurons), relative to control-viral GFP infected cultures.
Fig. 2. Exogenous triple mutant constitutive-active (CA) FoxO3a expression causally prevents non-amyloidogenic processing of APP coincidental with increased Aβ1-40 and Aβ1-42 contents in the conditioned medium of cortico-hippocampal Tg2576 neurons.
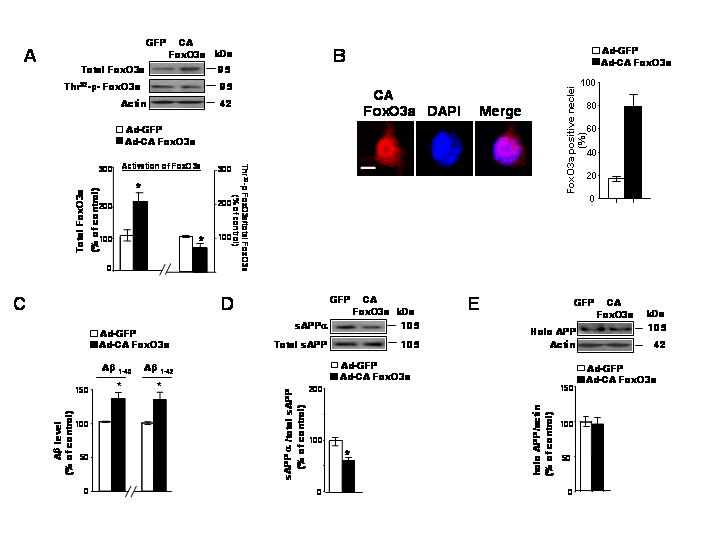
(A) Tg2576 neuron cultures were infected with adenovirus expressing GFP or CA FoxO3a for 48h. Resulting cell lysates were subjected to western blot analysis of adenovirus mediated-FoxO3a expression and Thr32-p-FoxO3a content using anti-FoxO3a antibody and Thr32-p-FoxO3a antibody, respectively. (B) Subconfluent Tg2576 neurons were cultures on Lab-Tek chamber slides and infected with CA FoxO3a adenovirus for 24 h. The cells were then fixed and immunofluorescence staining for FoxO3a (a) using anit-FKHRL-1 antibody, which is visualized using Texas Red. In (B) nuclei were identified using DAPI staining (blue). Scale bar: 20μM. Data are expressed as mean ± SEM for three separate experiments (n= 100 cells per experiment). (C) Adenovirus-mediated over-expression of CA FoxO3a in Tg2576 neuron cultures lead to increased contents of Aβ1-40 and Aβ1-42 released into the culture media. Aβ contents were assessed by ELISA 48 hr post-infection (10 MOI). (D) Assessment of changes in sAPPa concentration and (E) full-length APP (expressed as % of total sAPP and actin immunoreactivity, respectively) in response to adenoviral CA FoxO3a expression in Tg2576 neurons. Results are expressed as a % of control (adeno-GFP) infection; values represent means ± SEM of determinations made in three separate culture preparations; n=3 per culture preparation. *p<0.05 versus control group.
Most interestingly, we found that increased exogenous CA FoxO3a protein expression in the nucleus of Tg2576 neuron coincided with significantly increased Aβ1-40 and Aβ1-42 peptide contents in the culture medium (Fig. 2C), coincident with significantly lower content of sAPPα (Fig. 2D), reflecting reduced non-amyloidogenic processing of APP by α-secretase. No detectable change in total full-length APP (O-442 immunoreactive) content in response to CA FoxO3a expression was found (Fig. 2E). Thus, our in vitro evidence tentatively indicates that CA FoxO3a could causally increase Aβ level by inhibition of non-amyloidogenic α-secretase activity, suggesting that FoxO3a control of APP processing sets up an inverse relationship between FoxO3a activity and Alzheimer’s disease-type amyloidosis. Collectively, our evidence in vitro suggests that conditions resulting in the translocation of FoxO3a transcription factor to the nucleus (e.g. in response to exogenous CA FoxO3a expression) may ultimately promote Aβ generation through mechanisms that inhibit the non-amyloidogenic processing of APP. On the contrary, our in vivo studies suggest that conditions promoting cytoplasmic sequestration of phosphorylated (inactivated) FoxO3a, for example in response to CR (Fig. 1) promotes non-amyloidogenic processing of APP and decrease Aβ peptide generation.
3.3. SIRT1-mediated FoxO3a deacetylation is involved in FoxO3a modulation of Aβ generation
One mechanism controlling cell response to stress is through regulation of FoxO3a deacetyaltion by mammalian SIRT120. Based on this consideration, we tested the hypothesis that a parallel mechanism to FoxO3a exclusion from the nucleus involved in CR-mediated prevention of Alzheimer’s disease- type amyloid neuropathology in the brain of CR Tg2576 mice is through inhibition of FoxO3a activity by SIRT1 mediated deacetylation in nucleus. Interestingly, we found that CR treatment inhibits acetylation of FoxO3a at lysine residues in the brain of CR Tg2576 mice (P<0.05) (Fig. 3A), relative to AL-fed Tg2576 mice. The CR-mediated decreased acetylation of FoxO3a coincided with elevation in nuclear SIRT1 expression/activity in the brain of CR Tg2576 mice relative to AL-fed Tg2576 mice10. Moreover, in a parallel in vitro study, we found that viral expression of SIRT1 similarly inhibits FoxO3a acetylation (p<0.05) in the Tg2576 neurons, 48 hr after infection relative to viral Lac Z infected neurons (Fig. 3B). Thus there exists cross communications between FoxO3a and SIRT1 in vivo and in vitro.
Fig. 3. SIRT1-mediated FoxO3a deacetylation is involved in FoxO3a modulation of Aβ generation.
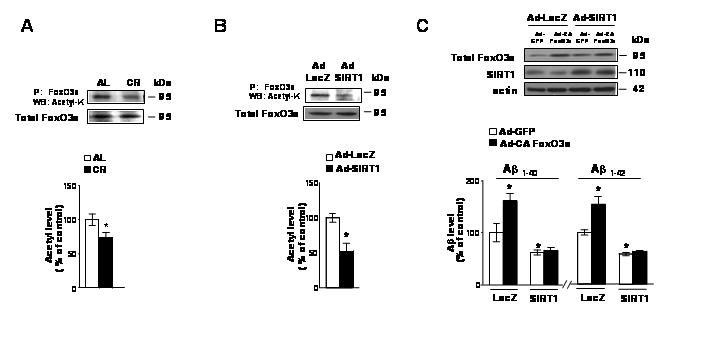
(A) Acetylation of FoxO3a decreases in the brain of CR treated Tg2576 transgenic mice. Endogenous FoxO3a was immunoprecipitated by anit-FKHRL-1 antibody followed by western blot using acetyl- lysine antibody. Total levels of FoxO3a were detected by western blot with anit-FKHRL-1 antibody. (B) SIRT1 deacetylates FoxO3a in cultured Tg2576 neurons. Tg2576 neurons were infected with SIRT1 adenovirus (MOI=10) for 48h. The acetylation of immunoprecipitated FoxO3a and total levels of FoxO3a were analyzed by western blot as described in (A). (C) Tg2576 neurons were infected with adeno-GFP or CA FoxO3a virus in combination with Ad-lacZ or Ad-WT SIRT1 viral infection. The resulting conditioned medium and cell lysates 48 hr post-transfection/infection were assessed for Aβ1-40 and Aβ1-42 concentrations. (C) Inset. Western blot analysis of SIRT1 and FoxO3a expressions. In (A) values represent means ± SEM, n = 4–5 per group. In (B) and (C), values represent means ± SEM of determinations made in three separate culture preparations; n=3 per culture preparation. In (A-C), *p<0.05 versus control group (2-tailed Student’s t test).
Next, we continued to determine the effect of cross communications between SIRT1 and FoxO3a on FoxO3a-mediated β-amyloidogenesis. To address this question Tg2576 neurons were virally infected with CA FoxO3a or GFP in combination with viral WT SIRT1, or control Lac Z infection. As expected, viral expression of SIRT1 significantly lowered Aβ1-40 and Aβ1-42 content in the conditioned medium of Tg2576 mouse neurons 48 hr after treatment as previously found10 (Fig. 3C). Interestingly, we found that the expression of SIRT1 resulting in significant deacetylation of FoxO3a (Fig. 3B) prevented CA FoxO3a mediated generation of Aβ1-40 and Aβ1-42 peptide levels (p<0.05) in the conditioned medium, relative to control viral Lac Z/GFP infected Tg2576 mouse neurons (Fig. 3C). These findings suggest that CA FoxO3a-mediated promotion of Aβ generation in neurons is also mediated by a SIRT1 dependent mechanism involving deacetylation.
3.4. FoxO3a activates mouse ROCK1 promoter activity, which is inhibited by viral SIRT1 expression
Recent evidence strongly suggests that inhibition of Rho kinase ROCK1 activity promotes non-amyloidogenic processing of APP21, precluding the generation of Aβ peptides. Based on this evidence and the fact the mammalian ROCK1 gene promoter contains 13 ((A/G)TAAA(C/T)A)-FoxOs consensus binding sites4 within ~6kb upstream of the ROCK1 coding sequence, we hypothesized that FoxO3a might be a critical ROCK1 transcriptional activator, and that inactivation of FoxO3a in response to CR-treatment (e.g. by sequestration in the cytoplasm or by deacetylation of FoxO3a via SIRT1 in nucleus) could lead to down-regulation of ROCK1 in the brain, thus promoting non-amyloidogenic processing of APP.
To test whether FoxO3a could activate the ROCK1 promoter, we constructed a ROCK1-promoter reporter gene (herein referred to as the ROCK1-luc reporter gene construct) in which expression of the luciferase reporter gene is under the control of a 500 bp mouse ROCK1 promoter sequence that contains three consensus FoxO3a binding sites (Fig. 4A). We transfected CHO-APPswe cells with the ROCK1-luc reporter construct while simultaneously over-expressing CA FoxO3a, or SIRT1 by infecting the cells with CA FoxO3a and/or SIRT1 adenoviruses; infection with a green fluorescent protein (GFP) adenovirus controlled for the specificity of CA FoxO3a and SIRT1 responses. Consistent with our hypothesis, we found that over-expression of CA FoxO3a significantly increased ROCK1 promoter activity by ~3.5-fold, as reflected by a commensurate induction of luciferase reporter gene expression (Fig. 4B). We consistently found significant induction of ROCK1 protein expression in CHO-APPswe cells 24 hr after viral CA FoxO3a infection, relative to control viral infected cells (Fig. 4C). Most interestingly, consistent with our evidence that SIRT1 may inactivate FoxO3a through a SIRT1-mediated deacetylation of FoxO3a (Fig. 3B), we found that the CA FoxO3a-mediated induction of luciferase activity was abolished by SIRT1 expression (Fig. 4B). Thus, this evidence suggests that FoxO3a may directly promote ROCK1 gene expression through interaction with the ROCK1 promoter, possibly via the FoxO3a consensus sequences in the ROCK1 promoter sequence. Moreover, our data strongly indicates that SIRT1 activation in response to CR may promote non-amyloidogenic processing of APP through inhibition of FoxO3a-mediated ROCK1 gene expression.
Fig. 4. FoxO3a activates mouse ROCK1 promoter, which is inhibited by viral SIRT1 expression.
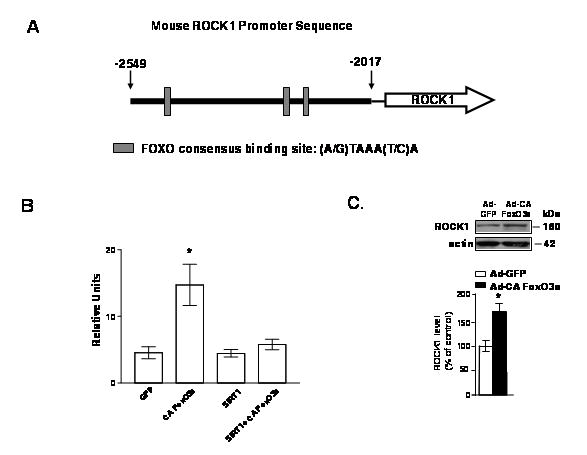
(A) Schematic representation of the ROCK1-luc reporter gene construct in which expression of the reporter luciferase gene is under the control of a ~500 bp mouse ROCK1 promoter sequence encompassing −2549 nt to −2017 nt of the ROCK1 promoter region. (B) CHO-APPswe cells were seeded at a density of 1×105 cells/well in 24-well-plates. 18 hours later, cells were transiently transfected with the ROCK-luc reporter construct using Lipofectamine, following the manufacturer’s instructions. Media were changed 5 hours later and then cells were infected with CA FoxO3a or SIRT1, or both FoxO3a and SIRT1 adenoviruses. Control cultures were transfected with the ROCK1-luc reporter gene construct while simultaneously infected with a green fluorescent protein adenovirus (GFP). Luciferase activities were measured 24h after transfection using Dual-Luciferase Reporter Assay System. (C) Viral CA FoxO3a in CHO-APPswe cells cultures results in increased levels of ROCK1 protein expression. Resulting cell lysates were separated by SDS-PAGE and probed with a rabbit polyclonal antibody against ROCK1; β-actin immunoreactivity served as a loading control. Value are expressed as mean ± SEM from two independent studies, n=3 repetitions per study. * p<0.05 (student t-test) vs. GFP infection.
4. Discussion
The present study suggests that a novel mechanism by which CR prevents Alzheimer’s disease-type amyloid neuropathology is, in part, through modulation of forkhead transcription factor FoxO3a activity. We show that CR activates the IR signaling pathway (IR-PI3k-Akt), which leads to hyperphosphorylation of and exclusion of FoxO3a from nucleus resulting in inactivation of FoxO3a activity in the brain of Tg2576 mice (Fig. 1). The changes in IR signaling cascades are functionally associated with CR-mediated prevention of Alzheimer’s disease- type amyloid neuropathology. We note that while the disruption of insulin/IGF-1 signaling and activation of DAF-16 result in formation of dauer larvae contributes CR extension of lifespan in C. Elegans, the main events leading to CR-mediated prevention of Alzheimer’s disease-type amyloid neuropathology in our mouse model are the CR sensitizing of insulin signaling pathway, the activation of IR signaling cascades and inactivation of FoxO3a. This apparent paradox is consistent with previous findings suggesting that mammals with genetic or acquired defects in IR signaling pathway are at risk for age-related diseases and increased mortality but not a beneficial effect2,22,23. This contradiction can be explained by the acquisition of more complicated metabolic pathways in mammalians over evolution. Mammals have insulin/IGF-1 receptors in many organs, but their functions are opposite if they are located in the central nervous system or in the periphery; whereas lower species have insulin/IGF-1 receptors signaling mainly through the nervous system. Furthermore, mammalians have different and very specific receptors for insulin and IGF-1, with distinct pathways and diverse functions. Striking evidence suggests that decreased IGF-1 levels and signaling during early development, but not the insulin signaling may modulate longevity in many species. Thus, paradoxical outcomes follow the decrease of insulin and/or IGF-1 signal pathway in invertebrates and in mammals, prolonging life in the former and shortening it in the latter2. We also note a potential apparent discrepancy between our studies and a recent study in C. elegans showing that reductions in IR expression (daf-2 RNAi), on the contrary, protects against Aβ toxicity through mechanisms dependent upon the expression of daf-16, and that daf-16 prevents the accumulation of toxic Aβ oligomers by promoting their aggregation into high molecular weight non-toxic aggregates24. The apparent discrepancy could be explained by the following facts: 1) by the acquisition of more complex metabolic pathways in mammal (e.g. the current study in mice vs. the study of Cohen et al in C. Elegans) over evolution; 2) by the fact that in the Cohen et al study, worms expressed a “human Aβ1-42 minigene” lacking a reasonable platform to address an intrinsic mechanism regarding APP processing in the C. Elegans model.
Our findings reveal that FoxO3a-mediated amyloidosis may also be modulated by SIRT1-mediated deacetylation. This mechanism is in parallel to the reduced amyloidosis as a consequence of hyperphosphorylation of FoxO3a via CR-mediated activation of the IR signaling pathway. We found that CR treatment inhibits acetylation of FoxO3a in the brain of CR Tg2576 mice (Fig. 3A), which coincided with elevation in nuclear SIRT1 expression/activity in the brain of CR Tg2576 mice10. We also found that viral expression of SIRT1 similarly inhibits FoxO3a acetylation in the Tg2576 neurons (Fig. 3B). More interestingly, we found that viral expression of SIRT1 prevent CA FoxO3a-mediated promotion of Aβ generation (Fig. 3C), suggesting that the cross communication between SIRT1 and FoxO3a in nucleus resulting in inhibition of nuclear FoxO3a activity by SIRT1-mediated deacetylation is involved in FoxO3a-mediated APP processing and Aβ generation in response to CR. Therefore, FoxO3a and SIRT1 might possibly have molecular cross communication in the mechanism modulating the prevention of Alzheimer’s disease- type amyloid neuropathology by CR.
Mammalian FoxO factors control various biological functions, including apoptosis, cell cycle, differentiation, or the expression of genes involved in DNA repair and oxidative stress resistance1,3. For example, the activation of FoxOs in haematopoietic and neuronal cells can result in the induction of apoptosis by up-regulation of the pro-apoptotic Bcl-2 family member bim25. Consensus DNA binding sites for FKHRL1 (FoxO3a) are present in the promoter region of bim26. FKHRL1 activation (via dephosphorylation) has been shown to increase bim expression after cytokine withdrawal in lymphocytes25 and IGF-I withdrawal in cerebellar granule neurons27. In this context, we postulate that one of the mechanisms by which FoxO3a may influence α-secretase activity and Aβ generation is through direct impact of FoxO3a on ROCK1 promoter activity. Consistent with this hypothesis, we found that CA FoxO3a activates ROCK1 promoter activity and ROCK1 protein expression, which can be prevented by SIRT1, possibly through SIRT1-mediated deacetylation of FoxO3a (Fig. 4). This evidence indicates that FoxO3a is a positive regulator of ROCK1 at a transcriptional level. Blockade of FoxO3a activity either by CR activation of IR signaling pathway (Fig. 1) or by SIRT1 deacetylation (Fig. 3) in response to CR can inhibit ROCK1 protein expression28 resulting in promotion of non-amyloidogenic APP processing and reduction in Aβ generation28. Thus, FoxO3a plays a central role in CR-mediated prevention of Alzheimer’s disease- type amyloid neuropathology as illustrated in Scheme 1. It is worth noting that a novel molecular mechanism by which SIRT1 activation in response to CR might influence ROCK1 expression/α-secretase activity10, is through SIRT1-mediated deacetylation of nuclear FoxO3a resulting in inhibition of ROCK1 gene expression.
Scheme 1.
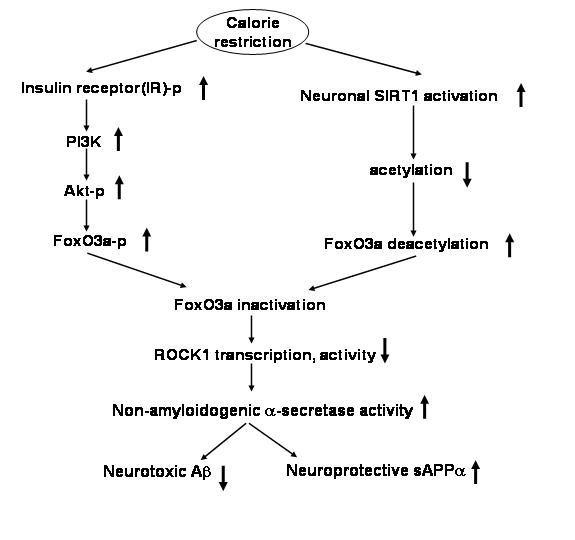
Scheme illustrates the potential relationship for CR, FoxO3a inactivation/SIRT1 activation and promotion of non-amyloidogenic processing of APP.
Collectively, the present study provides new insights into the mechanism through which CR beneficially prevents Alzheimer’s disease-type amyloid neuropathology. The present study strengthens the possibility that CR may exert beneficial effects on delaying the onset of Alzheimer’s disease-type brain amyloid neuropathology in humans similar to those observed in rodent model of Alzheimer’s disease and in squirrel monkeys9,10,28. However, if the underlying pathways can be dissected, then drug targets will be revealed. Thus the development of pharmacological agents “mimetic” of CR, such as insulin sensitizers and SIRT1 activators in modulating specific cellular processes leading to Aβ-lowering activities in the brain appear to offer attractive novel pharmacological directions for future AD treatment and/or prevention.
Acknowledgments
These studies were supported by Dr. Robert C. Atkins Foundation, NIH AG14766, Dana Foundation for Brain Research Initiative, and Merit Review to GMP; SM was supported by NIH PPG AG10491. We thank Drs. W. Todd Penberthy and Soumya Chari for their invaluable editorial comments.
Abbreviations
- CR
calorie restriction
- AL
ad libitum
- WT
wild-type
- Aβ
β-amyloid
- APP
amyloid precursor protein
- sAPPα
soluble APP
- CA
constitutively active
- IR
insulin receptor
- ROCK
serine/threonine Rho kinase
References
- 1.Katic M, Kahn CR. The role of insulin and IGF-1 signaling in longevity. Cell Mol Life Sci. 2005;62:320–343. doi: 10.1007/s00018-004-4297-y. [DOI] [PubMed] [Google Scholar]
- 2.Rincon M, Muzumdar R, Atzmon G, Barzilai N. The paradox of the insulin/IGF-1 signaling pathway in longevity. Mech Ageing Dev. 2004;125:397–403. doi: 10.1016/j.mad.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 3.Birkenkamp KU, Coffer PJ. Regulation of cell survival and proliferation by the FOXO (Forkhead box, class O) subfamily of Forkhead transcription factors. Biochem Soc Trans. 2003;31:292–297. doi: 10.1042/bst0310292. [DOI] [PubMed] [Google Scholar]
- 4.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 5.Giannakou M, Partridge L. The interaction between FOXO and SIRT1: tipping the balance towards survival. Trends Cell Biol Trends Cell Biol. 2004;14:408–412. doi: 10.1016/j.tcb.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 6.Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- 7.van der Horst A, Tertoolen LG, de Vries-Smits LM, Frye RA, Medema RH, Burgering BM. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2 (SIRT1) J Biol Chem. 2004;279:28873–28879. doi: 10.1074/jbc.M401138200. [DOI] [PubMed] [Google Scholar]
- 8.Daitoku H, Hatta M, Matsuzaki H, Aratani S, Ohshima T, Miyagishi M, Nakajima T, Fukamizu A. Silent information regulator 2 potentiates FoxO1-mediated transcription through its deacetylase activity. Proc Natl Acad Sci USA. 2004;101:10042–10047. doi: 10.1073/pnas.0400593101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang J, Ho L, Qin W, Rocher AB, Seror I, Humala N, Maniar K, Dolios G, Wang R, Hof PR, Pasinetti GM. Caloric restriction attenuates beta-amyloid neuropathology in a mouse model of Alzheimer’s disease. FASEB J. 2005;19:659–661. doi: 10.1096/fj.04-3182fje. [DOI] [PubMed] [Google Scholar]
- 10.Qin W, Yang T, Ho L, Zhao Z, Wang J, Chen L, Zhao W, Thiyagarajan M, MacGrogan D, Rodgers JT, Puigserver P, Sadoshima J, Deng H, Pedrini S, Gandy S, Sauve AA, Pasinetti GM. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J Biol Chem. 2006a;281:21745–21754. doi: 10.1074/jbc.M602909200. [DOI] [PubMed] [Google Scholar]
- 11.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 12.Ho L, Qin W, Pompl PN, Xiang Z, Wang J, Zhao Z, Peng Y, Cambareri G, Rocher A, Mobbs CV, Hof PR, Pasinetti GM. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J. 2004;18:902–904. doi: 10.1096/fj.03-0978fje. [DOI] [PubMed] [Google Scholar]
- 13.Barber AJ, Nakamura M, Wolpert EB, Reiter CE, Seige GM, Antonetti DA, Gardner TW. Insulin rescues retinal neurons from apoptosis by aphosphatidylinositol 3-kinase/Akt-mediated mechanism that reduces the activation of caspase-3. J Biol Chem. 2001;276:32814–32821. doi: 10.1074/jbc.M104738200. [DOI] [PubMed] [Google Scholar]
- 14.Kelley KA, Ho L, Winger D, Freire-Moar J, Borelli CB, Aisen PS, Pasinetti GM. Potentiation of excitotoxicity in transgenic mice overexpressing neuronal cyclooxygenase-2. Am J Pathol. 1999;155:995–1004. doi: 10.1016/S0002-9440(10)65199-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 16.Qin W, Ho L, Pompl PN, Peng Y, Zhao Z, Xiang Z, Robakis NK, Shioi J, Suh J, Pasinetti GM. Cyclooxygenase (COX)-2 and COX-1 potentiate beta-amyloid peptide generation through mechanisms that involve gamma-secretase activity. J Biol Chem. 2003;278:50970–50977. doi: 10.1074/jbc.M307699200. [DOI] [PubMed] [Google Scholar]
- 17.Barnes CA. Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psycho. 1979;193:74–104. doi: 10.1037/h0077579. [DOI] [PubMed] [Google Scholar]
- 18.Pompl PN, Mullan MJ, Bjugstad K, Arendash GW. Adaptation of the circular platform spatial memory task for mice: use in detecting cognitive impairment in the APPsw transgenic mouse model for Alzheimer’s disease. J Neurosci Methods. 1999;87:87–95. doi: 10.1016/s0165-0270(98)00169-1. [DOI] [PubMed] [Google Scholar]
- 19.Skurk C, Maatz H, Kim HS, Yang J, Abid MR, Aird WC, Walsh K. The Akt-regulated forkhead transcription factor FoxO3a controls endothelial cell viability through modulation of the caspase-8 inhibitor FLIP. J Biol Chem. 2004;279:1513–1525. doi: 10.1074/jbc.M304736200. [DOI] [PubMed] [Google Scholar]
- 20.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 21.Pedrini S, Carter TL, Prendergast G, Petanceska S, Ehrlich ME, Gandy S. Modulation of statin-activated shedding of Alzheimer APP ectodomain by ROCK. PLoS Med. 2005;2:e18. doi: 10.1371/journal.pmed.0020018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reaven GM. Role of insulin resistance in human disease. Banting lecture. Diabetes. 1988;37:1595–15607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 23.Facchini FS, Hua N, Abbasi F, Reaven GM. Insulin resistance as a predictor of age-related diseases. J Clin Endocrinol Metab. 2001;86:3574–3578. doi: 10.1210/jcem.86.8.7763. [DOI] [PubMed] [Google Scholar]
- 24.Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313(5793):1604–10. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- 25.Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol. 2000;10:1201–1204. doi: 10.1016/s0960-9822(00)00728-4. [DOI] [PubMed] [Google Scholar]
- 26.Yin KJ, Hsu CY, Hu XY, Chen H, Chen SW, Xu J, Lee JM. Protein phosphatase 2A regulates bim expression via the Akt/FKHRL1 signaling pathway in amyloid-beta peptide-induced cerebrovascular endothelial cell death. J Neurosci. 2006;26:2290–2299. doi: 10.1523/JNEUROSCI.5103-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Linseman DA, Phelps RA, Bouchard RJ, Le SS, Laessig TA, McClure ML, Heidenreich KA. Insulin-like growth factor-I blocks Bcl-2 interacting mediator of cell death (Bim) induction and intrinsic death signaling in cerebellar granule neurons. J Neurosci. 2002;22:9287–9297. doi: 10.1523/JNEUROSCI.22-21-09287.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qin W, Chachich M, Lane M, Roth G, Bryant M, de Cabo R, Ottinger MA, Mattison J, Ingram D, Gandy S, Pasinetti GM. Calorie restriction attenuates Alzheimer’s disease type brain amyloidosis in Squirrel monkeys (Saimiri sciureus) J Alzheimers Dis. 2006b;10(4):417–422. doi: 10.3233/jad-2006-10411. [DOI] [PubMed] [Google Scholar]